
Abstract
ISG15 (ISG15 ubiquitin-like modifier), a ubiquitin-like protein, is one of the major type I IFN (interferon) effector systems. ISG15 can be conjugated to target proteins (ISGylation) via the stepwise action of E1, E2, and E3 enzymes. Conjugated ISG15 can be removed (deISGylated) from target proteins by USP18 (ubiquitin-specific peptidase 18). Here we investigated the role of deISGylation by USP18 in regulating autophagy and EGFR degradation in cells treated with type I IFNs. We show that type I IFN induced expression of ISG15 leads to ISGylation of BECN1 at Lys117, as well as Lys263, Lys265, and Lys266 which competes with Lys63 ubiquitination of BECN1. We demonstrate that ISGylation of BECN1 at Lys117, as well as Lys263, Lys265, and Lys266 serve an important role in negative regulation of intracellular processes including autophagy and EGFR degradation that are critically dependent upon the activity of class III PtdIns 3-kinase. Our studies provide fundamental new mechanistic insights into the innate immunity response implemented by type I IFNs.
Keywords:
Abbreviations:
- ATG, autophagy-related
- AKT/PKB, v-akt murine thymoma viral oncogene homolog
- BECN1, Beclin 1, autophagy-related
- EGF, epidermal growth factor
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- HERC5, HECT, and RLD domain containing E3 ubiquitin protein ligase 5
- IFN, interferon
- ISG15, ISG15 ubiquitin-like modifier
- LC3, microtubule-associated protein 1 light chain 3
- MTOR, mechanistic target of rapamycin
- MX1, MX dynamin-like GTPase 1
- PIK3C3, phosphatidylinositol 3-kinase, catalytic subunit type 3
- RPS6KB, ribosomal protein S6 kinase, 70kDa, polypeptide
- SQSTM1/p62, sequestosome 1
- USP18, ubiquitin specific peptidase 18
- UBA7, ubiquitin-like modifier activating enzyme 7
- Ub, ubiquitin
Introduction
Type I interferons (IFNs) serve a critical role in innate immunity response in defense against viral infections through the upregulation of numerous interferon stimulated genes (ISGs). The coordinated actions of ISGs provide a highly regulated program to antagonize viral replication.Citation1 Understanding the mechanism by which the ISGs function in mediating the action of IFNs is critical for us to develop effective antiviral therapies. ISG15, a member of the ubiquitin family, is a highly inducible ISG and one of the major type I interferon effector systems.Citation2 ISG15 encodes and expresses as a 165 amino acid polypeptide (17 kDa) precursor protein that is immediately proteolytically processed at its C terminus to expose a C-terminal LRLRGG amino acid sequence, identical with the C-terminal sequence of ubiquitin.Citation3 ISG15 contains 2 ubiquitin-like domains, each with approximately 30% amino acid sequence similarity to ubiquitin.Citation2,4,5 Similar to that of ubiquitin, ISG15 can be conjugated to target proteins (ISGylation) via the stepwise action of E1, E2, and E3 enzymes. Conjugated ISG15 can be removed (deISGylated) from target proteins by USP18/UBP43.Citation6 All components of the ISG15 system are induced by type I interferons. ISG15 serves an important antiviral function, as mice deficient for ISG15 are highly susceptible to infections by viral infections such as influenza, herpes, or Sindbis virus.Citation7
Autophagy is an ancient mechanism involved in the degradation of intracellular organelles and misfolded proteins through lysosomal pathway. Autophagy serves important functions in regulating intracellular recycling, cellular energy homeostasis and host defense response.Citation8 On the other hand, the function of autophagy machinery is also exploited by numerous types of viruses, including poliovirus, rotavirus, HIV, coronaviruses, Dengue virus, and the hepatitis B and C viruses.Citation9 In particular, autophagosomes may act as a scaffold for intracellular membrane-associated replication of certain cytoplasmic RNA viruses. However, it is not yet understood how genes regulated by type I IFNs, which serve as an important host defense mechanism against viral infection, are involved in regulating autophagy. In this manuscript, we examine the roles of ISG15 and deISGylating enzyme USP18 in regulating autophagy. We found that type I IFN-induced expression of ISG15 led to ISGylation of BECN1 at Lys117, as well as Lys263, Lys265, and Lys266, which competes for Lys63 ubiquitination of BECN1. ISGylation of BECN1 led to inhibition of PIK3C3 kinase activity and suppression of autophagy as well as EGFR degradation, which also depends critically on the levels of phosphatidylinositol-3-phosphate (PtdIns3P). Our studies provide fundamental mechanistic insights into the cellular responses activated by type I IFNs as a part of innate immunity.
Results
Regulation of autophagy by USP18
USP18/UBP43 was identified as a gene when its expression was knocked down, leading to inhibition of autophagy in a siRNA screen targeting 127 human deubiquitinating enzymes (data not shown). We first confirmed the effect of USP18 by transiently transfecting H4 cells with siRNAs targeting 3 different regions of USP18. As shown in Figure S1A, transfection of siUSP18-b and siUSP18-c, but not siUSP18-a, in H4 cells led to a reduction in the levels of USP18 and LC3-II (the lipidated, phagophore, and autophagosome-associated form of MAP1LC3/LC3), suggesting inhibition of autophagy after reduction of USP18 expression. To further verify this result, we established a stable USP18 knockdown H4 cell line infected with a lentivirus carrying an shRNA targeting the 3′ region of USP18 gene. Consistent with inhibition of autophagy, the levels of LC3-II were lower and the levels of SQSTM1/p62 were higher in this USP18 knockdown cell line compared to that of control under basal conditions as well as the induced condition after the treatment with rapamycin (Fig. S1B). To further determine the impact of USP18 on autophagy, we transiently transfected an expression vector of USP18 into H4 cells. Overexpression of USP18 led to upregulation of LC3-II and downregulation of SQSTM1 in a dose-dependent manner (Fig. S1C). We also generated an H4 line stably expressing USP18 to further characterize the role of USP18 in autophagy induction. We found that H4 cells stably expressing USP18 showed elevated LC3-II and decreased SQSTM1 under basal conditions (Fig. S1D). Notably, USP18 knockdown cell line showed less accumulation of LC3-II with rapamycin stimulation in the presence of NH4Cl, a lysosomal inhibitor that blocks autophagic flux (Fig. S1E). Taken together, our results suggest that USP18 is a positive regulator of autophagy and autophagic flux.
USP18, a member of the ubiquitin-specific protease family, is involved specifically in cleaving conjugation of ISG15, a homolog of ubiquitin, to Lys residues,Citation6 and plays an important role in regulating IFN signaling.Citation10 To determine whether the catalytic activity of USP18 is important for its effect on autophagy, we transfected expression vectors of wild-type USP18 (WT) or a catalytically inactive USP18 mutant, USP18C64S into H4 cells (Fig. S1F). The expression of WT, but not catalytically inactive USP18 mutant, in H4 cells upregulated the levels of LC3-II compared to that of vector alone (Fig. S1G), suggesting that the catalytic activity of USP18 is important for its ability to regulate autophagy.
Induction of ISGylation by type I IFN inhibits autophagy
Since type I IFN regulates the expression of USP18 and ISG15,Citation10 we next focused our investigation on the physiological roles of USP18 in regulating autophagy in cells stimulated by type I IFN. Type I IFN has been reported to induce autophagy.Citation11 However, we found that the increases of autophagy induced by type I IFN in both H4 cells () and HepG2 cells (Fig. S2A) were transient: prolonged treatment with type I IFN led to a reduction in LC3-II/TUBA ratio when the expression of ISG15 was induced. Moreover, prolonged treatment with type I IFN decreased the levels of autophagy induced by rapamycin in a time- and dose-dependent manner as determined by both western blotting and GFP-LC3B assay (, S2B and C). Furthermore, the treatment with type I IFN inhibited the accumulation of LC3-II in the presence of NH4Cl (). Thus, prolonged treatment with type I IFN leads to inhibition of autophagy and autophagy flux.
Figure 1. Induction of ISGylation by type I IFN inhibits autophagy. (A) H4 cells were treated with 1 nM IFNA2 for the indicated periods and then harvested for western blotting. Ratios of LC3-II/TUBA were calculated and are shown below. (B) H4-LC3-GFP cells were treated with 1 nM IFNA2 for the indicated periods in the presence of 200 nM rapamycin. Images of the cells were collected using an ArrayScan HCS 4.0 Reader. Representative cells are shown. The average spot intensity in 500 cells from each indicated sample was determined. Data are displayed as means ± SD of the spot intensity per cell (below). RLU, relative leight unit. (C) Type I IFN inhibits autophagic flux. H4 cells were treated with 2 nM IFN1@ for 24 h in the presence or absence of 10 mM NH4Cl. Then cells were harvested for western blotting analysis. Ratios of LC3-II/TUBA were calculated and are shown below. (D, E, F) ISGylation inhibits autophagy. H4 cells were transfected with control siRNA or 2 unique siRNAs targeting USP18 (D), ISG15 (E) and UBA7 (F), respectively, cultured for 48 h, and then stimulated with 2 nM IFNA2 for another 24 h. The cells were harvested and subjected to western blotting analysis. Ratios of LC3-II/TUBA were calculated and are shown below.
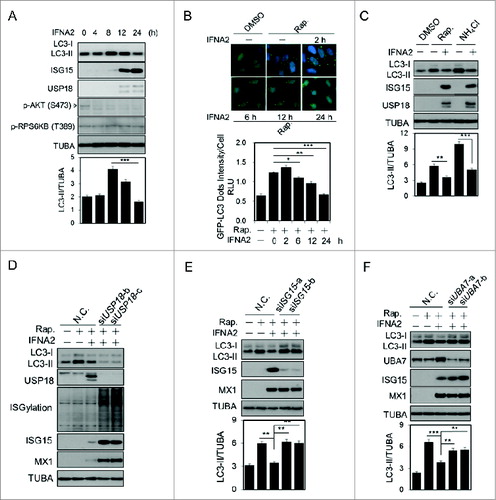
Consistent with the role of USP18 as a negative regulator of type I IFN signaling,Citation6 knockdown of USP18 further potentiated the reduction of LC3-II induced by type I IFN (). To characterize the role of ISGylation in regulating autophagy, we knocked down the expression of ISG15 by introducing siRNAs targeting 2 different regions of ISG15. As shown in , autophagy in type I IFN-treated cells could be restored with knockdown of ISG15. Furthermore, autophagy in type I IFN treated cells can also be restored by knocking down the expression of UBA7, the E1 enzyme involved in mediating ISGylation (). Taken together, these results suggest that ISGylation may negatively regulate autophagy mediated by type I IFN.
ISGylation suppresses PIK3C3 activity
Since type I IFN has been reported to regulate PI3K-AKT signalingCitation12 and the MTOR-RPS6KB kinase pathway,Citation13 we first tested whether these pathways were involved in modulation of autophagy by ISGylation. We found that type I IFN treatment had no effect on the Ser473 phosphorylation of AKT, a marker for its activation, nor did it affect the inhibition of RPS6KB phosphorylation mediated by rapamycin (Fig. S3A). Thus, AKT or MTOR-RPS6KB signaling is not likely involved in modulation of autophagy by type I IFN-mediated ISGylation.
We next tested the possible involvement of apoptosis, as type I IFN has been reported to have proapoptotic function.Citation14 However, we found that inhibition of caspase activation by zVAD.fmk, a caspase inhibitor, failed to restore inhibition of rapamycin induced autophagy by IFN (Fig. S3B). In addition, treatment with MG132, an inhibitor of proteasomal degradation, also had no effect on inhibition of rapamycin-induced autophagy by IFN (Fig. S3C). Thus, caspases or the proteasome pathway are not likely involved in autophagy inhibition mediated by type I IFN. In addition, the treatment with type I IFN had no effect on the levels of major regulators of autophagy, including BECN1, PIK3C3, ATG14, UVRAG, ATG12–ATG5, ATG7, or ATG3 (Fig. S3D).
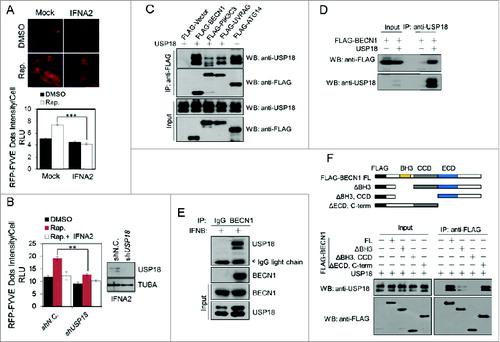
We next examined the effect of type I IFN and USP18 on the levels of PtdIns3P, a key lipid signaling molecule involved in regulating autophagy. The recruitment of FYVE-RFP to PtdIns3P located on endosomes has been used as an assay for measuring the cellular levels of PtdIns3P.Citation15 We found that the treatment with type I IFN strongly inhibited the formation of FYVE-RFP dots induced by rapamycin (). Furthermore, the levels of FYVE-RFP dots in H4-USP18-stable knockdown cells with rapamycin were significantly lower than that of control cells, suggesting that USP18 promotes the levels of PtdIns3P stimulated by rapamycin (). These results suggest that ISGylation may negatively regulate PIK3C3 activity critical for the production of PtdIns3P.
Figure 2. USP18 specifically interacts with BECN1. (A) IFNA2 suppresses PIK3C3 activity. H4-FYVE-RFP cells were treated with 2 nM IFNA2 for 24 h in the presence or absence of 200 nM rapamycin. Images of the cells were collected using an ArrayScan HCS 4.0 Reader. Representative cells are shown. The average FYVE-RFP spot intensity in 500 cells from each indicated sample was determined. The data are displayed as the means ± SD of the spot intensity per cell (below). (B) USP18 knockdown inhibits PIK3C3 activity. H4-FYVE-RFP cells with USP18 stable knockdown were treated with 2 nM IFNA2 for 24 h in the presence or absence of 200 nM rapamycin and then imaged and quantified as in (A). (C, D) USP18 associates with PIK3C3 complexes. The expression vectors of USP18 and FLAG-tagged BECN1, PIK3C3, ATG14, or UVRAG were cotransfected into 293T cells, respectively, and immunoprecipitations (IP) were performed at 36 h post-transfection. (E) H4 cells were treated with 1 nM IFNB for 24 h, which induced the expression of USP18. The cell lysates were analyzed by immunoprecipitation with anti-BECN1 antibody and the immunocomplexes were analyzed with the indicated antibodies by western blotting. (F) Mapping the interacting regions of BECN1 with USP18. The expression vectors of FLAG-tagged BECN1 truncations encoding the indicated regions were cotransfected with that of full-length USP18 into 293T cells and analyzed by immunoprecipitation and western blotting.
USP18 specifically interacts with BECN1
To explore the mechanism by which USP18 regulates the production of PtdIns3P, we screened for the components of PIK3C3 complexes that can interact with USP18. Interestingly, we found that overexpressed USP18 showed a strong interaction with BECN1 and ATG14 ( and D). Since the expression of USP18 affects both autophagy (Fig. S1) and EGFR degradation (see below), we focused our analysis on the interaction of USP18 and BECN1 which is involved in both autophagy and EGFR degradation. Strikingly, we found that there is interaction of endogenous BECN1 and USP18 after treatment with type I IFN ().
To characterize the interaction of USP18 with BECN1, we determined the domains of BECN1 that interact with USP18. Expression vectors of truncated BECN1 mutants were coexpressed with that of USP18 in 293T cells and the interaction of USP18 with BECN1 mutants was analyzed by coimmunoprecipitation. The binding of USP18 with BECN1 was significantly reduced upon deletion of its BH3 domain (BECN1BH3Δ) and completely eliminated after further deletion of the CC domain (BECN1BH3Δ,CCDΔ) (), suggesting that both the BH3 and the CC domain of BECN1 are important for the interaction with USP18. Since the BH3 domain of BECN1 is involved in the interaction with BCL2, and the CC domain is necessary for the interaction with ATG14,Citation16 we tested whether the interaction of USP18 with BECN1 had any effect on the interaction of BCL2 with BECN1 and that of BECN1 with ATG14. However, since overexpression of USP18 did not have any effect on the interaction of BCL2 and BECN1 (Fig. S3E) or that of BECN1 and ATG14 (Fig. S3F), we conclude that USP18 does not directly compete for binding with BECN1 with BCL2 or that of ATG14.
Modification of BECN1 by ISGylation
To examine the possibility that USP18 regulates BECN1 by ISGylation, we stimulated H4 cells with type I IFN to induce ISGylation. Immunoprecipitation analysis revealed that type I IFN markedly induced ISGylation of endogenous BECN1 and led to the presence of ∼100 kDa BECN1 species (). To further examine the possibility that BECN1 is a potential target for ISG15 modification, we tested whether HERC5, an E3 ligase for ISG15,Citation17 interacted with BECN1. 293T cells were transfected with expression vectors of FLAG-HERC5 and HA-BECN1. As shown in , FLAG-HERC5 strongly interacted with HA-BECN1. On the other hand, HERC5 did not interact with other components of PIK3C3 complex, including PIK3C3, ATG14, or UVRAG (Fig. S3G). These results provided further support for BECN1 as a potential target for ISG15 modification.
Figure 3. Modification of BECN1 by ISGylation. (A) ISGylation of endogenous BECN1 is induced by type I IFN. H4 cells were stimulated by 2 nM IFNA2 for 48 h. The cell lysates were subjected to immunoprecipitation with anti-BECN1 antibody and western blotting analysis using the indicated antibodies. (B) BECN1 interacts with HERC5. HA-BECN1 and FLAG-HERC5 were cotransfected into 293T cells for 36 h and analyzed by immunoprecipitation. (C) ISGylation of BECN1. 293T cells were transfected with the expression vectors of FLAG-BECN1, HA-HERC5, UBA7, UBE2L6, and His6-ISG15 as indicated. Thirty-six h after the transfection, the cell lysates were subjected to Ni-NTA pulldown and western blotting. (D) 293T cells were transfected as in (C) in the presence or absence of USP18 expression plasmid. The cell lysates were analyzed by western blotting using the indicated antibodies.
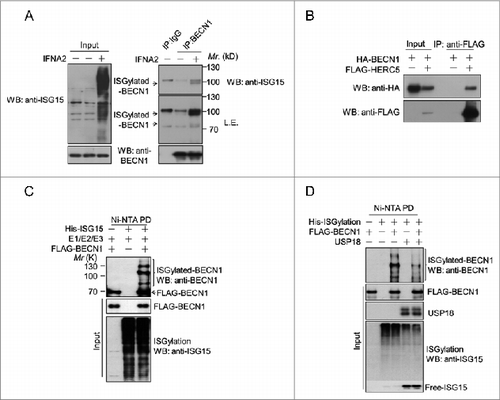
To further characterize whether BECN1 may be subject to ISGylation, we transfected expression vectors of FLAG-BECN1 and ISG15 conjugation system including HIS-ISG15, UBA7 (E1), UBE2L6 (E2), and HERC5 (E3) into 293T cells.Citation18 The cells were lysed in strong detergent 8 M urea and the products of ISGylation were purified by Ni-NTA for HIS-tagged ISG15. Western blotting using anti-BECN1 demonstrated 3 ISGylated BECN1 species around 90 kDa, 110 kDa and 130 kDa in cells expressing the ISGylation system, suggesting that BECN1 may be modified by multiple ISG15 conjugates (). Consistent with the ability of USP18 to promote de-ISGylation, the modification of BECN1 was significantly reduced upon overexpression of USP18 (). Taken together, these data suggest that BECN1 can be modified by ISGylation.
Identification of Lys117 and Lys263 in BECN1 as the main ISGylation sites
ISG15 forms a monomeric conjugate with its targeting Lys residues without multimerization as occurs with ubiquitination.Citation18 Given the MW of ISGylated endogenous BECN1, we hypothesized that there might be at least 2 Lys residues on BECN1 that can be subject to modification by ISG15. To determine the targeting site of ISGylation in BECN1, we coexpressed 5 different truncation mutants of BECN1 with ISG15-conjugating system. We found that expression of full-length BECN1 (amino acids 1 to 450, FL) with the ISGylation system led to 3 ISGylation bands (, S4A); whereas the expression of a C-terminal truncated BECN1 (amino acids 1 to 270, F1) with the ISGylation system still led to an upshift about 30 kDa, suggesting the N terminal half of the protein may have 2 ISGylation sites. The major effect of deleting the N-terminal 60 amino acids (F2) was the disappearance of the 3 species of BECN1 that differ slightly in MW, suggesting that these are products of differential translation of BECN1 from the N terminus. On the other hand, the deletion constructs F3 and 4, that encode truncated BECN1 (amino acids 60 to 150 and amino acids 210 to 270, respectively), appear to have each lost one possible ISGylation species (Fig. S4B), suggesting that there might be 2 possible ISGylation sites located in the amino acids 60 to 150 and amino acids 210 to 270 regions, respectively.
Figure 4. ISG15 modification of BECN1 inhibits PIK3C3 activity. (A) The expression constructs that encode truncation mutants of BECN1 were generated. FL, full-length. (B, C) ISG15 modification of wild-type or mutant BECN1. The ISGylation system was expressed in 293T cells with F1 (B) and BECN1 mutants (C) as indicated. The cells were lysed in 8 M urea, collected by Ni-NTA pulldown and analyzed by western blotting using anti-BECN1, and anti-ISG15. Note that the 2 major ISGylation bands were not detected when Lys117, Lys263, Lys265, and Lys266 were mutated to arginine. [4KR], (K117R, K263R, K265R, K266R) mutation. (D) BECN1-silenced H4 cells were transfected with wild-type or mutant BECN1 and then stimulated by 2 nM IFNA2 for 48 h. The cell lysates were subjected to immunoprecipitation with anti-FLAG antibody and western blotting analysis using the indicated antibodies. (E) Schematic representation of ISG15-BECN1 fusion proteins. The C-terminal amino acid sequence of active ISG15 is –LRLRGG, and the last glycine residue was deleted when fused to the BECN1 N-terminal region to prevent unexpected cleavage. (F) The BECN1-associated PIK3C3 lipid kinase assay using wild-type or fusion proteins. 293T cells were transfected with expression vectors of HA-PIK3C3 and FLAG-tagged BECN1 as indicated. Two×106 cells were lysed and incubated with anti-FLAG antibody to precipitate FLAG-tagged BECN1-HA-PIK3C3 complex. The immunoprecipitates were separated into 2 equal parts, one for input detection (left) and the other for kinase assays (right). The production of PtdIns3P was detected by protein-lipid blot assay. Lipids were extracted and applied onto polyvinylidene fluoride membrane. The commercial PtdIns3P was spotted as indicated for controls. The levels of PtdIns3P were detected using GST-PX-p40 domain protein, which binds to PtdIns3P, and anti-GST antibody.
![Figure 4. ISG15 modification of BECN1 inhibits PIK3C3 activity. (A) The expression constructs that encode truncation mutants of BECN1 were generated. FL, full-length. (B, C) ISG15 modification of wild-type or mutant BECN1. The ISGylation system was expressed in 293T cells with F1 (B) and BECN1 mutants (C) as indicated. The cells were lysed in 8 M urea, collected by Ni-NTA pulldown and analyzed by western blotting using anti-BECN1, and anti-ISG15. Note that the 2 major ISGylation bands were not detected when Lys117, Lys263, Lys265, and Lys266 were mutated to arginine. [4KR], (K117R, K263R, K265R, K266R) mutation. (D) BECN1-silenced H4 cells were transfected with wild-type or mutant BECN1 and then stimulated by 2 nM IFNA2 for 48 h. The cell lysates were subjected to immunoprecipitation with anti-FLAG antibody and western blotting analysis using the indicated antibodies. (E) Schematic representation of ISG15-BECN1 fusion proteins. The C-terminal amino acid sequence of active ISG15 is –LRLRGG, and the last glycine residue was deleted when fused to the BECN1 N-terminal region to prevent unexpected cleavage. (F) The BECN1-associated PIK3C3 lipid kinase assay using wild-type or fusion proteins. 293T cells were transfected with expression vectors of HA-PIK3C3 and FLAG-tagged BECN1 as indicated. Two×106 cells were lysed and incubated with anti-FLAG antibody to precipitate FLAG-tagged BECN1-HA-PIK3C3 complex. The immunoprecipitates were separated into 2 equal parts, one for input detection (left) and the other for kinase assays (right). The production of PtdIns3P was detected by protein-lipid blot assay. Lipids were extracted and applied onto polyvinylidene fluoride membrane. The commercial PtdIns3P was spotted as indicated for controls. The levels of PtdIns3P were detected using GST-PX-p40 domain protein, which binds to PtdIns3P, and anti-GST antibody.](/cms/asset/768f02b4-6009-44b3-aa0d-6049ec802274/kaup_a_1023982_f0004_b.gif)
We next attempted to determine the Lys (K) residues in BECN1 that might be subject to ISGylation. Replacement of Lys117, a highly conserved K residue, with Arg (R) led to the reduction of one ISGylation band (). Conversely, mutating any single Lys residue among the 6 Lys residues in amino acids 210 to 270 to Arg failed to have any effect on ISGylation (Fig. S4D). Since neighboring Lys residues might provide alternative sites for ISGylation, we generated a triple mutant, K263R, K265R, K266R [3KR], and found that this triple mutant lost an alternative species of ISGylation (). Furthermore, the quadruple mutant K117R, K263R, K265R, K266R [4KR] encoded in F1 and full-length BECN1 lost all ISGylation events ( and C). Taken together, we conclude that Lys117, as well as Lys263, Lys265, and Lys266 are the major alternative ISGylation sites in BECN1. Consistent with this conclusion, the treatment with type I IFN can only induce ISGylation of WT BECN1 after complementation of WT, but not the [4KR] mutant BECN1, in BECN1-silenced H4 cells ().
ISG15 modification of BECN1 inhibits PIK3C3 activity
Since ISGylation suppresses PIK3C3 activity and autophagy, we hypothesized that the kinase activity of BECN1-PIK3C3 complex may be regulated by ISG15 modification of BECN1. To investigate the functional role of ISG15 modification of BECN1, ISG15 was fused to the N terminus of BECN1, since the native ISGylation sites of BECN1 are close to the N terminus (). Such fusion proteins have been found to mimic the ISG15-modified state of a protein and are particularly useful when the native ISG15 modification site is close to the N or C termini of a protein.Citation18,19 We overexpressed HA-PIK3C3 with FLAG-ISG15, FLAG-BECN1, or FLAG-ISG15-BECN1 in 293T cells and performed lipid kinase assays to study the impact of ISGylation on its interaction with PIK3C3 and PIK3C3 kinase activity. Interestingly, the interaction of FLAG-ISG15-BECN1 with PIK3C3 was slightly reduced compared to that of FLAG-BECN1 (). However, the production of PtdIns3P in vitro by the FLAG-ISG15-BECN1+PIK3C3 complexes was strongly reduced compared to that of FLAG-BECN1+PIK3C3 complex (), suggesting that ISGylation of BECN1 inhibits PIK3C3 activity.
ISGylation competes for BECN1 Lys63 ubiquitination to inactivate PIK3C3 activity
Since Lys63 ubiquitination of BECN1 is involved in regulating BECN1 and autophagy,Citation20 we hypothesize that ISGylation on selected Lys residues may compete for Lys63 ubiquitination of BECN1. Interestingly, we found that the treatment of H4 cells with type I IFN significantly inhibited the Lys63-linked ubiquitination of BECN1 (), suggesting that type I IFN may negatively regulate Lys63 ubiquitination of BECN1. To directly verify this possibility, we tested the requirement of Lys117, as well as Lys263, Lys265, and Lys266 for Lys63 ubiquitination. Interestingly, we found that Lys63 ubiquitination of BECN1 could be significantly reduced by the [4KR] mutation with the K117R and K263R, K265R, K266R mutations, but not by any single mutations ().
Figure 5 (See previous page). ISGylation competes for Lys63-linked ubiquitination in BECN1 to inactivate PIK3C3 activity. (A) ISGylation inhibits the Lys63-linked ubiquitination of BECN1. H4 cells were treated with 2 nM IFNA2 for 24 h in the presence or absence of 200 nM rapamycin. The cell lysates were analyzed by immunoprecipitation with antibody against BECN1 and western blotting with anti-Lys63-ubiquitin-specific antibody. (B) The [4KR] mutation abrogates Lys63-linked ubiquitination of BECN1. 293T cells were transfected with an expression vector of HA-Lys63-only-ubiquitin and that of either WT or mutant FLAG-BECN1 as indicated. The cell lysates were analyzed by immunoprecipitation with anti-FLAG, and western blotting with the indicated antibodies. (C) WT BECN1, but not the [4KR] mutant, could complement the deficiency of BECN1 in mediating autophagy induction. BECN1-silenced H4 cells were transfected with control vector or that of WT BECN1 or the [4KR] mutant BECN1 and cultured for 36 h. The cell lysates were analyzed by western blotting with the indicated antibodies. (D) BECN1-silenced H4-LC3-GFP cells were transfected as in (C) and imaged by fluorescence microscopy. Representative cells are shown (left panel). The average of GFP-LC3 spot intensity in 500 cells from each indicated sample was determined. The data are displayed as means ± SD of the spot intensity per cell (right panel). (E) Lys63 ubiquitination of BECN1 is required for autophagy induction. BECN1-silenced H4 cells were transfected with control vector or that of WT BECN1 or the [4KR] mutant of BECN1 in the presence of Lys63-only ubiquitin and cultured for 36 h. The cell lysates were analyzed by western blotting with the indicated antibodies. (F) BECN1-silenced H4-GFP-LC3 cells were transfected as in (E) and imaged by fluorescence microscopy. The average of GFP-LC3 spot intensity in 500 cells from each indicated sample was determined. The data are displayed as means ± SD of the spot intensity per cell. (G, H) Lys63 ubiquitination of BECN1 is required for PIK3C3 activation. H4-FYVE-RFP cells were transfected as in (E) and imaged by fluorescence microscopy. The average of RFP-FYVE spot intensity in 500 cells from each indicated sample was determined. The data are displayed as means ± SD of the spot intensity per cell (G). PIK3C3 kinase activity is impaired by the expression of the BECN1 [4KR] mutant. 293T cells were transfected with the indicated vectors and cultured for 24 h. The cell lysates were analyzed by immunoprecipitation using anti-FLAG and followed by a PIK3C3 kinase assay and protein-lipid blot assay. The immunoprecipitates were separated into 2 parts, one for the kinase assay and the other for input detection (H). (I, J) ISGylation impairs Lys63-linked ubiquitination of BECN1. 293T cells were transfected with expression vectors of HA-Lys63-only ubiquitin and FLAG-tagged BECN1 as indicated with or without the ISGylation system and cultured for 24 h. The cell lysates were immunoprecipitated with anti-FLAG antibody and the immunocomplexes were analyzed by western blotting with anti-HA.
![Figure 5 (See previous page). ISGylation competes for Lys63-linked ubiquitination in BECN1 to inactivate PIK3C3 activity. (A) ISGylation inhibits the Lys63-linked ubiquitination of BECN1. H4 cells were treated with 2 nM IFNA2 for 24 h in the presence or absence of 200 nM rapamycin. The cell lysates were analyzed by immunoprecipitation with antibody against BECN1 and western blotting with anti-Lys63-ubiquitin-specific antibody. (B) The [4KR] mutation abrogates Lys63-linked ubiquitination of BECN1. 293T cells were transfected with an expression vector of HA-Lys63-only-ubiquitin and that of either WT or mutant FLAG-BECN1 as indicated. The cell lysates were analyzed by immunoprecipitation with anti-FLAG, and western blotting with the indicated antibodies. (C) WT BECN1, but not the [4KR] mutant, could complement the deficiency of BECN1 in mediating autophagy induction. BECN1-silenced H4 cells were transfected with control vector or that of WT BECN1 or the [4KR] mutant BECN1 and cultured for 36 h. The cell lysates were analyzed by western blotting with the indicated antibodies. (D) BECN1-silenced H4-LC3-GFP cells were transfected as in (C) and imaged by fluorescence microscopy. Representative cells are shown (left panel). The average of GFP-LC3 spot intensity in 500 cells from each indicated sample was determined. The data are displayed as means ± SD of the spot intensity per cell (right panel). (E) Lys63 ubiquitination of BECN1 is required for autophagy induction. BECN1-silenced H4 cells were transfected with control vector or that of WT BECN1 or the [4KR] mutant of BECN1 in the presence of Lys63-only ubiquitin and cultured for 36 h. The cell lysates were analyzed by western blotting with the indicated antibodies. (F) BECN1-silenced H4-GFP-LC3 cells were transfected as in (E) and imaged by fluorescence microscopy. The average of GFP-LC3 spot intensity in 500 cells from each indicated sample was determined. The data are displayed as means ± SD of the spot intensity per cell. (G, H) Lys63 ubiquitination of BECN1 is required for PIK3C3 activation. H4-FYVE-RFP cells were transfected as in (E) and imaged by fluorescence microscopy. The average of RFP-FYVE spot intensity in 500 cells from each indicated sample was determined. The data are displayed as means ± SD of the spot intensity per cell (G). PIK3C3 kinase activity is impaired by the expression of the BECN1 [4KR] mutant. 293T cells were transfected with the indicated vectors and cultured for 24 h. The cell lysates were analyzed by immunoprecipitation using anti-FLAG and followed by a PIK3C3 kinase assay and protein-lipid blot assay. The immunoprecipitates were separated into 2 parts, one for the kinase assay and the other for input detection (H). (I, J) ISGylation impairs Lys63-linked ubiquitination of BECN1. 293T cells were transfected with expression vectors of HA-Lys63-only ubiquitin and FLAG-tagged BECN1 as indicated with or without the ISGylation system and cultured for 24 h. The cell lysates were immunoprecipitated with anti-FLAG antibody and the immunocomplexes were analyzed by western blotting with anti-HA.](/cms/asset/59f46a15-5bc9-4656-a1ed-5322800cbfb6/kaup_a_1023982_f0005_c.jpg)
To further characterize the role of the Lys117, Lys263, Lys265, Lys266, and Lys63-linked ubiquitinations of BECN1 in autophagy induction, we expressed WT and the [4KR] mutant BECN1 in BECN1-silenced H4 cells. Strikingly, expression of WT BECN1, but not the [4KR] mutant, could complement BECN1 deficiency to mediate autophagy induction ( and D). Furthermore, overexpression of Lys63-only ubiquitin in H4 cells induces autophagy but not in BECN1-silenced cells (Fig. S6A). The defect in this Lys63-only ubiquitin response in BECN1-silenced cells could be complemented by expression of WT BECN1, but not the [4KR] mutant ( and), suggesting that Lys177, Lys263, Lys265, and Lys266 of BECN1 are important for inducing autophagy by Lys63 ubiquitination.
To test whether the Lys117, Lys263, Lys265, and Lys66 residues in BECN1 are important for PIK3C3 activity, we overexpressed Lys63-only ubiquitin together with WT or the [4KR] mutant BECN1 in H4-FYVE-RFP cells. As shown in, the levels of FYVE-RFP dots in cells transfected with WT BECN1 were higher than that of cells transfected with the [4KR] mutant. To further verify this result, we determined the PtdIns3P lipid kinase activity of PIK3C3 complexes isolated from 293T cells overexpressing HA-PIK3C3 together with either WT BECN1 or the [4KR] mutant BECN1 and Lys63-only ubiquitin. As shown in and Figure S6B, WT BECN1-PIK3C3 complexes showed significantly higher activity in vitro in producing PtdIns3P compared with that of the [4KR] BECN1-PIK3C3 complexes. These results suggest that the Lys117, Lys263, Lys265 and Lys266 residues of BECN1 are important for the activation of BECN1-PIK3C3 complexes by Lys63 ubiquitination.
To test whether ISGylation of BECN1 competes for the Lys63 ubiquitination of BECN1, we overexpressed vectors that encode either WT BECN1 or the fusion of ISG15 with BECN1 together with Lys63-only ubiquitin in 293T cells. As shown in , the expression of ISG15-fused BECN1 led to reduced Lys63 ubiquitination compared to that of WT BECN1. Furthermore, coexpression of ISGylation system also reduced Lys63 ubiquitination of BECN1 ().
Taken together, these results suggest that the ISGylation of BECN1 competes for Lys63-linked polyubiquitination at Lys117, Lys263, Lys265, and Lys266 of BECN1 to negatively regulate PIK3C3 activity to inhibit autophagy.
Type I IFN inhibits EGFR degradation through ISG15 modification of BECN1
Because the production of PtdIns3P by BECN1-PIK3C3 complexes also plays a critical role in regulating EGFR degradation, the negative regulation of class III PtdIns 3-kinase by ISGylation of BECN1 might affect EGFR degradation, which involves PtdIns3P.Citation21,22 To directly test this possibility, we examined the roles of ISGylation and USP18 in regulating the internalization and degradation of epidermal growth factor receptor (EGFR) upon EGF stimulation, which involves EGFR degradation.Citation23 Compared to that of control H4 cells, treatment with type I IFN led to significant increases in the levels of ISG15 and deceleration of EGFR degradation (). Furthermore, stable knockdown of USP18 also decelerated the degradation of EGFR in EGF stimulated H4 cells (). To further validate the role of ISGylation in type I IFN-inhibited EGFR degradation, we generated a stable ISG15 knockdown H4 cell lines. As shown in , ISG15 knockdown completely rescued the deceleration of EGFR degradation in the presence of IFN, as compared to that of control cells.
Figure 6. Type I IFN inhibits EGFR degradation through ISG15 modification of BECN1. (A) Type I IFN inhibits the EGFR degradation of EGFR. H4 cells were serum starved for 24 h in the presence or absence of 2 nM IFNA2, stimulated with 100 ng/mL EGF for the indicated periods, and then harvested for western blotting analysis. The ratios of EGFR/TUBA were calculated and are shown at the right panel. Band intensity was normalized to t = 0 of control. Results are shown as means ± SD of 3 independent sets of experiments (B) Knockdown of USP18 inhibits the EGFR degradation of EGFR. H4 cells with USP18 stable knockdown were serum starved for 24 h and harvested for western blotting analysis. The ratios of EGFR/TUBA were calculated and are shown in the right panel. (C) Knockdown of ISG15 rescues the deceleration of EGFR degradation induced by IFNΑ. H4 cells with ISG15 stable knockdown were treated as in (A) and harvested for western blotting. The ratios of EGFR/TUBA were calculated and are shown at the right panel. (D) EGFR degradation is impaired by ISG15 modification of BECN1. BECN1-silenced H4 cells expressing vectors encoding BECN1 or ISG15 modified BECN1 were serum starved for 24 h and harvested for western blotting analysis. The ratios of EGFR/TUBA were calculated and are shown at the right panel. (E) WT BECN1, but not the [4KR] mutant, can complement BECN1 deficient cells for inhibition of EGFR degradation induced by type I IFN. BECN1-silenced H4 cells were transfected with expression vectors of WT BECN1 or the [4KR] mutant BECN1 and cultured for 24 h, then treated as in (A) and harvested for western blotting analysis. The ratios of EGFR/TUBA were calculated and are shown below. (F) A working model for regulation of PIK3C3 complex by ISGylation and deISGylation. Induction of BECN1 ISGylation by type I IFN competes for Lys63-linked ubiquitination of BECN1, which is important for BECN1 activation, and leads to inactivation of PIK3C3 activity. Reduction of PIK3C3 activity in turn leads to inhibition of autophagy and EGFR degradation. USP18 mediates deISGylation of BECN1 to promote autophagy and EGFR degradation. Ub, ubiquitin.
![Figure 6. Type I IFN inhibits EGFR degradation through ISG15 modification of BECN1. (A) Type I IFN inhibits the EGFR degradation of EGFR. H4 cells were serum starved for 24 h in the presence or absence of 2 nM IFNA2, stimulated with 100 ng/mL EGF for the indicated periods, and then harvested for western blotting analysis. The ratios of EGFR/TUBA were calculated and are shown at the right panel. Band intensity was normalized to t = 0 of control. Results are shown as means ± SD of 3 independent sets of experiments (B) Knockdown of USP18 inhibits the EGFR degradation of EGFR. H4 cells with USP18 stable knockdown were serum starved for 24 h and harvested for western blotting analysis. The ratios of EGFR/TUBA were calculated and are shown in the right panel. (C) Knockdown of ISG15 rescues the deceleration of EGFR degradation induced by IFNΑ. H4 cells with ISG15 stable knockdown were treated as in (A) and harvested for western blotting. The ratios of EGFR/TUBA were calculated and are shown at the right panel. (D) EGFR degradation is impaired by ISG15 modification of BECN1. BECN1-silenced H4 cells expressing vectors encoding BECN1 or ISG15 modified BECN1 were serum starved for 24 h and harvested for western blotting analysis. The ratios of EGFR/TUBA were calculated and are shown at the right panel. (E) WT BECN1, but not the [4KR] mutant, can complement BECN1 deficient cells for inhibition of EGFR degradation induced by type I IFN. BECN1-silenced H4 cells were transfected with expression vectors of WT BECN1 or the [4KR] mutant BECN1 and cultured for 24 h, then treated as in (A) and harvested for western blotting analysis. The ratios of EGFR/TUBA were calculated and are shown below. (F) A working model for regulation of PIK3C3 complex by ISGylation and deISGylation. Induction of BECN1 ISGylation by type I IFN competes for Lys63-linked ubiquitination of BECN1, which is important for BECN1 activation, and leads to inactivation of PIK3C3 activity. Reduction of PIK3C3 activity in turn leads to inhibition of autophagy and EGFR degradation. USP18 mediates deISGylation of BECN1 to promote autophagy and EGFR degradation. Ub, ubiquitin.](/cms/asset/754565e3-9188-4cb8-92ce-3c689288a269/kaup_a_1023982_f0006_c.jpg)
To further assess the impact of ISG15 modification of BECN1 in the degradation of EGFR, we established lines of endogenous BECN1-silenced H4 cells stably expressing WT BECN1 and BECN1 fused to ISG15, and then stimulated cells with EGF (). The degradation of EGFR in the cells expressing ISG15 fused to the N terminus of BECN1 was strongly decelerated compared to that of WT BECN1, consistent with reduced PIK3C3 activity in these cells ().
Since we found that the [4KR] mutant BECN1 could not be ISGylated upon type I IFN treatment (), we hypothesized that the [4KR] mutant BECN1 might be defective in blocking EGFR degradation induced by type I IFN. To test this hypothesis, we expressed either WT BECN1 or the [4KR] mutant in BECN1-silenced H4 cells, then cells were prestarved for 24 h in the presence or absence of IFNA2 following treatment with EGF (). Interestingly, the cells expressing the [4KR] mutant BECN1 were defective in responding to the treatment with type I IFN in blocking the degradation of EGFR degradation. Taken together, our results suggest that ISGylation of BECN1 at Lys117, as well as Lys263, Lys265, and Lys266 serve an important role in negative regulation of intracellular processes including autophagy and EGFR degradation that are critically dependent upon the activity of class III PtdIns 3-kinase.
Discussion
In this study, we demonstrate that ISGylation of BECN1 induced by type I IFN leads to the inhibition of class III PtdIns 3-kinase activity, which is required for the activation of autophagy and EGFR degradation. We show that ISGylation of BECN1 can compete and block Lys63 ubiquitination of BECN1, which is involved in promoting the activation of PIK3C3 complexes.Citation20 Furthermore, we demonstrate that deISGylation by USP18 promotes the activity of class III PtdIns 3-kinase and autophagy. Our findings reveal a key mechanistic connection between the action of type I IFN and autophagy, 2 important cellular mechanisms involved in innate immunity.
ISG15 serves as a primary effector system for type I IFN that is critical for antiviral response as mice deficient for ISG15 are highly susceptible to infections by viral infections such as influenza, herpes, or Sindbis virus.Citation7 Since prolonged treatment with type I IFN, coincident with the induction of ISG15, leads to inhibition of autophagy, our study suggests that the inhibition of autophagy by type I IFN might serve to restraint viral replication. Consistently, a number of recent studies have demonstrated the accumulation of autophagy vesicles in hepatitis C virus (HCV) infected cells and that autophagy proteins can function as proviral factors required for HCV replication.Citation24-26 Inhibition of autophagy reduces the production of HCV viral particles.Citation24 Furthermore, inhibition of autophagy enhances the innate immune response in HCV-infected hepatocytes.Citation27 Thus, the ability of type I IFN to inhibit autophagy is consistent with its role as a first-line defense against viral infection.
ISG15 has been shown to mediate protection in a number of different viral infection models.Citation28 The expression of ISG15 is robustly induced upon the treatment with type I IFN and can modify a board range of protein targets upon their synthesis.Citation29 The sequence specificity required to lead to ISGylation is currently unknown and may occur immediately upon their synthesis depending upon the availability of ISGylation system. Our data suggest that the induction of ISG15 and ISGylation system upon type I IFN treatment may compete for Lys63 ubiquitination by mediating ISGylation of selected newly synthesized proteins. Thus, blocking Lys63 ubiquitination of selected target proteins by ISGylation may provide a general mechanism for type I IFN and ISG15 to modulate target activities.
Our study demonstrates that USP18 positively regulates autophagy and EGFR degradation by promoting de-ISGylation of BECN1. Knockdown of USP18 leads to inhibition of autophagy, which can be rescued upon knockdown of ISG15 and UBA7, the E1 enzyme involved in mediating ISGylation. Consistent with the role of ISGylation in inhibiting autophagy and promoting antiviral response, usp18−/− mice show resistance to lymphocytic choriomeningitis virus and vesicular stomatitis virus and enhanced interferon-mediated resistance.Citation30 This is also consistent with our hypothesis that inhibition of autophagy by ISGylation of BECN1 mediated by type I IFN promotes innate immunity against viral infection. Taken together, our study suggests that promoting ISGylation of BECN1 by type I IFN may provide an important mechanism to inhibit the activity of class III PtdIns 3-kinase to lead to inhibition of autophagy as a part of antiviral response.
Materials and Methods
Cell culture
293T and HepG2 cells were cultured in DMEM (Gibco, C11995500BT) with 10% (vol/vol) fetal bovine serum (Gibco, 10099–141) and 1% penicillin/streptomycin. H4, H4-LC3-GFP, and H4-FYVE-RFP cells were maintained in DMEM supplemented with 10% (vol/vol) fetal bovine serum, 1% penicillin/streptomycin and 1×sodium pyruvate (Gibco, 11360–070).
Antibodies and reagents
The commercial antibodies used for western blotting analysis include the following: anti-LC3B (2775, 1:1000 dilution), anti-ISG15 (2743, 1:1000 dilution), anti-USP18 (4813, 1:1000 dilution), anti-EGFR (2646, 1:1000 dilution), anti-FLAG (2368, 1:1000 dilution) were from Cell Signaling Technology. Anti-BECN1 (sc-11427 for WB, 1:1000 dilution; sc-48341 for IP, 1:200 dilution) and anti-ISG15 (sc-50366, 1:1000 dilution) were from Santa Cruz Biotechnology. Anti-PIK3C3 (12452–1-AP, 1:1000 dilution), anti-MYC (16286–1-AP, 1:1000 dilution), anti-HA (51064–2-AP, 1:1000 dilution) and anti-GAPDH (60004–1-Ig, 1:10000 dilution) were from Proteintech. Anti-ATG3 (PM034, 1:1000 dilution), anti-ATG5 (PM050, 1:1000 dilution), anti-ATG7 (PM039, 1:1000 dilution) and anti-tubulin, α (TUBA; PM054, 1: 10000 dilution) were from MBL. Anti-ubiquitin, Lys63-specific antibody (2210353, 1:2000 dilution) was from Milllipore. Mouse monoclonal anti-FLAG (F1804; 1:500 for IP), IFNA2 (SRP4595) and EGF (E9644) were from Sigma-Aldrich. Rapamycin (S1039) and MG132 (S2619) were from Selleckchem.
Plasmids and siRNA transfection
cDNAs for BECN1 and its deletion mutants were cloned into pcDNA3.1 using Phanta®Max Super-Fidelity DNA Polymerase (Vazyme Biotech Co., Ltd, P505) and ClonExpressTM II cloning kit (Vazyme Biotech Co., Ltd, C112). Mutagenesis was performed using MutExpressTM II mutagenesis kit (Vazyme Biotech Co., Ltd, C212). Cells were transfected with plasmid DNA using PolyJet DNA In Vitro Transfection Reagent (Signagen Laboratories, SL100688) according to the manufacturer's instructions. The sequences of siRNAs used in this study were as follows: siUSP18-a: 5′-CUGCAUAUCUUCUGGUUUA-3′; siUSP18-b: 5′-GGAAGAAGACAGCAACAUG-3′; siUSP18-c: 5′- GGACUACCCUCAUGGCCUG-3′; siISG15-a: 5′-UGAGCACCGUGUUCAUGAA-3′; siISG15-b: 5′-GGCAACGAAUUCCAGGUGU-3′; siUBA7-a: 5′-CAUCUUUGCUAGUAAUCUA-3′; siUBA7-b: 5′-AUAGAGCGCUCCAAUCUCA-3′. siRNA transient transfections were performed using HiPerFect Transfection Reagent (Qiagen, 1029975) according to the manufacturer's instructions.
Generation of knockdown and reconstitution of cell lines
H4 cells were stably infected with shRNA against USP18 (GGAAGAAGACAGCAACATG), ISG15 (TGAGCACCGTGTTCATGAA), BECN1 3′-UTR (CTCTGTGTTAGAGATATGA) or scramble control in the pLVX lentiviral background. Lentiviral particles were made according to the manufacturer's instructions (Clontech, 632177). Viral supernatant fractions were collected from 36 to 48 h after the transfection. Cleared supernatant fraction was filtered through a 0.45-μm filter (CORNING, 431220). Polybrene (8 μg/ml; Sigma, H9268) was supplemented to viral supernatant fractions. Twenty-four h after infection, cells stably expressing shRNA were obtained by selection with 1 μg/ml puromycin (Sangon Biotech, PJ593). BECN1 3′-UTR shRNA expressing H4 cells were then infected with lentiviral particles expressing FLAG-BECN1 (WT or mutant). Polyclonal populations were screened until WT and mutant lines were generated that had near endogenous BECN1 reconstitution levels.
Immunoprecipitation and Ni-NTA pulldown analysis
293T cells were transfected with relevant expressing vectors for 24 h. The cells were harvested and treated with lysis buffer (150 mM NaCl, 50 mM Tris–Cl, 1% NP-40 [Sangon Biotech, NDB0385], protease inhibitor cocktail [Millipore, 539134], 100 μg/ml PMSF [AMRESCO, 0754], 1mM Na3VO4 [Sigma, S6508], 50 mM NaF [Sigma, S6776], pH 7.4). Supernatant fractions were generated by centrifugation (13000g, 15 min, 4°C), and incubated with the indicated antibodies for 6 h at 4°C followed by immunoprecipitation with 40 μl protein G agarose (Invitrogen, 15900–010). The precipitates were washed with lysis buffer and tested by western blotting. For Ni-NTA pull down analysis, cells were lysed in 8 M urea. Equal amounts of cell extracts (1 mg) and 40 μl Ni-NTA-agarose beads (Qiagen, catalog number) were incubated overnight at room temperature. Precipitates were washed 3 times with the same buffer and subjected to SDS-PAGE.
PIK3C3 lipid kinase assay and protein-lipid blot assay
HA-PIK3C3 and FLAG-BECN1 complexes were immunoprecipitated by antibody against FLAG-BECN1. The immunoprecipitated beads were washed 3 times and added with PI:3PS (Promega, V1711, 0.1 mg/ml) and 25 μM ATP in 50 μl reaction buffer (50 mM HEPES [pH 7.5], 50 mM NaCl, 3 mM MgCl2, 0.025 mg/ml BSA [Genebase, 0332]) for 1 h at 25°C. The production of PtdIns3P was detected by protein-lipid blot assay as reported.Citation32,33 Briefly, lipids extracted from the reaction was spotted onto Hybond C-extra membrane (Amersham, RPN303E) using a BIO-DOTTM APPARATUS (Bio-Rad, 170–6545). The membrane was allowed to dry overnight in the dark. Then the membrane was incubated with blocking buffer (5% BSA in TBST [Sangon Biotech, PW009]) for 1 h, washed once in TBST for 30 min, and incubated with protein buffer (1 mg purified GST-tagged PX-p40 domain protein,Citation31 which binds to PtdIns3P, per 1 ml TBST with 5% BSA) overnight at 4°C. Then the membrane was washed in TBST 3 times at 30 min each, incubated with anti-GST (Sigma, G7781) in 5% BSA buffer for 4 h, washed in TBST for 3 changes with 5 min each, incubated with secondary antibody (Pierce, 31210) for 1 h, and washed in TBST for 3 changes with 5 min each. All incubations were at room temperature unless noted otherwise. The signals were visualized with ECL.
Cell imaging and statistical analysis
Cells were fixed with 4% paraformaldehyde (Sigma, 16005) and stained with 3 μg/ml DAPI (Sigma, D9542). Image data were collected with an ArrayScan HCS 4.0 Reader with a 203 objective (Cellomics ArrayScan VTI, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032) for DAPI-labeled nuclei and GFP or RFP-tagged intracellular proteins.
Statistical method
The Student t test was used as statistical analysis by using GraphPad Prism. Error bars for microscopy are presented as the standard deviation of triplicate samples. Error bars for western blot analysis represent the standard deviation between densitometry data from 3 unique experiments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the pub-lisher's website.
1023982_Supplemental_Figures.pdf
Download PDF (749.9 KB)Acknowledgments
We thank Dr. Jon Huibregtse of University of Texas at Austin for HA-HERC5 expression vector, Dr. Chin Ha Chung of Seoul National University for UBA7 and UBE2L6 expression vectors, Dr. Chen Wang of Shanghai Institute of Biochemistry and Cell Biology for His-ISG15 expression vector.
Funding
This work was supported in part by grants (to JY) from the Chinese Academy of Sciences and grants from the National Institute on Aging (US) (R37-AG012859 and R01-AG047231).
References
- Stetson DB, Medzhitov R. Antiviral defense: interferons and beyond. J Exp Med 2006; 203:1837-41; PMID:16880263; http://dx.doi.org/10.1084/jem.20061377
- Haas AL, Ahrens P, Bright PM, Ankel H. Interferon induces a 15-kgdalton protein exhibiting marked homology to ubiquitin. J Biol Chem 1987; 262:11315-23; PMID:2440890
- Potter JL, Narasimhan J, Mende-Mueller L, Haas AL. Precursor processing of pro-ISG15/UCRP, an interferon-β-induced ubiquitin-like protein. J Biol Chem 1999; 274:25061-8; PMID:10455185; http://dx.doi.org/10.1074/jbc.274.35.25061
- Dao CT, Zhang DE. ISG15: a ubiquitin-like enigma. Front Biosci 2005; 10:2701-22; PMID:15970528; http://dx.doi.org/10.2741/1730
- Narasimhan J, Wang M, Fu Z, Klein JM, Haas AL, Kim JJ. Crystal structure of the interferon-induced ubiquitin-like protein ISG15. J Biol Chem 2005; 280:27356-65; PMID:15917233; http://dx.doi.org/10.1074/jbc.M502814200
- Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem 2002; 277:9976-81; PMID:11788588; http://dx.doi.org/10.1074/jbc.M109078200
- Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, García-Sastre A., et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci U S A 2007; 104:1371-6; PMID:17227866; http://dx.doi.org/10.1073/pnas.0607038104
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID:21801009; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005
- Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle 2010; 9:1295-307; PMID:20305376; http://dx.doi.org/10.4161/cc.9.7.11109
- Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, Shuai K, Zhang DE. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J 2006; 25:2358-67; PMID:16710296; http://dx.doi.org/10.1038/sj.emboj.7601149
- Schmeisser H, Fey SB, Horowitz J, Fischer ER, Balinsky CA, Miyake K, Bekisz J, Snow AL, Zoon KC. Type I interferons induce autophagy in certain human cancer cell lines. Autophagy 2013; 9:683-96; PMID:23419269; http://dx.doi.org/10.4161/auto.23921
- Kaur S, Sassano A, Dolniak B, Joshi S, Majchrzak-Kita B, Baker DP, Hay N, Fish EN, Platanias LC. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc Natl Acad Sci U S A 2008; 105:4808-13; PMID:18339807; http://dx.doi.org/10.1073/pnas.0710907105
- Lekmine F, Uddin S, Sassano A, Parmar S, Brachmann SM, Majchrzak B, Sonenberg N, Hay N, Fish EN, Platanias LC. Activation of the p70 S6 kinase and phosphorylation of the 4E-BP1 repressor of mRNA translation by type I interferons. J Biol Chem 2003; 278:27772-80; PMID:12759354; http://dx.doi.org/10.1074/jbc.M301364200
- Ambjorn M, Ejlerskov P, Liu Y, Lees M, Jaattela M, Issazadeh-Navikas S. IFNB1/interferon-β-induced autophagy in MCF-7 breast cancer cells counteracts its proapoptotic function. Autophagy 2013; 9:287-302; PMID:23221969; http://dx.doi.org/10.4161/auto.22831
- Kutateladze TG, Ogburn KD, Watson WT, de Beer T, Emr SD, Burd CG, verduin M. Phosphatidylinositol 3-phosphate recognition by the FYVE domain. Mol Cell 1999; 3:805-11; PMID:10394369; http://dx.doi.org/10.1016/S1097-2765(01)80013-7
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360-72; PMID:18843052; http://dx.doi.org/10.1091/mbc.E08-01-0080
- Wong JJ, Pung YF, Sze NS, Chin KC. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc Natl Acad Sci U S A 2006; 103:10735-40; PMID:16815975; http://dx.doi.org/10.1073/pnas.0600397103
- Okumura F, Okumura AJ, Uematsu K, Hatakeyama S, Zhang DE, Kamura T. Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J Biol Chem 2013; 288:2839-47; PMID:23229543; http://dx.doi.org/10.1074/jbc.M112.401851
- Okumura F, Zou W, Zhang DE. ISG15 modification of the eIF4E cognate 4EHP enhances cap structure-binding activity of 4EHP. Genes Dev 2007; 21:255-60; PMID:17289916; http://dx.doi.org/10.1101/gad.1521607
- Xia P, Wang S, Du Y, Zhao Z, Shi L, Sun L, Huang G, Ye B, Li C, Dai Z., et al. WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. EMBO J 2013; 32:2685-96; PMID:23974797; http://dx.doi.org/10.1038/emboj.2013.189
- Thoresen SB, Pedersen NM, Liestol K, Stenmark H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Expl Cell Res 2010; 316:3368-78; PMID:20643123; http://dx.doi.org/10.1016/j.yexcr.2010.07.008
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S., et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253-7; PMID:18097414; http://dx.doi.org/10.1038/nature06421
- Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol 2014; 24:26-34; PMID:24295852; http://dx.doi.org/10.1016/j.tcb.2013.11.002
- Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A 2009; 106:14046-51; PMID:19666601; http://dx.doi.org/10.1073/pnas.0907344106
- Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol 2008; 82:2241-9; PMID:18077704; http://dx.doi.org/10.1128/JVI.02093-07
- Guevin C, Manna D, Belanger C, Konan KV, Mak P, Labonte P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010; 405:1-7; PMID:20580051; http://dx.doi.org/10.1016/j.virol.2010.05.032
- Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011; 53:406-14; PMID:21274862; http://dx.doi.org/10.1002/hep.24073
- Morales DJ, Lenschow DJ. The antiviral activities of ISG15. J Mol Biol 2013; 425:4995-5008; PMID:24095857; http://dx.doi.org/10.1016/j.jmb.2013.09.041
- Durfee LA, Lyon N, Seo K, Huibregtse JM. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol Cell 2010; 38:722-32; PMID:20542004; http://dx.doi.org/10.1016/j.molcel.2010.05.002
- Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nat Med 2004; 10:1374-8; PMID:15531891; http://dx.doi.org/10.1038/nm1133
- Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T., et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011; 147:223-34; PMID:21962518; http://dx.doi.org/10.1016/j.cell.2011.08.037
- Dowler S, Kular G, Alessi DR. Protein lipid overlay assay. Sci STKE 2002; 2002:pl6; PMID:11972359
- Gozani O, Karuman P, Jones DR, Ivanov D, Cha J, Lugovskoy AA, Baird CL, Zhu H, Field SJ, Lessnick SL., et al. The PHD finger of the chromatin-associated protein ING2 functions as a nuclear phosphoinositide receptor. Cell 2003; 114:99-111; PMID:12859901; http://dx.doi.org/10.1016/S0092-8674(03)00480-X