
Abstract
Autophagy is closely related to tumor cell sensitivity to anticancer drugs. The HDAC (histone deacetylase) inhibitor valproic acid (VPA) interacted synergistically with chemotherapeutic agents to trigger lymphoma cell autophagy, which resulted from activation of AMPK (AMP-activated protein kinase) and inhibition of downstream MTOR (mechanistic target of rapamycin [serine/threonine kinase]) signaling. In an HDAC-independent manner, VPA potentiated the effect of doxorubicin on lymphoma cell autophagy via reduction of cellular inositol 1,4,5 trisphosphate (IP3), blockade of calcium into mitochondria and modulation of PRKAA1/2-MTOR cascade. In murine xenograft models established with subcutaneous injection of lymphoma cells, dual treatment of VPA and doxorubicin initiated IP3-mediated calcium depletion and PRKAA1/2 activation, induced in situ autophagy and efficiently retarded tumor growth. Aberrant genes involving mitochondrial calcium transfer were frequently observed in primary tumors of lymphoma patients. Collectively, these findings suggested an HDAC-independent chemosensitizing activity of VPA and provided an insight into the clinical application of targeting autophagy in the treatment of lymphoma.
Abbreviations
| 3-MA | = | 3-methyladenine |
| ATG5 | = | autophagy-related 5 siRNA |
| CI | = | combination index |
| CNV | = | copy number variation |
| DLBCL | = | diffuse large B-cell lymphoma |
| DOX | = | doxorubicin |
| EIF4EBP1 | = | eukaryotic translation initiation factor 4E binding protein 1 |
| ELISA | = | enzyme-linked immunosorbent assay |
| HDAC | = | histone deacetylase |
| IP3 | = | inositol 1,4,5 trisphosphate |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 β |
| LRR | = | Log R Ratio |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| PPID/CyPD | = | peptidylprolyl isomerase D |
| PRKAA1/2 | = | protein kinase, AMP-activated, α catalytic subunit |
| RPS6KB | = | ribosomal protein S6 kinase, 70kDa |
| SAHA | = | suberoylanilide; hydroxamic acid |
| siRNA | = | small-interfering RNA |
| SQSTM1/p62 | = | sequestosome 1 |
| TCL | = | T-cell lymphoma |
| ULK1 | = | unc-51 like autophagy activating kinase |
| VDAC | = | voltage-dependent anion channel |
| VPA | = | valproic acid |
| XeC | = | Xestospongin C. |
Introduction
Despite remarkable progress in lymphoma treatment over the past decades, chemotherapy resistance and disease relapse become the main obstacles to further improve the prognosis of the patients.Citation1 Autophagy, which can become an independent form of cell death, is essential for the homeostasis of tumor cell growth.Citation2 Induction of autophagy plays an important role in tumor cell sensitivity to anticancer drugs.Citation3 Therefore, how autophagy modulates the effect of chemotherapeutic agents on lymphoma cells remains of great interest.
Autophagy is tightly controlled by MTOR (mechanistic target of rapamycin [serine/threonine kinase]) signaling pathway.Citation4 Previous studies demonstrated that HDAC (histone deacetylase) inhibitors induced autophagy via MTOR cascade, translating into promising clinical efficiency in various cancer types including lymphoma.Citation5,6 Valproic acid (VPA), a widely used oral antiepilepsy drug, is a class I HDAC inhibitor of fatty acid and possesses antilymphoma activity with the levels achieved in clinical practice.Citation7 Recently, we have reported that VPA potentiates the effect of MTOR inhibition to induce lymphoma cell autophagy.Citation8 However, the interaction of VPA with chemotherapeutic agents, particularly its direct action on MTOR pathway and cell autophagy, has not yet been fully understood in lymphoma.
AMPK (AMP-activated protein kinase) acts as an upstream inhibitor of MTOR signaling and a key inducer of autophagy.Citation9 Inositol 1,4,5 trisphosphate (IP3) regulates mitochondrial uptake of IP3 receptor-released calcium and is fundamentally required to activate PRKAA.Citation10 Here we showed that VPA, in addition to HDAC inhibition, induced IP3-mediated PRKAA1/2 activation and autophagy induction, sensitizing lymphoma cells to chemotherapy.
Results
VPA interacted with chemotherapeutic agents to induce synergistic cytotoxicity in lymphoma cells
T-lymphoma cell lines (Jurkat and H9) and B lymphoma cell lines (SU-DHL-4 and Nalmawa) were treated with different concentrations of VPA and/or doxorubicin. Dose-response curves are shown in . Compared with each agent alone, a significant increase in cell growth inhibition was observed with combined treatment. For example, 0.5 mM VPA and 15 nM doxorubicin alone induced approximately 10% reduction in cell viability; however, in combination they achieved more than a 40% cell reduction. The combination index (CI) curve yielded most of the data points to the area <1, denoting synergistic interactions in all cell lines (). Accordingly, VPA exhibited a significant inhibition of HDAC1 and HDAC3 activity from the concentration of 0.5 mM (Fig. S1). This clinically achievable concentration was then used for the following experiments.
Figure 1. Valproic acid interacted synergistically with chemotherapeutic agents to inhibit lymphoma cell growth. (A and B) Cell growth determined by MTT assay (A) and Combination Index (CI) curve calculated by compusyn software (B) in T-lymphoma cell lines (Jurkat, H9) and B-lymphoma cell lines (SU-DHL-4, Nalmawa) treated with valproic acid (VPA) and/or doxorubicin (DOX) at 48 h. (C) Colony formation assay of Jurkat and SU-DHL-4 cells treated with VPA and DOX, either alone or in combination. Representative results shown in cells treated with VPA (0.5 mM) and/or DOX (15 nM) at 48 h. (D) CI curve of Jurkat and SU-DHL-4 cells treated with VPA and chemotherapeutic agents like cyclophosphamide, cisplatin, or gemcitabine at 48 h.
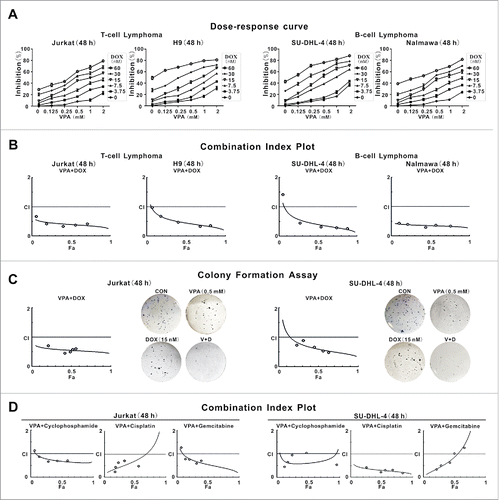
Similar results were obtained by colony formation assay in Jurkat and SU-DHL-4 cells (). Synergistic cytotoxicity was also observed in lymphoma cells when treated with VPA and cyclophosphamide, cisplatin or gemcitabine, chemotherapeutic agents regularly applied in lymphoma treatment ().
VPA combined with doxorubicin synergistically triggered lymphoma cell autophagy through PRKAA1/2 activation and MTOR inhibition
In T- and B-lymphoma cells cotreated with VPA (0.5 mM) and doxorubicin (15 nM), a significantly increased MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β) intensity was observed, compared with those of the control cells and cells treated with VPA or doxorubicin alone for 48 h (). Moreover, VPA in conjunction with doxorubicin induced higher expression of autophagosome-associated LC3B-II and BECN1 than each agent alone. Also the autophagic flux was confirmed in the combination group by a corresponding decrease in SQSTM1/p62 (sequestosome 1) expression (), which is degraded during autophagy.Citation11
Figure 2. Valproic acid combined with doxorubicin induced lymphoma cell autophagy. (A) LC3B intensity determined by flow cytometry in T-lymphoma cell lines (Jurkat, H9) and B-lymphoma cell lines (SU-DHL-4, Nalmawa) treated with valproic acid (VPA, 0.5 mM) and/or doxorubicin (DOX, 15 nM) at 48 h. (B) The protein expression levels of LC3B-II detected by western blot in lymphoma cells treated with VPA (0.5 mM) and DOX (15 nM), either alone or in combination at 24 h (upper panels) and at 48 h (with BECN1 and SQSTM1 expression, lower panels). ACTB was used to assess equivalent protein loading. (C) Representative autophagic structures and quantitative data obtained by transmission electron microscopy in Jurkat and SU-DHL-4 cells treated with VPA (0.5 mM) and/or DOX (15 nM) at 48 h. Scale bars: 0.5 μm. **, P < 0.01, *, P < 0.05 compared with the CON (untreated) cells. (D) Growth inhibition determined by MTT assay in Jurkat cells treated with the autophagy inhibitor 3-methyladenine (0.5 mM, left panels) and BafA1 (10 nM, right panels). **, P < 0.01, *, P < 0.05 compared with the absence of inhibitor. (E) Growth inhibition determined by MTT assay in Jurkat cells treated with ATG5 siRNA (left panels) and BECN1 siRNA (right panels). **, P < 0.01, *, P < 0.05 compared with the CON siRNA.
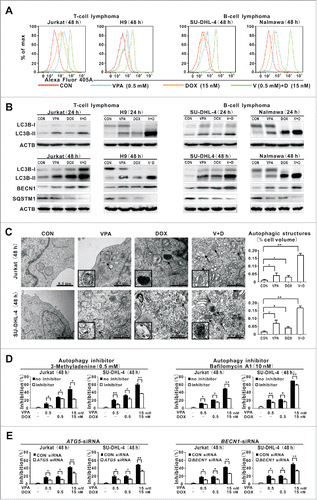
Cell cycle analysis revealed a significant elevation of G0/G1-phase cells in the combination group, compared with those in the single-agent group, indicating that VPA and doxorubicin synergistically inhibited lymphoma cell growth through cell cycle arrest (Fig. S2A). However, no obvious change in the percentage of ANXA5/annexin V-positive cells was found among the groups (Fig. S2B).
To confirm whether the growth inhibition was caused by other type of cell death, namely autophagy-associated cell death, the ultrastructure of tumor cells was studied in Jurkat and SU-DHL-4 cells. Typical autophagosomes, occurred in cells treated with VPA or doxorubicin alone, were more frequently observed in those cotreated with VPA and doxorubicin, with a corresponding increased volume occupied by autophagic structures (). Synergistic cytotoxicity was significantly diminished by pharmacological (3-methyladenine [3-MA] and bafilomycin A1 [BafA1], ) and molecular inhibition of autophagy (autophagy-related 5 small-interfering RNA, ATG5 siRNA, and BECN1 siRNA, ), but not by pan-CASP/caspase inhibitor ZVAD-FMK (Fig. S2C). Moreover, increased G0/G1 phase and LC3B-II induced by cotreatment with VPA and doxorubicin were abrogated by 3-MA (Fig. S2D and S2E), further indicating that this cytotoxic effect was autophagy-dependent.
PRKAA is a key inducer of autophagy.Citation12 VPA in combination with doxorubicin significantly induced phosphorylation of PRKAA1/2, whereas single-drug treatments exerted minimal effects (). Meanwhile, dual treatment resulted in decreased expression of p-MTOR, as well as the downstream effectors phospho(p)-RPS6KB (ribosomal protein S6 kinase; 70 kDa) and p-EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1), while their total levels remained constant. PRKAA1/2 activation specifically induced phosphorylation of ULK1 (unc-51 like autophagy activating kinase 1) at Ser555.Citation13 Cotreatment of VPA and doxorubicin was accompanied by increased expression of p-ULK1 Ser555, representing a PRKAA1/2-associated induction of lymphoma cell autophagy. Moreover, synergistic cytotoxicity of VPA combined with doxorubicin was significantly diminished by molecular inhibition of ULK1 using the specific siRNA (Fig. S2F).
Figure 3. Valproic acid combined with doxorubicin induced PRKAA1/2 activation and MTOR inhibition though reducing cellular IP3 and mitochondrial calcium. (A) Phosphorylated and total protein expression of PRKAA1/2, MTOR, RPS6KB, EIF4EBP1, and phosphorylated ULK1 (p-ULK1 Ser555) detected by western blot in Jurkat and SU-DHL-4 cells treated with valproic acid (VPA, 0.5 mM) and/or doxorubicin (DOX, 15 nM) at 48 h. (B and C) Expression of HDAC1, HDAC3, p-PRKAA1/2, p-ULK1 Ser555 as well as LC3B-II and SQSTM1 by western blot in Jurkat cells transfected with HDAC1 and HDAC3 siRNA (B), or overexpression vectors (C), followed by treatment with VPA (0.5 mM) and DOX (15 nM) for 48 h. ACTB was used to monitor equivalent protein loading. (D) IP3 levels assessed by ELISA in Jurkat cells treated with VPA (0.5 mM) and/or DOX (15 nM) at 48 h. **P < 0.01,*P < 0.05 compared with the untreated cells. (E) IP3 levels in Jurkat cells transfected with HDAC1 and HDAC3 siRNA, followed by treatment with VPA (0.5 mM) and/or DOX (15 nM) at 48 h. (F) Representative immunofluorescence images of mitochondrial (green) and calcium (red) in Jurkat cells treated with VPA (0.5 mM) and/or DOX (15 nM) at 48 h. Cells were counterstained with DAPI (blue).
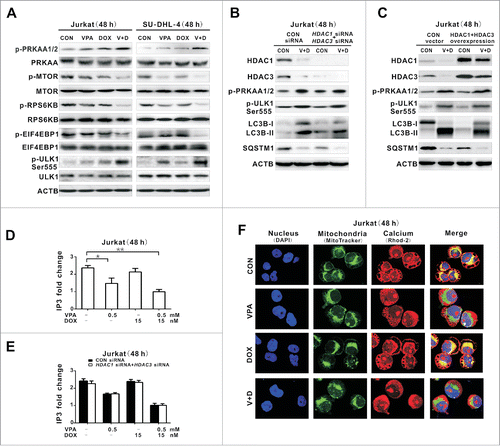
VPA potentiated the effect of doxorubicin on lymphoma cell autophagy in an HDAC-independent manner via reduction of cellular IP3 and blockade of calcium into mitochondria
Functioning as a class I HDAC inhibitor, VPA inhibited HDAC1 and HDAC3 enzymatic activity in Jurkat cells (Fig. S3). To determine whether the chemosensitizing effect of VPA depended on HDAC1 and HDAC3, Jurkat cells were transfected with the specific siRNA targeting HDAC1 and HDAC3. Compared with those transfected with control siRNA, the autophagy-inducing effect of VPA combined with doxorubicin on HDAC1 siRNA- and HDAC3 siRNA-transfected lymphoma cells was maintained, as revealed by increased expression of p-PRKAA1/2, p-ULK1 on Ser555, LC3B-II, as well as decreased expression of SQSTM1 (). Similarly, autophagy induced by VPA-doxorubicin cotreatment was not altered in Jurkat cells overexpressing HDAC1 and HDAC3 (). Therefore, VPA enhanced the effect of doxorubicin on lymphoma cell autophagy independent of HDAC modulation.
Based on the fact that VPA can exert an autophagy-inducing effect via IP3 depletion,Citation14 cellular IP3 was assessed in Jurkat cells treated with VPA and/or doxorubicin (). IP3 level of the VPA-treated lymphoma cells, either alone or combined with doxorubicin, was significantly lower than that of the control cells and doxorubicin-treated cells. The IP3-depleting effect by VPA remained constant in Jurkat cells transfected with HDAC1 and HDAC3 siRNA, as well as those transfected with the control siRNA (), in agreement with the above results that lymphoma cell autophagy was not affected by molecular silencing of HDAC1 and HDAC3 (). IP3 depletion promotes cell autophagy by interrupting mitochondrial uptake of IP3 receptor-released calcium.Citation10 The mitochondrial calcium signal was then measured in Jurkat cells. Calcium, which colocalized with mitochondria in the control cells, was diminished by VPA, but not by doxorubicin ().
To confirm that the HDAC-independent autophagy-inducing effect was IP3-mediated and VPA-specific, Jurkat cells were treated with Xestospongin C (XeC), a specific IP3R inhibitor, and suberoylanilide hydroxamic acid (SAHA), an hydroxamate-type HDAC inhibitor, respectively. An IP3-depleting effect was observed upon treatment with VPA or XeC, but not with SAHA. Correspondingly, XeC mimicked the effect of VPA, resulting in reduced mitochondrial calcium, an effect that was not present in SAHA-treated cells (). XeC increased expression of p-PRKAA1/2, p-ULK1 Ser555 and LC3B-II without changing p-MTOR expression (), as observed in VPA-treated cells (). To determine whether VPA acted on lymphoma cell growth via mitochondrial calcium signaling, Jurkat cells were transduced with the gene encoding the calcium quencher CALB1/calbindin D28 (calbindin 1, 28kDa). Compared with those transduced with the control vector, VPA-mediated calcium transfer to mitochondria was blocked in CALB1-overexpressing cells (), in association with reduction of lymphoma cell autophagy () and cell growth inhibition ().
Figure 4. IP3-depleting effect of valproic acid mimicked by the IP3R inhibitor but counteracted by the calcium inhibitor. (A) IP3 levels assessed by ELISA in Jurkat cells treated with valproic acid (VPA, 0.5 mM), specific IP3R inhibitor Xestospongin C (XeC, 300 nM) or suberoylanilide hydroxamic acid (SAHA, 2 μM) at 48 h. **P < 0.01 compared with the CON (untreated) cells. (B) Representative immunofluorescence images of mitochondria (green) and calcium (red) in Jurkat cells treated with XeC (300 nM) or (SAHA, 2 μM) at 48 h. Cells were counterstained with DAPI (blue). (C) Expression of p-PRKAA1/2, p-ULK1 Ser555, LC3B-II and p-MTOR by western blot in Jurkat cells treated with XeC (300 nM) at 48 h. ACTB was used to monitor equivalent protein loading. (D) Representative immunofluorescence images of mitochondria (green) and calcium (red) in Jurkat cells transduced with the specific calcium inhibitor CALB1 vector and control vector, followed by treatment with VPA (0.5 mM) and DOX (15 nM) for 48 h. Cells were counterstained with DAPI (blue). (E) Expression of LC3B-II by western blot (Upper panel) and growth inhibition by MTT assay in Jurkat cells transduced with CALB1 overexpression vector and control vector, followed by treatment with VPA (0.5 mM) and DOX (15 nM) for 48 h. ACTB was used to assess equivalent protein loading. **, P < 0.01, *, P < 0.05 compared with the control vector.
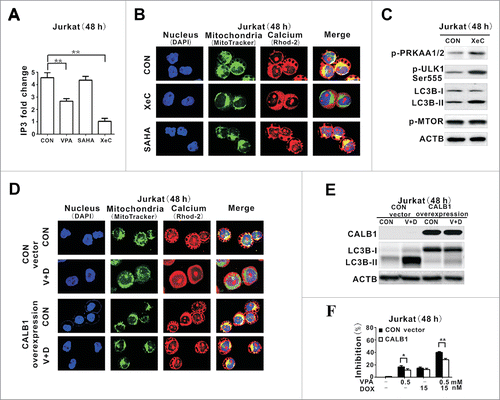
Together, independent of HDAC inhibition, VPA initiated specific reduction of IP3, blockade of mitochondrial calcium transferring, activation of PRKAA1/2 and induction of autophagy, which may contribute to the chemosensitizing effect of VPA on lymphoma cells.
Cotreatment of VPA and doxorubicin induced in situ autophagy in murine xenograft models through IP3-mediated PRKAA1/2 activation
In vivo chemosensitizing activity of VPA on T- and B-lymphoma cells was further evaluated in murine xenograft models. Subcutaneous inoculation of Jurkat and SU-DHL-4 cells into nude mice resulted in tumor formation at the site of injection in all mice. The size of tumors formed in mice cotreated with VPA and doxorubicin was significantly smaller than those of the control and single-agent group since 15 d of treatment ().
Figure 5. Valproic acid synergized with doxorubicin in murine xenograft lymphoma models. (A) Tumor volume of xenograft nude mice (injected subcutaneously with Jurkat and SU-DHL-4 cells) treated with valproic acid (VPA), doxorubicin (DOX), either alone or in combination. ***, P < 0.001 compared with the CON (untreated) group. (B) IP3 levels assessed by ELISA assay in xenograft tumors of the VPA group, DOX group and combination group. **, P < 0.01 compared with the CON (untreated) group. (C) Representative autophagic structures and quantitative data obtained by transmission electron microscopy in xenograft tumors of the VPA group, DOX group and combination group. Scale bars: 0.5 μm. **, P < 0.01, *, P < 0.05 compared with CON (untreated) group. (D) Immunohistochemical staining and semiquantitative data of p-PRKAA1/2 and BECN1 in xenograft tumors of the VPA group, DOX group and combination group. *, P < 0.05 compared with the CON (untreated) group.
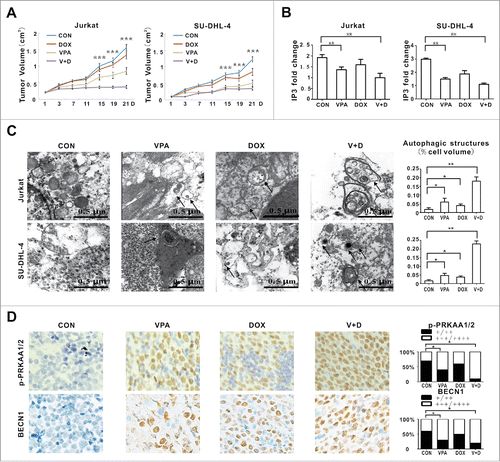
In agreement with in vitro data of lymphoma cells, the most significantly reduced IP3 level was found in xenograft tumors treated with VPA and doxorubicin (). Ultrastructure study on mice tumor sections revealed that tumor cells in the combination group exhibited significantly increased autophagosomes and autophagic structures, compared with those treated with each agent alone (). Increases of p-PRKAA1/2 and BECN1 were more perceptibly detected in the combination group than in the single treatment group ().
Genes related to mitochondrial calcium transfer were frequently altered in primary tumors of lymphoma patients
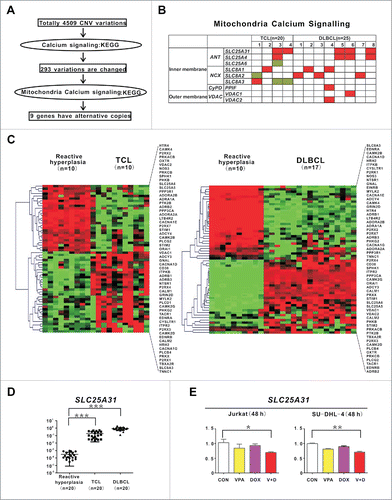
To search for more clinical evidence of dysregulation of mitochondria calcium signaling in lymphoma, genome-wide copy number variation (CNV) analysis was performed on tumor samples of 20 T-cell lymphoma (TCL) and 25 diffuse large B-cell lymphoma (DLBCL) patients. As illustrated in , compared with reactive hyperplasia, CNV data revealed a total of 4509 CNV variations. Enriched by KEGG, 293 CNV variations were involved in 155 genes associated with calcium signaling pathway. Among them, 9 genes related to mitochondria calcium signaling were identified, including adenine nucleotide translocator-family genes (SLC25A31, SLC25A4 and SLC25A6), sodium/calcium exchanger-family genes (SLC8A1, SLC8A2 and SLC8A3), PPID (peptidylprolyl isomerase D)-family genes including PPIF (the PPIF protein is located in the inner mitochondrial membrane), and voltage-dependent anion channel (VDAC)-family genes (VDAC1 and VDAC2) encoding proteins that reside in the outer mitochondrial membrane. Alterations of these genes were present in 4 of 20 TCL (20%) and 8 of 25 DLBCL (32%) cases, respectively (). Correspondingly, by gene expression profiling (NCBI, accession number GSE47357), the calcium signaling pathway was dysregulated in both TCL and DLBCL (). Moreover, the most frequently observed gene (SLC25A31), was assessed by quantitative PCR. Overexpressed in lymphoma patients compared with reactive hyperplasia (), SLC25A31 was significantly downregulated by VPA combined with doxorubicin in Jurkat cells and SU-DHL-4 cells ().
Figure 6. Genes involved in mitochondrial calcium transferring were altered in T- and B-cell lymphoma. (A) Schematic of CNV data analysis on tumor samples of 20 T-cell lymphoma (TCL) and 25 diffuse large B-cell lymphoma (DLBCL) patients. (B) Distribution of mitochondria calcium signaling gene aberrations in TCL (left panel) and DLBCL (right panel) cases. (C) Calcium signaling pathway revealed by gene expression profile in TCL and DLBCL cases. (D) Quantitative PCR results of SLC25A31 in TCL and DLBCL cases. ***, P < 0.001 compared with reactive hyperplasia. A relative quantification calculated using the ΔΔCT method based on the expression value of H9 cells. (E) Quantitative PCR results of SLC25A31 in Jurkat and SU-DHL-4 cells treated with valproic acid (VPA, 0.5 mM) and/or doxorubicin (DOX, 15 nM) at 48 h. **, P < 0.01, *, P < 0.05 compared with the CON (untreated) cells.
Discussion
Dysregulation of autophagy plays a pivotal role in lymphoma cell resistance to chemotherapy,Citation15 suggesting that autophagy can be a potential therapeutic target.Citation16 Growing evidence has suggested that autophagy is modulated by PRKAA, a key energy sensor in the regulation of cellular metabolism and maintenance of energy homeostasis.Citation9 Here we showed that VPA, at a clinically achievable concentration,Citation17 interacted with chemotherapeutic agents to induce caspase-independent autophagic cell death through PRKAA activation. This was observed not only in T- and B-lymphoma cell lines, but also in nude-mouse xenograft lymphoma models. As previously reported, VPA synergizes with anticancer drugs, involving cellular processes like proliferation, differentiation, and apoptosis.Citation18-20 Based on our results, in addition to the above well-recognized biological effects, VPA may function as an active inducer of autophagy and a potential chemosensitizing agent in lymphoma therapy.
Due to an HDAC-dependent mechanism, VPA induced tumor cell autophagy through multiple signaling pathways, particularly the MTOR cascade.Citation8,21-23 However, it has recently been proposed that VPA can modulate autophagy via other targets than HDACs.Citation24 In the present study, induction of autophagy was observed in lymphoma cells cotreated with VPA and doxorubicin, irrespective of HDAC1 and HDAC3 activity, thus indicating an HDAC-independent effect of VPA on lymphoma cell autophagy when combined with chemotherapeutic agents. As mechanisms of action, VPA can reduce cellular IP3 level and stimulates PRKAA1/2-mediated cell autophagy, without affecting phosphorylation of MTOR. Indeed, aberrant IP3-IP3 receptor signaling is related to resistance to chemotherapeutic agents in lymphoid malignancies like chronic lymphocytic leukemia.Citation25,26 Lithium, another mood stabilizer, also decreases IP3 and activates autophagy through a MTOR-independent pathway.Citation27 To our knowledge, we provided the first evidence that VPA potentiated the effect of doxorubicin in lymphoma cell autophagy by an IP3-mediated PRKAA1/2 activation, suggestive of an alternative mechanism underlying the chemosensitizing activity of VPA.
Mitochondrial calcium signaling controls autophagy in cancer cells.Citation28 Regulating the mitochondrial permeability transition has the potential to bypass the tumor cell resistance to conventional chemotherapy.Citation29 IP3-induced blockade of mitochondrial calcium transfer results in enhanced phosphorylation of pyruvate dehydrogenase, followed by activation of PRKAA and induction of autophagy.Citation10 Here in lymphoma, the function of VPA as a modulator of calcium release to mitochondria was associated with its activity on tumor cell autophagy and chemosensitivity, further highlighting the role of regulating cellular calcium to improve clinical efficiency of anticancer drugs.
Studies have identified pronounced alterations in expression of proteins involved in calcium-permeable ion channels in cancers.Citation30,31 The mitochondrial permeability transition pore (mPTP), includes the ANT, PPID, and VDAC proteins, the specific group of calcium channel modulators that determine mitochondrial calcium transfer and are overexpressed in hematological malignancies.Citation32 Targeting mPTP sensitizes both T- and B-cell acute lymphoblastic leukemia cells to the anticancer agent resveratrol.Citation33 Alterations of the genes encoding the inner and outer membrane components of mPTPCitation32 were found in primary tumors of lymphoma patients, indicating that lymphoma cell chemoresistance might be caused by the abnormal function of mPTP. For example, the adenine nucleotide translocator family, the most abundant protein family of the mitochondrial inner membrane, regulates ADP/ATP exchanges across cytosol and matrix of mitochondria.Citation34 SLC25A31, one of the main isoforms of the adenine nucleotide translocator family,Citation34 can stabilize the mitochondrial transmembrane potential (ΔΨm), in turn preventing nuclear chromatin fragmentation and tumor cell death.Citation35 VDAC1 is upregulated in chronic lymphocytic leukemia and sensitizes acute lymphoblastic leukemia cells to chemotherapeutic agents.Citation36,37 Other reports also indicate PPID overexpression in many tumor types.Citation38 In addition to aberrations of these genes in lymphoma, our results showed that expression of SLC25A31 could be regulated by VPA treatment. Although further studies need to be carried out, abnormal expression of these genes might possibly contribute to the mitochondria calcium overload of lymphoma cells and suggested a new indicator of lymphoma cell sensitivity to VPA.
In summary, the chemosensitizing effect of VPA was linked to an HDAC-independent, IP3-mediated activation of PRKAA1/2 and induction of autophagy. Targeting autophagy could thus represent a promising therapeutic strategy in treating lymphoma.
Materials and Methods
Cells and reagents
T-lymphoma cell lines (Jurkat and H9) and B-lymphoma cell lines (SU-DHL-4 and Nalmawa) were available from American Type Culture Collection. Cells were grown in RPMI-1640 medium, supplemented with 10% heat-inactivated fetal bovine serum in a humidified atmosphere containing 95% air-5% CO2 at 37°C. VPA (V3640) was obtained from Sigma. 3-MA (189490), ZVAD-FMK (219007) and XeC (682160) were from Merck KgaA. Bafilomycin A1 (BafA1) (sc-201550) was from Santa Cruz Biotechnology.
Cell proliferation assay
Cells were seeded at a density of 5 × 105 cells per well in 96-well plates and incubated at 37°C with VPA, doxorubicin, alone or in combination. After 48-h incubation, 0.1 mg MTT was added to each well and the absorbance was measured at 490 nm by spectrophotometry.
Isobolographic analysis
To determine the synergistic effect of VPA combined with chemotherapeutic agents, the combination index (CI) method described by Chou and TalalayCitation39 and the Calcusyn software program (Biosoft, Cambridge, UK) for automated analysis were applied. This method allows quantitative determination of drug interactions, where CI<1, =1, and >1 indicate synergism, additive effect, and antagonism, respectively. When the drugs were combined, the CI values were calculated for each experiment and for combination experiment at a fixed concentration ratio. When at least 80% of CI values for a combination were <1, the drug combination was considered to be synergistic.
Colony formation assay
Cells were incubated with various concentrations of VPA and doxorubicin for 48 h. Single-cell suspensions were then prepared and mixed with an equal volume of 0.7% soft agar (Biowest, 111860) and plated in 6-well plates (3500 cells/well) for 21 d. After staining with MTT, colonies with a diameter of more than 0.1 mm were counted to calculate the colony formation rate as number of colonies/number of cells×100%. Isobolographic analysis was performed as mentioned above.
Flow cytometry
Cell autophagy was assessed using rabbit anti-human LC3B (Cell signaling Technology, 4108) as the primary antibody and DyLight 405 labeled anti-rabbit antibody (KPL, KPL072-08-15-06) as the secondary antibody. For cell cycle analysis, cells were collected, washed in phosphate-buffered saline (GeneMark, GB07-1) and fixed overnight in 75% ethanol at 20°C, treated with 1% RNaseA (Beyotime, ST578) for at least 15 min at 37°C and stained with 50 μg/ml propidium iodide (Beyotime, ST511). Cell apoptosis was analyzed using an ANXA5/Annexin V-Phycoerythrin kit (BD Bioscience, 559763). The flow cytometry data were collected by a FACS Calibur cytometer (Becton-Dickinson, San Jose, CA, USA) and analyzed by FlowJo software.
Western blot
Cells were lysed in 200 μl lysis buffer (0.5 M Tris-HCl, pH 6.8, 2 mM EDTA, 10% glycerol, 2% SDS and 5% β-mercaptoethanol). Protein extracts (20 μg) were electrophoresed on 10% SDS polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked with 5% nonfat dried milk in Tris-buffered saline (CWBIO, CW0042) and incubated for 2 h at room temperature with the appropriate primary antibody, followed by a horseradish peroxidase-conjugated secondary antibody. The immunocomplexes were visualized using a chemiluminescence phototope-horseradish peroxidase kit. Antibodies against LC3B-I/-II (4108), phospho-PRKAA1/2 (p-PRKAA1/2, Thr172, 2535), PRKAA1/2 (2532), phospho-MTOR (p-MTOR, 2971), MTOR (2972), phospho-RPS6KB (p-RPS6KB, 9205), RPS6KB (2708), phospho-EIF4EBP1 (p-EIF4EBP1, 9456), EIF4EBP1 (9644), phospho-ULK1 Ser555 (p-ULK1 Ser555, 5869), ULK1 (8054), CALB1/calbindin D28 (2173), ACTB/β-actin (4970) and the chemiluminescence phototope-horseradish peroxidase kit (7003) were obtained from Cell Signaling Technology. Anti-BECN1 antibody (ab51031) was from Abcam. Anti-SQSTM1 antibody (BML-PW9860) was from Enzo Life Sciences, Inc. Horseradish peroxidase-conjugated goat anti-mouse-IgG (sc-2005) and goat anti-rabbit IgG (sc-2004) antibodies were from Santa Cruz Biotechnology. ACTB was used to monitor equivalent protein loading.
Transmission electron microscopy
Cells and tissue samples were fixed overnight in 2% glutaraldehyde at 4°C, washed in 0.1 M cacodylate buffer, postfixed in 1% osmium tetroxide for 2 h at 4°C, dehydrated in graded ethanol and embedded in Epon 812 (TAAB Laboratories). Ultrathin sections were prepared, collected on copper grids, stained with uranyl acetate and lead citrate, and examined with an electron microscope (Philips CM120, Amsterdam, the Netherlands). Ultrastructural studies were focused on double-membrane autophagic vesicles named autophagosomes, a gold standard for autophagy and autolysosomes. Cell autophagy was quantified by estimating the volume occupied by autophagic structures (as percent of cellular volume using imageJ software in 20 cell profiles per sample), according to the guidelines for the use and interpretation of assays for monitoring autophagy.Citation11
Small-interfering RNA (siRNA) transfection
Cells were transfected with ON-TARGET plus human ATG5 siRNA SMARTpool (004374), ON-TARGET plus human BECN1 siRNA SMARTpool (010552), ON-TARGET plus human ULK1 siRNA SMARTpool (005049), ON-TARGET plus human HDAC1 siRNA SMARTpool (3065) and ON-TARGET plus human HDAC3 siRNA SMARTpool (130102) or ON-TARGET plus Non-Targeting pool (D-001810) as a negative control, using DharmaFECT2 transfection reagent (Dharmacon, T-2001-01) following the manufacturer's instructions.
Lentivirus packaging and transduction
To overexpress HDAC1, HDAC3, and CALB1, purified plasmids pGV358-HDAC1, pGV320-HDAC3, and pGV287-CALB1 were transfected into HEK-293T cells with package vectors using lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer's protocol. The supernatant fraction of HEK-293T cell cultures was then condensed to a viral concentration of approximately 3 × 10Citation8 transducing units/ml. The lentiviral particles were incubated with Jurkat cells for 72 h. The stably transduced cells were selected by EGFP or mCherry fluorescence protein after transduction.
Enzyme-linked immunosorbent assay (ELISA)
IP3 levels of cell lysates and tissue homogenates were measured with a human IP3 ELISA kit (Cusabio, CSB-E12636h) and a mouse IP3 ELISA kit (Cusabio, CSB-E13410m), respectively.
HDACs enzyme activity assay
Enzymatic activity of HDAC1 and HDAC3 were quantified using a nonisotopic HDAC colorimetric KIT (BioVision, K331-100) according to the manufacturer's instructions.
Confocal microscopy
Cells were simultaneously loaded with mitochondrion-selective MitoTracker Green FM (Life Technologies, M-7514, 20 nM) and calcium-selective Rhod-2 AM (Life Technologies, R1244, 2 µM) for 20 min at 37°C. Nuclei were counterstained with DAPI. Confocal laser-scanning microscopy was applied to observe colocalization of fluorescent staining by MitoTracker Green FM (505 to 530 nm) and Rhod-2 AM (585 nm) using 488 nm excitation and spectrally resolved detection.
Immunohistochemistry
Immunohistochemistry was performed on 5-µm paraffin sections with an indirect immunoperoxidase method using antibodies against p-PRKAA1/2 and BECN1. Expression levels were scored semiquantitatively based on the percentage of positive cells: +/++ <50%; +++/++++ 50 to 100%.
Genome-wide copy number variation (CNV) analysis
Genomic DNA was extracted using a Wizard Genomic DNA Purification kit (Promega). Genome-wide CNV genotyping was performed on frozen tumor samples of de novo patients with TCL (n = 20), DLBCL (n = 25) and reactive hyperplasia cases (n = 8), using Human 610-Quad_v1 (610 k SNP probes) or 660 W-Quad_v1 (660 k SNP probes) DNA Analysis BeadChips. The study was approved by the institutional review board with informed consent obtained in accordance with Helsinki Declaration. Regions were determined based on the Log R Ratio (LRR) of the signal intensity and B allele frequency of genotyping call from the sample using platform of GenomeStudio V2011.1 with CnvPartition 3.1.6 (Illumina). All the data were available on NCBI (accession number GSE47357).
Gene network and pathway analysis
The Human Genome U133 Plus 2.0 Array GeneChip microarray (Affymetrix) was performed on tissue samples of reactive hyperplasia patients, tumor samples of 10 TCL and 17 DLBCL patients. The data were analyzed by Expression Console software (Partek GS 6.5, Affymetrix). The data are available on NCBI (accession number GES47355). Genes of calcium pathway involved in both TCL and DLBCL were hierarchical clustered using MeV v4.8.1 (Dana-Farber Cancer Institute).
Real-time PCR
Total RNA was extracted using Trizol reagent and reverse transcribed using a PrimeScript RT Reagent kit with gDNA Eraser for Real Time PCR (TaKaRa, RR047A). Real-time PCR was performed on frozen samples of lymphoma and reactive hyperplasia cases. The primers were as follows: SLC25A31, Forward 5′-3′ TGGTGAGGCTAAACGGCAAT and Reverse 5′-3′ CCCTGTACCGCGAAGAACAT; GAPDH, Forward 5′-GAAGGTGAAGGTCGGAGTC-3′ and Reverse 5′-GAAGATGGTGATGGGATTTC-3′. A relative quantification was calculated using the 2-ΔΔCT method.
Murine model
Nude mice (5-to 6-wk-old) were obtained from Shanghai Laboratory Animal Center and injected subcutaneously with 4 × 107 Jurkat or SU-DHL-4 cells into the right flank. Treatments (10 mice per group) were started after tumor became about 0.5 cm × 0.5 cm in surface (d 0). The control group received dimethyl sulfoxide, while the other 3 groups received for 21 d oral VPA (0.2% w/v in the drinking water daily), intraperitoneal doxorubicin (1.5 mg/kg twice weekly), or both in combination, respectively. Tumor volumes were calculated as 0.5 × a × b2, where ‘a’ is the length and ‘b’ is the width.
Statistical analysis
Data were calculated as the mean ±SD from 3 separate experiments. The student t test was applied to compare 2 normally distributed groups and the Mann-Whitney U test to compare 2 groups which did not conform to normal distribution. Bonferroni was used to perform multiple comparisons. All the statistical analyses were evaluated using the Statistical Package for the Social Sciences (SPSS) 16.0 software (SPSS Inc.). P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1082024_supplemental_files.zip
Download Zip (782.9 KB)Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported, in part, by the National Natural Science Foundation of China (81520108003, 81325003, 81201862, 81172254 and 81101793), the Shanghai Commission of Science and Technology (11JC1407300), the Program of Shanghai Subject Chief Scientists (13XD1402700), Collaborative Innovation Center of Systems Biomedicine and the Samuel Waxman Cancer Research Foundation.
Related Research Data
References
- Vose JM, Chiu BC, Cheson BD, Dancey J, Wright J. Update on epidemiology and therapeutics for non-Hodgkin's lymphoma. Hematology Am Soc Hematol Educ Program 2002; 1:241-62; PMID:12446426; http://dx.doi.org/10.1182/asheducation-2002.1.241
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/10.1038/ncb0910-814
- Klionsky DJ, Thorburn A. Clinical research and Autophagy. Autophagy 2014; 10:1357-8; PMID:24991837; http://dx.doi.org/10.4161/auto.29159
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981-91; PMID:19211835; http://dx.doi.org/10.1091/mbc.E08-12-1248
- Schrump DS. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: mechanisms and potential clinical implications. Clin Cancer Res 2009; 15:3947-57; PMID:19509170; http://dx.doi.org/10.1158/1078-0432.CCR-08-2787
- Banreti A, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy 2013; 9:819-29; PMID:23466676; http://dx.doi.org/10.4161/auto.23908
- Brodie SA, Brandes JC. Could valproic acid be an effective anticancer agent? The evidence so far. Exp Rev Anticancer Ther 2014; 14:1097-100; PMID:25017212; http://dx.doi.org/10.1586/14737140.2014.940329
- Dong LH, Cheng S, Zheng Z, Wang L, Shen Y, Shen ZX, Chen SJ, Zhao WL. Histone deacetylase inhibitor potentiated the ability of MTOR inhibitor to induce autophagic cell death in Burkitt leukemia/lymphoma. J Hematol Oncol 2013; 6:53; PMID:23866964; http://dx.doi.org/10.1186/1756-8722-6-53
- Mack HI, Zheng B, Asara JM, Thomas SM. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012; 8:1197-214; PMID:22932492; http://dx.doi.org/10.4161/auto.20586
- Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270-83; PMID:20655468; http://dx.doi.org/10.1016/j.cell.2010.06.007
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Meijer AJ, Codogno P. Autophagy: regulation by energy sensing. Curr Biol 2011; 21: R227-9; PMID:21419990; http://dx.doi.org/10.1016/j.cub.2011.02.007
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Williams RS, Cheng L, Mudge AW, Harwood AJ. A common mechanism of action for three mood-stabilizing drugs. Nature 2002; 417:292-5; PMID:12015604; http://dx.doi.org/10.1038/417292a
- Levine B. Unraveling the role of autophagy in cancer. Autophagy 2006; 2:65-6; PMID:16874090 http://dx.doi.org/10.4161/auto.2.2.2457
- Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy 2006; 2:85-90; PMID:16874083 http://dx.doi.org/10.4161/auto.2.2.2463
- Shimizu R, Kikuchi J, Wada T, Ozawa K, Kano Y, Furukawa Y. HDAC inhibitors augment cytotoxic activity of rituximab by upregulating CD20 expression on lymphoma cells. Leukemia 2010; 24:1760-8; PMID:20686505; http://dx.doi.org/10.1038/leu.2010.157
- Scherpereel A, Berghmans T, Lafitte JJ, Colinet B, Richez M, Bonduelle Y, Meert AP, Dhalluin X, Leclercq N, Paesmans M, et al. Valproate-doxorubicin: promising therapy for progressing mesothelioma. A phase II study. Eur Respir J 2011; 37:129-35; PMID:20530048; http://dx.doi.org/10.1183/09031936.00037310
- Cimino G, Lo-Coco F, Fenu S, Travaglini L, Finolezzi E, Mancini M, Nanni M, Careddu A, Fazi F, Padula F, et al. Sequential valproic acid/all-trans retinoic acid treatment reprograms differentiation in refractory and high-risk acute myeloid leukemia. Cancer Res 2006; 66:8903-11; PMID:16951208; http://dx.doi.org/10.1158/0008-5472.CAN-05-2726
- Munster P, Marchion D, Bicaku E, Lacevic M, Kim J, Centeno B, Daud A, Neuger A, Minton S, Sullivan D. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC. Clin Cancer Res 2009; 15:2488-96; PMID:19318486; http://dx.doi.org/10.1158/1078-0432.CCR-08-1930
- Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro-oncology 2010; 12:328-40; PMID:20308311; http://dx.doi.org/10.1093/neuonc/nop005
- Ouyang DY, Xu LH, He XH, Zhang YT, Zeng LH, Cai JY, Ren S. Autophagy is differentially induced in prostate cancer LNCaP, DU145 and PC-3 cells via distinct splicing profiles of ATG5. Autophagy 2013; 9:20-32; PMID:23075929; http://dx.doi.org/10.4161/auto.22397
- Botrugno OA, Robert T, Vanoli F, Foiani M, Minucci S. Molecular pathways: old drugs define new pathways: non-histone acetylation at the crossroads of the DNA damage response and autophagy. Clin Cancer Res 2012; 18:2436-42; PMID:22512979; http://dx.doi.org/10.1158/1078-0432.CCR-11-0767
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; http://dx.doi.org/10.1038/nrd3802
- Akl H, Bultynck G. Altered Ca2+ signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochimica et biophysica acta 2013; 1835:180-93; PMID:23232185; http://dx.doi.org/10.1016/j.bbcan.2012.12.001
- Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, Rong YP, Molitoris JK, Lam M, Ryder C, et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood 2011; 117:2924-34; PMID:21193695; http://dx.doi.org/10.1182/blood-2010-09-307405
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170:1101-11; PMID:16186256; http://dx.doi.org/10.1083/jcb.200504035
- Rasola A, Bernardi P. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium 2014; 56:437-45; PMID:25454774; http://dx.doi.org/10.1016/j.ceca.2014.10.003
- Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov 2010; 9:447-64; PMID:20467424; http://dx.doi.org/10.1038/nrd3137
- Kondratskyi A, Yassine M, Kondratska K, Skryma R, Slomianny C, Prevarskaya N. Calcium-permeable ion channels in control of autophagy and cancer. Front Physiol 2013; 4:272; PMID:24106480; http://dx.doi.org/10.3389/fphys.2013.00272
- Szatkowski C, Parys JB, Ouadid-Ahidouch H, Matifat F. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Mol Cancer 2010; 9:156; PMID:20565939; http://dx.doi.org/10.1186/1476-4598-9-156
- Bonora M, Pinton P. The Mitochondrial Permeability Transition Pore and Cancer: Molecular Mechanisms Involved in Cell Death. Front Oncol 2014; 4:302; PMID:25478322; http://dx.doi.org/10.3389/fonc.2014.00302
- Zunino SJ, Storms DH. Resveratrol-induced apoptosis is enhanced in acute lymphoblastic leukemia cells by modulation of the mitochondrial permeability transition pore. Cancer Lett 2006; 240:123-34; PMID:16226372; http://dx.doi.org/10.1016/j.canlet.2005.09.001
- Clemencon B, Babot M, Trezeguet V. The mitochondrial ADP/ATP carrier (SLC25 family): pathological implications of its dysfunction. Mol Aspects Med 2013; 34:485-93; PMID:23506884; http://dx.doi.org/10.1016/j.mam.2012.05.006
- Gallerne C, Touat Z, Chen ZX, Martel C, Mayola E, Sharaf el dein O, Buron N, Le Bras M, Jacotot E, Borgne-Sanchez A, et al. The fourth isoform of the adenine nucleotide translocator inhibits mitochondrial apoptosis in cancer cells. Int J Biochem Cell Biol 2010; 42:623-9; PMID:20060930; http://dx.doi.org/10.1016/j.biocel.2009.12.024
- Shoshan-Barmatz V, Ben-Hail D, Admoni L, Krelin Y, Tripathi SS. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochimica et biophysica acta 2014; Nov 4. pii: S0005-2736(14)00375-7. PMID:25448878; http://dx.doi.org/10.1016/j.bbamem.2014.10.040
- Jiang N, Kham SK, Koh GS, Suang Lim JY, Ariffin H, Chew FT, Yeoh AE. Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (ALL). J Proteomics 2011; 74:843-57; PMID:21396490; http://dx.doi.org/10.1016/j.jprot.2011.02.034
- Suh DH, Kim MK, Kim HS, Chung HH, Song YS. Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front Oncol 2013; 3:41; PMID:23483560; http://dx.doi.org/10.3389/fonc.2013.00041
- Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006; 58:621-81; PMID:16968952; http://dx.doi.org/10.1124/pr.58.3.10