
Abstract
Antigen-presenting cells survey their environment and present captured antigens bound to major histocompatibility complex (MHC) molecules. Formation of MHC-antigen complexes occurs in specialized compartments where multiple protein trafficking routes, still incompletely understood, converge. Autophagy is a route that enables the presentation of cytosolic antigen by MHC class II molecules. Some reports also implicate autophagy in the presentation of extracellular, endocytosed antigen by MHC class I molecules, a pathway termed “cross-presentation.” The role of autophagy in cross-presentation is controversial. This may be due to studies using different types of antigen presenting cells for which the use of autophagy is not well defined. Here we report that active use of autophagy is evident only in DC subtypes specialized in cross-presentation. However, the contribution of autophagy to cross-presentation varied depending on the form of antigen: it was negligible in the case of cell-associated antigen or antigen delivered via receptor-mediated endocytosis, but more prominent when the antigen was a soluble protein. These findings highlight the differential use of autophagy and its machinery by primary cells equipped with specific immune function, and prompt careful reassessment of the participation of this endocytic pathway in antigen cross-presentation.
Abbreviations:
- 3-MA, 3-methyladenine
- Atg7-DC CKO, Atg7 DC conditional knockout
- BafA, bafilomycin A1
- CD, cluster of differentiation
- CTL, cytotoxic T lymphocyte
- DC, dendritic cell
- DALIS, dendritic cell aggresome-like inducible structures
- green fluorescent protein, GFP
- IFC imaging flow cytometry
- LAP, LC3 associated phagocytosis
- LC3B, microtubule-associated protein 1 light chain 3 β
- MHC II, major histocompatibility complex class II
- MHC I, major histocompatibility complex class I
- OVA, ovalbumin
- OT-I, OVA-specific CD8+ T cell
- OT-II, OVA-specific CD4+ T cell; SIM, structured illumination microscopy.
Introduction
Understanding of macroautophagy (referred herein as autophagy) has evolved from recognizing its function in the survival of starved cells to the realization that autophagy is a pathway that can be co-opted for specialized cell functions.Citation1 One of the major pathways in which a novel role for autophagy has been described is antigen presentation by major histocompatibility complex class II molecules (MHC II).Citation2 This activity involves the degradation of foreign proteins (antigens) in endo-lysosomal compartments, binding of the resulting peptides to MHC II, and delivery of the MHC II-peptide complexes to the cell surface for presentation to helper T lymphocytes.Citation3 Material present in endosomal compartments including plasma membrane proteins, components of the endosomal route and extracellular pathogens that are captured by endocytosis is thus readily presented via MHC II. Autophagy enables cells to expand the repertoire of antigens presented via this pathway by delivering cytosolic material to endo-lysosomal compartments, where it can be used as a source of MHC II peptide ligands.Citation4-6
Another route for presentation of endocytosed antigens, in which autophagy has been suggested to participate, is cross-presentation. This is a pathway whereby antigens captured from the extracellular environment are trafficked to the cytosol for degradation, chiefly by the proteasome. Resulting peptides are transported to the endoplasmic reticulum (ER) for binding to major histocompatibility complex class I molecules (MHC I). The MHC I-peptide complexes are delivered to the plasma membrane for activation of cytotoxic T lymphocytes.Citation3 Cross-presentation is carried out primarily, if not only, by specific types of dendritic cells (DC). DC are highly specialized cells of the immune system that are dedicated to surveillance of the extracellular environment, the capture of pathogens and the initiation of T cell immunity. Cross-presentation plays a critical role in immunity, as it enables DC to elicit cytotoxic T cell immunity against viruses and tumors. Consequently, it is a pathway targeted by synthetic vaccines to induce protective immunity.Citation7 How autophagy, a pathway that transports cytosolic material to the endocytic route, participates in MHC I cross-presentation, which requires antigen transport in the opposite direction (from endosomes to cytosol), is unknown. Indeed, the very notion that autophagy partakes in cross-presentation is controversial. While it has been suggested by some studies to play a role,Citation8-10 others have concluded that cross-presentation is autophagy-independent.Citation11 Resolving this is important given the therapeutic potential of targeting cross-presentation with drugs that manipulate autophagy.
Primary mouse and human DC represent a heterogeneous network comprised of defined subtypes with specialized functions.Citation12 Notably, not all of these DC subtypes are equally capable of cross-presentation. It is reasonable to expect that if autophagy is required for cross-presentation, it may operate in DC capable of cross-presentation differently to DC that are not able to cross-present. This has not been investigated. Most studies to date have examined autophagy in monocyte-derived DC generated in culture from bone marrow precursors. This is a model that does not resemble the complex functional specializations exhibited by their natural counterparts produced in vivo.Citation13 Furthermore, cross-presentation by monocyte-derived DC does not involve the canonical endosome-to-cytosol route required for cross-presentation of viral and tumor antigens in vivo.Citation14 Therefore, the contribution of autophagy to specialized DC biology remains largely unknown. Here, we have investigated the use of autophagy by cross-presenting and non-cross-presenting primary DC contained in mouse and human lymphoid organs, and assessed the role of this endosomal trafficking pathway in cross-presentation.
Results
Enhanced autophagy in primary mouse DC with cross-presentation capacity
While DC are suggested to use autophagy constitutively,Citation4 they do not undergo significant cell death in its absence,Citation11 unlike other primary cell types (for example T cells).Citation15 This suggests that DC are less reliant on autophagy for intracellular homeostasis and consequently represent an ideal primary cell type in which to study mechanisms of autophagy-mediated intracellular trafficking, and the role of this pathway in antigen cross-presentation.
First, we tested the hypothesis that if autophagy contributes to cross-presentation, it should be more prominent in mouse primary DC specialized in this function. Mouse CD8+ DC are the dominant cross-presenters in vivo,Citation16,17 so we compared their use of autophagy to that in CD8− DC, that are not capable of cross-presentation. Both populations were purified from spleens. In the steady state, all splenic DC are in a so-called “immature” state characterized by high antigen uptake and poor antigen presentation capacity.Citation18 This is the phenotype of freshly purified cells. Culture overnight causes activation of isolated DC in vitro.Citation19 Activation mimics the DC encounter with pathogen and triggers dramatic changes that endow activated DC with high capacity to present antigens to, and stimulate, T cells. We compared expression of the common autophagy marker Map1lc3b/Lc3b (microtubule-associated protein 1 light chain 3 β) by 2 DC subsets. CD8+ DC clearly upregulated Lc3b transcription following activation, but that was not apparent for CD8− DC (). To directly measure autophagosomes, the 2 DC populations were isolated from the spleen of green fluorescent protein (GFP)-LC3 transgenic mice.Citation20 GFP-LC3+ vesicles were detected in immature and activated CD8+ and CD8− DC by imaging flow cytometry (IFC). IFC is a highly useful methodology for autophagy measurement given that it combines flow cytometry for cell type discrimination and high throughput analysis with high-resolution fluorescent microscopy for visualization of each individual cell. IFC analysis showed very few GFP-LC3+ vesicles in resting CD8+ or CD8− DC. In contrast, activation elicited a significant increase in the proportion of cells containing GFP-LC3+ vesicles in the 2 subsets. The frequency of activated CD8+ DC with GFP-LC3+ vesicles was significantly higher relative to activated CD8− DC (). To confirm IFC data, we examined the cells by confocal microscopy. Again activated CD8+ DC possessed significantly more GFP-LC3 vesicles per cell, than activated CD8− DC, where very few were detected (). These data suggested activated CD8+ DC were undergoing enhanced autophagy, however, while autophagosome accumulation can be consistent with increased autophagy, it can also indicate a failure of autophagosomal turnover due to an impaired fusion of autophagosomes with lysosomes. To distinguish between these scenarios, we analyzed autophagy-mediated degradation of endogenous LC3 that provides a measure of how much autophagy-mediated degradation a specific cell type is undertaking. To do this, we monitored the accumulation of LC3-II in the presence of lysosomal protease inhibitor bafilomycin A1 (BafA). Activated CD8+ DC underwent significantly more autophagic flux relative to CD8− DC as evidenced by their significantly increased LC3-II accumulation in the presence of BafA (). Therefore, cross-presenting CD8+ DC exhibit a striking increase in autophagy following activation, with accumulated GFP-LC3+ vesicles and increased autophagic flux, relative to CD8− DC.
Figure 1. For figure legend, see page 909.Figure 1 (See previous page). Enhanced autophagy in primary mouse DC with cross-presentation capacity. (A) Splenic CD8+ DC and CD8− DC were purified by flow cytometry. Cells were examined directly ex vivo (immature) or following activation by culture at 37°C overnight (activated). RNA was isolated and quantitative real-time PCR performed to measure Lc3b transcription, relative to the housekeeping gene Tbp (TATA box binding protein). Data are mean ± 1 SEM, pooled from 3 independent experiments, *P ≤ 0.05, one-way ANOVA with Tukey multiple comparison test. (B) Spleen GFP-LC3 DC were cultured at 4°C (immature) or 37°C overnight (activated) and CD8+ DC and CD8− DC analyzed by imaging flow cytometry. Data were pooled from 2 or 3 independent experiments, mean ± 1 SEM, *P ≤ 0.05, one-way ANOVA with Tukey multiple comparison test. (C) Spleen GFP-LC3 CD8+ DC and CD8− DC were cultured overnight (activated). Cells were counterstained with DAPI and imaged by confocal microscopy; scale bar: 10 μm, arrows indicate GFP-LC3 vesicles. Data are from 3 or 4 independent experiments from a total of 126 immature CD8+ DC, 129 immature CD8− DC, 122 activated CD8+ DC, 120 activated CD8 DC, mean ± 1 SEM **P ≤ 0.001, one-way ANOVA with Tukey multiple comparison test. (D) Spleen CD8+ DC and CD8− DC were isolated and cultured overnight (activated). Activated DC were incubated in the presence or absence of 100 nM BafA for 2 h. Cell lysates were subjected to SDS-PAGE and immunoblotted for LC3. Data were pooled from 4 independent experiments, mean ± 1 SEM *P ≤ 0.05, Mann Whitney t test. (E) GFP-LC3 CD8+ DC or CD4+ DC were pulsed with zymosan-Alexa 594 and cultured overnight. Cells were imaged by confocal microscopy; scale bar: 10 μm.
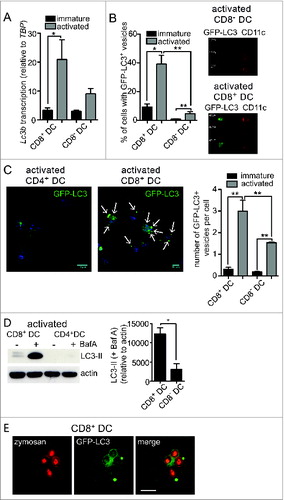
While these data clearly demonstrate cross-presenting DC utilize autophagy, of interest was whether compartments utilized for cross-presentation and autophagy machinery directly intersect. LC3-associated phagocytosis (LAP) involves the recruitment of LC3 to phagosomes that contain captured exogenous antigen.Citation21,22 We examined whether LC3 was recruited to phagosomes in cross-presenting DC by feeding fluorescently labeled yeast (zymosan) particles to GFP-LC3+ CD8+ DC. GFP-LC3 was indeed recruited to phagosomes containing exogenous zymosan particles, indicative of LAP (). Therefore, autophagy is upregulated in activated cross-presenting CD8+ DC and its machinery intersects with phagosomes containing exogenous antigens.
Increased ubiquitin and SQSTM1/p62 aggregates following activation of mouse and human cross-presenting DC
Given the presence of an active autophagy pathway in cross-presenting CD8+ DC, we next looked for evidence of autophagic substrates. Ubiquitinated protein aggregates are known to be targeted by autophagyCitation23,24 and are of particular interest given that dendritic cell aggresome-like inducible structures (DALIS) have been previously detected in bone marrow-derived and epidermal sheet DC following activation.Citation25 DALIS function as a reservoir for MHC I antigens and are putative substrates of autophagy.Citation26 Here, we detected ubiquitinated aggregates in the primary DC subsets using an antibody that recognizes poly- and mono-ubiquitinated proteins, but not free ubiquitin. Few ubiquitinated aggregates were present in either the immature DC subsets, or activated CD8− DC as measured by IFC. In contrast, a high frequency of activated CD8+ DC possessed ubiquitinated aggregates (). IFC data were verified by structured illumination microscopy (SIM), where examination of activated CD8+ DC revealed large ubiquitin+ vesicular aggregates that were not present in activated CD8− DC (). Therefore, primary CD8+ DC, but not CD8− DC, accumulate ubiquitinated aggregates, also termed DALIS, following activation.
Figure 2. Increased ubiquitin and SQSTM1 receptor aggregates following activation of mouse and human cross-presenting DC. (A) Ubiquitinated proteins were detected in freshly isolated (immature) spleen CD8+ DC and CD8− DC, or following overnight activation (activated) by imaging flow cytometry. Data were pooled from 2 or 3 independent experiments, mean ± 1 SEM, *P ≤ 0.05, one-way ANOVA with Tukey multiple comparison test. (B) Splenic CD8+ DC and CD8− DC were activated by culture overnight. DC were stained with an anti-mono and polyubiquitin specific antibody, nuclei were counterstained with DAPI and cells imaged by structured illumination microscopy (SIM). Scale bar: 5 μm. Number of ubiquitinated aggregates per cell was scored from SIM images for 19 activated CD8+ DC and 17 activated CD8− DC, mean ± 1 SEM, ***P ≤ 0.001, Mann Whitney t test. (C) Spleen wild-type or GFP-LC3 CD8+ DC were purified by flow cytometry and cultured overnight (activated). Cells were stained for ubiquitin or SQSTM1, counterstained with DAPI and imaged by confocal microscopy; scale bar: 10 μm. (D) Spleen CD8+ DC and CD8− DC were cultured at 4°C (immature) or 37°C overnight (activated), stained for SQSTM1 and analyzed by imaging flow cytometry. Data were pooled from 2 or 3 independent experiments, mean ± 1 SEM, **P ≤ 0.01, one-way ANOVA with Tukey multiple comparison test. (E) Spleen CD8− DC and CD8+ DC were activated overnight and stained for SQSTM1 and nuclear dye DAPI and imaged by confocal microscopy; scale bar: 10 μm. Graph displays number of cells containing SQSTM1 aggregates. Data are from 96 CD8+ DC, 111 CD8− DC scored in 4 independent experiments, mean ± 1 SEM, ***P ≤ 0.001, one-way ANOVA with Tukey multiple comparison test. SQSTM1 aggregate size (μm2) measured from 291 aggregates in activated CD8− DCs and 232 aggregates in activated CD8+ DCs. (F) Splenic CD8+ DC and CD8− DC were purified by flow cytometry. Cells were examined directly ex vivo (immature) or following activation by culture at 37°C overnight (activated). RNA was isolated and quantitative real time PCR performed to measure Sqstm1 transcription, relative to the housekeeping gene Tbp. Data are mean ± 1 SEM, pooled from 3 independent experiments, *P ≤ 0.05, one-way ANOVA with Tukey multiple comparison test. (G) Spleen CD8+ DCs were purified by flow cytometry and activated by culture overnight. Cells were stained for SQSTM1 and examined by SIM; scale bar: 6 μm. (H) CD8+ DC, CD103+ DC and CD103− DC isolated from skin-draining lymph nodes were stained for SQSTM1, counterstained with DAPI and imaged by confocal microscopy; scale bar: 10 μm. Graph represents data from 27 CD8+ DC, 32 CD103+ DC and 26 CD103− DC, mean ± 1 SEM ***P ≤ 0.001. (I) CD1a+ and CD14+ human DC were analyzed immediately after isolation (immature) or following overnight culture (activated) for mono and polyubiquitinated proteins by confocal microscopy; scale bar: 10 μm. Graph displays proportion of CD1a+ and CD14+ DC containing ubiquitin aggregates for DC that were freshly isolated, culture overnight (act) or stimulated overnight in presence of 1 μg/ml Pam3-CSK4 (Pam3) (n > 350 cells). (J) Human CD1c+ and CD141+ DC were imaged for SQSTM1 aggregates by confocal microscopy; scale bar: 16 μm. Left graph: represents 59 CD1c+ DC and 33 CD141+ DC, mean ± 1 SEM *P ≤ 0.05, Mann Whitney t test. Right graph: human CD1c+ and CD141+ DC were pulsed with cytomegalovirus pp65 protein and MHC I cross-presentation of the pp65495–503 epitope measured by IFNG secretion by pp65495–503-specific CD8+ T cells. Data were pooled from 5 individual donors.
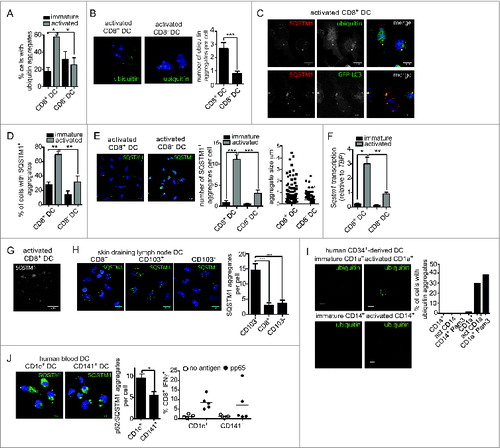
Ubiquitinated aggregates are recognized by SQSTM1 (sequestosome 1), an autophagy cargo receptor that also binds LC3.Citation27 SQSTM1 colocalized with both ubiquitinated aggregates and GFP-LC3 in activated cross-presenting DC (). Further analysis of SQSTM1 localization in immature and activated CD8+ and CD8− DC was performed by IFC. Less than 30% and 20% of the 2 immature cell types, respectively, contained SQSTM1 aggregates (). This was confirmed by confocal microscopy, where strikingly activation caused a large increase in the number of cells that contained SQSTM1 aggregates, together with an increase in aggregate size, in CD8+ DC (). Increased SQSTM1 aggregation was also accompanied by CD8+ DC upregulation of Sqstm1 transcription with activation, with these cells displaying significantly increased levels relative to activated CD8− DC (). SIM analysis of ubiquitin-SQSTM1 aggregates identified a vesicular staining pattern (), supporting previous observations of DALIS structure.Citation28
We extended our analysis to mouse primary DC that undergo activation naturally in vivo. To do this, we analyzed DC isolated from lymph nodes. Lymph nodes contain the same “resident” CD8+ and CD8− DC as the spleen, together with the 2 counterparts of these populations that have migrated from peripheral tissues via the afferent lymph.Citation12 The migrating equivalents of the “resident” CD8+ DC are distinguished by their high ITGAE/CD103 (integrin, α E, [antigen CD103, human mucosal lymphocyte antigen 1; α polypeptide]) expression (CD103+ DC), whereas the equivalent of the CD8− DC are CD103−.Citation29 Importantly, both of the lymph node immigrant subsets are activated upon entering the lymph node, most likely in response to stimuli encounter in the peripheral tissue.Citation30 This enables examination of autophagy components in cross-presenting (CD103+) and noncross-presenting (CD103−) DC activated without external intervention. CD103+ DC freshly isolated from skin-draining lymph nodes displayed many more SQSTM1 aggregates per cell than CD103− DC or CD8+ DC isolated from the same lymph nodes (). This indicates that formation of SQSTM1 aggregates is elicited in response to DC activation stimuli in vivo, not only as a result of in vitro culture, and only in DC subtypes capable of cross-presentation (CD8+ DC and CD103+ DC).
Human DC display a similar complexity to the mouse DC network. An array of different human DC subtypes have recently been identified that can be directly isolated from blood and lymphoid tissue.Citation12 These subsets are only beginning to be characterized and have not been studied in detail. In terms of cross-presentation, it is not clear that human DC subtypes display the same level of specialization observed among mouse DC. Effective cross-presentation can be achieved by numerous subtypes depending on their activation status, their origin, and the type of antigen involved. For example, human CD1C/BDCA1 (CD1c molecule) (CD1c+ DC) can exert superior,Citation31,32 inferiorCitation31,33-35 or equivalentCitation36 cross-presentation capacity compared to THBD/BDCA3/CD141+ (thrombomodulin) (CD141+ DC), depending on the experimental setting. Notably, a role for autophagy in the specific functions of specialized human DC subsets has not been investigated. First, we examined DC where a differential in cross-presentation capacity has previously been described. CD1A-expressing DC (CD1a+ DC) and CD14+ DC are equivalent to human skin DC and are grown from human CD34+ progenitors. CD1a+ DC are the dominant cross-presenting subset.Citation36 Examination of CD1a+ and CD14+ DC by confocal microscopy revealed cross-presenting CD1a+ DC, but not CD14+ DC, displayed ubiquitinated aggregates. Again, similar to mouse cross-presenting DC, significant numbers of ubiquitinated aggregates were detected only when the cells were activated by overnight culture or following culture with Pam3-CSK4, a bacterial lipopeptide (). Second, we examined SQSTM1 receptor localization in CD1c+ and CD141+ DC isolated from human blood and activated by in vitro culture overnight.Citation37 Activated CD1c+ DC and CD141+ DC both displayed SQSTM1 aggregates, but these were more abundant in CD1c+ DC (). This correlated with our previous observations that CD1c+ DC are slightly more efficient than CD1c+ DC at cross-presenting soluble human cytomegalovirus pp65 (glycoprotein 64 or UL83) ().Citation31
In summary, mouse and human DC types adept at cross-presentation show enhanced cytosolic ubiquitinated substrates of the autophagy pathway, in line with the observation that autophagosomes and autophagic flux are also more prominent in these DC.
Autophagy-deficient DC are less efficient at cross-presenting soluble OVA, but cross-present cell-associated and DEC-205 targeted antigen
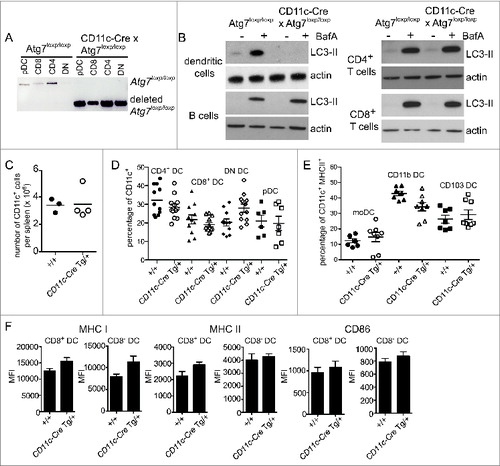
Next we assessed the role of autophagy in cross-presentation by CD8+ DC. We used DC isolated from the spleens of ITGAX/CD11C (integrin, α X [complement component 3 receptor 4 subunit]) (CD11c)-Cre x Atg7loxp/loxp mice (Henceforth described as Atg7-DC conditional knockout or Atg7-DC CKO).Citation38,39 These mice express Cre-recombinase under the control of the Itgax/CD11c promoterCitation39 and therefore delete the loxp-flanked Atg7 gene in CD11c+ DC. Deletion of Atg7 was confirmed in all splenic DC populations: CD8+ DC, CD8− DC (further segregated into CD4+ CD8 and CD8− CD4− or double-negative, DN), and plasmacytoid DC (pDC) (). Analysis of autophagic flux showed that, as expected, Atg7 deletion in primary DC, but not in B cells, CD4+ T cells or CD8+ T cells, impaired their ability to degrade LC3-II (). DC-specific Atg7 deletion did not impair the recovery of total CD11c+ DC from the spleen (), or alter the proportion of splenic DC subsets (). This was also the case for monocyte-derived DC, CD11b+ DC expressing ITGAM/CD11B (integrin, α M [complement component 3 receptor 3 subunit]) and CD103+ DC isolated directly from lung parenchyma (). Splenic DC showed no major differences in their expression levels of MHC I, MHC II or the activation marker CD86 in the absence of autophagy (). Atg7-deficient DC displayed no significant alterations in the proportions CD8+ and CD8− DC expressing either ubiquitin or SQSTM1 aggregates (Fig. S1). In summary, autophagy is not required for steady-state DC homeostasis.
Figure 3. Autophagy is not required for DC development or homeostasis. (A) Atg7-WT or Atg7-DC CKO CD8+ DC, CD4+ DC, CD8− CD4− double-negative (DN) DC and plasmacytoid DC (pDC) were isolated from spleen. Genomic DNA was isolated and PCR performed for the intact loxp-flanked Atg7 allele and the deleted Atg7 allele. (B) Purified Atg7-WT or Atg7-DC CKO DC, B cells, CD4+ T cells and CD8+ T cells were cultured overnight and incubated in the presence or absence of BafA for 2 h. Cell lysates were subjected to SDS-PAGE and immunoblotted for LC3 and actin. (C) CD11c+ DC were purified from Atg7-WT or Atg7-DC CKO spleens. Bar indicates mean. Data pooled from 3 independent experiments. (D) Proportions of CD11c CD4+ DC (CD4 DC), CD11c+ CD8+ DC (CD8 DC), CD11c+ CD4− CD8− DC (DN DC) and CD11c+ CD45RA+ plasmacytoid DC (pDC) present in Atg7-WT or Atg7-DC CKO spleens. Bar indicates mean, each circle represents an individual mouse. (E) Proportions of monocyte-derived DC (moDC), CD11c+ CD11b+ DC (CD11b DC) and CD11c+ CD103+ DC (CD103 DC) present in Atg7-WT or Atg7-DC CKO lung parenchyma. Gating strategies for respiratory tract DC are described in Materials and Methods. Bar indicates mean, each circle represents an individual mouse (F) Expression levels of MHC I, MHC II, and CD86 for immature CD8+ DC and immature CD8− DC isolated from Atg7-WT or Atg7-DC CKO spleens. Data are mean ± 1 SEM, n = 3 or 4 mice per group.
Next we assessed the contribution of autophagy to MHC I cross-presentation of antigen. Cross-presentation capacity of Atg7-deficient DC was determined following in vivo immunization.Citation40 Atg7-DC CKO or Atg7loxp/loxp wild-type mice (henceforth described as Atg7-WT) were intravenously immunized with ovalbumin (OVA)-coated irradiated H-2bm1 splenocytes (termed OVA-coated spleen) in the presence of lipopolysaccharide. Atg7-WT mice generated strong cytotoxic T lymphocytes (CTL) as evidenced by the elimination of carboxyfluorescein succinimidyl ester (fluorescent cell staining dye) CFSEhigh OVA257-264 (MHC I OVA epitope) -pulsed target cells, relative to unpulsed CFSElow control cells. In contrast, Atg7 DC CKO mice displayed an impaired ability to generate efficient CTL killers (Fig. S2A). At face value, this suggested autophagy is required for efficient cross-presentation of cell-associated antigen in vivo. To further address this, we examined cross-presentation in vivo following adoptive transfer of Atg7 wild-type OT-I T cells. No impairment in OT-I division in response to cell-associated antigen was detected (Fig. S2B). Therefore, this suggests the impaired CTL generation following immunization of Atg7-DC CKO mice most likely results from deletion of Atg7 in CD8+ T cells, that can express CD11c when activated,Citation41 rather than a true requirement for autophagy in DC-mediated cross-presentation of cell-associated antigen.
To more directly assess the role of autophagy in cross-presentation, we shifted to antigen presentation assays with directly isolated primary cross-presenting CD8+ DC in vitro. Initial experiments examined wild-type CD8+ DC treated with the autophagy inhibitor 3-methyladenine (3-MA). Treatment with 3-MA inhibited the cross-presentation of soluble OVA antigen to OT-I T cells. Notably, 3-MA treatment likely caused CD8+ DC toxicity as evidenced by impaired presentation of OVA257–264 peptide in the presence of this drug (Fig. S3). Given this, and the high likelihood for off-target effects of 3-MA on DC endocytosis and antigen uptake, the use of this inhibitor in the context of primary DC analysis is limited. Subsequent antigen presentation assays were performed with CD8+ DC isolated directly from spleens of Atg7-DC CKO mice.
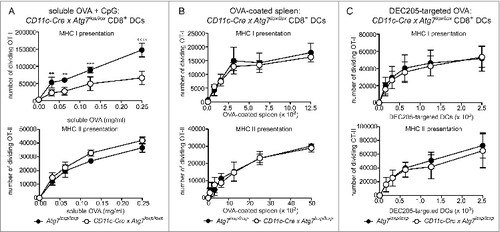
Atg7-sufficient or deficient CD8+ DC were purified and assessed for their ability to cross-present OVA in 3 different forms: soluble, cell-associated and targeted to the DC receptor DEC-205 with a monoclonal antibody. Presentation of OVA antigen by MHC I was measured by coculture with wild-type OT-I CD8+ OVA-specific T cells. Transgenic T cells were labeled with Cell Trace Violet dye to track their proliferation. To control for antigen uptake, we also examined presentation of antigen by MHC II to OT-II CD4+ OVA-specific T cells. First, purified CD8+ DC from Atg7-WT or Atg7-DC CKO mice were cultured in the presence of soluble OVA antigen, together with TLR9 agonist CpG. Atg7-deficient DC exhibited impaired MHC I cross-presentation of soluble OVA relative to wild-type DC as evidenced by the reduced ability of these DC to stimulate OVA-specific OT-I T cell division. MHC II antigen presentation of soluble OVA was similar for wild-type and Atg7-deficient DC (). This suggests the efficiency of cross-presentation of soluble antigen is dependent on an intact autophagy pathway. In contrast to soluble OVA antigen, cross-presentation of cell-associated OVA (OVA-coated spleen) was not altered by the absence of autophagy. Atg7-deficient CD8+ DC efficiently presented cell associated OVA by MHC I or MHC II, similar to wild-type DC (). A similar response was also observed for OVA-coated spleen in the presence of CpG (Fig. S4). This confirmed outcomes of in vivo antigen presentation assays using OT-I to monitor cross-presentation of OVA-coated spleen in immunized Atg7-DC CKO mice (Fig. S2B). Next, we examined cross-presentation of OVA following its delivery via targeting antigen to surface DEC205, a C-type lectin receptor. Delivering antigen by targeting DC surface receptors is an emerging mode of antigen delivery that is of intense interest in settings of vaccination.Citation42 More importantly, delivering antigen via DEC205 enabled us to analyze autophagy and antigen presentation outcomes in CD8+ DC following activation. The ability to examine activated DC was important, given that we detected increased autophagy in CD8+ DC under these conditions. Purified CD8+ DC from Atg7-WT or Atg7-DC CKO mice was targeted with a biotinylated antibody specific for DEC205, followed by an anti-biotin antibody conjugated to OVA. Notably, DEC205 expression did not differ between wild-type and Atg7-deficient CD8+ DC (Fig. S5). DEC205-targeted antigen was cross-presented independently of autophagy, with similar OT-I division elicited by wild-type and Atg7 -deficient CD8+ DC. This was also the case for MHC II presentation of DEC205-targeted OVA (). Finally, OVA257–264-pulsed wild-type of Atg7-deficient CD8+ (and CD8− DC) displayed similar capacity to stimulate OT-I in the presence or absence of CpG (Fig. S6). In summary, autophagy does not play a major role in the CD8+ DC cross-presentation and/or MHC II presentation of exogenous cell-associated antigen and/or in antigen delivered to activated CD8+ DC via DEC205 receptor-mediated endocytosis. In contrast, autophagy participates in the efficient cross-presentation of exogenous soluble antigen by CD8+ DC.
Figure 4. Autophagy is required for efficient cross-presentation of soluble antigen, but not cell-associated antigen or antigen delivered via DEC205. Purified Atg7-WT or Atg7-DC CKO splenic CD8+ DC were incubated with (A) OVA protein (+ 0.5 μM CpG) for 45 min or (B) irradiated H-2bm1 splenocytes coated with OVA protein (OVA-coated spleen). (C) For DEC205 targeting, purified CD8+ DC were activated by culture at 37°C overnight. Activated CD8+ DC were incubated with biotinylated anti-DEC205 antibodies followed by anti-biotin antibodies conjugated to OVA. Presentation of OVA by MHC I (cross-presentation) was assessed by monitoring the division of Cell Trace Violet-labeled OT-I T cells, while presentation of OVA by MHC II was assessed by monitoring the division of Cell Trace Violet-labeled OT-II T cells. In all cases, experiment was performed in triplicate (mean ± 1 SD) with data normalized and pooled from 2 independent experiments. Data were analyzed by 2-way ANOVA followed by Bonferroni multiple comparison test ****P ≤ 0.0001, ***P ≤ 0.001, **P ≤ 0.01.
While CD8− DC are very poor at cross-presenting either cell-associated or soluble antigen,Citation16,17 these cells can present DEC205-targeted antigen in the context of MHC I, albeit with lower efficiency than CD8+ DC.Citation43 We examined whether autophagy participates in the cross-presentation and/or MHC II presentation of DEC205-targeted antigen by CD8− DC. Similar to activated CD8+ DC, DEC205-targeted antigen was cross-presented, and presented by MHC II, with the same efficiency by wild-type and Atg7-deficient activated CD8− DC (Fig. S7). Therefore, autophagy does not play a major role in the presentation of antigen delivered to activated CD8 DC via targeting antigen for delivery by DEC205-mediated endocytosis.
Discussion
DC survive in the absence of functional autophagy, but use this pathway to control intracellular antigen trafficking.Citation2 Here, we have examined the contribution of autophagy to cross-presentation, a pathway that traffics antigen to the cytosol for presentation by MHC I. We delineated the association between autophagy and DC cross-presentation by comparing primary DC subsets with different cross-presentation function. Previous studies dissecting autophagy in cross-presentation have provided conflicting results. Our study aimed to shed light on this controversy being the first to examine this pathway in the primary cells responsible for cross-presentation activity in vivo. Autophagy was evident only in cross-presenting DC. Despite this, autophagy was not strictly required for cross-presentation of antigen to CD8+ T cells, with the requirement for this pathway being dependent on the form of the antigen acquired.
Surprisingly, differential use of autophagy by cross-presenting DC, relative to non-cross-presenting DC, does not manifest as a critical role for autophagy in the major function of this cell type, that is, the presentation of exogenous antigen by MHC I (cross-presentation). We conducted a comprehensive analysis of cross-presentation using purified Atg7-deficient primary CD8+ DC, the first assays to be undertaken with primary cross-presenting cells. Autophagy was dispensable for CD8+ DC cross-presentation of cell-associated antigen, in addition to antigen delivered via the surface C-type lectin DEC205. Autophagy-independent cross-presentation of cell-associated antigen supports findings by Lee et al.Citation11 in assays of bulk primary DC populations. In contrast, Ravindran et al.Citation10 report a significant role for autophagy in the cross-presentation of cell-associated antigen using cultures with bone marrow-derived DC. The discrepancy in these outcomes most likely arise from the DC populations studied. We have previously reported cultured bone marrow-derived DC use alternative pathways of cross-presentation to primary CD8+ DC.Citation14 Our findings stress the importance of using primary DC, if results are to be extrapolated to settings of vaccination in vivo.
Soluble antigen was less efficiently cross-presented by Atg7-deficient DC. This is specific to cross-presentation, as soluble OVA presentation by MHC II was not impaired. Therefore, autophagy contributes to efficient cross-presentation of soluble antigen that is acquired by macropinocytosis or pinocytosis, but is not required following antigen uptake by phagocytosis or receptor-mediated endocytosis. How would autophagy, a pathway designed to degrade cytosolic material, contribute to effective cross-presentation of exogenous soluble antigen? One possibility is that autophagy contributes to the creation of a compartment where antigen is preserved, rather than degraded. Prolonged antigen storage favors MHC I cross-presentation as it facilitates antigen transfer to the cytosol, rather than its rapid degradation in the lysosomes.Citation44,45 Indeed, LAP, the process by which LC3 is recruited to phagosomes, has recently been implicated in promoting antigen stability in DCCitation46 unlike in other cell types where it is proposed to enhance antigen degradation.Citation21,22,47 This autophagy-dependent mechanism could be of more importance for soluble antigen that is likely to be more rapidly degraded than cell-associated antigen.
In summary, our findings are the first to demonstrate the differential use of autophagy by primary DC specialized in distinct antigen presentation pathways. We show that autophagy is not a pathway that is active in all DC, but functions only in some subsets and under specific conditions. Despite this, we do not detect a critical role for autophagy in cross-presentation. Rather, autophagy contributes to the efficiency of this pathway under specific conditions. Furthermore, autophagy does not participate in MHC II responses to exogenous antigen. Therefore, participation of autophagy in antigen presentation trafficking, and in particular in cross-presentation, may be more limited than previously considered.
Materials and Methods
Mice
C57BL/6, GFP-LC3 (provided by Noboru Mizushima, RIKEN BioResource Center),Citation20 CD11c-Cre,Citation39 Atg7loxp/loxp (provided by Masaaki Komatsu, Tokyo Metropolitan Institute of Medical Science),Citation38 OT-I, OT-II and bm1 mice were bred under specific pathogen-free conditions at the Walter and Eliza Hall Institute or Bio21 Institute Animal Facility. All experiments were conducted in accordance with guidelines provided by National Health and Medical Research Council of Australia. Experimental procedures were approved by the Animal Ethics Committee, Melbourne Health Research Directorate, and the University of Melbourne.
Murine dendritic cells
DC were isolated from spleens as previously described.Citation48 In brief, spleens were digested with DNase (Roche Applied Science, 10104159001) and type 3 collagenase (Worthington Biochemicals, CLS3) and enriched for light density cells by centrifugation in 1.077 g/cm3 Nycodenz (Progen Biotechnik, 1002424). DC were isolated after depletion using antibodies (generated in house) against CD3 (clone KT3–1.1), THY1 (clone T24/31.7), LY76/Ter119, LY6G (clone RB68C5) and PTPRC/CD45R (clone RA36B2), followed by incubation with anti-rat immunoglobulin-coupled magnetic beads (Qiagen Biomags, 310107). For isolation of pDC, anti-PTPRC antibody was omitted from the depletion cocktail. DC isolation yielded preparations with approximately 70% to 85% CD11c+ purity. When necessary, cells were then sorted to purity by flow cytometry. DC were activated by culture overnight in media supplemented with 10 ng/ml granulocyte macrophage colony stimulating factor (GM-CSF, Peprotech, 315–03).
For respiratory DC analysis, lung tissue was digested with DNase I (Sigma-Aldrich, AMPD1) and 0.1% w/v type 3 collagenase. Erythrocytes were lysed (BD FACS Lysing Solution, BD Biosciences, 349202). Lung DC were phenotyped with antibodies specific for FCER1A/Fc epsilon receptor 1 α (eBioscience, clone MAR-1, 14–5898), CD64 (BD Biosciences, clone X54–5/7.1, 558455), ITGAM/CD11b (eBioscience, clone M1/70, 50–0112), ITGAX/CD11c (eBioscience, clone N418, 53–0114), MHC-II (BD Biosciences, clone M5/114.15.2, 53236), PTPRC/CD45 (eBioscience, clone 30-F11, 58–0451), Siglec-F (BD Biosciences, clone E50–2440, 55212) and ITGAE/CD103 (eBioscience, clone 2E7, 1031). Monocyte DC were defined as CD11c+ CD103+ MHC-II+ CD64+ FcϵRI+ Siglec F−, CD11b DC as CD11c+ CD11b+ CD103− MHC-II+ CD64− FcϵRI− Siglec F− and CD103 DC as CD11c+ CD11b− CD103+ MHC-II+ CD64− FcϵRI− Siglec F−.
Human dendritic cells
CD1a+ and CD14+ DC were generated from peripheral blood mononuclear cells that were isolated from healthy donor buffy coats obtained from Etablissement Français du Sang, France as previously described.Citation36 CD1c+ DC and CD141+ DC were isolated directly from human blood as previously described.Citation37 Human blood and leukapheresis products were obtained from healthy donors after approval by Mater Health Services Australia Human Research Ethics Committee. CD1a+ and CD14+ DC were stimulated with Pam3-CSK4 (Invivogen, tlrl-pms).
Quantitative real-time PCR
RNA was isolated from sorted cell populations (Qiagen, 74134). A total of 100 ng of RNA was used to generate cDNA (Qiagen, 205310). LightCycler 480 SYBR green I master mix (Roche, 04707516001) was used in quantitative PCR reactions on the Roche LightCycler 480 (University of Melbourne, Department of Microbology and Immunology). Map1lc3b/Lc3b and Sqstm1 mRNA expression was normalized to housekeeping gene Tata-binding protein (Tbp). Lc3b mRNA was identified by forward 5′-CGGAGCTTTGAACAAAGACTG-3′ and reverse TCTCTCACTCTCGTACACTTC primers. Sqstm1 mRNA was identified by forward 5′-CTACATTAAAGAGAAGAA
GGAGTGC-3′ and reverse 5′-GAAAGATGAGCTTGCTG-TGTAC-3′ primers. Tbp was identified by forward 5′-TCCGCAGTGCCCAGCATCACTAT-3′ and reverse 5′-GAGTCATGGCGCCCTGTGGGG-3′ primers.
Autophagic flux
Cells incubated for 2 h in the presence or absence of 100 nM BafA (Sigma Aldrich, B1793). Cells were lysed in 2% Triton X-100 lysis buffer (laboratory grade, Sigma Aldrich, X100), separated by SDS-PAGE and proteins transferred to PVDF membranes by iBlot (Invitrogen, IB4010). Membranes were probed for LC3 (Cell Signaling Technology, 3868) and actin (Sigma Aldrich, A2668).
Imaging flow cytometry
Cells were analyzed with an Amnis ImageStream imaging flow cytometer (Melbourne Materials Institute, University of Melbourne). Images were acquired for 5,000 cells, excluding clusters and out of focus cells. Imaging flow cytometry was performed at the Advanced Fluorescence Imaging Facility, University of Melbourne.
Flow cytometry
Cells were analyzed and sorted by flow cytometry using the following markers (antibody clones); ITGAX/CD11c (N418), CD8 (YTS169) or CD4 (GK1.5), CD45RA (14.8), B220 (RA36B2), CD3 (KT3–1.1), ITGAE/CD103 (2E7), MHC II (M5114), ITGAM/CD11b (M170), EPCAM/CD326 (G8.8), TCRVα2 (B20.1). All antibodies were generated in house. Samples were analyzed using a BD Biosciences LSRFortessa (University of Melbourne, Department of Biochemistry and Molecular Biology) and sorted using a Becton Dickinson FACSDIVA, DakoCytomation Moflo (Murdoch Childrens Research Institute Flow Cytometry Facility) or BD Biosciences FACSAria (Murdoch Childrens Research Institute Flow Cytometry Facility).
Antigen presentation assays
MHC I cross-presentation assays and MHC II antigen presentation assays with mouse spleen DC involved DC isolation and incubation with antigen. For soluble protein antigen, DC were pulsed with OVA protein (Worthington Biochemicals, LS003054) in the presence of fully phosphorothioated 0.5 μM CpG 1668, type B (custom made, Geneworks) for 45 min at 37°C. For cell-associated antigen, DC were incubated with H-2bm1 splenocytes that were irradiated at 1500 rad. Irradiated splenocytes were pulsed with 10 mg/ml OVA protein at 37°C for 10 min. For DEC205-targeted OVA, DC were labeled with anti-DEC205-biotin (in house, clone NLDC145) on ice for 30 min, washed twice and labeled with anti-biotin OVA (Miltenyi Biotec, 130094663) for 15 min at 4°C. In all cases, antigen-pulsed DC were incubated with Cell Trace Violet (Invitrogen, C34557) labeled OT-I or OT-II transgenic T cells for 64 h in media supplemented with 10 ng/ml GM-CSF (Peprotech, 315–03). T cell division was assessed by flow cytometry. Cell number was determined by inclusion of calibration particles (BD Biosciences, 556296).
MHC I cross-presentation assays with human CD1c+ and CD141+ DC involved DC culture with recombinant pp65 protein (Miltenyi Biotec, 130091824) and cross-presentation of the pp65495–503 epitope measured by IFNG secretion by pp65495–503 -specific CD8+ T cells as previously described.Citation31
Intracellular confocal microscopy
For phagocytosis assays, DC were pulsed with zymosan-Alexa-594 (Invitrogen, Molecular Probes, Z-23374) prior to overnight activation. DC were attached to coverslips with anti-MHC II antibody (N22, in house) for 30 min at room temperature. Coverslips were washed in phosphate-buffered saline (1.37 M NaCl, 27 mM KCl, 100 mM Na2HPO4, 18 mM KH2PO4), samples were fixed in 4% paraformaldehyde (Santa Cruz Biotechnology, sc-281692) and permeabilized with 0.3% Triton X-100 prior to staining for intracellular markers LC3 (Novus Biologicals, NB100–2220), SQSTM1 (Sigma Aldrich, P0067), ubiquitin (FK2, Enzo Life Sciences, BML-PW8810) and LAMP1 (1D4B, in house). Secondary antibodies were Alexa Fluor 488 or Alexa Fluor 555 conjugated (Moleclular Probes, Life Technologies, A-21208, A-31572). Nuclei were stained with 5 μg/ml DAPI (Life Technologies, D1306). Coverslips were mounted with fluorescence mounting media (DAKO, S3023). Images were acquired on a Zeiss LSM700 confocal microscope (University of Melbourne, Department of Microbiology and Immunology) and analyzed with ImageJ software.
Human DC were coated on poly-L-lysine (Sigma Aldrich, P4707) treated glass slides, fixed with 2% paraformaldehyde and permeabilized in phosphate-buffered saline containing 0.05% saponin (Sigma Aldrich, 84510) and 10% bovine serum (Sigma Aldrich, A2153). Cells were stained with FK2 antibody (Enzo Life Sciences, BML-PW8810) followed by staining with Alexa Fluor 488-coupled anti-mouse immunoglobulin antibody (Molecular Probes, Life Technologies, A-11029). Slides were analyzed on a Zeiss LSM 510 confocal microscope (Institut Curie).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1045178_Supplemental_files.zip
Download Zip (7.1 MB)Acknowledgements
We acknowledge B. Hibbs at the Advanced Fluorescence Imaging Platform for technical assistance.
Funding
This work was funded by NHMRC, Australia. JDM. is a Career Development Fellow, NHMRC. ES is supported by Association pour la Recherche sur le Cancer and INSERM. SB is a Career Development Fellow, NHMRC. MH is supported the German Research Council. JV is a Senior Research Fellow, NHMRC.
References
- Bestebroer J, V'Kovski P, Mauthe M, Reggiori F. Hidden behind autophagy: the unconventional roles of ATG proteins. Traffic 2013; 14:1029-41; PMID:23837619; http://dx.doi.org/ 10.1111/tra.12091
- Munz C. Enhancing immunity through autophagy. Annu Rev Immunol 2009; 27:423-49; PMID:19105657; http://dx.doi.org/10.1146/annurev.immunol.021908.132537
- Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol 2013; 31:443-73; PMID:23298205; http://dx.doi.org/10.1146/annurev-immunol-032712-095910
- Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007; 26:79-92; PMID:17182262; http://dx.doi.org/10.1016/j.immuni.2006.10.018
- Chicz RM, Urban RG, Gorga JC, Vignali DA, Lane WS, Strominger JL. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med 1993; 178:27-47; PMID:8315383; http://dx.doi.org/10.1084/jem.178.1.27
- Dongre AR, Kovats S, deRoos P, McCormack AL, Nakagawa T, Paharkova-Vatchkova V, Eng J, Caldwell H, Yates JR 3rd, Rudensky AY. In vivo MHC class II presentation of cytosolic proteins revealed by rapid automated tandem mass spectrometry and functional analyses. Eur J Immunol 2001; 31:1485-94; PMID:11465105; http://dx.doi.org/10.1002/1521-4141(200105)31:5%3c1485::AID-IMMU1485%3e3.0.CO;2-A
- Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol 2012; 12:557-69; PMID:22790179; http://dx.doi.org/10.1038/nri3254
- Li H, Li Y, Jiao J, Hu HM. Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat Nanotechnol 2011; 6:645-50; PMID:21926980; http://dx.doi.org/10.1038/nnano.2011.153
- De Luca A, Iannitti RG, Bozza S, Beau R, Casagrande A, D'Angelo C, Moretti S, Cunha C, Giovannini G, Massi-Benedetti C, et al. CD4(+) T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J Clin Invest 2012; 122:1816-31; PMID:22523066; http://dx.doi.org/10.1172/JCI60862
- Ravindran R, Khan N, Nakaya HI, Li S, Loebbermann J, Maddur MS, Park Y, Jones DP, Chappert P, Davoust J, et al. Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science 2014; 343:313-7; PMID:24310610; http://dx.doi.org/10.1126/science.1246829
- Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, Mizushima N, Grinstein S, Iwasaki A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010; 32:227-39; PMID:20171125; http://dx.doi.org/10.1016/j.immuni.2009.12.006
- Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 2013; 31:563-604; PMID:23516985; http://dx.doi.org/10.1146/annurev-immunol-020711-074950
- Xu Y, Zhan Y, Lew AM, Naik SH, Kershaw MH. Differential development of murine dendritic cells by GM-CSF versus Flt3 ligand has implications for inflammation and trafficking. J Immunol 2007; 179:7577-84; PMID:18025203; http://dx.doi.org/10.4049/jimmunol.179.11.7577
- Segura E, Albiston AL, Wicks IP, Chai SY, Villadangos JA. Different cross-presentation pathways in steady-state and inflammatory dendritic cells. Proc Natl Acad Sci U S A 2009; 106:20377-81; PMID:19918052; http://dx.doi.org/10.1073/pnas.0910295106
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 2007; 204:25-31; PMID:17190837; http://dx.doi.org/10.1084/jem.20061303
- Pooley JL, Heath WR, Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J Immunol 2001; 166:5327-30; PMID:11313367; http://dx.doi.org/10.4049/jimmunol.166.9.5327
- den Haan JM, Bevan MJ. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(−) dendritic cells in vivo. J Exp Med 2002; 196:817-27; PMID:12235214; http://dx.doi.org/10.1084/jem.20020295
- Wilson NS, El-Sukkari D, Belz GT, Smith CM, Steptoe RJ, Heath WR, Shortman K, Villadangos JA. Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood 2003; 102:2187-94; PMID:12791652; http://dx.doi.org/10.1182/blood-2003-02-0513
- Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, Whitney JA, Connolly J, Banchereau J, Mellman I. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity 2007; 27:610-24; PMID:17936032; http://dx.doi.org/10.1016/j.immuni.2007.08.015
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/10.1091/mbc.E03-09-0704
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253-7; PMID:18097414; http://dx.doi.org/10.1038/nature06421
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A 2011; 108:17396-401; PMID:21969579; http://dx.doi.org/10.1073/pnas.1113421108
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/10.1038/nature04723
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/10.1038/nature04724
- Lelouard H, Gatti E, Cappello F, Gresser O, Camosseto V, Pierre P. Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 2002; 417:177-82; PMID:12000969; http://dx.doi.org/10.1038/417177a
- Wenger T, Terawaki S, Camosseto V, Abdelrassoul R, Mies A, Catalan N, Claudio N, Clavarino G, de Gassart A, Rigotti Fde A, et al. Autophagy inhibition promotes defective neosynthesized proteins storage in ALIS, and induces redirection toward proteasome processing and MHCI-restricted presentation. Autophagy 2012; 8:350-63; PMID:22377621; http://dx.doi.org/10.4161/auto.18806
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/10.1016/j.cell.2007.10.035
- Lelouard H, Ferrand V, Marguet D, Bania J, Camosseto V, David A, Gatti E, Pierre P. Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol 2004; 164:667-75; PMID:14981091; http://dx.doi.org/10.1083/jcb.200312073
- Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR, et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol 2009; 10:488-95; PMID:19349986; http://dx.doi.org/10.1038/ni.1724
- Wilson NS, Young LJ, Kupresanin F, Naik SH, Vremec D, Heath WR, Akira S, Shortman K, Boyle J, Maraskovsky E, et al. Normal proportion and expression of maturation markers in migratory dendritic cells in the absence of germs or Toll-like receptor signaling. Immunol Cell Biol 2008; 86:200-5; PMID:18026177; http://dx.doi.org/10.1038/sj.icb.7100125
- Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med 2010; 207:1247-60; PMID:20479116; http://dx.doi.org/10.1084/jem.20092140
- Segura E, Valladeau-Guilemond J, Donnadieu MH, Sastre-Garau X, Soumelis V, Amigorena S. Characterization of resident and migratory dendritic cells in human lymph nodes. J Exp Med 2012; 209:653-60; PMID:22430490; http://dx.doi.org/10.1084/jem.20111457
- Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 2010; 207:1273-81; PMID:20479115; http://dx.doi.org/10.1084/jem.20100348
- Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med 2010; 207:1283-92; PMID:20479118; http://dx.doi.org/10.1084/jem.20100223
- Poulin LF, Reyal Y, Uronen-Hansson H, Schraml BU, Sancho D, Murphy KM, Håkansson UK, Moita LF, Agace WW, Bonnet D, et al. DNGR-1 is a specific and universal marker of mouse and human Batf3-dependent dendritic cells in lymphoid and nonlymphoid tissues. Blood 2012; 119:6052-62; PMID:22442345; http://dx.doi.org/10.1182/blood-2012-01-406967
- Segura E, Durand M, Amigorena S. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J Exp Med 2013; 210:1035-47; PMID:23569327; http://dx.doi.org/10.1084/jem.20121103
- Kassianos AJ, Hardy MY, Ju X, Vijayan D, Ding Y, Vulink AJ, McDonald KJ, Jongbloed SL, Wadley RB, Wells C, et al. Human CD1c (BDCA-1)+ myeloid dendritic cells secrete IL-10 and display an immuno-regulatory phenotype and function in response to Escherichia coli. Eur J Immunol 2012; 42:1512-22; PMID:22678905; http://dx.doi.org/10.1002/eji.201142098
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/10.1083/jcb.200412022
- Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med 2007; 204:1653-64; PMID:17591855
- Li M, Davey GM, Sutherland RM, Kurts C, Lew AM, Hirst C, Carbone FR, Heath WR. Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J Immunol 2001; 166:6099-103; PMID:11342628; http://dx.doi.org/10.4049/jimmunol.166.10.6099
- Lin Y, Roberts TJ, Sriram V, Cho S, Brutkiewicz RR. Myeloid marker expression on antiviral CD8+ T cells following an acute virus infection. Eur J Immunol 2003; 33:2736-43; PMID:14515257; http://dx.doi.org/10.1002/eji.200324087
- Kastenmuller W, Kastenmuller K, Kurts C, Seder RA. Dendritic cell-targeted vaccines - hope or hype? Nat Rev Immunol 2014; 14:705-11; PMID:25190285; http://dx.doi.org/10.1038/nri3727
- Moffat JM, Segura E, Khoury G, Caminschi I, Cameron PU, Lewin SR, Villadangos JA, Mintern JD. Targeting antigen to bone marrow stromal cell-2 expressed by conventional and plasmacytoid dendritic cells elicits efficient antigen presentation. Eur J Immunol 2013; 43:595-605; PMID:23303646; http://dx.doi.org/10.1002/eji.201242799
- Spadaro F, Lapenta C, Donati S, Abalsamo L, Barnaba V, Belardelli F, Santini SM, Ferrantini M. IFN-alpha enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood 2012; 119:1407-17; PMID:22184405; http://dx.doi.org/10.1182/blood-2011-06-363564
- van Montfoort N, Camps MG, Khan S, Filippov DV, Weterings JJ, Griffith JM, Geuze HJ, van Hall T, Verbeek JS, Melief CJ, et al. Antigen storage compartments in mature dendritic cells facilitate prolonged cytotoxic T lymphocyte cross-priming capacity. Proc Natl Acad Sci U S A 2009; 106:6730-5; PMID:19346487; http://dx.doi.org/10.1073/pnas.0900969106
- Romao S, Gasser N, Becker AC, Guhl B, Bajagic M, Vanoaica D, Ziegler U, Roesler J, Dengjel J, Reichenbach J, et al. Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. J Cell Biol 2013; 203:757-66; PMID:24322427; http://dx.doi.org/10.1083/jcb.201308173
- Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 2011; 13:1335-43; PMID:22002674; http://dx.doi.org/10.1038/ncb2363
- Moffat JM, Cheong WS, Villadangos JA, Mintern JD, Netter HJ. Hepatitis B virus-like particles access major histocompatibility class I and II antigen presentation pathways in primary dendritic cells. Vaccine 2013; 31:2310-6; PMID:23473776; http://dx.doi.org/10.1016/j.vaccine.2013.02.042