
Abstract
Oxygen (O2) is an essential substrate in cellular metabolism and signaling and as such is linked to the survival and normal function of metazoans. Central to the molecular mechanisms underlying O2 homeostasis are hypoxia-inducible factors (HIFs), heterodimeric transcription factors composed of O2-regulated α subunits (HIF1A/HIF-1α or EPAS1/HIF-2α), and a constitutively expressed ARNT/HIF-1β subunit, that serve as master regulators of the adaptive response to hypoxia. HIF1A and EPAS1 have both unique and overlapping functions in the regulation of diverse cellular processes, but so far there has been no evidence linking HIF signaling to peroxisomes. In a recent study we identified a unique function of EPAS1 as promoter of pexophagy in hepatocytes. Here we summarize our findings and discuss potential mechanisms by which EPAS1 might trigger pexophagy.
Peroxisomes are ubiquitous and highly dynamic organelles whose number, size, and function are dependent on metabolic needs and cell type. They play essential roles in reactive oxygen species and lipid metabolism. Their importance is stressed by the existence of genetic disorders in which the biogenesis of the organelle is defective, leading to complex developmental and metabolic phenotypes. A general rule applicable for both yeast and mammalian cells is that environmental conditions that require peroxisomal metabolism cause peroxisome proliferation, followed by degradation of the organelles via pexophagy when they are no longer required.
Since peroxisomal function depends highly on molecular O2 due to the various oxidative reactions, we hypothesized that regulation of peroxisome homeostasis may be directly linked to O2 availability and the regulatory system of O2 signaling. The HIF-α subunits are targeted by the VHL protein for ubiquitination and subsequent proteasomal degradation under normoxia. Hypoxia or loss of VHL function results in the stabilization of the HIF-α subunits and activation of their signaling, providing an experimental model system to study peroxisome homeostasis in the context of HIF signaling. Specifically, we examined the effect of HIF signaling on hepatic peroxisome abundance in control and liver-specific vhl−/−, vhl−/− hif1a−/−, and vhl−/− epas1−/− mice. Peroxisome abundance is significantly decreased in livers of vhl−/− mice, whereas ER and mitochondrial protein levels are not affected. Reduction of peroxisome abundance is mediated by EPAS1, because, with respect to the peroxisomal phenotype, we observed a striking rescue in vhl−/− epas1−/− but not vhl−/− hif1a−/− mice. EPAS1 promotes pexophagy because reduction of peroxisome abundance is suppressed after inhibition of autophagy with 3-methyladenine (3-MA) in vhl−/− mice and in liver-specific, autophagy-deficient atg7−/− mice expressing a constitutively active EPAS1 variant. In support of this finding super-resolution and electron microscopy demonstrated that both single and multiple peroxisomes, but no other cytoplasmic organelles, are sequestered in autophagosomes in vhl−/− livers.
NBR1 and SQSTM1/p62 are autophagy receptors of ubiquitinated targets and they play a role in pexophagy in vitro. Peroxisome abundance and protein levels of NBR1 and SQSTM1 are concomitantly decreased in vhl−/− and vhl−/− hif1a−/− livers. Neither peroxisome abundance nor NBR1 and SQSTM1 levels decline in 3-MA-treated vhl−/− mice, showing that the abundance of these receptors and peroxisomes are interconnected. NBR1 and SQSTM1 colocalize with peroxisomes in vhl−/− livers, but surprisingly NBR1 already localizes to peroxisomes in control livers. NBR1 might bind to an as-yet unidentified ubiquitinated peroxisomal membrane protein (PMP) or is recruited to peroxisomes independently of ubiquitin via its amphipathic α-helical membrane-interacting domain (J domain), capable of binding to the peroxisomal lipid bilayer.
Reduced peroxisome abundance and the ensuing deficiency in peroxisomal function encompass major changes in the lipid profile that are reminiscent of peroxisomal disorders and include the depletion of docosahexaenoic acid and the accumulation of very long-chain (VLC) fatty acids, VLC-polyunsaturated fatty acids, and C27-bile acid intermediates. VLC fatty acids are activating ligands for the transcription factor PPARA, a promoter of peroxisome proliferation; however, we showed that EPAS1 restrains ligand-induced PPARA-mediated peroxisome proliferation. Thus, by simultaneously inducing pexophagy and counteracting PPARA, EPAS1 ensures efficient depletion of the peroxisome pool.
Loss of VHL function occurs in up to 90% of sporadic human clear cell renal cell carcinomas (ccRCC), and EPAS1 is considered to be a driver oncoprotein for ccRCC. Indeed, peroxisome abundance is reduced in VHL-deficient human ccRCC characterized by high EPAS1 levels. Whether reduction in peroxisome abundance represents a critical aspect of ccRCC progression remains to be explored, but one could envision that changes in lipid composition contribute to alterations in lipid signaling that contribute to the malignant phenotype.
We propose 2 alternative models for how EPAS1 might trigger pexophagy. Overexpression of NBR1 induces clustering and subsequent degradation of peroxisomes in cell lines. Hence, the most likely possibility would be that EPAS1 induces the expression of an autophagy receptor and subsequent clustering of peroxisomes via oligomerization of receptor-bound organelles, however, neither Nbr1 nor Sqstm1 are EPAS1 target genes. Artificial mono-ubiquitination of PMPs in mammalian cells causes peroxisome turnover by pexophagy in a NBR1- and SQSTM1-dependent manner. Since ubiquitination of cargos prone for selective autophagic degradation is the most prevalent autophagy-targeting signal in mammals, EPAS1 might also induce an E3 ubiquitin ligase that mediates the ubiquitination of a PMP (). In this manner, EPAS1 signaling would enhance NBR1 accumulation on peroxisomes, which in turn serves as a platform for the recruitment of SQSTM1 to achieve a critical mass of autophagy receptors on peroxisomes required for pexophagy. An intriguing feature of autophagy receptors is their tendency to oligomerize, which facilitates sequestration and clustering of the autophagic cargo. Indeed, treatment of vhl−/− mice with 3-MA leads to a significant clustering of NBR1- and SQSTM1-positive peroxisomes.
Figure 1. Two alternative models illustrating how EPAS1 might trigger pexophagy. (A) EPAS1 might induce an E3 ubiquitin ligase that specifically ubiquitinates a peroxisomal membrane protein that enhances the recruitment of the autophagy receptor NBR1 to the peroxisome surface. Accumulation of NBR1 on peroxisomes likely recruits SQSTM1, which was suggested to act as pexophagy co-receptor, and subsequently leads to clustering of peroxisomes via oligomerization of receptor-bound organelles. Accumulation of a critical mass of autophagy receptors at peroxisomes might concentrate sufficient ubiquitin-like modifiers (e.g., LC3 and GABARAPs) in close proximity to peroxisomes and prime phagophore assembly. (B) NBR1 could also be recruited to peroxisomes independently of ubiquitin via its membrane-interacting amphipathic α-helical J domain. In fact, NBR1 already localizes to peroxisomes in wild-type livers where pexophagy is not induced. EPAS1 might induce or inhibit a kinase/phosphatase that leads to a change in the posttranslational modification of peroxisome-bound NBR1 and thereby triggers pexophagy.
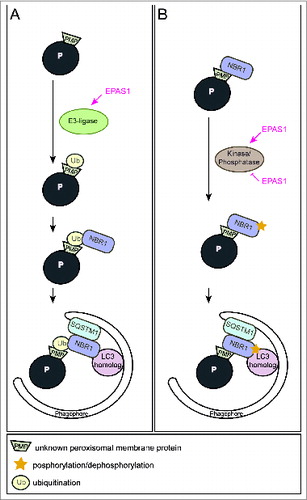
Why does NBR1 localize to peroxisomes in wild-type livers although pexophagy is not induced? We propose that posttranslational modifications such as phosphorylation and dephosphorylation of autophagy proteins are crucial for induction, inhibition, cargo-recognition, and fine-tuning of autophagy. For example, the yeast pexophagy receptors Atg30 and Atg36 depend on phosphoregulation for their interactions with components of the autophagy machinery during pexophagy conditions. Hence, additional protein modifications are very likely necessary to ultimately drive pexophagy by recruiting and tailoring the autophagic machinery to peroxisomes. We propose that EPAS1 governs pexophagy by promoting posttranslational modifications of PMPs and/or autophagy receptors that enhance interactions of receptor-labeled peroxisomes with the autophagic machinery (). Furthermore, one could envision that EPAS1-dependent activation of pexophagy is a 2-step process that involves interplay between ubiquitination of a PMP(s) as well as phosphoregulation of autophagy receptors or a PMP(s) as a trigger of pexophagy. Remarkably, a similar interplay promotes mitophagy whereby phosphorylation of ubiquitin contributes to a feedforward mechanism for ubiquitination events on dysfunctional mitochondria.
Finally, a receptor protein complex model has been proposed that encompasses the receptor protein as the key player that establishes interactions with ligands, scaffold, and phagophore proteins. The question remains which components of the receptor protein complex are involved in EPAS1-mediated pexophagy and if there is an Atg11 homolog in the mammalian liver that would act as a scaffold.
In summary, the identification of EPAS1 as an inducer of pexophagy opens new avenues for studying the underlying molecular mechanism and to further characterize the functional consequences of EPAS1-driven pexophagy and associated lipid alterations.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.