
ABSTRACT
Although autophagy is most critical for survival of cancer cells, especially in fast-growing tumors, the mechanism remains to be fully characterized. Herein we report that PSMD10/gankyrin promotes autophagy in hepatocellular carcinoma (HCC) in response to starvation or stress through 2 complementary routes. PSMD10 was physically associated with ATG7 in the cytoplasm, and this association was enhanced by initial nutrient deprivation. Subsequently, PSMD10 translocated into the nucleus and bound cooperatively with nuclear HSF1 (heat shock transcription factor 1) onto the ATG7 promoter, upregulated ATG7 expression in the advanced stage of starvation. Intriguingly, the type of PSMD10-mediated autophagy was independent of the proteasome system, although PSMD10 has been believed to be an indispensable chaperone for assembly of the 26S proteasome. A significant correlation between PSMD10 expression and ATG7 levels was detected in human HCC biopsies, and the combination of these 2 parameters is a powerful predictor of poor prognosis. The median survival of sorafenib-treated HCC patients with high expression of PSMD10 was much shorter than those with low expression of PSMD10. Furthermore, PSMD10 augmented autophagic flux to resist sorafenib or conventional chemotherapy, and inhibition of autophagy suppressed PSMD10-mediated resistance. We conclude that these results present a novel mechanism involving modulation of ATG7 by PSMD10 in sustaining autophagy, promoting HCC cell survival against starvation or chemotherapy. Targeting of PSMD10 might therefore be an attractive strategy in HCC treatment by suppressing autophagy and inducing HCC cell sensitivity to drugs.
Introduction
Autophagy is a process in which intracellular membrane structures sequester proteins and intracellular components to degrade and recycle these materials in response to nutrient starvation or environmental stress.Citation1 Autophagy often takes place under stress conditions, resulting in either adaptation and survival, or death. Increasing evidence implies the significance of autophagy in cancer.Citation2 However, the role of autophagy in cancer is controversial, especially due to its opposing functions in tumor initiation and development.Citation3 For example, deletion of autophagy-related genes, such as Becn1, Atg7, and Atg5, enhances liver carcinogenesis,Citation4,5 suggesting that autophagy acts as a tumor suppressor at the early stage of hepatocarcinogenesis. Conversely, at advanced stages of tumor development, induction of autophagy promotes tumor progression under low-oxygen, nutrient-deprivation, and stressed conditions.Citation6,7 Moreover, accumulating data argue that autophagy could protect some cancer cells against anticancer treatment by blocking the apoptotic pathway,Citation8-10 indicating its emergence as a therapeutic target for cancer prevention.
PSMD10/gankyrin/p28GANK/p28 is one of the nonATPase subunits of the regulatory complex of the human 26S proteasome. Interestingly, PSMD10 has been identified as an oncoprotein frequently overexpressed in HCC,Citation11 and plays complicated roles in HCC, which are yet to be fully elucidated. For example, PSMD10 controls the balance between cell cycle and apoptosis through degradation of RB1 and TP53.Citation11,12 Overexpression of PSMD10 accelerates HCC invasiveness and metastasis, serving as a valuable predicting factor for recurrence and survival.Citation13 Increasing evidence indicates that the contribution of PSMD10 to HCC progression is closely associated with its resistance to chemotherapeutic agents.Citation12,14 However, it is not clear whether autophagy contributes to the oncogenic role of PSMD10, especially in nutrient deprivation and drug resistance. Here we show that PSMD10 promotes autophagy through 2 complementary mechanisms in response to environmental stressors to promote HCC progression.
Results
PSMD10 promotes autophagy in response to starvation or stress conditions
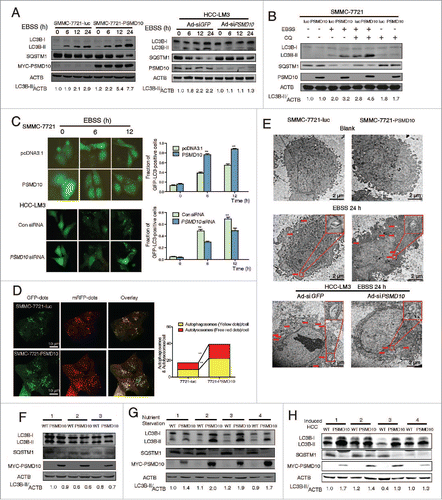
To assess the effect of PSMD10 on autophagy in HCC cells,Citation15 we used Earle's Balanced Salt Solution (EBSS) treatment to mimic starvation conditions. Overexpression of PSMD10 caused increased autophagic flux as evidenced by conversion from MAP1LC3B-I/LC3B-I to LC3B-II and degradation of SQSTM1 in SMMC-7721 cells, while knockdown of PSMD10 in HCC-LM3 cells reversed it (). After blockage of the autophagosomal-lysosomal fusion process with chloroquine (CQ), overexpression of PSMD10 resulted in more accumulation of autophagosomes (). To confirm the effect of PSMD10 on autophagic flux, we examined GFP-LC3B and mRFP-GFP-LC3B puncta formation with a fluorescence microscope. Consistently, PSMD10 promoted the autophagosomal-lysosomal fusion process (). Furthermore, autophagosomes increased by PSMD10 were directly visualized using transmission electron microscope (). To confirm regulation of autophagy by PSMD10 in vivo, we used transgenic overexpression of Psmd10 in mouse hepatocytes, which was driven by the albumin (Alb) promoter (Fig. S1A). As shown in , no effect of PSMD10 on the basal level of autophagy was observed in the liver tissues of littermates. However, PSMD10 significantly enhanced autophagic flux upon fasting (). Given a possible protective role of autophagy against stress conditions during tumor progression, we further examined the effect of oncogenic PSMD10 on autophagy in liver tumors from diethylnitrosamine (DEN) plus TCPOBOP-induced HCC models. At 20 wk after DEN plus TCPOBOP-treatment, 100% of littermates or Psmd10-transgenic mice developed hepatocarcinomas. Interestingly, PSMD10 overexpression increased LC3B-II amounts and decreased SQSTM1 in tumors compared to the control (). Thus, PSMD10 promoted autophagy in response to starvation.
Figure 1. PSMD10 enhances autophagy in starvation. (A, B) Evaluation for LC3B-II and SQSTM1 alteration indicative of autophagy induction in SMMC-7721-luc, SMMC-7721-PSMD10, or HCC-LM3 cells infected with adenovirus-mediated siRNA against GFP (Ad-siGFP) or PSMD10 (Ad-siPSMD10) treated with Earle's balanced salt solution (EBSS) for up to 24 h (A) or EBSS plus chloroquine (CQ) for 12 h (B). (C) SMMC-7721 cells were transiently cotransfected with pcDNA3.1 or PSMD10, or HCC-LM3 cells with siCon or siPSMD10. After 18 h transfection, cells were retransfected with pGFP-LC3B plasmid and subjected to EBSS treatment for the indicated time before observation for GFP-LC3B puncta under a fluorescence microscope. The percentage of cells with accumulation of GFP-LC3B in puncta was calculated in 3 random fields. (D) Representative images of LC3B staining in SMMC-7721-luc or SMMC-7721-PSMD10 cells infected with adenovirus-delivering mRFP-GFP-LC3B and treated with EBSS for 12 h. Quantification of autophagosome and autolysosome formation representing puncta staining sites per cell of 10 independent images. (E) Representative electron micrographs of autophagic vesicles or autophagosomes of (A) for up to 24 h. Arrows denote autophagosomes. Magnified image is shown. (F, G) Immunoblot for the indicated molecules in liver tissues isolated from 8-wk-old Psmd10-transgenic mice and littermates in a fed (F) or a fasted state for 24 h (G). (H) Immunoblot in liver tumors isolated from diethylnitrosamine (DEN) plus TCPOBOP-induced HCC in Psmd10-transgenic mice and littermates. Data represent the mean ±SD of 3 independent experiments (*P < 0.05, **P < 0.01).
Some evidence has shown the linkage of autophagy to cancer-associated dysregulation of TP53.Citation16 Several stimuli of proteasome-mediated degradation of TP53 are also inducers of autophagy. PSMD10 plays its oncogenic role via promoting ubiquitylation of TP53 by MDM2 and their subsequent degradation by the 26S proteasome.Citation12 In this study, we further explored the possibility of TP53 involvement in PSMD10-mediated autophagy. Intriguingly, PSMD10 did not alter EBSS-induced TP53 degradation in SMMC-7721 and HCC-LM3 cells (Fig. S1B). This effect was further confirmed in HepG2 cells with wild-type TP53 (Fig. S1C). Accordingly, even in TP53-deficient Hep3B cells, PSMD10 overexpression augmented EBSS-stimulated LC3-II and autophagosome formation (Fig. S1D, E). Taken together, these results revealed that PSMD10 enhances autophagy in HCC independent of TP53 status.
Association of PSMD10 with ATG7 is required for PSMD10-induced autophagy
PSMD10 plays its oncogenic role mainly through its ankyrin repeats to mediate protein-protein interaction,Citation11,12,14,17,18 implicating the possible involvement of this structure in autophagy regulation. The autophagy interaction network reveals the significance of interacting proteins during the autophagy process.Citation19 We assessed the possibility that PSMD10 associated with a series of ATG family members in the autophagic vacuole process. In the SMMC-7721-PSMD10 stable cell lines, the MYC-tagged PSMD10 was used to perform coimmunoprecipitation with the typical markers of autophagy after EBSS treatment, including BECN1, the ATG12–ATG5 complex and ATG7 (Fig. S2A). We found that PSMD10 and ATG7, an E1-like enzyme for LC3-conjugation systems,Citation20,21 are present in a single complex (Fig. S2B). Furthermore, their interaction increased with the starvation stimuli (). Their endogenous interaction in SMMC-7721 cells was also detected in the precipitates (). Confocal microscopy analysis revealed colocalization of PSMD10 with ATG7 mainly in the cytoplasm (). These exogenous and endogenous studies and results therefore clearly showed the interaction of PSMD10 with ATG7 under physiological conditions. To determine which region of PSMD10 is required for its interaction with ATG7, a series of PSMD10 deletion mutants containing different ankyrin repeats were used (Fig. S2C).Citation18 As shown in , removing the last 3 ankyrin repeats or C-terminal tail domain impaired PSMD10 interaction with ATG7, implicating these motifs essential for both associations.
Figure 2. PSMD10 interacts with ATG7 to promote autophagy in EBSS starvation. (A) SMMC-7721-PSMD10 cells were EBSS-starved for the indicated time, and the cytosolic lysate was extracted for coimmunoprecipitation with anti-MYC, followed by probing with anti-ATG7. (B) The cell lysate from SMMC-7721 treated with EBSS for 12 h was extracted for coimmunoprecipitation with anti-PSMD10 or ATG7, followed by probing with anti-ATG7 or PSMD10. (C) HCC-LM3 cells treated with EBSS for 12 h were immunostained for endogenous PSMD10 and ATG7 to observe their colocalization, DNA was stained by 4',6-diamidino-2-phenylindole (DAPI). Representative confocal microphotographs are shown together with profiles of colocalization within the area of interest. (D) Different MYC-tagged PSMD10-expressing plasmids (mutants with each of the ankyrin repeats [dA1 to dA5], the N terminus [dN, deleted for amino acids 1 to 38], or the C terminus [dC, deleted for amino acids 204 to 226] deleted), were transfected into SMMC-7721 cells, and immunoprecipitated and immunoblotted with ATG7 or MYC-tag antibodies. (E) HEK-293 cells cotransfected with MYC-tagged PSMD10 and HA-tagged ATG7 (WT), ATG571S, ATG7CΔ, or a combination of the 2 ATG571S, CΔ, were immunoprecipitated and immunoblotted with the indicated antibodies. (F) LC3B-II accumulation was detected in SMMC-7721 cells transfected with MYC-tagged PSMD10 mutants (dA3 to 5, dC) or a PSMD10 complete plasmid after EBSS treatment for 12 h. (G) Coimmunoprecipitation with anti-HA was performed in SMMC-7721 cells transfected with HA-tagged ATG7, MYC-tagged PSMD10 mutants (dA3 to 5, dC), or a PSMD10 complete plasmid, and EBSS treated for 12 h, followed by probing for the indicated molecules.
![Figure 2. PSMD10 interacts with ATG7 to promote autophagy in EBSS starvation. (A) SMMC-7721-PSMD10 cells were EBSS-starved for the indicated time, and the cytosolic lysate was extracted for coimmunoprecipitation with anti-MYC, followed by probing with anti-ATG7. (B) The cell lysate from SMMC-7721 treated with EBSS for 12 h was extracted for coimmunoprecipitation with anti-PSMD10 or ATG7, followed by probing with anti-ATG7 or PSMD10. (C) HCC-LM3 cells treated with EBSS for 12 h were immunostained for endogenous PSMD10 and ATG7 to observe their colocalization, DNA was stained by 4',6-diamidino-2-phenylindole (DAPI). Representative confocal microphotographs are shown together with profiles of colocalization within the area of interest. (D) Different MYC-tagged PSMD10-expressing plasmids (mutants with each of the ankyrin repeats [dA1 to dA5], the N terminus [dN, deleted for amino acids 1 to 38], or the C terminus [dC, deleted for amino acids 204 to 226] deleted), were transfected into SMMC-7721 cells, and immunoprecipitated and immunoblotted with ATG7 or MYC-tag antibodies. (E) HEK-293 cells cotransfected with MYC-tagged PSMD10 and HA-tagged ATG7 (WT), ATG571S, ATG7CΔ, or a combination of the 2 ATG571S, CΔ, were immunoprecipitated and immunoblotted with the indicated antibodies. (F) LC3B-II accumulation was detected in SMMC-7721 cells transfected with MYC-tagged PSMD10 mutants (dA3 to 5, dC) or a PSMD10 complete plasmid after EBSS treatment for 12 h. (G) Coimmunoprecipitation with anti-HA was performed in SMMC-7721 cells transfected with HA-tagged ATG7, MYC-tagged PSMD10 mutants (dA3 to 5, dC), or a PSMD10 complete plasmid, and EBSS treated for 12 h, followed by probing for the indicated molecules.](/cms/asset/cd3ab45f-7ed1-4390-b34c-d9e4205e9c79/kaup_a_1034405_f0002_c.gif)
The E1-like ubiquitin-conjugating activity of ATG7 is crucial for ATG7-mediated autophagy, which could be blocked by ATG7 mutants with a Cys-to-Ser replacement at the active site (ATG7C571S), a deletion of the C terminus (ATG7CΔ), or combination of the 2 (ATG7C571S, CΔ) (Fig. S2D).Citation22 However, the full-length ATG7, as well as the enzymatically deficient ATG7 constructs, all appeared to bind to PSMD10 (). The E1-like activity of ATG7 is correlated with LC3-II formation or its recognition of LC3 and ATG12.Citation22,23 In our present study, depletion of the PSMD10 motifs, indispensible for the interaction with ATG7, would never facilitate starvation-induced LC3B-II formation unlike full-length PSMD10 (). Moreover, coimmunoprecipitation experiments revealed that these motifs are also essential for PSMD10 to regulate the interaction of ATG7 with LC3, but not for ATG12-conjugation (). Therefore, these data implicated that the interaction of PSMD10 with ATG7 partially influences the E1-like activity, although the whole catalytic domain of ATG7 seems dispensable for their interaction. Together, these results strongly suggested that PSMD10-mediated autophagy relies on its interaction with ATG7, which is inseparable from the region of the last 3 ankyrin repeats and C-terminal tail.
PSMD10 upregulates ATG7 expression
With EBSS treatment for 24 h, the interaction between PSMD10 and ATG7 gradually increased within the first 12 h and maintained at steady levels at later time points, whereas PSMD10 enhanced LC3B-II formation with prolonged starvation (). Thus, it is possible that other mechanisms are also involved in PSMD10-mediated autophagy. Given that PSMD10, as a chaperone, usually interacts with target proteins, and promotes or blocks their degradation by the 26S proteasome, we further evaluated the effect of PSMD10 on ATG protein expression. Remarkably, overexpression of PSMD10 caused gradual elevation of ATG7 within 24 h of starvation, while knockdown of PSMD10 had the opposite effect (), implying that PSMD10 promotes ATG7 expression. Consistently, increased ATG7 protein levels were detected in liver tissues of Alb promoter-driven Psmd10-transgenic mice after food deprivation (), or in tumors induced by DEN plus TCPOBOP (). However, PSMD10 affected neither the expression of BECN1 nor the formation of the ATG12–ATG5 conjugate in vitro and in vivo (). Pattison et al. and Lee et al. report that increased ATG7 expression can induce LC3-II formation.Citation22,24 Herein, overexpression of ATG7 stimulated autophagic flux upon starvation in SMMC-7721 or HCC-LM3 cells, represented by an increase in LC3B-II formation, autophagosomes, and autolysosomes' puncta (Fig. S3A, B). We further determined whether the effect of PSMD10 on autophagy is dependent on ATG7. As anticipated, downregulation of ATG7 by siRNA impaired PSMD10-induced autophagy ( and Fig. S3C, D). Unexpectedly, PSMD10 still partially promoted LC3B-II formation even ATG5 downregulation (Fig. S4A and B). Thus, our data indicate the main requirement of ATG7 for PSMD10-mediated autophagy.
Figure 3. PSMD10 promotes ATG7 transcriptional activation. (A) Protein contents of ATG7, BECN1, ATG12–ATG5 conjugation, and PSMD10 were examined by immunoblot in SMMC-7721-PSMD10 versus SMMC-7721-luc cells, and HCC-LM3-Ad-siPSMD10 vs. Ad-siGFP cells after EBSS treatment for the indicated times. (B, C) The effect of PSMD10 on ATG7 expression was analyzed by immunoblot in vivo, including 24-h fasting (B) or DEN-TCPOBOP-induced HCC models (C). (D) Representative micrographs of autophagic vesicles or autophagosomes are shown in SMMC-7721 cells after transient cotransfection with pcDNA3.1 or MYC-PSMD10 and a nonspecific siRNA control or siATG7 RNA #1. (E) SMMC-7721-luc or SMMC-7721-PSMD10 cells were exposed to EBSS and cycloheximide (CHX, 1 μg/ml), actinomycin D (ActD, 5 μg/ml), or MG132 (5 μM) for 12 h. ATG7 was determined by immunoblot. (F) Quantitative PCR was performed to measure the transactivity of ATG7 in stable SMMC-7721-luc or SMMC-7721-PSMD10 cells exposed to EBSS for 24 h. Data represent the mean ± SD of 3 independent experiments (*P < 0.05).
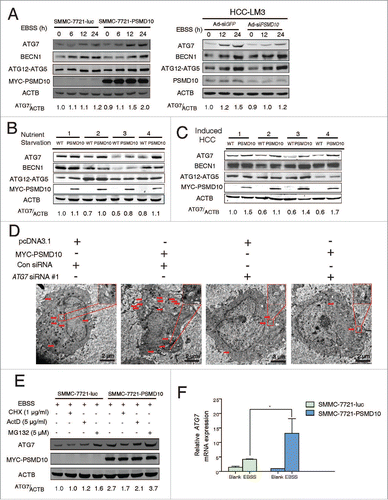
Next, we explored the mechanisms about PSMD10 regulating ATG7 at transcriptional or post-transcriptional levels. As shown in , upregulation of ATG7 by PSMD10 under EBSS stimulation was blocked by inhibitors of protein synthesis, cycloheximide (CHX) or actinomycin D (ActD), but not by the proteasome inhibitor MG132, implying that PSMD10 modulates ATG7 protein synthesis rather than degradation. In addition, overexpression of PSMD10 increased mRNA expression of ATG7 (). Thus, these results indicate that PSMD10 also transactivates ATG7 expression, resulting in enhanced autophagy.
PSMD10 enters the nucleus and promotes HSF1 binding to the ATG7 promoter
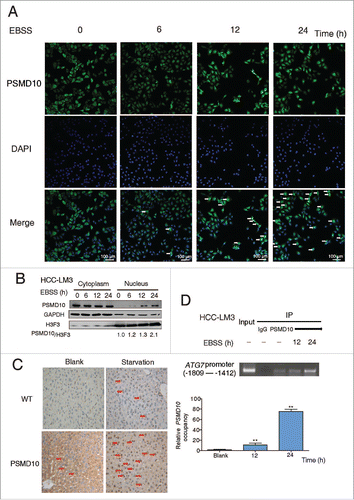
We next examined how PSMD10 upregulates ATG7 transcription. Our previous studies demonstrate that PSMD10, as a nuclear-cytoplasmic shuttling protein, can accelerate NFKB1-RELA nuclear export.Citation18 Here we observed that exogenously expressed tagged-PSMD10 was diffusely present in the cytoplasm and nucleus in SMMC-7721 cells, with increasing translocation into the nucleus after starvation (Fig. S5A). Both immunostaining and immunoblotting showed increased nuclear relocation of endogenous PSMD10 in HCC-LM3 cells following starvation (). Consistently, upon fasting, Psmd10-transgenic mice also presented more nuclear immunostaining of PSMD10 in hepatocytes (). Given the nuclear translocation of PSMD10 and its transcriptional activation of ATG7, we reasoned that PSMD10 might bind to the ATG7 promoter directly. A series of primers flanking the ATG7 promoter from −2074 to +100 were used for ChIP assay followed by quantitative PCR analysis. A significant increase of PSMD10 binding to a fragment of the ATG7 promoter from −1809 to −1412 was observed, and this binding further increased upon EBSS starvation (, Fig. S5B).
Figure 4. PSMD10 translocates into the nucleus and binds to the promoter of ATG7. (A) . PSMD10 translocates into the nucleus and binds to the promoter of ATG7. (A) Nuclear translocation of endogenous PSMD10 in HCC-LM3 was detected by immunostaining with anti-PSMD10 antibody after EBSS starvation for different time points as indicated. Arrows indicate nuclear localization of PSMD10. (B) Cell fractionation from HCC-LM3 cells was performed to analyze the cellular localization of endogenous PSMD10. Histone H3 and GAPDH were assessed as loading controls for nuclear and cytoplasmic proteins, respectively. (C) Liver tissues from fasting Psmd10-transgenic mice or littermates were immunostained for PSMD10. Representative pictures are shown. Arrows indicate nuclear expression of PSMD10. (D) ChIP assay covering the region of the ATG7 promoter from −1809 to −1412 was performed to measure the binding activity of PSMD10 to the ATG7 promoter in HCC-LM3 cells after EBSS starvation for 12 or 24 h (upper panel). Quantification of the ratio of PSMD10 binding to IgG (lower panel). Data represent the mean ± SD of 3 independent experiments (**P < 0.01).
Interestingly, the region of the ATG7 promoter from −1570 and −1602 has been identified to bind HSF1,Citation25 a master regulator of tumorigenesisCitation2628 that plays a critical role in controlling cytoprotective autophagy. ChIP assay showed that overexpression of PSMD10 improved the binding of HSF1 to the ATG7 promoter following starvation (). To further validate whether the potential HSF1 binding is required for upregulation of ATG7 by PSMD10, we used the 2 ATG7 promoter deletion constructs lacking the HSF1 binding sites.Citation25 The induction of ATG7 promoter activation by PSMD10 was delayed by removing the HSF1 binding sites (). Furthermore, PSMD10-mediated ATG7 production was counteracted by silencing HSF1 during starvation ( and Fig. S6A). These results suggest that HSF1 is involved in the PSMD10-mediated promotion of ATG7.
Figure 5. PSMD10 associates with HSF1 to cooperate in the expression of ATG7. (A) ChIP assay of the ATG7 promoter containing HSF1 binding sites from −1809 to −1412 was performed to measure the effect of PSMD10 on the binding activity of HSF1 under EBSS starvation for 24 h (left panel). Quantification of the ratio of HSF1 binding to IgG (lower panel). (B) The effect of PSMD10 on ATG7 reporter activity. SMMC-7721 cells were transiently cotransfected with pcDNA3.1/MYC-PSMD10 and ATG7 WT/mut1/mut2 luciferase plasmids. Luciferase activities were detected at 24 h after transfection and EBSS starvation. Data represent mean (±SD) from 3 independent experiments (*P < 0.05). (C) SMMC-7721 cells were transiently cotransfected with pcDNA3.1 or MYC-PSMD10 and a nonspecific RNAi control or siHSF1 RNA #3, and the indicated molecules were detected after EBSS starvation for 24 h. Data represent mean (±SD) from 3 independent experiments (*P < 0.05). (D) Cytoplasmic and nuclear fractionation from SMMC-7721-PSMD10 cells was performed for coimmunoprecipitation with MYC-tag antibody, followed by probing with anti-HSF1 or MYC-tag antibody. (E) HCC-LM3 cells treated with EBSS for 24 h were immunoprecipitated and immunoblotted for the indicated molecules. (F) HCC-LM3 cells were immunostained for endogenous PSMD10 and HSF1 to observe their colocalization in the presence or absence of EBSS for 24 h. DNA was stained by DAPI. Representative confocal microphotographs are shown together with profiles of colocalization within the area of interest (yellow signals in the nucleus).
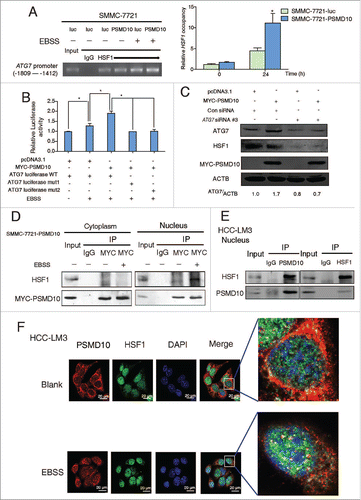
Since PSMD10 translocates into the nucleus and modulates HSF1 binding to ATG7 promoter under starvation, we hypothesized that the association of PSMD10 with HSF1. As expected, the interaction of exogenous or endogenous proteins was detected in the nucleus in response to 24 h of starvation ( and Fig. S6B). Confocal laser scanning microscopy assay demonstrated that most of the endogenous PSMD10 were normally localized in the cytoplasm, while HSF1 was centralized in the nucleus. Upon starvation, both of the proteins were colocalized in the nucleus (). Interestingly, overexpression of PSMD10 enhanced phosphorylation of HSF1 at Ser326 (Fig. S6C), which is indispensable for activation of HSF1.Citation29 Therefore, PSMD10 interacts and coordinates with HSF1 to activate ATG7, leading to autophagy in advanced starvation.
PSMD10 is involved in impairment of the proteasome system-induced autophagy
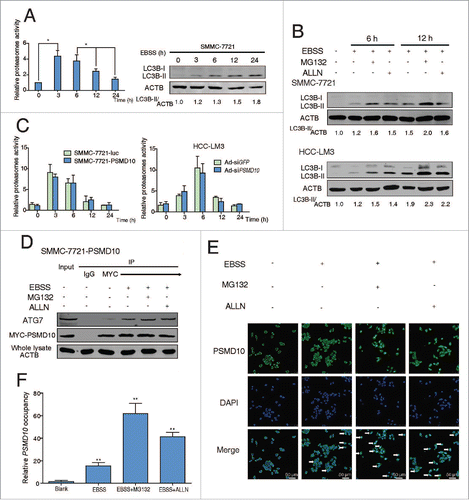
Autophagy has been reported to act as a compensatory degradation system when the proteasome system is impaired in neurodegenerative diseases.Citation30,31 How the 2 complementary degradative systems communicate and coordinate in HCC remains poorly understood. Here we observed that, during starvation for 24 h, the proteasome activity dramatically rose to the top within the first 3 h and fell to the basal level after, whereas autophagy sustained activation (). Moreover, even when the proteasome system is suppressed by the 26S proteasome inhibitor MG132 or calpain inhibitor ALLN, autophagy still elevated in response to starvation (). Upon nutrient deprivation, the autophagic machinery of the cell is primarily mobilized and utilized for the challenges of eliminating the bulk of an entire organelle, which is beyond the capacity of the proteasome system.Citation32 Nas6, the yeast ortholog of PSMD10, has been verified as one of the bona fide regulatory particle chaperones essential for the assembly of 26S proteasomes.Citation33 However, no alteration of the proteasome activity was detected whenever overexpression or downregulation of PSMD10 (), indicating that a small part of PSMD10 is possibly enough for proteasome assembly due to recyclable utilization. Interestingly, in the presence of MG132 or ALLN, the interaction between PSMD10 and ATG7 was improved (). Meanwhile, increased nuclear translocation of PSMD10 and enhanced occupancy of PSMD10 on the ATG7 promoter was also detected in HCC (). Collectively, our data implied that PSMD10, despite the chaperone for the 26S proteasome, is also involved in regulating autophagy to degrade the aggregated proteins especially when proteasome is powerless.
Figure 6. PSMD10 connects the proteasome and the autophagic pathway. (A) Proteasome activity and autophagy level were detected in SMMC-7721 with or without EBSS for the indicated time. (B) Proteasome activity was measured in the indicated cells treated with EBSS at the indicated times. (C) Expression of LC3B-II was detected after EBSS treatment combined with or without proteasome inhibitors (MG132 10 μM, ALLN 10 μM) for 6 or 12 h in SMMC-7721 and HCC-LM3 cells. (D) SMMC-7721-PSMD10 cells were treated as in (C), immunoprecipitated with MYC-tag antibodies, and then immunoblotted with anti-ATG7 or anti-MYC-tag antibodies. (E) Localization of PSMD10 in HCC-LM3 cells treated as in (C) was detected. (F) ChIP assay of the binding capability of PSMD10 to the ATG7 promoter in HCC-LM3 cells treated as in (C).
PSMD10 expression and ATG7 levels are correlated in human HCC tissues, and a combination of both has a more accurate prognostic value for HCC
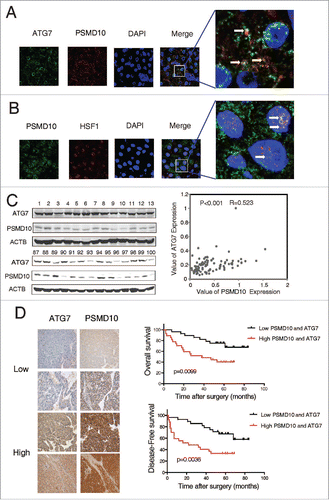
We collected 100 HCC patient specimens, and evaluated a possible correlation of PSMD10 with ATG7. First, colocalization of PSMD10 and ATG7 was observed mainly in the cytoplasm of HCC biopsies (), whereas colocalization of PSMD10 and HSF1 primarily in the nucleus (). Immunoblotting analysis of 100 HCC patient specimens revealed a strong correlation between PSMD10 expression and ATG7 level in tumor tissues ( and Fig. S7A). Consistently, the results from another 59 paired HCC samples, along with adjacent normal tissues, showed a similar association (Fig. S7B). Tissue microarrays (TMAs) of 119 clinical HCC specimens by immunohistochemistry further confirmed their association (, left panel). For patients whose tumors had above-average levels of both PSMD10 and ATG7, both overall and disease-free survivals were markedly reduced (, right panel and Table S1). Thus, the combination of these 2 parameters is a powerful predictor of poor prognosis, further supporting a critical role of PSMD10 in modulating ATG7, resulting in autophagy and thus HCC progression.
Figure 7. The relationship between PSMD10 and ATG7 in the clinical samples. (A, B) The liver tumor frozen sections from 2 patients were performed by immunofluorescence analysis to detect the localization of PSMD10 and ATG7 (A) or HSF1 (B). DNA was stained by DAPI. Representative confocal microphotographs are shown together with profiles of colocalization within the area of interest (yellow signals merge), and higher-magnification images of the outlined areas are also shown. (C) Proteins were extracted from 100 patients with HCC for immunoblot with anti-PSMD10 or anti-ATG7 antibody. The protein bands were analyzed by ImageJ2x software and the relative band intensity was normalized to ACTB band. Representative samples are shown (left panel). Correlation of expression level of PSMD10 and ATG7 is shown (right panel). (D) Representative PSMD10 and ATG7 expression in TMA sections of 117 patients with HCC (original magnification 200 ×; left panel). The median value for PSMD10 and ATG7 expression in HCCs was used to divide the patients into high (above median) and low (below median) PSMD10 and ATG7 groups. The disease-free and overall survival rates were compared between the low PSMD10 and ATG7 expression group (n = 28) and the high PSMD10 and ATG7 expression group (n = 27; right panel).
PSMD10-mediated autophagy facilitates HCC resistance to sorafenib or chemotherapeutic agents
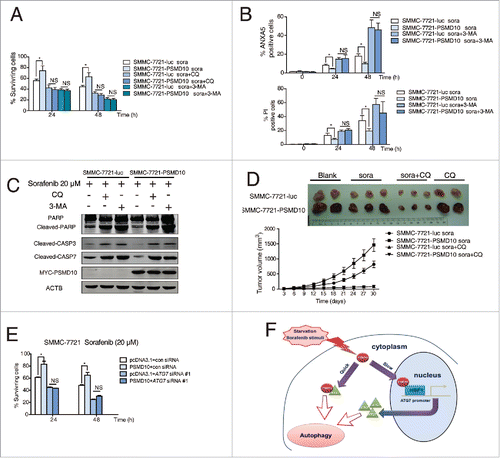
The multikinase inhibitor sorafenib has been widely used for patients with advanced HCC.Citation34 Apart from its application in patients with unresectable HCC, it has been also used for patients following surgical tumor resection to prevent recurrence. However, some evidence shows that sorafenib may induce autophagy and cause drug resistance.Citation9,35,36 We evaluated whether PSMD10-mediated autophagy influences the efficacy of sorafenib on HCC. A tissue microarray from 70 patients with HCC who underwent liver resection and sorafenib therapy revealed that high expression of PSMD10 was associated with a poorer prognosis (P = 0.024) ( and Table S1). To determine the significance of the above clinical data, we used the SMMC-7721 cell line with stable PSMD10-overexpression to evaluate the effect of PSMD10 on sorafenib treatment. Intriguingly, overexpression of PSMD10 indeed promoted HCC survival in the sorafenib treatments ( and Fig. S8A, B). Moreover, PSMD10 further improved sorafenib-stimulated autophagy, which was diminished by PSMD10 knockdown (). However, PSMD10-mediated resistance to sorafenib was completely blocked by CQ or 3-methyladenine (3-MA) (). More importantly, the subcutaneous xenograft model based on SMMC-7721 cells showed that overexpression of PSMD10 still promoted tumor growth even under sorafenib or the CQ-alone treatment (). Only if both were combined together, were PSMD10-driven tumors dramatically shrunk (). A similar result was observed after the conventional chemotherapeutic agent treatment including cisplatin and epirubicin (Fig. S8C). Correspondingly, knockdown of ATG7 markedly impeded PSMD10-mediated resistance to sorafenib ( and Fig. S8D). Hence, these findings revealed that PSMD10-mediated autophagy contributes to HCC resistance to sorafenib or conventional chemotherapeutics.
Figure 8. PSMD10 resisted apoptosis and enhanced autophagy under sorafenib treatment. (A) Representative PSMD10 expression in TMAs from 70 patients with HCC who underwent liver resection and sorafenib therapy (upper panel). Overall survival rates were compared between the low-PSMD10 (below the median value, n = 35) and high-PSMD10 groups (above the median value, n = 35) (lower panel). (B) SMMC-7721-luc or SMMC-7721-PSMD10 cells were treated with sorafenib (20 μM) for the indicated time or with the indicated dosage for 24 h. (C) LC3B-II was immunoblotted in SMMC-7721 or HCC-LM3 cells after sorafenib (20 μM) treatment for the indicated time. (D) The formation of LC3 puncta was detected in the indicated cells exposed to 20 μM sorafenib for 12 or 24 h. Quantification of the ratio of LC3 punctate cells to overall cells was shown in right panel. (E) Electron microscopy analysis showed autophagic vesicles or autophagosomes in cells from (D).
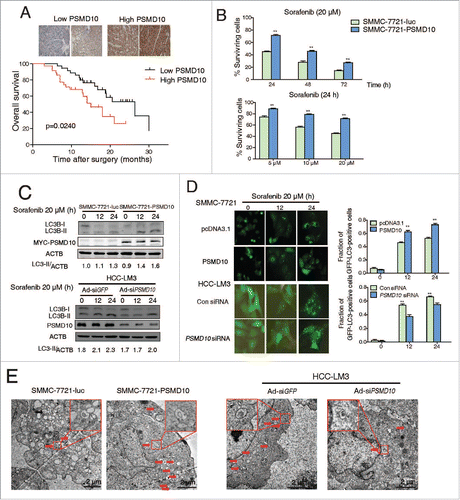
Figure 9. PSMD10-mediated autophagy caused drug resistance to sorafenib. (A) SMMC-7721-luc, SMMC-7721-PSMD10 cells were subjected to sorafenib (20 μM) for 24 or 48 h in the presence or absence of either CQ (10 μM) or 3-MA (10 mM) before performing CCK-8 assays. (B) Cells from (A) were analyzed for apoptosis. The percentage of cells was determined using the ANXA5 and propidium iodide (PtdIns) staining assay. (C) Immunoblot for apoptosis markers including PARP, CASP3, and CASP7 in cells from (B). (D) Effects of sorafenib and CQ on subcutaneous PSMD10-overexpressing tumors. SMMC-7721-luc or SMMC-7721-PSMD10 cells were injected into the right flanks of nude mice. Two weeks after injection, mice were randomly assigned into 4 groups, and set for sorafenib or CQ alone or both combination treatments (intraperitoneally, i.p.). Tumors from mice in each group, after tumor therapy, are presented, and average tumor volume for each group is shown. (E) SMMC-7721 cells were transiently cotransfected with pcDNA3.1 or MYC-PSMD10 and a nonspecific RNAi control or siATG7 RNA #1, then CCK-8 assays were performed. (F) A model for PSMD10, as a recyclable molecule to modulate autophagy in 2 different and complementary mechanisms. Upon the stressed condition, PSMD10 will first take part in an autophagic vacuole-forming process, through an interaction with cytosolic ATG7. Subsequently, PSMD10 will translocate into the nucleus and interact with HSF1 bound onto the ATG7 promoter to upregulate ATG7 expression, leading to further increased autophagy. Data represent mean (±SD) from 3 independent experiments (*P < 0.05).
Discussion
Autophagy is a catabolic process to maintain cellular homeostasis and mitigate metabolic stress. The role of autophagy in tumorigenesis is complicated. It has been proposed that autophagy suppresses cancer initiation while enabling growth of aggressive cancers.Citation2 In the established tumors, autophagy maintains mitochondrial metabolic function essential for tumor development, creating “autophagy addiction.”Citation37 Tumor growth in a stressed microenvironment may cause dependency on autophagy for survival in aggressive cancer.Citation38,39 Some oncoproteins have been found to activate autophagy for tumor growth, but the mechanism remains unclear. For example, autophagy is required for activated Ras to maintain oxidative metabolism and tumorigenesis.Citation37,40 In this study, we found that oncogenic PSMD10 upregulates stress-induced but not basal level of autophagy, to promote tumor cell survival in exposure to nutrient withdrawal or therapeutic agents during HCC progression. This process involves 2 complementary mechanisms: interaction between PSMD10 and ATG7 in the cytoplasm, and nuclear entry of PSMD10 coordinated with HSF1 to transactivate ATG7. Moreover, PSMD10 could augment autophagic flux to resist sorafenib or chemotherapy.
Autophagy-related genes are required for various types of autophagy whose functions in cancer are context-dependent. The E1-like enzyme ATG7 coordinates with the E2-like enzyme ATG10 to mediate conjugation of ATG12 to ATG5, and subsequently promote lipid phosphatidylethanolmine formation.Citation41 Atg7 deletion in mouse liver promotes HCC development.Citation4 In contrast, inhibition of ATG7 suppresses oncogenic KRAS-induced breast tumor growth,Citation37 and high levels of ATG7 expression predict poor prognosis for breast cancer.Citation25 In addition, upregulation of ATG7 is detected in most HCC samples and facilitates tumor cell survival during metabolic stress.Citation42 Our current study showed that PSMD10 regulated ATG7 to affect autophagy in 2 different manners. The direct association of PSMD10 with ATG7 in the cytoplasm occurred earlier upon starvation. Subsequently, PSMD10 entered into the nucleus, and bound to the ATG7 promoter in coordination with HSF1, to transactivate ATG7, leading to sustained autophagy. The interaction between PSMD10 and ATG7 depends on the last 3 ankyrin repeats and the C-terminal region of PSMD10, but not on the E1-like catalytic domain of ATG7. These dual roles of PSMD10 in regulation of autophagy activation contribute to tumor cell adaptation and tolerance to complex stresses. Remarkably, a strong correlation between PSMD10 and ATG7 was observed in HCC patient specimens, and combination of these 2 parameters increased prognostic value, strongly suggesting that the concerted activities of PSMD10 and ATG7 detected in our study are recapitulated in clinical patients with HCC. Our findings point to an oncogenic role of ATG7 in advanced HCC, and suggest that PSMD10 induces autophagy by directly targeting ATG7. This also explained why PSMD10 can stimulate autophagy without affecting MTOR phosphorylation status (Fig. S8F), because ATG7 is downstream of MTOR in the autophagic process but also a direct executor of autophagy. Therefore, we propose that PSMD10 promotes autophagy by dual control of ATG7 and desensitizes tumor cells to stressed conditions. To our knowledge, this is the first time to reveal a novel molecular mechanism for differential regulation of ATG7 by PSMD10 in autophagy.
In addition to cytoplasm, the nuclear events are significant for autophagy, but the mechanism is unclear.Citation43,44 Our group has previously shown that PSMD10, as a nuclear-cytoplasmic shuttling protein, controls some nuclear proteins, such as NFKB1, OCT4, CEBPB, or HNF4A,Citation14,18,45,46 Nuclear localization of PSMD10 was also detected in some clinical HCC samples (data not shown). However, it is still unclear what drives nuclear transport of PSMD10 and how it functions in the nucleus. Our current studies showed the nuclear colocalization of PSMD10 with HSF1 to modulate ATG7. Thus, it is very likely that PSMD10 simultaneously regulates other unknown nuclear transcription factors. HSF1, a major transactivator of stress proteins, has been implicated in the pathogenesis of cancer, including controlling hepatic steatosis, hepatic insulin resistance, and HCC development.Citation26,27 Here we found that PSMD10 increased phospho-HSF1. Given the complex HSF1 pathways, whether PSMD10 affects HSF1's other functions requires further clarification. This study clearly demonstrates the crucial role for PSMD10, as a recyclable shuttling molecule, in modulating autophagy through 2 interdependent ways ().
Autophagy and the ubiquitin-proteasome system are the 2 major intracellular degradation pathways, and both have largely distinct clients. Some evidence has emerged to imply active crosstalk between these protein degradation pathways.Citation30,47 Inhibition of the proteasome will lead to autophagy.Citation31,48 Nevertheless, how the 2 complementary degradative systems communicate and coordinate, especially in cancer cells under metabolic stress, remains poorly understood. Our study showed a fast elevation of both proteasome activity and autophagy at the first short time of starvation, the former then declined rapidly but the latter continued (). Despite no obvious effect of PSMD10 on proteasome activity (), PSMD10 can still activate autophagy even when the proteasome is impaired (). Given its chaperone molecule character, we postulate that a certain level of PSMD10 is enough to maintain the assembly of the 26S proteasome. Once the proteasome is defective, the original PSMD10 involved in assemblage of the proteasome will be discharged to take part in the autophagic vacuole assembly-process through interaction with cytosolic ATG7. Meanwhile, the unused and the rest of PSMD10 translocates into the nucleus, and cooperates with HSF1 to promote the transcription of ATG7 and sustain autophagy for compensating the degradative function.
The combination of autophagy inhibitors with sorafenib or conventional chemotherapeutic agents to treat tumors might be more effective in the clinical treatment of liver cancer.Citation49 However, it remains unclear when and how to take this combinatorial therapy. One urgent issue is to determine the mechanism by which sorafenib interacts with intracellular molecules in order to increase its efficacy. In the current study, those HCC patients with high expression of PSMD10 had worse prognosis for sorafenib therapy. Overexpression of PSMD10 indeed profoundly improved HCC cells survival following sorafenib treatment, which was accompanied by increased autophagy. Furthermore, the combination of autophagy inhibition with sorafenib significantly blocked PSMD10-enhanced tumor growth and caused apoptosis in vitro or in vivo, suggesting that PSMD10-induced autophagy is one vital element in HCC response to the efficacy of sorafenib. Collectively, this is the first report to demonstrate that PSMD10 may be a potential molecular marker in the classification of HCC patients who are suitable for sorafenib or chemotherapy in combination with autophagy inhibition.
Materials and methods
Cell lines, recombinant adeno- or lenti-virus, and small interfering RNA
Liver cancer cell lines SMMC-7721 and Hep3B were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (TCHu52, TCHu106). MHCC-LM3 was from American type culture collection (ATCC, JN-CL-0834). The SMMC-7721-luciferase (luc), SMMC-7721-PSMD10 stable cell lines, adenovirus-delivered siRNA against PSMD10 and adenovirus-delivered PSDM10 overexpression were obtained from our lab.Citation13,14 The sequences of siRNA against PSMD10, ATG7, HSF1 and scrambled siCon (Genepharma, A10001) are diplayed as following.
Antibody
The antibodies used in this study, and their source are indicated in the following table:
Transgenic mouse model
Hepatocyte-specific PSMD10-transgenic mice, carrying the human PSMD10 cDNA after the Alb promoter, in C57BL/6 background were generated in the Model Animal Research Center of Nanjing University. After 24 h fasting, 8-wk-old male mice were sacrificed to test the level of LC3B in liver. Liver cancer was induced by DEN (Sigma, 73861) plus TCPOBOP (Sigma t1443). In brief, DEN (25 mg/kg intraperitoneally [i.p.]) was given at d 15 postpartum to initiate tumor growth. Beginning at 28 d, mice were injected with TCPOBOP (3 mg/kg i.p.) once every 2 wk for a total of 8 times to promote hepatocarcinoma growth.
Tissue microarrays
Human liver tissues were obtained from surgical resection specimens of HCC patients in Eastern Hepatobiliary Surgery Hospital (Shanghai, China). Immunohistochemistry was performed following the routine protocol. The slides were incubated with the following primary antibodies: anti-PSMD10 (Santa Cruz Biotechnology, sc-101498), 1:50; anti-ATG7 (Santa Cruz Biotechnology, sc-376212), 1:50. The density of positive staining was measured with the use of a computerized image system (Leica Microsystems Imaging Solutions Ltd, Cambridge, United Kingdom). Under high-power magnification (× 200), photographs of 3 representative fields were captured by the Leica QWin Plus v3 software; identical settings were used for each photograph. The density was counted by Image-Pro Plus v6.2 software (Media Cybernetics Inc., Bethesda, MD). For the reading of each antibody staining, a uniform setting for all the slides was applied. Integrated optical density of all the positive staining in each photograph was measured, and its ratio to total area of each photograph was calculated as density. The patients were divided between the low and high expression groups by the median of the density as calculated before.
Immunofluorescence staining
Briefly, samples were fixed with 4% paraformaldehyde for 10 min. Subsequently, the cells were permeabilized with 0.1% Triton X-100 (Bio-light Inc., BL-SJ-0749) for 10 min at room temperature, washed with phosphate-buffered saline (PBS; Beyotime Inc, C0221A) and blocked with PBS containing 1% (w/v) bovine serum albumin (BSA; Bio-light Inc., BL-SJ-0135) and 10% (w/v) goat serum (Fan-ke-shi-ye Inc., FK0124; in BSA buffer) for 0.5 h at room temperature. Samples were incubated with the indicated antibodies overnight at 4°C, washed with PBS, and incubated with corresponding secondary IgG for 1 h at room temperature. After rinsing in PBS, the samples were counterstained with diamidino phenylindole (DAPI) (Sigma, 32670). Representative images were photographed using a confocal microscope (Leica BMI-6000, Cambridge, UK).
Quantitative real-time PCR analysis
Real-time PCR was performed using SYBR Green PCR Kit (Applied Biosystems 4367659) and ABI 7900HT Fast Real-Time PCR System (Applied Biosystems USA). The expression of specific transcripts was normalized against ACTB, and the experiments were performed in triplicate. ATG7 Forward Primer: 5′-TGCTATCCTGCCCTCTGTCTT-3′; Reverse Primer: 5′-TGCCTCCTTTCTGGTTCTTTT-3′.
Assessment of apoptosis
Apoptotic cells were evaluated by ANXA5 and PI staining (Invitrogen, A13201) according to the manufacturer's instructions, and analyzed by flow cytometry (catalog ID: MoFlo XDP 1051628, Beckman Coulter, USA).
Nude mice xenograft
All animal experiments meet the requirement of Second Military Medical University Animal Care Facility and the National Institutes of Health guidelines. SMMC-7721-luc and SMMC-7721-PSMD10 cells were injected subcutaneously into nude mice. CQ (Sigma c6628 60 mg/kg/d) and sorafenib (30 mg/kg/d) were administered in 100 μl by i.p. injections one wk after tumor implantation. The mice were sacrificed at 4 wk after tumor implantation. The volumes of tumors were measured and calculated as V = a × b2 × π/6.
Cell growth monitored by CCK-8 viability assay
The cancer cells were seeded in 100 μl of growth medium at a density of 8 × 103 cells per well in 96-well plates. Following an overnight incubation, cells were treated with indicated regents or not. Every 24 h until 72 h, 10 μl CCK-8 solution (Dojindo Laboratories, CK04) was added per well for 2 h before the end of incubation at 37°C. Cell viability was measured with a microplate reader (BioTek USA) at an absorbance of 450 nm. Viability is given as a percent of the control value.
Autophagy analysis
Autophagy was assessed using LC3 mobility shift. GFP- LC3B redistribution and subcellular localization were analyzed by fluorescence and electron microscopy. Redistribution was detected with an inverted fluorescence microscope after GFP-LC3B transfection for at least 24 h. The fraction of GFP-LC3B-positive cells (> 5 punctate staining sites per cell) was determined in 3 independent experiments. Eight random fields representing 200 cells were counted. For the LC3 mobility shift assay, cells were lysed and then subjected to immunoblot analysis with antibody against LC3 (MBL, M152-3). Liver cancer cells or mice samples were fixed in ice-cold 2% glutaraldehyde and finally examined with a H800 transmission electron microscope (Hitachi Japan).
mRFP-GFP-LC3B system
The mRFP-GFP-LC3B adenovirus construct was obtained from Hanbio Inc. (autophagy-adv-GFP-RFP-LC3B-1000). This construct fluorescence depends on the difference in pH between the acidic autolysosome and the neutral autophagosome, and the exhibited red/green (yellow) or red fluorescence makes it possible to monitor progression of autophagic flux. Briefly, the cells infected by the adenovirus are imaged by confocal fluorescence microscopy.
ChIP (chromatin immunoprecipitation) assay
Cells were processed and analyzed with a ChIP assay kit (Beyotime, P2078). The region of the ATG7 promoter (−2074 – +100) was divided into 7 parts, and the primers are the following:
Statistical analysis
Continuous variables were expressed as mean ± SD and compared using the Student t test or Mann-Whitney U test. Categorical variables were compared using the χ2 test or Fisher exact test. We calculated the correlation coefficient between PSMD10 and ATG7 using Pearson statistic. Survival curves were calculated using the Kaplan-Meier method and were compared using the log-rank test.
Abbreviation
| ActD | = | actinomycin D |
| ChIP | = | chromatin immunoprecipitation |
| CHX | = | cycloheximide |
| CQ | = | chloroquine |
| DEN | = | diethylnirtosamine |
| EBSS | = | Earle's balanced salt solution |
| HCC | = | hepatocellular carcinoma |
| HSF1 | = | heat shock transcription factor 1 |
| TCPOBOP | = | 1,4-bis-[2-(3,5-dichloropyridyloxy)]benzene 3,3′,5,5′-tetrachloro-1,4-bis(pyridyloxy)benzene |
| 3-MA | = | 3-methyladenine |
| TMAm | = | tissue microarrays |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Ming Tan (University of South Alabama), for the gift of ATG7 luciferase plasmids. We thank Dr. Toren Finkel (National Heart, Lung, and Blood Institute, Bethesda) for the ATG7 and mutant plasmids. We thank Dr. Zhengqing Lei (Eastern Hepatobiliary Surgery Hospital) for statistic support.
Funding
Research was supported by the projects from National Natural Science Foundation of China (30921006, 81372206, 91029732, 30900770, 81101714, 81471768), the State Key Project for Infectious Diseases (2012ZX10002–009, 011, 2013ZX10002-010-002), National Key Basic Research Program (2012CB316503), the foundation of economy and information technology of Shanghai (12CH-03).
Related Research Data
References
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726–34; PMID:16148885; http://dx.doi.org/10.1038/nrc1692
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:1845–6; PMID:23656658; http://dx.doi.org/10.1056/NEJMra1205406
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961–7; PMID:17972889; http://dx.doi.org/10.1038/nrc2254
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011; 25:795–800; PMID:21498569; http://dx.doi.org/10.1101/gad.2016211
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809–20; PMID:14638851; http://dx.doi.org/10.1172/JCI20039
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717–29; PMID:21406549; http://dx.doi.org/10.1101/gad.2016111
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51–64; PMID:16843265; http://dx.doi.org/10.1016/j.ccr.2006.06.001
- Song J, Qu Z, Guo X, Zhao Q, Zhao X, Gao L, Sun K, Shen F, Wu M, Wei L. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy 2009; 5:1131–44; PMID:19786832; http://dx.doi.org/10.4161/auto.5.8.9996
- Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, Wang XY, Dai Z, Peng YF, Gu CY, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011; 7:1159–72; PMID:21691147; http://dx.doi.org/10.4161/auto.7.10.16818
- Sun WL, Chen J, Wang YP, Zheng H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011; 7:1035–44; PMID:21646864; http://dx.doi.org/10.4161/auto.7.9.16521
- Higashitsuji H, Itoh K, Nagao T, Dawson S, Nonoguchi K, Kido T, Mayer RJ, Arii S, Fujita J. Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in he patomas. Nat Med 2000; 6:96–9; PMID:10613832; http://dx.doi.org/10.1038/71600
- Higashitsuji H, Itoh K, Sakurai T, Nagao T, Sumitomo Y, Masuda T, Dawson S, Shimada Y, Mayer RJ, Fujita J. The oncoprotein gankyrin binds to MDM2/HDM2, enhancing ubiquitylation and degradation of p53. Cancer Cell 2005; 8:75–87; PMID:16023600; http://dx.doi.org/10.1016/j.ccr.2005.06.006
- Fu J, Chen Y, Cao J, Luo T, Qian YW, Yang W, Ren YB, Su B, Cao GW, Yang Y, et al. p28GANK overexpression accelerates hepatocellular carcinoma invasiveness and metastasis via phosphoinositol 3-kinase/AKT/hypoxia-inducible factor-1alpha pathways. Hepatology 2011; 53:181–92; PMID:21254169; http://dx.doi.org/10.1002/hep.24015
- Qian YW, Chen Y, Yang W, Fu J, Cao J, Ren YB, Zhu JJ, Su B, Luo T, Zhao XF, et al. p28(GANK) prevents degradation of Oct4 and promotes expansion of tumor-initiating cells in hepatocarcinogenesis. Gastroenterology 2012; 142:1547–58 e14; PMID:22387393; http://dx.doi.org/10.1053/j.gastro.2012.02.042
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445–544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10:676–87; PMID:18454141; http://dx.doi.org/10.1038/ncb1730
- Sun W, Ding J, Wu K, Ning BF, Wen W, Sun HY, Han T, Huang L, Dong LW, Yang W, et al. Gankyrin-mediated dedifferentiation facilitates the tumorigenicity of rat hepatocytes and hepatoma cells. Hepatology 2011; 54:1259–72; PMID:21735473; http://dx.doi.org/10.1002/hep.24530
- Chen Y, Li HH, Fu J, Wang XF, Ren YB, Dong LW, Tang SH, Liu SQ, Wu MC, Wang HY. Oncoprotein p28 GANK binds to RelA and retains NF-kappaB in the cytoplasm through nuclear export. Cell Res 2007; 17:1020–9; PMID:18040287; http://dx.doi.org/10.1038/cr.2007.99
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68–76; PMID:20562859; http://dx.doi.org/10.1038/nature09204
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 20 06; 441:880–4; PMID:16625205; http://dx.doi.org/10.1038/nature04723
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425–34; PMID:15866887; http://dx.doi.org/10.1083/jcb.200412022
- Lee IH, Kawai Y, Fergusson MM, Rovira, II, Bishop AJ, Motoyama N, Cao L, Finkel T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012; 336:225–8; PMID:22499945; http://dx.doi.org/10.1126/science.1218395
- Tanida I, Yamasaki M, Komatsu M, Ueno T. The FAP motif within human ATG7, an autophagy-related E1-like enzyme, is essential for the E2-substrate reaction of LC3 lipidation. Autophagy 2012; 8:88–97; PMID:22170151; http://dx.doi.org/10.4161/auto.8.1.18339
- Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res 2011; 109:151–60; PMID:21617129; http://dx.doi.org/10.1161/CIRCRESAHA.110.237339
- Desai S, Liu Z, Yao J, Patel N, Chen J, Wu Y, Ahn EE, Fodstad O, Tan M. Heat shock factor 1 (HSF1) controls chemoresistance and autophagy through transcriptional regulation of autophagy-related protein 7 (ATG7). J Biol Chem 2013; 288:9165–76; PMID:23386620; http://dx.doi.org/10.1074/jbc.M112.422071
- Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007; 130:1005–18; PMID:17889646; http://dx.doi.org/10.1016/j.cell.2007.07.020
- Jin X, Moskophidis D, Mivechi NF. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab 2011; 14:91–103; PMID:21723507; http://dx.doi.org/10.1016/j.cmet.2011.03.025
- Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012; 150:549–62; PMID:22863008; http://dx.doi.org/10.1016/j.cell.2012.06.031
- Guettouche T, Boellmann F, Lane WS, Voellmy R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem 2005; 6:4; PMID:15760475; http://dx.doi.org/10.1186/1471-2091-6-4
- Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 2010; 584:1393–8; PMID:20040365; http://dx.doi.org/10.1016/j.febslet.2009.12.047
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447:859–63; PMID:17568747; http://dx.doi.org/10.1038/nature05853
- Lamark T, Johansen T. Autophagy: links with the proteasome. Curr Opin Cell Biol 2010; 22:192–8; PMID:19962293; http://dx.doi.org/10.1016/j.ceb.2009.11.002
- Saeki Y, Toh EA, Kudo T, Kawamura H, Tanaka K. Multiple proteasome-interacting proteins assist the assembly of the yeast 19S regulatory particle. Cell 2009; 137:900–13; PMID:19446323; http://dx.doi.org/10.1016/j.cell.2009.05.005
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R, Kelley S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006; 5:835–44; PMID:17016424; http://dx.doi.org/10.1038/nrd2130
- Shimizu S, Takehara T, Hikita H, Kodama T, Tsunematsu H, Miyagi T, Hosui A, Ishida H, Tatsumi T, Kanto T, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer 2012; 131:548–57; PMID:21858812; http://dx.doi.org/10.1002/ijc.26374
- Gauthier A, Ho M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: an update. Hepatol Res 2013; 43:147–54; PMID:23145926; http://dx.doi.org/10.1111/j.1872-034X.2012.01113.x
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25:460–70; PMID:21317241; http://dx.doi.org/10.1101/gad.2016311
- Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, Stevens C, Brunton VG, Langdon WY, Vidal M, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol 2012; 14:51–60; PMID:22138575; http://dx.doi.org/10.1038/ncb2386
- Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF, Gallo CA, Plas DR, Biesiada J, Meller J, et al. VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 2012; 21:532–46; PMID:22516261; http://dx.doi.org/10.1016/j.ccr.2012.02.019
- Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 2012; 122:4621–34; PMID:23143306; http://dx.doi.org/10.1172/JCI62973
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102–9; PMID:17909521; http://dx.doi.org/10.1038/ncb1007-1102
- Chang Y, Yan W, He X, Zhang L, Li C, Huang H, Nace G, Geller DA, Lin J, Tsung A. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology 2012; 143:177–87 e8; PMID:22504094; http://dx.doi.org/10.1053/j.gastro.2012.04.009
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429–33; PMID:21617040; http://dx.doi.org/10.1126/science.1204592
- Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, Joseph B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 2013; 500:468–71; PMID:23863932; http://dx.doi.org/10.1038/nature12313
- Jiang Y, Iakova P, Jin J, Sullivan E, Sharin V, Hong IH, Anakk S, Mayor A, Darlington G, Finegold M, et al. Farnesoid X receptor inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology 2013; 57:1098–106; PMID:23172628; http://dx.doi.org/10.1002/hep.26146
- Her GM, Cheng CH, Hong JR, Sundaram GS, Wu JL. Imbalance in liver homeostasis leading to hyperplasia by overexpressing either one of the Bcl-2-related genes, zfBLP1 and zfMcl-1a. Dev Dyn 2006; 235:515–23; PMID:16273521; http://dx.doi.org/10.1002/dvdy.20624
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472–83; PMID:18054316; http://dx.doi.org/10.1016/j.cmet.2007.11.004
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171:513–24; PMID:17620365; http://dx.doi.org/10.2353/ajpath.2007.070188
- Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev 2011; 25:1999–2010; PMID:21979913; http://dx.doi.org/10.1101/gad.17558811