
Abstract
Hexokinases (HKs) catalyze the first step of glucose metabolism, phosphorylating glucose to glucose 6-phosphate (G6P). HK2/hexokinase-II is a predominant isoform in insulin-sensitive tissues such as heart, skeletal muscle, and adipose tissues and is also upregulated in many types of tumors associated with enhanced aerobic glycolysis (the Warburg effect). Accumulating evidence indicates that HK2 plays an important role not only in glycolysis but also in cell survival. Although there is increasing recognition that cellular metabolism and cell survival are closely related, the molecular link between metabolism and autophagic pathways has not been fully elucidated. We recently discovered that HK2 facilitates autophagy in response to glucose deprivation (HK substrate deprivation) to protect cardiomyocytes, and suggest that HK2 functions as a molecular switch from glycolysis to autophagy to ensure cellular energy homeostasis under starvation conditions.
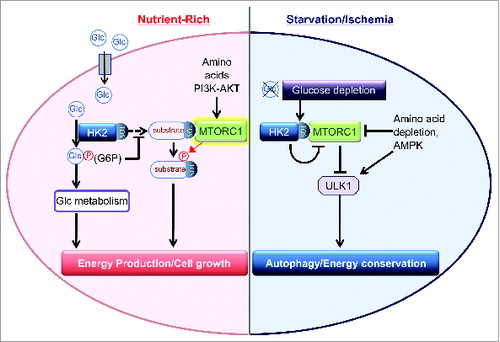
In the face of limited energy availability, cells initiate a process of catabolic self-digestion termed macroautophagy (hereafter referred to as autophagy), supplying energy substrates. Under nutrient-rich conditions, the mechanistic target of rapamycin complex 1 (MTORC1) supports cellular growth and serves as a brake on autophagy through inhibition of ULK1. When cellular energy is low, however, AMPK is activated and it suppresses the activity of MTORC1 while activating ULK1, thus inducing autophagy. Depletion of amino acids also inhibits MTORC1 activity. Although glucose deprivation leads to inhibition of MTORC1 activity, it is unclear whether glucose depletion directly regulates MTORC1 activity.
Examining the protective role of HK2, we found that 2-deoxy-D-glucose (2-DG), an analog of glucose that is phosphorylated by HKs but not metabolized further and used as a HK inhibitor, decreases autophagy development and augments cardiomyocyte death induced by glucose deprivation. This observation implied that HK2 acts to stimulate autophagy in the absence of its substrate and this was further supported by our observations that autophagy induced by glucose deprivation is attenuated by HK2 knockdown but potentiated by its overexpression. In contrast, knockdown of HK1, which is ubiquitously expressed, fails to affect the progression of autophagy, suggesting a specific role of HK2.
The autophagic effect of HK2 is not mediated by regulation of AMPK because the activity of AMPK is unaffected by manipulation of HK2 expression in the presence or absence of glucose. Instead, a series of experiments demonstrated that HK2 directly inhibits MTORC1 activity in response to glucose withdrawal. The autophagic effect of HK2 is independent of its catalytic activity, suggesting a previously unrecognized scaffold function of HK2. We found that HK2 binds to MTORC1 to inhibit its function and this binding is increased under glucose deprivation. A TOS motif (MTOR signaling motif) present in MTORC1 substrates is critical for their binding to RPTOR/raptor, the defining component of MTORC1, and subsequent MTOR-mediated phosphorylation. HK2, but not HK1, contains a TOS motif at positions 199–203 (FDIDI) that is conserved in mouse, rat, and human and we demonstrated that HK2 interacts with MTORC1 via binding to RPTOR through its TOS motif, functioning as a decoy substrate.
What triggers the switch between the glycolytic and autophagic effects of HK2? We found that kinase-dead HK2 expression, but not wild-type HK2, in endogenous HK2-depleted cells induces autophagy in the presence of glucose, suggesting that the autophagic effect of HK2 is suppressed by its catalytic activity. We further observed that, in contrast to 2-DG, 5-thio-glucose, another glucose analog that binds to HKs but cannot be phosphorylated, does not inhibit autophagy induced by glucose deprivation. Thus, phosphorylation of substrate, but not binding, is the key event responsible for suppressing the autophagic effect of HK2 in the presence of glucose. These observations lead us to conclude that G6P generated by HK2 catalytic activity inhibits HK2 binding to MTORC1 and that HK2 serves as a sensor to changes in cellular glucose levels to control the balance between cellular growth mediated by glycolysis and MTORC1, and preservation of energy status mediated by autophagy ().
Figure 1. HK2 detects glucose depletion to bind and inhibit MTORC1, enhancing autophagy. In the presence of glucose, the binding is inhibited by glucose 6-phosphate (G6P) generated by HK2 catalytic activity. Glc, glucose.
It remains to be elucidated how G6P inhibits HK2 binding to MTORC1. We speculate that G6P changes the intracellular localization of HK2, regulating binding of HK2 to MTORC1. A fraction of cellular HK2 is associated with mitochondria via a mitochondrial-binding motif at the N terminus, providing facilitation of glycolysis as well as mitochondrial protection against mitochondrial death pathways. G6P accumulation decreases the mitochondrial distribution of HK2. Our results using a mitochondrial binding-deficient mutant HK2, however, suggest that mitochondria are not the target site for the HK2-MTORC1 interaction. It would be of interest to determine if HK2 translocates to the lysosome, a site of MTORC1 activation, to bind and inhibit MTORC1. Additional studies will be required to determine the precise localization of the interaction.
It is intriguing that G6P suppresses the ability of HK2 to bind to MTORC1 and to facilitate autophagy. Importantly, G6P inhibits the activity of HKs providing a negative feedback regulation. As mentioned above, the binding to mitochondria is also subjected to G6P-dependent feedback inhibition. Thus, accumulation of G6P leads to negative regulation of HK2 kinase activity, mitochondrial distribution, and autophagic effect. Furthermore, G6P serves as a precursor not only for glycolysis, but also for several metabolic pathways: the pentose phosphate pathway, glycogenesis, and hexosamine biosynthetic pathway. Therefore, HK2 detects glucose availability through G6P-mediated regulation to serve as a critical nexus of integration of metabolic status, ensuring cellular energy homeostasis and survival. This signaling axis may have implications for the treatment of ischemic heart disease. In addition, HK2 expression undergoes dynamic changes in various diseases. As mentioned earlier, the expression of HK2 is increased in many tumors, whereas it is remarkably decreased in type-I diabetes. For example, the expression of the HK2 gene is nearly silent in hepatocytes, but it is highly induced in hepatocellular carcinoma. Interestingly, we observed that overexpression of WT HK2, but not the F199A TOS motif mutant, increases autophagy induced by glucose deprivation in a hepatocyte cell line. The switch in isoform expression to HK2 also occurs in activated T cells. Therefore, the integrative role of the glucose sensing mechanism of HK2 in energy balance may play an important role not only in heart disease but also in other diseases including cancer, diabetes, and inflammation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
SM is supported by NIH (HL097037) and AHA Grant (15GRNT22970009).