
Abstract
An accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER) leads to stress conditions. To mitigate such circumstances, stressed cells activate a homeostatic intracellular signaling network cumulatively called the unfolded protein response (UPR), which orchestrates the recuperation of ER function. Macroautophagy (hereafter autophagy), an intracellular lysosome-mediated bulk degradation pathway for recycling and eliminating wornout proteins, protein aggregates, and damaged organelles, has also emerged as an essential protective mechanism during ER stress. These 2 systems are dynamically interconnected, and recent investigations have revealed that ER stress can either stimulate or inhibit autophagy. However, the stress-associated molecular cues that control the changeover switch between induction and inhibition of autophagy are largely obscure. This review summarizes the crosstalk between ER stress and autophagy and their signaling networks mainly in mammalian-based systems. Additionally, we highlight current knowledge on selective autophagy and its connection to ER stress.
Abbreviations
| AKT1/AKT | = | v-akt murine thymoma viral oncogene homolog 1 |
| ALS | = | familial amyotrophic lateral sclerosis |
| AMPK | = | AMP-activated protein kinase |
| ATF4 | = | activating transcription factor 4 |
| ATF6 | = | activating transcription factor 6 |
| ATG | = | autophagy related |
| BAX | = | BCL2-associated X protein |
| BCL2/Bcl-2 | = | B-cell CLL/lymphoma 2 |
| ERAD | = | ER-associated degradation |
| BECN1 | = | Beclin 1, autophagy related |
| CASP4 | = | caspase 4, apoptosis-related cysteine peptidase |
| CASP12 | = | caspase 12 (gene/pseudogene) |
| DDIT3/CHOP | = | DNA-damage-inducible transcript 3 |
| DDIT4/REDD1 | = | DNA-damage-inducible transcript 4 |
| DAPK1 | = | death-associated protein kinase 1 |
| ECM | = | extracellular matrix |
| EIF2AK3/PERK | = | eukaryotic translation initiation factor 2-α kinase 3 |
| EIF2S1/eIF2α, eukaryotic translation initiation factor 2 | = | subunit 1 α, 35kDa |
| ER | = | endoplasmic reticulum |
| ERN1/IRE1 | = | endoplasmic reticulum to nucleus signaling 1 |
| FOXO | = | forkhead box O |
| HD | = | Huntington disease |
| HSPA5/GRP78 | = | heat shock 70kDa protein 5 (glucose-regulated protein, 78kDa) |
| Hyp-PDT | = | hypericin-mediated photodynamic therapy |
| ITPR/IP3R | = | Inositol 1,4,5-triphosphate receptor |
| LIR | = | LC3-interacting region |
| MAM | = | mitochondria-associated membrane |
| MAP3K5 | = | mitogen-activated protein kinase kinase kinase 5 |
| MAPK8/JNK | = | mitogen-activated protein kinase 8 |
| mtHTT | = | mutant HTT (huntingtin) |
| MTOR/mTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| NBR1 | = | neighbor of BRCA1 gene 1 |
| NFE2L2/NRF2 | = | nuclear factor, erythroid 2-like 2 |
| PRKAA1 | = | protein kinase, AMP-activated, α 1 catalytic subunit |
| PRKCQ | = | protein kinase C, theta |
| TRAF2 | = | TNF receptor-associated factor 2 |
| PI3K | = | phosphoinositide 3-kinase |
| PtdIns3K | = | class III phosphatidylinositol 3kinase complex |
| RAB7A/RAB7 | = | RAB7A, member RAS oncogene family |
| RIDD | = | regulated ERN1 dependent decay |
| RPS6KA3/RSK2 | = | ribosomal protein S6 kinase, 90kDa, polypeptide 3 |
| SESN2 | = | sestrin-2 |
| SQSTM1/p62 | = | sequestosome 1 |
| TRAF2 | = | TNF receptor-associated factor 2 |
| TRIB3/TRB3 | = | tribbles pseudokinase 3 |
| TSC1/TSC | = | tuberous sclerosis 1 |
| TSC2 | = | tuberous sclerosis 2 |
| UBL | = | ubiquitin-like |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| UPR | = | unfolded protein response |
| XBP1 | = | X-box binding protein 1. |
Introduction
The ER is a vast membranous network and the major assembly site for almost all secretory and integral membrane proteins. Through translocation mechanisms, nascent proteins enter secretory pathways in the ER,Citation1 where they are subsequently folded and assembled into higher order complexes through covalent modifications. The availability of molecular chaperones in the packed molecular domain of the ER makes it an ideal and unique milieu for proper protein folding as well as for identifying and marking improperly folded proteins for destruction. Indeed, in this way, only accurately folded proteins that pass ER quality control checkpoints are allowed to exit. In addition, co-translational and post-translational modifications of proteins in the ER finalize their 3-dimensional native structures,Citation2 facilitating their movement across the ER to endocytotic and exocytotic pathways. However, those quality control systems can be influenced by various extracellular and intracellular stimuli.
Because protein folding is a complex and error-prone process, the protein-folding capacity of the ER can easily be saturated under a number of physiological and pathological insults such as glucose deprivation, aberrant Ca2+ regulation, viral infection, environmental toxins, hypoxia, oxidative injury, hypoglycemia, mutant protein expression, aging, and simple increases in secretory protein synthesis. To buffer ER stress and orchestrate the recovery of ER function, cells use 4 different strategies. First, cells focus on translation attenuation for a few hours, thereby lessening the freshly prepared protein load into the ER until mRNAs encoding unfolded protein response (UPR) proteins are processed.Citation3 In a second attempt, the UPR upregulates the folding machinery by inducing ER chaperone genes. Third, the ER compartment proliferates to accommodate the high protein load and then begins ER-associated degradation (ERAD) of unfolded or misfolded proteins. ERAD mainly consists of 2 mechanisms: ubiquitin-proteasome-dependent ERAD (type I) and autophagy-lysosome dependent ERAD (type II). Type II ERAD represents an autophagic pathway in which both soluble and insoluble forms of misfolded proteins are targeted, whereas type I ERAD targets only soluble misfolded proteins.Citation4 Initially, it was thought that ER stress initiates autophagy only when aggregated proteins become excessive enough to overwhelm the canonical ubiquitin-proteasome-dependent ERAD. Current findings suggest, however, that ERAD-mediated partially processed proteins are also a target of autophagy, which includes all other unfolded proteins.Citation5 Finally, programmed cell execution is initiated when stress exceeds a given threshold and the ER is so severely impaired that compensatory mechanisms are no longer able to sustain its function.Citation6,7
Although ER stress and autophagy can function independently, they share a number of common features including protecting cells by relieving stress and inducing cell death under extreme conditions. Furthermore, altering the functions of one of these systems can influence the other. Therefore, ongoing work has attempted to integrate the signaling pathways responsible for the induction of autophagy upon ER stress and the cellular consequences in different cell types. Nevertheless, the relationship between autophagy and ER stress is not yet fully understood. This review provides a comprehensive overview of autophagy and its relationship to UPR signaling, and emphasizes recent advances in identifying the underlying mechanisms involved in ER-regulated autophagy that control several unanticipated pathways to cell fate regulation. In addition, we also review the role of ER stress in selective autophagy to provide a better understanding of the broad pathways by which ER stress can regulate autophagy.
UPR Signal Transduction Mechanism
In a perfectly functioning and “stress-free” ER, HSPA5/GRP78 (heat shock 70kDa protein 5 [glucose-regulated protein, 78kDa], a critical member of the HSP70 family, binds to and inhibits the 3 transmembrane ER stress sensors: ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1), EIF2AK3/PERK (eukaryotic translation initiation factor 2-α kinase 3) and ATF6 (activating transcription factor 6).Citation8 These 3 sensors transduce information regarding ER protein folding status to the nucleus via the cytosol to reestablish protein-folding capacity. Accumulated unfolded proteins in the ER bind to and recruit HSPA5 away from those complexes, thereby activating the sensors.Citation9 However, it has also been proposed that dissociation of HSPA5 alone is not always sufficient to activate ER stress sensors. Thus, direct interaction of the unfolded proteins with the sensors or other additional mechanisms might also regulate their activation.Citation10,11
ERN1/IRE1
ERN1/IRE1 is the most evolutionarily conserved bifunctional type I transmembrane protein of the UPR. It possesses both an endoribonuclease and a kinase domain that regulates XBP1 (X-box binding protein 1) processing and MAPK8/JNK1 (mitogen-activated protein kinase 8) activation, respectively.Citation12-14 Mammalian ERN1/IRE1 has 2 homologs, ERN1/IRE1α and ERN2/IRE1β, which differ with respect to their expression patterns and together compose a substantial part of the UPR signaling network ().Citation12
Table 1. Molecular mechanisms of UPR events and their connection to ER homeostasis and autophagy
EIF2AK3/PERK
Following ER stress, EIF2AK3, a type-I transmembrane protein with a kinase domain, causes phosphorylation of EIF2S1/EIF2α (eukaryotic translation initiation factor 2, subunit 1 α, 35kDa).Citation15 and NFE2L2/NRF2 (nuclear factor, erythroid 2-like 2).Citation16 and stimulates various downstream signaling pathways. Furthermore, transcription factor ATF4 is selectively translated under EIF2AK3 activation, which in turn activates the DDIT3/CHOP transcription factor and regulates stress-mediated cell death ().Citation9
Table 2. Involvement of the 3 axes of the UPR and ER Ca2+ and their molecular mechanisms in control of autophagy
Table 3. List of ER stress-regulated proteins and their role in autophagy inhibition
ATF6
ATF6, a type II transmembrane-activating transcription factor, has 2 homologs, ATF6/ATF6α and ATF6B/ATF6β. Under ER stress conditions, both of them move to the Golgi for processing and subsequent nuclear translocation. The ER lumenal domain of ATF6 is essential for stress sensing as it stably interacts with HSPA5 in unstressed cells and dissociates specifically during stress. However, the cytoplasmic domain of ATF6 is not necessary for its translocation to the Golgi apparatus ().Citation17
ER Stress and Cell Fate Determination
Mild to moderate ER stress-induced UPR signaling is seen as a compensatory mechanism, whereas severe and chronically prolonged ER stress deteriorates cellular functions and switches from an adaptation program to apoptosis to remove irreversibly injured cells.Citation18,19 Decreasing the ER protein load is the first attempt the ER makes to prevent such cell execution. ERN1 participates in the reduction of protein translation by degrading mRNAs in a process called regulated ERN1 dependent decay (RIDD), which is independent of the endoribonuclease function of ERN1. Specifically, RIDD reduces the production of membrane and secreted proteins to decrease the number of new proteins entering the ER.Citation20 In addition to RIDD, mRNAs that encode for proteins prone to accumulate in the ER undergo rapid degradation mediated by the ribonuclease domain of activated ERN1.Citation21 Similarly, ERN1 upregulation caused by XBP1 deficiency in pancreatic βcells enhances RIDD of cytosolic mRNAs through a negative feedback mechanism.Citation22 EIF2AK3 activation and EIF2S1 phosphorylation could also selectively inhibit the translation of proteins prone to aggregation, which has been attributed to induction of RIDD.Citation23 Furthermore, accumulation of proteins on the ER membrane leads to activation of the transcription factor NFKB, which initially stimulates the expression of cytoprotective genes, and this activity peaks when the kinase activity of ERN1 and the translation inhibitory activity of EIF2AK3 combine.Citation24,25
The exact mechanism controlling the transition of ER stress from “cytoprotective” to “cytotoxic” is not yet fully understood. In cases in which mild ER stress activates all UPR sensors, survival is favored as a consequence of increased instability of the mRNAs and proteins that promote apoptosis compared to those that facilitate protein folding and adaptation. In particular, prolonged ER stress attenuates ERN1 and ATF6 activity in mammalian cells.Citation26 Conversely, EIF2AK3 signaling for translational attenuation and pro-apoptotic transcription regulation remains active under the same conditions.Citation26
ER stress can stimulate at least 3 apoptotic signals, including both intrinsic and extrinsic pathways.Citation27 The first and most vital mechanism of ER stress-induced apoptosis is mediated through DDIT3, which regulates cell death through diverse mechanisms. Different branches of the UPR regulate DDIT3 at both the transcriptional and post-translational levels. Genes encoding DDIT3 contain promoter-binding sites for transcription factors from all 3 branches of the UPR, ATF4, ATF6, and XBP1.Citation28,29 However, the EIF2AK3-EIF2S1-ATF4 branch is dominant over the other branches. ER stress-induced death is absent in DDIT3−/− cells, possibly due to a decreased ER protein load. In addition, increasing biosynthesis of ER client proteins is caused mainly by dephosphorylation of EIF2S1 proteins by PPP1 (protein phosphatase 1), and DDIT3 regulates that pathway by activating PPP1R15A/GADD34.Citation30 Furthermore, DDIT3 promotes transcriptional activation of a myriad of pro-apoptotic factors: TNFRSF10B/DR5 (tumor necrosis factor receptor superfamily, member 10b), TRIB3/TRB3 (tribbles pseudokinase 3), CA6 (carbonic anhydrase VI), ERO1A (endoplasmic reticulum oxidoreductase α), and members of the BCL2 family of proteins.Citation31
ERN1 activation can also induce cell death, and that activation could partly converge into DDIT3-mediated pathways. Specifically, activated ERN1 recruits the adaptor protein TRAF2 (TNF receptor-associated factor 2), which in turn forms a complex with MAP3K5/ASK1 (mitogen-activated protein kinase kinase kinase 5).Citation32 The ERN1-TRAF2-MAP3K5 complex then goes on to activate MAPK/p38 (mitogen-activated protein kinase) and MAPK8. Among those targets, MAPK8-mediated phosphorylation of BCL2 (B-cell CLL/lymphoma 2) family of proteins such as pro-apoptotic BCL2 and BCL2L11/BIM promote cell death, whereas MAPK/p38 activation converges on a common cell death pathway induced by DDIT3.Citation32 Post-translational regulation of DDIT3 in the form of phosphorylation by MAPK/p38 results in the activation of DDIT3-mediated apoptosis.Citation33 In this way, DDIT3-induced cell death pathways can intersect both the ERN1 and EIF2AK3 corridors upon ER stress. Further downstream, caspases play a substantial role in cell demise caused by ER stress. Specifically, ER stress-inducing agents can activate the catalytic properties of ER membrane-localized human CASP4/caspase-4 and murine CASP12/caspase-12.Citation34 Interestingly, CASP4- and CASP12-mediated cell death is cytosolic Ca2+ dependent, evidenced by the inhibition of CASP4- and CASP12-depndent cell execution under concomitant treatment with the Ca2+ chelator BAPTA-AM and ER stress inducers.Citation35 Moreover, cell death signaling stimulated by causes other than ER stress cannot activate CASP4 and CASP12, suggesting that CASP4- and CASP12-dependent cell death is ER-stress specific.Citation34 However, Obeng et al. demonstrated that CASP4 and CASP12 are not necessarily important for apoptosis induction under ER stress conditions.Citation36 In addition to CASP4 and CASP12, other caspases such as CASP2 and CASP8 can trigger BID cleavage leading to loss of mitochondrial membrane potential and thereby connecting the signaling events from ER stress to the mitochondrial death machinery.Citation37
Salient Features of Cells and Animals with Knocked Out Upr Components
A compromised UPR caused by genetic manipulation in vivo and in vitro shows various specialized physiological and pathological outcomes in distinct organs.
Deletion of the EIF2AK3 axes of the UPR
Murine Eif2ak3 knockdown is not embryonic lethal; however, postnatal death is observed, probably due to severe cell degeneration, hyperglycemia, exocrine pancreatic impairment, and diabetes mellitus.Citation17 Strikingly, Eif2ak3 knockdown results in aberrant ER stress by stimulating global ER stress markers. In addition, severe osteopenia and spinal curvature, skeletal dysplasia, and compromised locomotor activity are also consequences of in vivo Eif2ak3 deletion.Citation38 Conditional mammary tissue-specific knockdown of Eif2ak3 can lead to a dramatic decrease in autophagy level.Citation39 Similarly, in vitro knockdown in cardiomyocytes causes compromised autophagy (furthers details in the following section).Citation40
A homozygous EIF2S1 S51A mutant results in postnatal death within 24 h of birth, which could be caused, at least in part, by severe hypoglycemia due to impaired gluconeogenesis and glycogen synthesis.Citation29 Although it remains to be determined whether in vivo Eif2s1 deficiency is associated with autophagy regulation, conditional nonphosphorylated in vitro knockin (S51A) is associated with compromised autophagy.Citation41 Mice that lack Atf4 are neither embryonic lethal nor vulnerable to postnatal death. However, various diseases such as microphthalmia,Citation42,43 growth retardation,Citation44 pancreatic hypotrophy,Citation45 and hematological defects, including severe anemia,Citation46 are observed during their life spans. DDIT3 ablation is not associated with embryonic death or developmental defects; rather it protects essential organs such as the lungs and kidneys from stress-associated injury. However, conditional knockdown results in liver injury and compromised autophagy, similar to the results seen in the in vitro knockdown model.
Deletion of the ATF6 axes of the UPR
Although no embryonic lethality or postnatal death results, genetic deficiency of Atf6 in mice results in intolerance to ER stress. Acute liver injury, kidney damage, and β-cell degeneration-associated diseases are aggravated in atf6−/− animals challenged with ER stress inducers compared with their wild-type counterparts.Citation47 Furthermore, combined knockdown of Atf6 and Atf6b is embryonic lethal.Citation48 Compromised autophagy in atf6−/− MEF cells suggests its involvement in the autophagy process.Citation49
Deletion of the ERN1 axes of the UPR
In terms of embryonic death, ern1−/− and xbp1−/− mice display distinguishing phenotypes compared to other UPR branches. Both types of animals die at the embryonic stage.Citation50-52 Ern1 knockdown can cause improper functioning of the placental blood vessels and reduce production of VEGF (vascular endothelial growth factor).Citation50 Similarly, embryonic lethality in xbp1−/− €mice is caused by hypoplastic fetal livers and depleted hematopoiesis that leads to severe anemia.Citation52 More interestingly, embryonic lethality can be reversed by reconstituting XBP1 and ERN1 expression.Citation50,53 Conditional adult knockdown of both of these proteins can lead to enhanced autophagy (further details in the following section). However, in vitro knockdown of ERN1 and XBP1 can jeopardize ER stress and starvation-induced autophagy.Citation54
Autophagy
Autophagy is a genetically programmed ancient catabolic system first described by Christian de Duve in the late 1950s after observing cytosolic vacuoles in mammalian cells under an electron microscope.Citation55 During the past 3 decades of autophagy research and discovery, comprehensive studies in yeast have shed significant light on its core molecular mechanism.Citation56-58 To date, numerous studies have been performed to understand the status of autophagy in cells, and scientists have found that cells maintain optimum activity by sustaining a minimum basal level of autophagy.Citation59 In this particular context, basal autophagy can be stimulated to play a crucial role in cellular adaptation to starvation and other cellular stress by endolysosomal degradation and elimination of long-lived and misfolded proteins, potentially detrimental cellular substances, defective organelles, and invading pathogens.Citation60,61 In addition, autophagy acts as a source of energy and building blocks for the biosynthesis of new macromolecules by recycling metabolites produced by lysosomal proteolysis. Likewise, autophagy can regulate the energy balance of not only single cells, but also entire organisms through the enhancement of metabolic activity. Furthermore, autophagy is crucial for cell growth and differentiation, tumor suppression, innate and adaptive immunity, life-span extension, and cell death.Citation62 In this way, autophagy plays a substantial role throughout the entire life span of an organism. For example, during pre-implantation processes, cytoplasmic components of the oocytes need to be cleared, whereas post-fertilization requires removal of paternal mitochondria; both processes are mediated by autophagy.Citation63,64 Furthermore, energy production in newborn mice substantially depends on autophagic processes. Specifically, prior to birth, the fetus uses maternal nutrients as an energy source, but that becomes unavailable just after birth due to placental cessation. Autophagy acts as an energy source at this critical stage by recycling metabolites.Citation65
At least 3 subtypes of autophagy have been documented in mammalian cells. They differ according to their physiological function and the mechanism of cargo transport for proteolytic degradation at common destination lysosomes. These subtypes are formally specified as chaperone-mediated autophagy, macroautophagy, and microautophagy.Citation61,66 Among those mechanisms, macroautophagy, the most widely studied mechanism, uses cytosolic double-membrane vesicles called autophagosomes to deliver cytosolic content to lysosomes.Citation67 In contrast, microautophagy, the least characterized mechanism, results in the isolation of cytoplasmic proteins and organelles by inward introversion or septation of lysosomal membranes.Citation68 In chaperone-mediated autophagy, targeted proteins reach the proteases of the lysosomal matrix by direct translocation across the lysosomal membrane with the help of LAMP2A (lysosomal-associated membrane protein 2A) and lysosomal HSPA8/hsc73/lys-HSC70.Citation69
Molecular mechanism of autophagy
Mechanistically, autophagy is a complex process that can be categorized into several sequential steps at the molecular level. Numerous cellular and external cues can initiate autophagy, which is followed by cargo selection and packaging, expansion of the phagophore membrane, closure to generate the completed autophagosome, fusion of the matured autophagosomes with lysosomes, degradation of the autophagosomal content by lysosomal hydrolases, and finally efflux of the breakdown products.Citation70-72 Molecularly, autophagy initiation is controlled by the ULK1/2 complex, which includes ULK1 (the mammalian homolog of yeast Atg1), ATG13, ATG101 (a novel autophagic factor), and RB1CC1/FIP200 (a functional ortholog of yeast Atg17),Citation73 which remains inhibited by MTOR (mechanistic target of rapamycin [serine/threonine kinase]). Starvation and other stress conditions inhibit MTOR and activate the ULK1/2 complex, which in turn facilitates the formation of the phagophore.Citation74 Phagophore formation involves a class III phosphatidylnositol 3kinase complex (PtdIns3K), composed of BECN1/BECLIN 1, PIK3C3/VPS34, PIK3R4/p150, ATG14, and UVRAG.Citation75-77 Anti-apoptotic BCL2 or BCL2L1/BCL-XL holds BECN1 at bay by directly binding to BECN1s BH3 domain, and in doing so restrains it from constructing an autophagy-inducing PtdIns3K ().Citation78
Figure 1. Schematic diagram highlighting ER stress signaling in the control of autophagy pathways. (A) MTORC1 regulation by ER stress: Autophagy initiation begins with MTOR inhibition. Following EIF2AK3 activation, transcription factor ATF4 leads to the upregulation of SESN2 and DDIT4. Further downstream, DDIT3 upregulates TRIB3. SESN2 and DDIT4 directly inhibit the kinase activity of MTORC1, whereas TRIB3 decreases AKT1 phosphorylation, which in turn inhibits MTORC1. (B) AMPK regulation by ER stress: ER stress integrates all UPR branches at AMPK by elevating cytoplasmic Ca2+, upregulating RPS6KA3, and depleting cellular ATP. ERN1 activation results in stimulation of RPS6KA3, which in turn activates AMPK and subsequently activates the ULK1 complex. EIF2AK3 and ATF6 activate AMPK via DDIT3-mediated ATP depletion. Elevated cytosolic Ca2+ activates CAMKK2, causing phosphorylation of AMPK and subsequent ULK1 activation. (C) BECN1 and ATG gene regulation by ER stress: MAPK8 (ERN1 dependent) and DAPK1 (Ca2+ dependent) activation have been implicated in the formation of PtdIns3K through the phosphorylation and dissociation of BCL2 and BECN1, respectively. Transcription factors JUN, XBP1, ATF4, DDIT3, and ATF6 from all UPR sensors induce expression of BECN1 and other ATG genes essential for autophagy. Ca2+-dependent phosphorylation of PRKCQ leads to its colocalization with LC3 on the elongating phagophore and facilitates autophagosome formation.
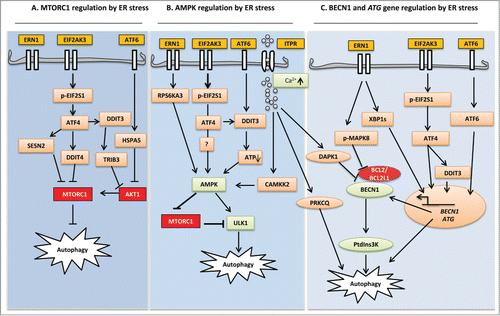
Phagophore expansion and closure require several ubiquitin-like (UBL) proteins that take part in 2 conjugation reactions.Citation79,80 ATG12-ATG5-ATG16L1, a product formed in the first conjugation reaction, stimulates the recruitment and conversion of proteolytically processed cytosolic MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), LC3-I, to the membrane-bound, lipidated form, LC3-II.Citation81-83 The nascent LC3 is cleaved by the cysteine protease ATG4 in the second reaction and this is followed by conjugation with membrane-bound phosphatidylethanolamine and subsequent incorporation into the phagophore membrane with the help of the ATG7 and ATG3 enzymes.Citation79,84 ATG4 can cause delipidation (deconjugation) and recycling of LC3-II on the outer surface of the autophagosome,Citation85 whereas inner autophagosomal membrane-bound LC3-II reflects the abundance of autophagosomes and their ability to reach autolysosomes for degradation. Hence, LC3-II is a marker protein for autophagy detection.Citation86
Cytosolic autophagosomes need to access lysosomes at the perinuclear region of the cells. Various cytosolic and lysosomal proteins are involved in this complex and incompletely understood process. Cytoskeletal microtubules and the motor protein dynein seem to play major roles in autophagosomes trafficking to lysosomes.Citation87 Similarly, autophagosomal transport and fusion is substantially mediated by the small GTPase RAB7A/RAB7 and its association with FYCO1 (FYVE and coiled-coil domain containing 1) and lysosomal membrane proteins LAMP1/2 accelerate the fusion machinery.Citation88,89 ATG9, an integral membrane protein, is also thought to be involved in the fusion machinery by transporting vesicle fusion proteins, SNAREs, and autophagic membranes to growing phagophores.Citation90 Furthermore, the class C vacuolar protein sorting-HOPS complex regulates autophagosome tethering with lysosomes by activating RAB7A.Citation91 Finally, autophagosomal-sequestered cargo is degraded by lysosomal/vacuolar acid hydrolases such as CTSB, CTSD, and CTSL immediately after accessing the lysosomal lumen.Citation92
Autophagy Induced by 3 UPR Axes
The 3 canonical branches of the UPR regulate autophagy in different ways during ER stress. Correlation between autophagosome formation and ER expansion caused by ER stress was first described in 2006.Citation93 Specifically, electron microscopy studies in yeast revealed that ER volume increases 5-fold under ER stress and that cells undergoing such stress exhibit concomitant increases in autophagosome abundance.Citation94 According to Ogata et al., tumor cells activate autophagy as a survival mechanism to escape ER stress-induced toxicity. ERN1-mediated MAPK8 phosphorylation appears to be a major regulator in this pathway. MAPK8 is considered a “stress-regulated protein kinase,” whereby stress-induced autophagy and apoptosis depend substantially on MAPK8 activation.Citation93,95 Specifically, ERN1 activation leads to MAPK8 phosphorylation, and that might initiate autophagy and allow cells to adapt to stress.Citation93 The autophagy-related gene BECN1 is the leading downstream regulator of MAPK8, and its activation is followed by direct phosphorylation of BCL2, which in turn disrupts the interaction between BECN1 and BCL2 and induces autophagy in tumor cells ().Citation96,97 Interestingly, BECN1 is transcriptionally upregulated by JUN/c-Jun, a MAPK8-dependent transcription factor, in a human cancer cell model treated with the ER stress inducers ceramide and topotecan.().Citation98 In addition to kinase activity, the endoribonuclease activity of ERN1 also participates in the induction of autophagy. Spliced XBP1 binds directly to the BECN1 promoter in the nucleus and promotes an autophagic response via transcriptional upregulation of BECN1 ().Citation54 Notably, MAPK8-mediated autophagy in rat aortic smooth muscle cells under oxidative stress is ERN1 independent,Citation99 suggesting an alternative pathway for activation of MAPK8. The ubiquitin proteasome system and autophagy degrade intracellular content independently. However, proteasome inhibition in a colorectal carcinoma model triggers autophagy via ER stress-mediated MAPK8 phosphorylation and is independent of ERN1s endoribonuclease activity.Citation100 That study suggests a functional linkage between 2 independent protein degradation pathways under which autophagy can be compensatory by enabling cells to cope with stress conditions that compromise the proteasome machinery.
Following ER stress, MAPK8 regulates both autophagy and apoptosis. Scientists have done kinetic studies to uncover the molecular switch that regulates the transition of MAPK8-dependent autophagy to apoptosis under stress conditions and discovered that the BH3 domain-containing proteins such as BECN1 and BAX are the main regulatory components.Citation101 Transient activation of MAPK8 activates autophagy by dissociating small amounts of BCL2 from BECN1 while leaving the interaction between BCL2 and BAX unchanged. In contrast, prolonged activation of MAPK8 induces apoptosis by fully dissociating BAX from BCL2, leading to BAX activation.Citation101
The main events of autophagy, such as phagophore formation and maturation, are substantially maintained by LC3-II and the ATG12-ATG5 conjugate.Citation102 To sustain autophagy flux, transcriptional upregulation of the corresponding autophagy genes is crucial. Evidence suggests that under ER stress conditions, the EIF2AK3 branch of the UPR helps to regulate those genes.Citation103 The association of EIF2AK3 in ER stress-mediated autophagy was first documented by Kouroku et al.Citation104 Specifically, they showed that aggregated polyglutamine (polyQ) proteins in the cytosol reduce proteasome activity, leading to the induction of autophagy through activation of the EIF2AK3 branch of the UPR. Under hypoxic conditions, transcriptional upregulation of LC3 and ATG5 depends on EIF2AK3-dependent ATF4 and DDIT3 induction ().Citation105 Similarly, ATG12 mRNA and protein levels are also enhanced by phosphorylated EIF2S1, a downstream regulator of EIF2AK3.Citation104 EIF2AK3-mediated activation of ATF4 is sufficient for upregulation of a dozen autophagy genes, including MAP1LC3B, BECN1, ATG3, ATG12, and ATG16L1 (), whereas the interaction of ATF4 and DDIT3 causes transcriptional upregulation of SQSTM1/p62, NBR1, and ATG7. In addition, ATG10, GABARAP, and ATG5 transcription is followed by activation of the DDIT3 transcription factor alone.Citation103 Furthermore, DDIT3 can stimulate autophagosome formation through downregulation of BCL2 expression.Citation106 Therefore, EIF2AK3 appears to be the most efficient UPR branch with respect to the regulation of autophagy genes under ER stress. EIF2AK3 maintains its autophagy regulatory activity in different pathological models, as well. For example, cardiomyocytes undergo an autophagy process as a protective mechanism upon ER stress triggered by lipopolysaccharides, and genetic ablation of EIF2AK3 leads to autophagy inhibition and the death of cardiomyocytes upon treatment with lipopolysaccharides.Citation40 Several mechanisms enable cancer cells to induce autophagy for their survival. Koumenis et al. demonstrated that in mouse and human lymphomas the proto-oncogene product MYC triggers ER stress by activating the EIF2AK3-EIF2S1-ATF4 branch and thereby inducing cytoprotective autophagy.Citation107 Similarly, autophagy enables ovarian cancer cells to survive under metformin-induced ER stress, and its inhibition results in cell death.Citation108 Alternatively, autophagic cell death can be a consequence of EIF2AK3 activation. For instance, nelfinavir and bortezomib, which are ER stress-inducing anticancer drugs, trigger autophagy by upregulating ATF4-mediated SESN2 (sestrin 2), an endogenous MTOR inhibitor ().Citation109 Through this process, both nelfinavir and bortezomib lead to autophagy-dependent growth arrest and the radiosensitization of cancer cells.Citation109 In summary, EIF2AK3-EIF2S1-ATF4 pathway-mediated autophagy can have both a cytotoxic and cytoprotective capacity; however, cytoprotective autophagy is predominant under conditions of ER stress.Citation107 The duration/intensity of the stress and cell types might be a vital and deciding factor for the contradictory function of the EIF2AK3-EIF2S1-ATF4 pathway.
Notably, it has been observed that proteasome inhibition stimulates ATF4-dependent LC3 conversion in breast cancer cells, which connects ER stress to autophagy in an EIF2AK3-independent manner.Citation110 Interestingly, the role of EIF2S1 in autophagy regulation extends beyond ER stress. For example, viral infections and amino acid starvation can cause EIF2S1 phosphorylation-dependent autophagy, regardless of ER stress.Citation41,111,112 Collectively, those findings indicate that in particular settings, EIF2S1-ATF4 signaling is enough to trigger autophagy, and the role of EIF2AK3 phosphorylation is negligible. It is also clear that downstream EIF2AK3 regulators are mainly associated with the enhancement of autophagy. Thus, it could be important for future work to focus on the threshold of the EIF2AK3-EIF2S1 branch required to switch between compensation and cell execution.
The ATF6 branch of the UPR is the least characterized branch in the context of ER stress and autophagy. Nevertheless, transcription activity of ATF6 is involved in autophagy induction through upregulation of HSPA5 expression and subsequent downregulation of AKT1/AKT (further details in the following section).Citation113 In addition, the ATF6-associated transcription factor CEBPB (CCAAT/enhancer binding protein [C/EBP], β) has been linked to IFNG-dependent autophagy through the expression of DAPK1 (death-associated protein kinase 1) ().Citation114 The subsequent phosphorylation of BECN1 results in diminished affinity for BCL2 and leads to the formation of a complex between the autophagy initiator BECN1 and PIK3C3.Citation115,116 Interestingly, genetic ablation of ATF6 causes repression of DAPK1 expression and ultimately failure of autophagy induction during treatment with IFNG.Citation114
Upr Branches Induce Autophagy by Regulating Akt1-Mtor Signaling
In mammals, growth factors, nutrients, ER stress inducers, and other stressors act as on-off switches for MTOR-regulated cell growth, survival, and energy balance. Among the 2 different forms of MTOR (MTOR complex 1 [MTORC1] and MTORC2), MTORC1 is known as the master regulator of nutrient signaling and autophagy.Citation117 The serine/threonine kinase AKT1 is a positive upstream regulator of MTOR, and initiation of autophagy depends substantially on the AKT1-MTOR pathway. Negative regulation of this pathway leads to the release of ULK1 from inactive MTOR and the subsequent formation of autophagosomes. One common consequence of ER stress is downregulation of AKT1, which in turn contributes to the induction of autophagy by decreasing MTOR activity.Citation118 Furthermore, it has been well established that the transcriptional activity of ATF6 upregulates the ER chaperone HSPA5 (), which triggers autophagy in placental choriocarcinoma cells by limiting AKT1 phosphorylation activity.Citation113 However, although ER stress could contribute to AKT1 inhibition, a more complicated scenario is likely that moderate and recoverable ER stress can activate 2 major cytoprotective signaling cascades, involving AKT1 and MAPK.Citation119 Indeed, rapid activation of phosphoinositide 3-kinase (PI3K)-AKT1-MTORC1 signaling and cell survival upon pharmacological induction of the UPR appears to be regulated by the ATF6 branch.Citation120 It is therefore plausible that autophagy can be negatively regulated under such conditions. Although the factors associated with ER stress-mediated switching of AKT1 activation to inhibition are not fully understood, stress intensity and duration are thought be crucial. Recent observations have also implicated an additional level of complexity: activated AKT1-dependent cell survival is due at least in part to upregulation of HSPA5.Citation121 Specifically, the failure of AKT1 activation in a HSPA5 knockout model could depend on upregulated PPP2/PP2A (protein phosphatase 2) activity that results in dephosphorylated AKT1.Citation122 Thus, it is surprising that although HSPA5 can downregulate AKT1, it can also promote cell survival in some situations.Citation122 This finding suggests that stimulation of HSPA5 signaling could be a novel strategy to induce autophagy and simultaneously promote AKT1 activation. Thus, the role of HSPA5 signaling in autophagy requires more comprehensive study.
Both the upstream and downstream signaling associated with MTOR can be regulated by ER stress, which can either stimulate or inhibit the anabolic activity of MTOR.Citation123 PI3K-AKT1 stimulates MTORC1 as a cell growth mechanism via phosphorylation and inhibition of TSC1 (tuberous sclerosis 1) and TSC2, which are negative regulators of MTOR,Citation124 whereas ER stress limits MTOR activity by stimulating TSC1/2.Citation125 Likewise, TSC deficiency impedes ER stress-mediated autophagy induction via constitutive activation of MTOR.Citation118 Moreover, cells with hyperactive MTORC1 due to TSC1/2 knockdown exhibit increased basal levels of ER stress that can be further augmented by treatment with stress inducers,Citation126 indicating that TSC2 is crucial for canonical feedback of ER stress. Conversely, MTOR deactivation under stress conditions is associated with downregulation of the AKT1-MTOR pathway in part via TSC1/2.Citation118 According to those studies, TSC1/2 is the common target for AKT1 and ER stress, and their effects are inversely related with respect to autophagy.
Following ER stress, ATF4 and CEBPB regulate the dephosphorylation of MTORC1 via DDIT4/REDD1 (DNA-damage-inducible transcript 4) expression,Citation127 which mainly acts on the TSC1/2 complex. DDIT4−/− breast cancer cells are unable to dephosphorylate MTOR under ATP depletion conditions or direct activation of AMPK.Citation128 Furthermore, EIF2AK3-EIF2S1-ATF4-dependent DDIT4 induction and subsequent inhibition of MTORC1 is also evident in in vivo models.Citation129 These data indicate that TSC-dependent MTOR regulation is primarily mediated by the activated EIF2AK3-EIF2S1-ATF4 branch of the UPR.
The ERN1-MAPK8 pathway appears to be the most abundant UPR branch downstream of activated MTORC1. Chronic activation of MTORC1, following genetic or pharmacological inhibition of TSC1/2, downregulates AKT1, which in turn selectively activates ERN1-MAPK8.Citation130 An apoptotic signal is initiated, probably caused by increased protein synthesis and high protein load in the ER, along with depleted cell survival-signaling AKT1 following activation of ER stress. It has also been suggested that constitutive activation of MTORC1 triggers an incomplete induction of the UPR, whereby all branches of the UPR other than the EIF2S1 branch are compromised.Citation131 Thus, it is intriguing to consider the possibility that existing studies describe shared pathological conditions whereby long-term activation of MTORC1 is linked to cellular demise caused by ER stress. This highlights a peculiar anomaly in that MTOR, a positive regulator of cell growth, can also stimulate cell death signaling in particular contexts.
TRIB3, an ER stress-associated protein, negatively regulates the AKT1-MTOR axis by directly binding to AKT1.Citation132 Various forms of ER stress can stimulate TRIB3 expression. The inability of ATF4-DDIT3-deficient malignant gliomas to induce TRIB3 under conditions of ER stress indicates that TRIB3 transcriptional activation is ATF4-DDIT3 dependent.Citation133 Δ9-tetrahydrocannabinol, an active component of marijuana, can induce TRIB3-dependent autophagy in a human glioblastoma multiforme model as well.Citation132 However, a negative feedback mechanism of hyperactivated TRIB3 degrades the transcriptional activity of ATF4 and DDIT3.Citation132,134 Thus, TRIB3-mediated stimulation of autophagy appears to be unnecessary for cell survival. Moreover, autophagy induction that relies on the transcription activity of ATF4 and DDIT3 reflects a cytotoxic condition, and that might happen only during the end stage of ER stress.
ER-localized MTORC2 is stress sensitive and interacts with the ER proteins PDIA3/GRP58 and HSP70.Citation135 AKT1 is also found at the ER and acts as a direct substrate of MTORC2. However, it is unclear how MTORC2 is regulated. ERstress-mediated phosphorylation of GSK3B/GSK-3β (glycogen synthase kinase 3 β), an AKT1 substrate, inhibits AKT1 signaling through RICTOR phosphorylation.Citation136 This finding supports the possibility that in addition to MTORC1-dependent AKT1 inhibition, MTORC2 could also regulate ER stress-mediated AKT1 activity, either by stimulating autophagy pathways or by triggering apoptotic signaling. Further studies are required to clarify this issue. However, the relationship between AKT1 signaling and GSK3B regulation appears to be complex. Specifically, in a model of AKT1 hyperactivation, apoptosis and ER stress are decreased by AKT1-mediated GSK3B inhibition.Citation137 It is therefore likely that under ER stress conditions, AKT1 and GSK3B act in contrasting fashion and dominate each other's roles in the cell. Taken together, multiple lines of evidence now indicate that prolonged ER stress activates apoptotic pathways by downregulating AKT1. However, at the early stage of ER stress, stimulated AKT1 acts as a survival kinase via MTORC1 activation. Thus, it is plausible to suggest that ER stress-regulated MTOR-dependent autophagy could be crucial for cell survival when AKT1 is inhibited (Table 2).
Upr Branches Induce Autophagy by Regulating Ampk Signaling
AMPK is an energy sensor that is mainly activated when the energy state of a cell is compromised, such as with increased levels of AMP and ADP. A wide array of metabolic stress stimuli can stimulate AMPK. Autophagy induction via AMPK stimulation is thought to be separated into 2 regulated pathways. Previously, it was shown that AMPK-dependent autophagy induction is based only on MTORC1 inactivation through TSC regulation, followed by dissociation and activation of ULK1.Citation138 However, elegant work from Kim et al. and Egan et al. revealed that AMPK induces autophagy by directly phosphorylating ULK1 at 6 different sites (S467, S555, T574, S637, S777, S317).Citation139,140 In addition, feedback signaling from active AMPK minimizes MTORC1 activation, resulting in an increase in activated ULK1 levels.
A key function of AMPK is to defend against ER stress-mediated cytotoxicity, primarily by inducing autophagy. The resulting bulk degradation of cytosolic cargo is beneficial for stressed cells. Supporting this notion, recent work from Lee et al. demonstrated that AMPK activation has suppressive effects on ER stress-mediated cellular toxicity upon albumin treatment.Citation141 In addition, pharmacological stimulation of AMPK reduces ER stress-stimulated DDIT3 and caspase activation, and genetic ablation of PRKAA1 renders cells susceptible to stress-mediated demise.Citation142 More recently, Cheng et al. demonstrated that RPS6KA3/RSK2 (ribosomal protein S6 kinase, 90kDa, polypeptide 3) (), a serine/threonine protein kinase, links ER stress and autophagy by activating AMPK. Specifically, silencing RPS6KA3 expression suppresses autophagy induction and aggravates ER stress in a human breast cancer model. It is plausible that an AMPK-mediated protective role might be deregulated, which in turn exacerbates the severity of stress. In support of this, ERN1 depletion blocks RPS6KA3 activation, and inhibition of RPS6KA3 expression results in reduced levels of phospho-AMPK. Thus, it is possible that aggravated ER stress results from PRKAA1 depletion through a process controlled by the upstream regulator RPS6KA3. Taken together, these data suggest that the ERN1-RPS6KA3-AMPK axis is a potential regulator of ER-stress-mediated autophagy.Citation143 ATP depletion under ER stress is regulated primarily by the EIF2S1-ATF4-DDIT3 branch of the UPR, which can cause rapid stimulation of AMPK ().Citation144,145 For instance, bufalin, an active component of Bufo gargarizan venom and a treatment for malignant glioma, causes ER stress-mediated ATP depletion and upregulation of AMPK activity with increased autophagy and overall survival of glioma cells.Citation146 EIF2AK3 involvement in AMPK-mediated cytoprotective autophagy has also been described in metabolic stress conditions caused by the extracellular matrix (ECM) detachment of mammary epithelial cells.Citation147 Adhesion to the ECM is essential for the proper function and homeostasis of epithelial tissues. ECM detached cells undergo rapid activation of the EIF2AK3 branch of the UPR, which in turn induces autophagy by activating AMPK and subsequently downregulating MTORC1.Citation147 This mechanism of autophagy induction is essential for the re-attachment and survival of the detached cells. It is well known that AMPK downregulates protein synthesis by inhibiting the cell growth regulator MTOR and subsequently phosphorylating EIF4EBP1/4E-BP1 and RPS6KB/p70S6K. However, the transcriptional activity of ATF4 and DDIT3 increases protein synthesis and cell death,Citation145 which has resulted in a controversy regarding the role of AMPK under ER stress. Further mechanistic insight is required to clarify how ATF4 and DDIT3 induce protein synthesis in parallel with AMPK activation. Based on all of these studies, it is our opinion that AMPK activation under ER stress conditions could represent a therapeutic target to protect against unwanted cellular demise (Table 2).
ER Ca2+ Changes Regulate Autophagy
An abundance of Ca2+ in the ER is essential for its proper functioning, including nascent protein folding and ER homeostasis. Interestingly, the ER is an intracellular Ca2+ reservoir that ensures regular communication with other organelles and effector proteins. Loss of lumenal Ca2+ is frequently accompanied by ER stress, which can activate various Ca2+-regulated pathways based on the degree and duration of stress. Alternatively, ER stress can cause loss of ER lumenal Ca2+. The influx and release of Ca2+ from the ER are tightly controlled by various regulatory systems. Cytosolic Ca2+ enters the ER through a Ca2+ pump called ATP2A/SERCA (ATPase, Ca2+ transporting) that is expressed on the ER surface.Citation148 Ca2+ storage in the ER is facilitated primarily by a number of intra-ER Ca2+ binding proteins and Ca2+-dependent chaperones, including CASQ/calsequestrin, CALR (calreticulin), CANX (calnexin), HSPA5, HSP90B1/GRP94, and PDI (protein disulfide isomerase).Citation149 Together, they ensure the primary quality control of proteins destined for Ca2+-dependent modification in the ER. Release of stored Ca2+ from the ER is mediated by 2 different receptor channels that reside on the membrane of the ER called RYR (ryanodine receptor) and ITPR/IP3R (inositol 1,4,5-trisphosphate receptor).Citation149
Optimum Ca2+ movement across the ER membrane ensures the proper functioning of various kinases and proteases. Indeed, perturbation of this dynamic Ca2+ movement can lead to activation of various Ca2+-regulated pathways, including autophagy. In a breast tumor model, the cytosolic Ca2+ mobilizing agents thapsigargin, ionomycin, and vitamin D activate CAMKK2/CAMKKβ (calcium/calmodulin-dependent protein kinase kinase 2, β), which in turn triggers autophagy via AMPK-dependent MTORC1 inhibition.Citation150 Those data are supported through sophisticated work by Jia et al.Citation151 They demonstrated that autophagy-deficient T lymphocytes have an expanded ER compartment due to excessive Ca2+ in the ER. Defective Ca2+ influx results in extensive Ca2+ stores in the ER, which could be caused by the inability of the Ca2+ stores to be depleted. This defect in Ca2+ influx can be repaired by treatment with the sarcoplasmic reticulum/ER Ca2+-ATPase pump inhibitor thapsigargin,Citation151 which indicates that autophagy can control Ca2+ movement across the ER. Taken together, these data suggest a relationship between Ca2+ mobilization and autophagy and indicate they can influence each other. ER stress can also be a consequence of extracellular Ca2+ influx; however, no direct evidence suggests that extracellular Ca2+ induction can cause ER stress-mediated autophagy. Furthermore, increasing cytosolic Ca2+ with exogenously introduced Ca2+ phosphate precipitates can induce autophagy at early time points without altering the ER condition.Citation152,153 Therefore, it is likely that ER Ca2+ is the main regulator of autophagy mediated by the UPR, whereas Ca2+ from other sources can induce autophagy that does not involve the UPR.
ITPR, a second messenger, makes the scenario more complex by playing a negative role in autophagy induction. Emerging evidence from various experimental systems suggests that pharmacological and genetic inhibition of ITPRs trigger autophagy independent of Ca2+.Citation154,155 This appears to contradict the role of ER-Ca2+ depleting agents (the ATP2A/SERCA antagonist thapsigargin) and lumenal ER Ca2+ store stimulators (the ITPR antagonist xestospongin B), both of which can induce autophagy. One possible explanation is that perturbation of typical ER Ca2+ levels can lead to autophagy. Conversely, autophagy signaling caused by ITPR inhibition might be mechanistically distinct from ER stress-induced autophagy. Moreover, blocking ITPRs and RYRs can attenuate thapsigargin-induced ER stress,Citation156 which might provide a better explanation for the negative role of ITPRs in autophagy induction. At basal levels, hyperactivation of autophagy is observed in cells in which all 3 ITPR isoforms are deleted.Citation157 In addition, the kinase activity of MTORC1 is significantly disrupted in such cells. Thus, it can be assumed that ITPR-dependent Ca2+ signaling ensures an elevated level of MTORC1 activity, which in turn negatively regulates basal levels of autophagy.Citation157 This notion is consistent with the fact that inhibition of new protein production and autophagy induction are mediated by the AMPK-induced Ca2+-calmodulin-dependent enzyme EEF2K and also suggests that basal levels of autophagy vary with changes in cytosolic Ca2+ levels and AMPK status.
Ca2+-mediated autophagy under ER stress conditions is regulated by the known tumor suppressor DAPK1 ().Citation7,115 Disruption of BECN1 and BCL2L1 interaction is mediated by activated DAPK1 through direct phosphorylation of BECN1 on Thr119, which allows autophagy to proceed.Citation116 Hypoxia stimulates extensive downregulation of protein synthesis through activation of the EIF2AK3-EIF2S1-ATF4 and AMPK-MTORC1 pathways.Citation158,159 Likewise, autophagy can also be triggered under hypoxic conditions, which can be attributed to hypoxia-induced Ca2+ influx and activation of CAMK1 (calcium/calmodulin-dependent kinase I) and CAMK4, which in turn can contribute to WIPI1 stimulation and autophagosome formation.Citation160,161
Sufficient evidence shows that activation of PRKCQ/PKCθ (protein kinase C, theta), a member of the novel-type PKC family, is required for ER stress-induced autophagy.Citation162-164 Interestingly, Ca2+ is an essential component of PRKCQ activation under ER stress conditions, which is supported by the observation of PRKCQ deactivation and the subsequent blockage of autophagy in cells treated with BAPTA-AM, an intracellular Ca2+ chelating agent.Citation163 This scenario mirrors the observation that cytosolic Ca2+ mobilization upon ER stress is mandatory for autophagy induction. Moreover, it has been reported that PRKCQ acts as a sensor for ER stress in skeletal muscle. Activation of PRKCQ alone can stimulate autophagy in the absence of ER stress, but it is augmented under ER stress conditions.
BAPTA-AM can also block autophagy flux following ER stress induced by proteasome inhibitors.Citation165 Specifically, BAPTA-AM-mediated Ca2+ chelation disrupts lysosomal function and subsequently inhibits autophagosome degradation.Citation165 Accordingly, it can be suggested that depletion of cytosolic Ca2+ might interrupt the lysosomal function essential for complete autophagy flux. In the same vein, other studies have shown that intracellular Ca2+ mobilization caused by plasma membrane damage results in increased interaction between lysosomes and SNAREs, which are crucial for membrane fusion.Citation166 Such elevated levels of Ca2+ could facilitate autophagosome and lysosomal fusion and thus enhance autophagy flux.
In addition to regulation of BECN1, it has been reported that ER-localized BCL2 is also liable for depleting ER Ca2+ and reduces agonist-induced Ca2+ leakage from the ER, possibly via an interaction through ITPR, in an attempt to negatively regulate autophagy.Citation150 Opposing the latter mechanism, Reed and colleagues demonstrated that TMBIM6/BI-1 promotes autophagy by reducing steady-state levels of ER Ca2+ through ITPRs.Citation167 Reduced ER Ca2+ is attributed to the depletion of mitochondrial Ca2+ uptake and subsequent ATP depletion, which in turn stimulates AMPK-dependent autophagy. Mechanistically, the ER and mitochondria are associated through the mitochondria-associated membranes (MAMs), and the efficiency of ITPRs in the MAM might be reduced in cells overexpressing TMBIM6, thereby decreasing Ca2+ transfer to the mitochondria through MAM-associated ITPRs. In the context of TMBIM6, it can be assumed that BCL2 modulates ER Ca2+ by regulating ITPRs outside of MAMs. Furthermore, Ca2+ leakage via ITPRs at distinct positions on the ER membrane could be followed by distinct outcomes (Table 2).
ER Stress Regulates Selective Autophagy
Autophagy was previously considered an essential pathway for random nonselective lysosomal bulk degradation of cytosolic components. However, accumulating evidence suggests that cells might consume part of their cytoplasm for self-nourishment and targeted removal of undesirable components, such as superfluous or damaged mitochondria through a process called mitophagy. Likewise, lipophagy, reticulophagy, ribophagy, xenophagy, pexophagy, zymophagy, and aggrephagy refer to autophagic removal of faulty lipid droplets, surplus endoplasmic reticulum, ribosomes, pathogens, peroxisomes, zymogen particles, and protein aggregates, respectively (; reviewed in referenceCitation168). It is also evident that selective autophagy peaks during development, pathogenic infections, and alteration of nutrient sources.Citation169 Interestingly, selective autophagy was first described in yeast, which is mainly based on the autophagy-like process called the cytoplasm to vacuole targeting (Cvt) pathway for delivering prApe1 (precursor aminopeptidase I), the precursor form of a vacuole-resident enzyme, from the cytosol to the vacuole.Citation170 Moreover, genetic screens in yeast have identified proteins essential for various forms of selective autophagy, and in mammalian cells a systematic procedure to identify the molecular determinants of selective autophagy is now emerging.
Figure 2. Schematic model of the speculative roles of ER stress components involved in selective autophagy in mammalian cells. During ER stress, EIF2AK3-dependent ATF4 translation and its interaction with DDIT3 in the nucleus stimulate expression of the autophagy receptor genes SQSTM1 and NBR1, which are ubiquitous. (A) ER-stress-mediated activation of ATF4 sustains the expression of PARK2. Damaged mitochondria recruit PARK2 as a degradation signal recognized by various autophagy receptors, including SQSTM1 and NBR1. (B) Following ER stress, upregulated SQSTM1 and NBR1 interacts with ubiquitin aggregates and enhance its selective clearance through aggrephagy. Autophagy receptor WDFY3 and BAG3 are also involved in aggrephagy.Citation185 (C) During pexophagy, SQSTM1 causes superfluous or damaged peroxisomes to cluster and subsequent be delivered into the growing selective phagophore.Citation220 (D) After cell division, midbody remnants are recognized by the autophagy receptors SQSTM1 and NBR1, which deliver them to the phagophore for degradation.Citation221 (E) The ubiquitin-binding protein SQSTM1 interacts with and traffics ubiquitinated zymogen granules to the nucleating phagophores.Citation222 (F) Ubiquitinated proteins interact with OPTN. The resulting autophagy receptor complex accelerates the selective autophagy flux of viruses, microbes, and other non-host entities.Citation223 (G) In mammals, the autophagy receptor FAM134B interacts with LC3 and GABARAP, and facilitates ER turnover by reticulophagy.Citation175 Similarly, yeast specific reticulophagy is Atg39 and Atg40 dependent.Citation224,225 However, the role of SQSTM1 and NBR1 in reticulophagy has not been clarified yet.
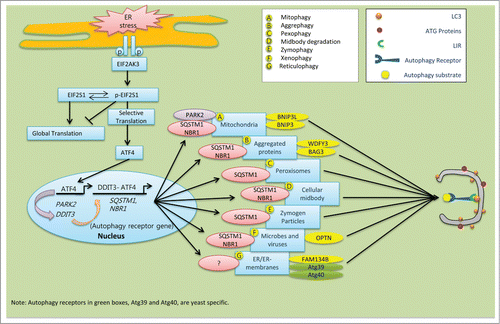
What differentiates selective autophagy from bulk autophagic degradation? To achieve selectivity, specific molecular markers are required for recognition of the autophagy cargos destined for degradation. Accumulating evidence suggests that Ub chains represent a general recognition signal.Citation171-173 Ubiquitinated cargos are then identified by special types of proteins called autophagy receptors, which can simultaneously interact with small UBL modifiers such as Atg8/LC3/GABARAP. Many of the autophagy receptor proteins possess an LC3-interacting region (LIR)Citation171 that enables them to bind with, and subsequently deliver, the autophagy cargo to the nucleating phagophore. There, UBLs facilitate their docking, which is followed by formation of the selective autophagosome.Citation174 Among the 2 dozen LIR-containing autophagy receptors, several in mammals can bridge cargo and phagophore membranes, including FAM134B (a functional homolog of yeast Atg40),Citation175 SQSTM1, NBR1, SMURF1, FUNDC1, STBD1, OPTN (optineurin), CALCOCO2/NDP52, BNIP3, BNIP3L/NIX, BAG3, WDFY3/ALFY and CBL/c-Cbl. (reviewed in reference.Citation171) Mounting evidence suggests that SQSTM1 and NBR1 can act as universal receptors for almost all types of ubiquitinated cargo.Citation172,174,176,177 Unfolded and ubiquitinated protein sequestration during selective autophagy is mainly mediated by the cooperative activity of both SQSTM1 and NBR1. These 2 proteins are identical in terms of LIR motif, an N-terminal PB1 domain, and a ubiquitin-interacting C-terminal UBA domain.Citation174 Notably, degradation and clearance of SQSTM1 and NBR1 proteins are mainly autophagy-specific and proteasome-independent. Manipulation of autophagy results in SQSTM1 and NBR1 aggregation under conditions of normal proteasomal activity. However, these 2 proteins differ in their size and primary sequence.Citation174 Conversely, the rest of the selective autophagy receptors in mammals are cargo specific. For example, CALCOCO2, SMURF1, and OPTN are required for invasive pathogens, BNIP3 and BNIP3L act as mitophagy-specific receptors,Citation178 and reticulophagy is FAM134B dependent.Citation175 Similarly, WDFY3/ALFY and BAG3 orchestrate aggrephagy,Citation179,180 and OPTN is specific for xenophagy, along with both SQSTM1 and NBR1 ().
Does ER stress also participate in selective autophagy? Limited evidence supports this idea, especially for mammalian cells. However, the ER is connected with autophagy in various ways, including ER stress-mediated autophagy activation and the formation of autophagosomes at the ER. In addition, the ER itself can be subject to autophagy, and thus it is possible that ER stress can regulate selective autophagy. A recent study in yeast by Schuck et al. demonstrated that ER stress can selectively degrade excess ER membranes by triggering a special type of autophagy called reticulophagy, in which the core autophagy machinery is not implicated.Citation181 Alternatively, ER stress leads to the formation of large ER whorls that can be subjected to autophagy by selective uptake into lysosomes. Instead of forming autophagosomes, ER whorls are sequestered by direct invagination by the vacuolar membrane, suggesting a topological similarity between microautophagy and reticulophagy. However, the proteins involved in microautophagy are not required for reticulophay.Citation181 These observations raise the question of the function of selective autophagy under ER stress. Reticulophagy regulates cell homeostasis by degrading excess ER membrane formed due to protein overload under stress.Citation181 Conversely, the UPR transcription factor XBP1s is responsible for the ER expansion necessary to accommodate more folding proteins during ER stress;Citation182 thus ERN1-XBP1 might serve as an upstream regulator of the reticulophagy machinery. Strikingly, accumulation of ER-containing autophagosomes under UPR activation suggests that the ER in mammalian cells can be subjected to reticulophagy.Citation93 Upon ERN1 deletion, compromised autophagosome formation and cell demise strengthens the speculation that the ERN1-XBP1 axis could serve as a potential mediator of ER stress-mediated reticulophagy.Citation93
With respect to cell homeostasis, following ER stress, elimination of damaged mitochondria is crucial, and it remains possible that mitophagy can be initiated to mitigate overall cellular stress. This notion is supported by mitophagy-mediated cell protection in a transient neuronal ischemia model under pharmacologically induced ER stress.Citation183 In that context, ER stress acts as a mitophagy-dependent neuroprotector by sustaining ATF4-mediated PARK2-expression. PARK2 forms a complex with PINK1 at the outer membrane of damaged mitochondria that causes recruitment of the autophagy receptor SQSTM1, which in turn may trigger mitophagy.Citation184 Furthermore, activated DDIT3 can interact with ATF4 in the nucleus to trigger stress-induced expression of the autophagy receptor genes SQSTM1 and NBR1 (). Thus, selective autophagy might be a potential outcome of ER stress. Together, these findings describe a complete autophagy pathway after EIF2AK3-EIF2S1 activation including expression of the genes necessary for phagophore formation, and elongation, and the autophagy receptors (SQSTM1 and NBR1) specific for selective degradation of intracellular components. In agreement with this model, Rubio et al. described and characterized the formation of SQSTM1 bodies and NBR1 in both normal cells and cancer cells following hypericin-mediated photodynamic therapy (Hyp-PDT).Citation185 Hyp-PDT triggers ER stress by generating ER-localized reactive oxygen species that mediates ATP2A2/SERCA2 loss of function and disrupts ER-Ca2+ homeostasis. Hyp-PDT treatment leads to the aggregation and sequestration of ubiquitinated, unfolded proteins that are then followed by SQSTM1- and NBR1-associated autophagic degradation.Citation185 This finding strongly supports the notion of selective autophagy-mediated degradation of aggregated proteins under stress conditions. Because the critical role of the UPR is to process aggregated proteins and re-establish cellular homeostasis, selective autophagy-mediated removal of misfolded proteins, protein aggregates, and protein complexes might also be part of ER stress-mediated autophagy. Indeed, it is likely that SQSTM1 and NBR1 constitute a complex under conditions of ER stress that is responsible for selective autophagic degradation of ubiquitinated proteins and other organelles. Studies in yeast and mammalian-based systems strongly suggest that reticulophagy can be enhanced during ER stress to minimize cell demise. However, to fully establish that scenario, further studies are required.
Negative Role of ER Stress in Autophagy
The UPR might not always support autophagic processes. In some pathological conditions, ER stress is aberrant and results in impaired autophagy. Particularly in neurodegenerative disease conditions, autophagy goes awry, and ER stress response regulators appear to be at the center of such compromised autophagy machinery. Indeed, a growing body of evidence has begun to shed light on the mechanisms that underlie ER stress-mediated inhibition of autophagy. For example, in Huntington disease (HD) and familial amyotrophic lateral sclerosis (ALS) disease models, knockdown of the ERN1-XBP1 axis elevates autophagy and heightens pathological conditions (), suggesting that UPR branches are molecular determinants of such compromised conditions.Citation186-188 Similarly, pharmacological induction of ER stress via thapsigargin or tunicamycin causes aggregation of mutant (mt) HTT (huntingtin) proteins and deteriorates the pathological condition of HD patients through ERN1-mediated autophagy inhibition.Citation189 Likewise, in HD, overexpression of USP14, a deubiquitinating enzyme, enhances autophagic clearance of mtHTT protein through proteasome-mediated degradation of nonphosphorylated ERN1.Citation176 USP14 assists in proteasome processing by deubiquitinating proteins destined for proteasomal degradation.Citation190 Phosphorylated ERN1 has less ability to interact with USP14, thus sustaining its level and accentuating mtHTT protein aggregation by inhibiting autophagy. Furthermore, in an ALS disease model, Xbp1 deficiency leads to the clearance of mutant SOD1 proteins by upregulating basal autophagy in the absence of any additional stimuli.Citation187 Mechanistically, XBP1 deficiency promotes high expression of FOXO (forkhead box O), which in turn increases the expression of several genes that positively regulate autophagy ().Citation147
Figure 3. Schematic representation of the molecular mechanisms specifically involved in ER stress-regulated compromised autophagy. UPR components are implicated in the pathogenesis of various diseases associated with inadequate protein clearance. The ERN1 axes of the UPR diminish autophagy flux in several neurodegenerative and inflammatory muscle diseases. In HD and ALS, the endoribonuclease activity of ERN1 undermines autophagy by decreasing FOXO levels by trafficking it to the 26S proteasomal pathway for degradation. XBP1 depletion reduces EDEM1 expression and ERAD, which in turn induces autophagy by causing the accumulation of unfolded protein. MAPK8 knockdown enhances several autophagy genes in neurons by translocating FOXO to the nucleus. Following ERN1 activation, the association of TRAF2 and MAP3K5 in HD reduces autophagy flux by preventing autophagosome and lysosomal fusion. ER stress upregulates the expression of SCAMP5 and disrupts lysosomes, which in turn downregulate autophagy flux. ER stress blocks lysosomal pH-dependent autophagy flux by depleting the VMA21 level essential for V-ATPase-mediated proton entry into the lysosome.
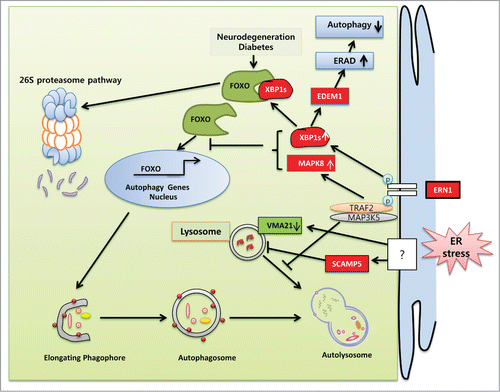
FOXO is one of the major intracellular hubs for the regulation of a variety of biological processes important for cell proliferation, development, metabolism, survival, and stress resistance.Citation191-194 Lately, FOXO has been identified as a critical regulator of autophagy that upregulates the transcription of several autophagy-related genes, such as ATG12, BECN1, BNIP3, GABARAPL1, and LC3. (reviewed in reference.Citation195) There are 4 FOXO family members in mammals: FOXO1/FKHR, FOXO3/FKHRL1, FOXO4/AFX, and FOXO6. Various intracellular signaling pathways, such as the cell proliferative PI3K-AKT pathway, the stress-dependent MAPK8 and MST1 (macrophage stimulating 1) pathways, and the AMPK and SIRT2 pathways, converge onto the FOXO signaling network.Citation196 These pathways regulate the subcellular localization, protein level, DNA binding, and transcriptional properties of FOXO.Citation196 The C-terminal DNA binding domain of FOXO proteins possesses both the nuclear localization and nuclear export sequences. Post-translational modifications such as phosphorylation and monoubiquitination regulate the effectiveness of the nuclear localization and export sequences, thereby providing the molecular basis of FOXO movement in and out of the nucleus. However, protein kinases AKT1 and serum and SGK (serum/glucocorticoid regulated kinase)-mediated phosphorylation lead to the association of FOXO with a chaperone protein named YWHA/14-3-3, which in turn retains FOXO in the cytosol.Citation197-200 Preferential binding of YWHA/14-3-3 proteins with phosphorylated FOXO also prevents its nuclear re-entry by blocking the nuclear localization signal.Citation201,202 Conversely, AMPK, MAPK8, MAPK/p38, MST1, and CDK1 (cyclin-dependent kinase 1) proteins can promote FOXO nuclear localization and transcriptional activity by disrupting the interaction between FOXO and YWHA/14-3-3.Citation200 Nuclear translocation of FOXO and subsequent induction of cytoprotective autophagy is observed in MAPK8 knockout neurons. In that condition, MAPK8 acts as a dominant negative regulator of FOXO. However, mutant fibroblasts with depleted MAPK8 expression exhibit compromised autophagy under serum withdrawal conditions (),Citation203 indicating that MAPK8 is essential for autophagy induction in cells other than neurons. From the existing information, it is clear that both upregulation and downregulation of the ERN1 branch of the UPR can modulate autophagy. Indeed, a recent study of pancreatic β cells demonstrated that overexpression of XBP1s downregulates FOXO expression through direct XBP1 binding and transferring of FOXO toward the 26S proteasome-mediated degradation pathway.Citation204 This finding supports the mouse model of HD in which Xbp1 deficiency in mouse brains leads to enhanced autophagy via FOXO accumulation (). Hence, it can be assumed that, among other regulatory components, FOXO remains at the core of autophagy when the UPR signaling network is compromised.
A pertinent question to be asked, in the context of neurodegenerative diseases, is how ER stress blocks autophagy flux. Lee et al. provided a partial explanation in their demonstration that ER stress impedes autophagy flux through the ERN1-TRAF2 pathway in HD, in which a mutant form of TRAF2 facilitates the clearance of aggregated proteins.Citation172 The results of that study suggest that the kinase activity of ERN1 leads to an accumulation of mtHTT protein by impairing autophagy flux, which in turn regulates neuronal cell death. Conversely, XBP1 splicing caused by endoribonuclease activity also inhibits autophagy by FOXO downregulation.Citation204 Given its importance to TRAF2 and ERN1 association, MAP3K5 might also be involved in autophagy regulation in neurodegenerative disorders. Indeed, MAP3K5 depletion prevents neuronal cells from stress-induced apoptosis by MAPK8 downregulation,Citation205 which is similar to MAPK8 knockdown models.Citation203 Whether such depletion of MAP3K5 preferentially activates autophagy remains to be explored.Citation206 However, the cumulative data regarding the ERN1 branch and its downstream regulation of the UPR are in conflict. Considering all the evidence, 2 possibilities are suggested. First, the ERN1 branch of the UPR is predominant during apoptosis, which might result from impaired autophagy. Second, ERN1-mediated autophagy manipulation is only conserved during pathological conditions. Mechanistically, prolonged ER stress-induced cell death pathways could converge into ERN1-mediated autophagy flux inhibition.Citation189 It is interesting that, despite having a potential role in autophagy induction, ERN1-MAPK8 inhibits autophagy in neuronal ER stress conditions. Thus, it remains to be determined which factors distinguish the effects of ERN1 in neurons from other types of cells with respect to autophagy. In this context, the duration of ER stress has been shown to be an important factor for the transition of complete autophagy flux to inhibition. In nonalcoholic fatty liver disease, mild ER stress leads to complete autophagy, and chronic stress causes cell death through the blockage of autophagic flux.Citation207 Interestingly, rapamycin can re-establish regular autophagy flux under chronic ER stress, suggesting that endocytosis pathways remain active and that AKT1-MTOR might be the main regulator of such autophagy blockage. Furthermore, under ER stress conditions, nuclear translocation of FOXO proteins seems to be absent in non-neuronal cells.Citation164 Notably, the induction of autophagy is a more predominant consequence of ER stress than inhibition; thus a negative interplay between the EIF2AK3-EIF2S1 and ERN1 pathways and chronic stress might activate a feedback loop that further aggravates protein accumulation by changing ER protein-folding capacity.Citation203
Autophagy flux is inhibited by imperfect signaling through autophagosomes and lysosomes. Various proteins integrate those signaling pathways and facilitate autophagosome and lysosomal fusion. Activated ERN1 does not inhibit the formation of autophagosomes, but rather leads to their accumulation, which might be caused by disruption of the endocytotic traffic machinery. In agreement with that possibility, Noh et al. showed that ER stress can impair endocytosis pathways by upregulating the expression of SCAMP5 (secretory carrier membrane protein 5) ().Citation208 Furthermore, SCAMP5 expression is significantly upregulated in the striatum of HD patients, and siRNA-mediated depletion of SCAMP5 increases endocytosis and autophagic clearance. However, the identity of the specific UPR branch that regulates SCAMP5 activity has not yet been clearly established. ERN1-mediated aggregation of mtHTT and autophagy flux inhibition suggest that SCAMP5 might be a potential target of the ERN1 branch. Further studies focusing on ERN1 and SCAMP5 in HD patients could represent a good opportunity to identify how to improve the pathological conditions associated with HD. Conversely; MAPK/p38 activation following ER stress can stimulate the RAB5 cycle and endocytosis.Citation209 Although it remains unclear whether endocytosis and autophagy follow the same pathway, it is well established that the 2 processes are closely related and converge upon common degradation through terminal lysosomes.
Thapsigargin undermines autophagy at multiple stages. It causes decreased autophagosome biosynthesis by activating calpains at an early stage of the autophagy mechanism and blocks autophagy flux at the end stage by inhibiting the fusion of matured autophagosomes with lysosomes.Citation210-212 The latter is independent of the endocytosis-mediated degradation machinery and might partially explain the distinction between autophagy and endocytosis. Mechanistically, thapsigargin blocks recruitment of RAB7A to autophagosomes while leaving endosomal RAB7A recruitment active.Citation212 These findings indicate that autophagy uses different machinery than endocytosis to recruit RAB7A to autophagosomes as well as different tethering complexes to bind to lysosomes. Although thapsigargin has proved to be a useful inhibitor of autophagy flux, the overall controversy remains obscure. Grotemeier et al., along with many other researchers, clearly showed that complete autophagy flux remains active under thapsigargin treatment.Citation213 Moreover, thapsigargin has recently been implicated in the inhibition of autophagy at the autophagosome maturation stage by preventing the closure of elongating phagophores.Citation214 According to that study, thapsigargin inhibits neither autophagy induction nor complete autophagy flux. Indeed, it causes immature autophagosome formation, as detected by the inability of prostate adenocarcinoma cells to sequester cytosolic LDH/lactate dehydrogenase.Citation214 Cells treated with the Ca2+ ionophore A23187 also exhibit a similar effect.Citation214 Strikingly, the role of ER stress in thapsigargin-mediated autophagy inhibition is not well established; rather ER-Ca2+ signaling has drawn the major focus. Together, these findings strongly suggest that autophagy modulation is predominantly a consequence of ER-Ca2+ perturbation rather than ER stress.
DDIT3 knockdown can accentuate autophagy in AR113Q knockin male mice with spinal and bulbar muscular atrophy. Interestingly, aberrant ER stress in the spinal and bulbar muscular atrophy model remains unchanged in the wild-type and knockout model, confirmed by expression of identical phosphorylated (p)-EIF2AK3 and p-EIF2S1 levels, suggesting a negative role for DDIT3 in autophagy regulation.Citation215 Furthermore, a negative interplay between ER stress and autophagy has also been identified after fatty liver graft preservation. Elevated levels of ER stress and impaired autophagy are usually found in steatotic livers in response to cold ischemia/reperfusion injury. Indeed, prolonged cold storage of steatotic livers can lead to the induction of graft injury. Steatotic liver storage in a solution of Institut Georges Lopez, melatonin and trimetazidine, instead of an Institut Georges Lopez solution alone, upregulates autophagy by reducing ER stress through high AMPK phosphorylation. Furthermore, a sharp increase in ER stress with a significant decrease in autophagy is observed when AMPK is inhibited.Citation216 Together, these data suggest that ER stress leads to impaired autophagy in steatotic livers in response to prolonged cold storage (Table 3).
Concluding Remarks
ER stress serves dual roles by favoring mechanisms of both autophagy induction and inhibition. In both mammalian and yeast cells, ER stress-mediated autophagy has been well characterized and is highly consistent. However, this is not the case for several disease models. During physiological ER stress, autophagy serves as an adaptive stress response that helps to sustain cell survival, whereas pathological ER stress can lead to the inhibition of autophagy. Surprisingly, genetic modulation of UPR branches appears to be effective in manipulating autophagy. Notably, it has been uncovered that under pathological ER stress (especially neurodegeneration), autophagy is substantially regulated by the ERN1 branch of the UPR and its downstream regulators. EDEM1 (ER degradation enhancer, mannosidase α-like 1), a target gene of XBP1 encoding an essential component of ERAD substrate signaling and recognition, might also play important undermining roles in ER stress-mediated autophagy. RNAi screening shows an elevated level of basal autophagy in an unstressed xbp1−/− fly model.Citation217 At the molecular level, Xbp1 deficiency results in depleted EDEM1 expression followed by impaired ERAD machinery and a subsequent aggregation of misfolded proteins. An autophagy signal might be triggered by those accumulated proteins.Citation217 XBP1 could suppress autophagy by clearing the aggregated protein through the EDEM1-dependent proteasome degradation pathway. Sporadic inclusion-body myositis has phenotypic similarities to Alzheimer and Parkinson diseases in terms of protein aggregation.Citation218 In sporadic inclusion-body myositis muscle fibers, ER stress enhances protein aggregation by significantly decreasing the proteolytic activity of CTSB and CTSD. This could be due to the decreased expression of an essential lysosomal V-ATPase assembly chaperone, VMA21.Citation218 Compromised autophagy is the consequence of decreased VMA21 because of increased lysosomal pH ().
Although it remains to be confirmed to what extent autophagy can remove misfolded proteins and damaged organelles, our review of the existing literature demonstrates the exciting possibility that ER stress can lead to the selective degradation/clearance of aggregated proteins and damaged organelles. However, the contribution of ER stress in selective autophagy has been reported very recently. The autophagy field is rapidly growing, and current research is converging into selective autophagy. It is therefore likely that future research focusing on signaling pathways regulating ER stress-induced selective autophagy will be of great importance. To elucidate such signaling pathways, many burning questions need to be answered. Here, in this review, we have related that the autophagy receptors SQSTM1 and NBR1 are ubiquitous in nature and are essential components for all types of selective autophagy. Furthermore, ER stress can stimulate both of those proteins significantly. However, those 2 cargo receptors work along with others that are organelle specific. Thus, identifying and characterizing more ER stress-specific cargo receptors will be an important focus in the near future. It has already been established that SQSTM1 and NBR1 together can sequester aggregated proteins and form SQSTM1/p62 bodies to deliver them into the degradation pathway and protect cells via sequestration, preventing aggregated proteins from affecting fresh proteins or normally functioning organelles. Thus it needs to be determined whether the UPR under mild or acute stress can produce SQSTM1/p62 bodies as a cell protective mechanism. Because cationic concentration and movement inside the subcellular organelles contribute to their self digestion and indirectly amplify degradation signaling, studies focusing on intracellular organelles' physiological environments will shed new light on stress-mediated selective autophagy.
Thapsigargin-induced autophagy remains an important unresolved issue, with numerous independent studies describing 3 different outcomes. Specifically, while some studies support the inhibition of autophagy at the early.Citation214 and mid stages,Citation219 others have observed complete autophagy flux.Citation213 following thapsigargin treatment. Experimental work from our lab (unpublished data) suggests that the protein levels of the autophagy markers LC3-II and SQSTM1 were significantly increased after thapsigargin treatment for up to 24 h, whereas bafilomycin A1 treatment did not increase the LC3-II level further. Importantly, these data strongly support the notion of incomplete autophagy flux following thapsigargin treatment. Although several studies have been done to identify the role of cytoplasmic or ER Ca2+ in autophagy regulation, it remains unclear which compartment's Ca2+ plays the pivotal role in the autophagy process following ER stress.
The concurrence between ER stress and autophagy is common in several human pathologies, including respiratory diseases, inflammatory diseases, cardiovascular diseases, neurodegenerative disorders, cancer, and diabetes. Hence, it is important to understand how autophagy can be manipulated via ER stress regulators to favor prosurvival or prodeath signaling. Development of new drugs that target the linkages between the UPR branches and autophagy will have significant therapeutic benefits in the treatment of such diseases.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by the National Research Foundation (2012R1A2A1A03001907).
References
- Wickner W, Schekman R. Protein translocation across biological membranes. Science 2005; 310:1452-6; PMID:16322447; http://dx.doi.org/10.1126/science.1113752
- Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev 2007; 87:1377-408; PMID:17928587; http://dx.doi.org/10.1152/physrev.00050.2006
- Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutation Res 2005; 569:29-63; PMID:15603751; http://dx.doi.org/10.1016/j.mrfmmm.2004.06.056
- Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum Mol Genet 2007; 16:618-29; http://dx.doi.org/10.1093/hmg/ddm002
- Houck SA, Ren HY, Madden VJ, Bonner JN, Conlin MP, Janovick JA, Conn PM, Cyr DM. Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol Cell 2014; 54:166-79; PMID:24685158; http://dx.doi.org/10.1016/j.molcel.2014.02.025
- Yoshida H. ER stress and diseases. FEBS J 2007; 274:630-58; PMID:17288551; http://dx.doi.org/10.1111/j.1742-4658.2007.05639.x
- Wu J, Kaufman RJ. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ 2006; 13:374-84; PMID:16397578; http://dx.doi.org/10.1038/sj.cdd.4401840
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2000; 2:326-32; PMID:10854322; http://dx.doi.org/10.1038/35014014
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519-29; PMID:17565364; http://dx.doi.org/10.1038/nrm2199
- Oikawa D, Kimata Y, Kohno K. Self-association and BiP dissociation are not sufficient for activation of the ER stress sensor Ire1. J Cell Sci 2007; 120:1681-8; PMID:17452628; http://dx.doi.org/10.1242/jcs.002808
- Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci USA 2006; 103:14343-8; PMID:16973740; http://dx.doi.org/10.1073/pnas.0606480103
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001; 107:881-91; PMID:11779464; http://dx.doi.org/10.1016/S0092-8674(01)00611-0
- Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 2002; 16:452-66; PMID:11850408; http://dx.doi.org/10.1101/gad.964702
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000; 287:664-6; PMID:10650002; http://dx.doi.org/10.1126/science.287.5453.664
- Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J Cell Biol 2006; 172:201-9; PMID:16418533; http://dx.doi.org/10.1083/jcb.200508099
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 2003; 23:7198-209; PMID:14517290; http://dx.doi.org/10.1128/MCB.23.20.7198-7209.2003
- Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 2002; 3:99-111; PMID:12110171; http://dx.doi.org/10.1016/S1534-5807(02)00203-4
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 2006; 7:880-5; PMID:16953201; http://dx.doi.org/10.1038/sj.embor.7400779
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol 2006; 4:e374; PMID:17090218; http://dx.doi.org/10.1371/journal.pbio.0040374
- Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006; 313:104-7; PMID:16825573; http://dx.doi.org/10.1126/science.1129631
- Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 2009; 457:736-40; PMID:19079237; http://dx.doi.org/10.1038/nature07641
- So JS, Hur KY, Tarrio M, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Lichtman AH, Iwawaki T, Glimcher LH, et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab 2012; 16:487-99; PMID:23040070; http://dx.doi.org/10.1016/j.cmet.2012.09.004
- Woehlbier U, Hetz C. Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochemical Sci 2011; 36:329-37; PMID:21482118; http://dx.doi.org/10.1016/j.tibs.2011.03.001
- Pahl HL, Sester M, Burgert HG, Baeuerle PA. Activation of transcription factor NF-kappaB by the adenovirus E3/19K protein requires its ER retention. J Cell Biol 1996; 132:511-22; PMID:8647884; http://dx.doi.org/10.1083/jcb.132.4.511
- Tam AB, Mercado EL, Hoffmann A, Niwa M. ER stress activates NF-kappaB by integrating functions of basal IKK activity, IRE1 and PERK. PloS One 2012; 7:e45078; PMID:23110043; http://dx.doi.org/10.1371/journal.pone.0045078
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318:944-9; PMID:17991856; http://dx.doi.org/10.1126/science.1146361
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004; 11:381-9; PMID:14685163; http://dx.doi.org/10.1038/sj.cdd.4401373
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 2003; 11:619-33; PMID:12667446; http://dx.doi.org/10.1016/S1097-2765(03)00105-9
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 2001; 7:1165-76; PMID:11430820; http://dx.doi.org/10.1016/S1097-2765(01)00265-9
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 2004; 18:3066-77; PMID:15601821; http://dx.doi.org/10.1101/gad.1250704
- Fribley A, Zhang K, Kaufman RJ. Regulation of apoptosis by the unfolded protein response. Methods Mol Biol 2009; 559:191-204; PMID:19609758; http://dx.doi.org/10.1007/978-1-60327-017-5_14
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000; 103:239-52; PMID:11057897; http://dx.doi.org/10.1016/S0092-8674(00)00116-1
- Maytin EV, Ubeda M, Lin JC, Habener JF. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp Cell Res 2001; 267:193-204; PMID:11426938; http://dx.doi.org/10.1006/excr.2001.5248
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol 2004; 165:347-56; PMID:15123740; http://dx.doi.org/10.1083/jcb.200310015
- Pallepati P, Averill-Bates DA. Activation of ER stress and apoptosis by hydrogen peroxide in HeLa cells: protective role of mild heat preconditioning at 40 °C. Biochimica Et Biophysica Acta 2011; 1813:1987-99; PMID:21875624; http://dx.doi.org/10.1016/j.bbamcr.2011.07.021
- Obeng EA, Boise LH. Caspase-12 and caspase-4 are not required for caspase-dependent endoplasmic reticulum stress-induced apoptosis. J Biol Chem 2005; 280:29578-87; PMID:15975932; http://dx.doi.org/10.1074/jbc.M502685200
- Mishra R, Karande AA. Endoplasmic reticulum stress-mediated activation of p38 MAPK, Caspase-2 and Caspase-8 leads to abrin-induced apoptosis. PloS One 2014; 9:e92586; PMID:24664279; http://dx.doi.org/10.1371/journal.pone.0092586
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 α kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 2002; 22:3864-74; PMID:11997520; http://dx.doi.org/10.1128/MCB.22.11.3864-3874.2002
- Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, Aguirre-Ghiso JA. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol 2011; 31:3616-29; PMID:21709020; http://dx.doi.org/10.1128/MCB.05164-11
- Zou X, Xu J, Yao S, Li J, Yang Y, Yang L. Endoplasmic reticulum stress-mediated autophagy protects against lipopolysaccharide-induced apoptosis in HL-1 cardiomyocytes. Exp Physiol 2014; 99:1348-58; PMID:24951501; http://dx.doi.org/10.1113/expphysiol.2014.079012
- Talloczy Z, Jiang W, Virgin HW, 4th, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 2002; 99:190-5; PMID:11756670; http://dx.doi.org/10.1073/pnas.012485299
- Tanaka T, Tsujimura T, Takeda K, Sugihara A, Maekawa A, Terada N, Yoshida N, Akira S. Targeted disruption of ATF4 discloses its essential role in the formation of eye lens fibres. Genes Cells 1998; 3:801-10; http://dx.doi.org/10.1046/j.1365-2443.1998.00230.x
- Hettmann T, Barton K, Leiden JM. Microphthalmia due to p53-mediated apoptosis of anterior lens epithelial cells in mice lacking the CREB-2 transcription factor. Dev Biol 2000; 222:110-23; PMID:10885750; http://dx.doi.org/10.1006/dbio.2000.9699
- Wang W, Lian N, Li L, Moss HE, Perrien DS, Elefteriou F, Yang X. Atf4 regulates chondrocyte proliferation and differentiation during endochondral ossification by activating Ihh transcription. Development 2009; 136:4143-53; PMID:19906842; http://dx.doi.org/10.1242/dev.043281
- Seo J, Fortuno ES, 3rd, Suh JM, Stenesen D, Tang W, Parks EJ, Adams CM, Townes T, Graff JM. Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes 2009; 58:2565-73; PMID:19690063; http://dx.doi.org/10.2337/db09-0335
- Masuoka HC, Townes TM. Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood 2002; 99:736-45; PMID:11806972; http://dx.doi.org/10.1182/blood.V99.3.736
- Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, Mori K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell 2010; 21:2975-86; PMID:20631254; http://dx.doi.org/10.1091/mbc.E09-02-0133
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell 2007; 13:365-76; PMID:17765680; http://dx.doi.org/10.1016/j.devcel.2007.07.018
- Gade P, Manjegowda SB, Nallar SC, Maachani UB, Cross AS, Kalvakolanu DV. Regulation of the death-associated protein kinase 1 expression and autophagy via ATF6 requires apoptosis signal-regulating kinase 1. Mol Cell Biol 2014; 34:4033-48; PMID:25135476; http://dx.doi.org/10.1128/MCB.00397-14
- Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 α in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA 2009; 106:16657-62; PMID:19805353; http://dx.doi.org/10.1073/pnas.0903775106
- Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest 2005; 115:268-81; PMID:15690081; http://dx.doi.org/10.1172/JCI200521848
- Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, et al. An essential role in liver development for transcription factor XBP-1. Genes Dev 2000; 14:152-7; PMID:10652269
- Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J 2005; 24:4368-80; PMID:16362047; http://dx.doi.org/10.1038/sj.emboj.7600903
- Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang Z, et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem 2013; 288:859-72; PMID:23184933; http://dx.doi.org/10.1074/jbc.M112.412783
- De Duve C, Wattiaux R. Functions of lysosomes. Ann Rev Physiol 1966; 28:435-92; PMID:5322983; http://dx.doi.org/10.1146/annurev.ph.28.030166.002251
- Devenish RJ, Klionsky DJ. Autophagy: mechanism and physiological relevance ‘brewed’ from yeast studies. Front Biosci (Schol Ed) 2012; 4:1354-63; PMID:22652877; http://dx.doi.org/10.2741/S337
- Klionsky DJ, Cregg JM, Dunn WA, Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell 2003; 5:539-45; PMID:14536056; http://dx.doi.org/10.1016/S1534-5807(03)00296-X
- Umekawa M, Klionsky DJ. The Cytoplasm-to-Vacuole Targeting Pathway: A Historical Perspective. Int J Cell Biol 2012; 2012:142634; PMID:22481942; http://dx.doi.org/10.1155/2012/142634
- Moretti L, Cha YI, Niermann KJ, Lu B. Switch between apoptosis and autophagy: radiation-induced endoplasmic reticulum stress? Cell Cycle 2007; 6:793-8; PMID:17377498; http://dx.doi.org/10.4161/cc.6.7.4036
- Settembre C, Ballabio A. Lysosome: regulator of lipid degradation pathways. Trends Cell Biol 2014; 24:743-50; PMID:25061009; http://dx.doi.org/10.1016/j.tcb.2014.06.006
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/10.1038/ncb0910-814
- Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutri 2007; 27:19-40; PMID:17311494; http://dx.doi.org/10.1146/annurev.nutr.27.061406.093749
- Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science 2008; 321:117-20; PMID:18599786; http://dx.doi.org/10.1126/science.1154822
- Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011; 334:1144-7; PMID:22033522http://dx.doi.org/10.1126/science.1211878
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/10.1038/nature03029
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/10.1016/j.ceb.2009.11.014
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823-30; PMID:20811354; http://dx.doi.org/10.1038/ncb0910-823
- Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012; 22:407-17; PMID:22748206; http://dx.doi.org/10.1016/j.tcb.2012.05.006
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010; 221:3-12; PMID:20225336; http://dx.doi.org/10.1002/path.2697
- Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med 2003; 9:65-76; PMID:12865942
- Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. Autophagy: for better or for worse. Cell Res 2012; 22:43-61; PMID:21912435; http://dx.doi.org/10.1038/cr.2011.152
- Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009; 5:649-62; PMID:19287211; http://dx.doi.org/10.4161/auto.5.5.8249
- Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, Fini M, McCubrey JA. Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget 2012; 3:371-94; PMID:22564882; http://dx.doi.org/10.18632/oncotarget.477
- Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009; 186:773-82; PMID:19797076; http://dx.doi.org/10.1083/jcb.200907014
- Sun Q, Zhang J, Fan W, Wong KN, Ding X, Chen S, Zhong Q. The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J Biol Chem 2011; 286:185-91; PMID:21062745; http://dx.doi.org/10.1074/jbc.M110.126425
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 1998; 72:8586-96; PMID:9765397
- Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci 2005; 118:7-18; PMID:15615779; http://dx.doi.org/10.1242/jcs.01620
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001; 2:211-6; PMID:11265251; http://dx.doi.org/10.1038/35056522
- Mizushima N, Yoshimori T, Ohsumi Y. Role of the Apg12 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2003; 35:553-61; PMID:12672448; http://dx.doi.org/10.1016/S1357-2725(02)00343-6
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298-302; PMID:17986448; http://dx.doi.org/10.1074/jbc.C700195200
- Matsushita M, Suzuki NN, Obara K, Fujioka Y, Ohsumi Y, Inagaki F. Structure of Atg5.Atg16, a complex essential for autophagy. J Biol Chem 2007; 282:6763-72; PMID:17192262
- Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. Embo J 2009; 28:1341-50; PMID:19322194; http://dx.doi.org/10.1038/emboj.2009.80
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004; 36:2503-18; PMID:15325588; http://dx.doi.org/10.1016/j.biocel.2004.05.009
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132-9; PMID:20056399; http://dx.doi.org/10.1016/j.ceb.2009.12.004
- Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct 2008; 33:109-22; PMID:18388399; http://dx.doi.org/10.1247/csf.08005
- Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjørkøy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol 2010; 188:253-69; PMID:20100911; http://dx.doi.org/10.1083/jcb.200907015
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 2004; 117:4837-48; PMID:15340014; http://dx.doi.org/10.1242/jcs.01370
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 2011; 80:125-56; PMID:21548784; http://dx.doi.org/10.1146/annurev-biochem-052709-094552
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nature Cell Biol 2008; 10:776-87; PMID:18552835; http://dx.doi.org/10.1038/ncb1740
- Tatti M, Motta M, Di Bartolomeo S, Scarpa S, Cianfanelli V, Cecconi F, Salvioli R. Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in Sap C-deficient fibroblasts. Hum Mol Genet 2012; 21:5159-73; PMID:22949512; http://dx.doi.org/10.1093/hmg/dds367
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220-31; PMID:17030611; http://dx.doi.org/10.1128/MCB.01453-06
- Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 2006; 4:e423; PMID:17132049; http://dx.doi.org/10.1371/journal.pbio.0040423
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995; 270:1326-31; PMID:7481820; http://dx.doi.org/10.1126/science.270.5240.1326
- Cheng X, Liu H, Jiang CC, Fang L, Chen C, Zhang XD, Jiang ZW. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int J Mol Med 2014; 34:772-81
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/10.1016/j.molcel.2008.06.001
- Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009; 28:886-98; PMID:19060920; http://dx.doi.org/10.1038/onc.2008.441
- Haberzettl P, Hill BG. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol 2013; 1:56-64; PMID:24024137; http://dx.doi.org/10.1016/j.redox.2012.10.003
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171:513-24; PMID:17620365; http://dx.doi.org/10.2353/ajpath.2007.070188
- Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008; 4:949-51; PMID:18769111; http://dx.doi.org/10.4161/auto.6788
- Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12˜ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol 2013; 20:59-66; PMID:23202584; http://dx.doi.org/10.1038/nsmb.2431
- B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013; 41:7683-99; PMID:23804767; http://dx.doi.org/10.1093/nar/gkt563
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230-9; PMID:16794605; http://dx.doi.org/10.1038/sj.cdd.4401984
- Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010; 120:127-41; PMID:20038797; http://dx.doi.org/10.1172/JCI40027
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001; 21:1249-59; PMID:11158311; http://dx.doi.org/10.1128/MCB.21.4.1249-1259.2001
- Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 2012; 122:4621-34; PMID:23143306; http://dx.doi.org/10.1172/JCI62973
- Moon HS, Kim B, Gwak H, Suh DH, Song YS. Autophagy and protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2 α kinase (eIF2alpha) pathway protect ovarian cancer cells from metformin-induced apoptosis. Mol Carcinog 2015; Forthcoming; PMID:25663310; http://dx.doi.org/10.1002/mc.22284
- Bruning A, Rahmeh M, Friese K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol Oncol 2013; 7:1012-8; PMID:23916134; http://dx.doi.org/10.1016/j.molonc.2013.07.010
- Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 2009; 69:4415-23; PMID:19417138; http://dx.doi.org/10.1158/0008-5472.CAN-08-2839
- Ramachandran N, Munteanu I, Wang P, Aubourg P, Rilstone JJ, Israelian N, Naranian T, Paroutis P, Guo R, Ren ZP, et al. VMA21 deficiency causes an autophagic myopathy by compromising V-ATPase activity and lysosomal acidification. Cell 2009; 137:235-46; PMID:19379691; http://dx.doi.org/10.1016/j.cell.2009.01.054
- Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{α} phosphorylation. J Biol Chem 2009; 284:5523-32; PMID:19114712; http://dx.doi.org/10.1074/jbc.M807440200
- Yung HW, Charnock-Jones DS, Burton GJ. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PloS one 2011; 6:e17894;
- Kalvakolanu DV, Gade P. IFNG and autophagy: a critical role for the ER-stress mediator ATF6 in controlling bacterial infections. Autophagy 2012; 8:1673-4; PMID:22874566; http://dx.doi.org/10.4161/auto.21403
- Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, Mizushima N, Yoshimori T, Kimchi A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ 2008; 15:1875-86; PMID:18806755; http://dx.doi.org/10.1038/cdd.2008.121
- Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/10.1038/embor.2008.246
- Knaevelsrud H, Simonsen A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett 2010; 584:2635-45; PMID:20412801; http://dx.doi.org/10.1016/j.febslet.2010.04.041
- Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010; 6:239-47; PMID:20104019; http://dx.doi.org/10.4161/auto.6.2.11062
- Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J Biol Chem 2004; 279:49420-9; PMID:15339911; http://dx.doi.org/10.1074/jbc.M407700200
- Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, Huang T, Nakajima S, Yao J, Paton AW, et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol 2009; 183:1480-7; PMID:19561103; http://dx.doi.org/10.4049/jimmunol.0900017
- Gray MJ, Mhawech-Fauceglia P, Yoo E, Yang W, Wu E, Lee AS, Lin YG. AKT inhibition mitigates GRP78 (glucose-regulated protein) expression and contribution to chemoresistance in endometrial cancers. Int J Cancer 2013; 133:21-30; PMID:23280503; http://dx.doi.org/10.1002/ijc.27994
- Chang YJ, Huang YP, Li ZL, Chen CH. GRP78 knockdown enhances apoptosis via the down-regulation of oxidative stress and Akt pathway after epirubicin treatment in colon cancer DLD-1 cells. PloS one 2012; 7:e35123; PMID:22529978; http://dx.doi.org/10.1371/journal.pone.0035123
- Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol 2012; 22:274-82; PMID:22444729; http://dx.doi.org/10.1016/j.tcb.2012.02.006
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002; 4:648-57; PMID:12172553; http://dx.doi.org/10.1038/ncb839
- Di Nardo A, Kramvis I, Cho N, Sadowski A, Meikle L, Kwiatkowski DJ, Sahin M. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci 2009; 29:5926-37; PMID:19420259; http://dx.doi.org/10.1523/JNEUROSCI.0778-09.2009
- Johnson CE, Hunt DK, Wiltshire M, Herbert TP, Sampson JR, Errington RJ, Davies DM, Tee AR. Endoplasmic reticulum stress and cell death in mTORC1-overactive cells is induced by nelfinavir and enhanced by chloroquine. Mol Oncol 2015; 9:675-88; PMID:25498902; http://dx.doi.org/10.1016/j.molonc.2014.11.005
- Jin HO, Seo SK, Woo SH, Kim ES, Lee HC, Yoo DH, An S, Choe TB, Lee SJ, Hong SI, et al. Activating transcription factor 4 and CCAAT/enhancer-binding protein-β negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radica Biol Med 2009; 46:1158-67; PMID:19439225; http://dx.doi.org/10.1016/j.freeradbiomed.2009.01.015
- Wu J, Wang ML, Qiu SL, Han JK, Zhu DX. IL-2 potentiates the IL-3 production by ConA activated mouse splenic T cells. Shi Yan Sheng Wu Xue Bao 1992; 25:25-30
- Kimball SR, Jefferson LS. Induction of REDD1 gene expression in the liver in response to endoplasmic reticulum stress is mediated through a PERK, eIF2alpha phosphorylation, ATF4-dependent cascade. Biochem Biophys Res Commun 2012; 427:485-9; PMID:23000413; http://dx.doi.org/10.1016/j.bbrc.2012.09.074
- Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ 2012; 19:310-20; PMID:21779001; http://dx.doi.org/10.1038/cdd.2011.98
- Kang YJ, Lu MK, Guan KL. The TSC1 and TSC2 tumor suppressors are required for proper ER stress response and protect cells from ER stress-induced apoptosis. Cell Death Differ 2011; 18:133-44; PMID:20616807; http://dx.doi.org/10.1038/cdd.2010.82
- Salazar M, Carracedo A, Salanueva IJ, Hernandez-Tiedra S, Lorente M, Egia A, Vázquez P, Blázquez C, Torres S, García S, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 2009; 119:1359-72; PMID:19425170http://dx.doi.org/10.1172/JCI37948
- Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J 2005; 24:1243-55; PMID:15775988; http://dx.doi.org/10.1038/sj.emboj.7600596
- Jousse C, Deval C, Maurin AC, Parry L, Cherasse Y, Chaveroux C, Lefloch R, Lenormand P, Bruhat A, Fafournoux P. TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J Biol Chem 2007; 282:15851-61; PMID:17369260; http://dx.doi.org/10.1074/jbc.M611723200
- Betz C, Hall MN. Where is mTOR and what is it doing there? J cell Biol 2013; 203:563-74; PMID:24385483; http://dx.doi.org/10.1083/jcb.201306041
- Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW, Wu J, Lin HK, Sarbassov dos D. ER stress inhibits mTORC2 and Akt signaling through GSK-3beta-mediated phosphorylation of rictor. Sci Signal 2011; 4:ra10; PMID:21343617; http://dx.doi.org/10.1126/scisignal.2001731
- Dong M, Hu N, Hua Y, Xu X, Kandadi MR, Guo R, Jiang S, Nair S, Hu D, Ren J. Chronic Akt activation attenuated lipopolysaccharide-induced cardiac dysfunction via Akt/GSK3beta-dependent inhibition of apoptosis and ER stress. Biochim Biophys Acta 2013; 1832:848-63; PMID:23474308; http://dx.doi.org/10.1016/j.bbadis.2013.02.023
- Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009; 196:65-80; PMID:19245654; http://dx.doi.org/10.1111/j.1748-1716.2009.01972.x
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Lee EK, Jeong JU, Chang JW, Yang WS, Kim SB, Park SK, Park JS, Lee SK. Activation of AMP-activated protein kinase inhibits albumin-induced endoplasmic reticulum stress and apoptosis through inhibition of reactive oxygen species. Nephron Exp Nephrol 2012; 121:e38-48; PMID:23108012; http://dx.doi.org/10.1159/000342802
- Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol 2005; 25:9554-75; PMID:16227605; http://dx.doi.org/10.1128/MCB.25.21.9554-9575.2005
- Martin MJ, Hayward R, Viros A, Marais R. Metformin accelerates the growth of BRAF V600E-driven melanoma by upregulating VEGF-A. Cancer Discov 2012; 2:344-55; PMID:22576211; http://dx.doi.org/10.1158/2159-8290.CD-11-0280
- Yao J, Bi HE, Sheng Y, Cheng LB, Wendu RL, Wang CH, Cao GF, Jiang Q. Ultraviolet (UV) and Hydrogen Peroxide Activate Ceramide-ER Stress-AMPK Signaling Axis to Promote Retinal Pigment Epithelium (RPE) Cell Apoptosis. Int J Mol Sci 2013; 14:10355-68; PMID:23685869; http://dx.doi.org/10.3390/ijms140510355
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 2013; 15:481-90; PMID:23624402; http://dx.doi.org/10.1038/ncb2738
- Shen S, Zhang Y, Wang Z, Liu R, Gong X. Bufalin induces the interplay between apoptosis and autophagy in glioma cells through endoplasmic reticulum stress. Int J Biol Sci 2014; 10:212-24; PMID:24550689; http://dx.doi.org/10.7150/ijbs.8056
- Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, Bardeesy N, Debnath J, Aguirre-Ghiso JA. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 2013; 32:4932-40; PMID:23160380; http://dx.doi.org/10.1038/onc.2012.512
- Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev 1994; 74:595-636; PMID:8036248
- Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol 2011; 3:a004317; http://dx.doi.org/10.1101/cshperspect.a004317
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol Cell 2007; 25:193-205; PMID:17244528; http://dx.doi.org/10.1016/j.molcel.2006.12.009
- Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol 2011; 186:1564-74; PMID:21191072; http://dx.doi.org/10.4049/jimmunol.1001822
- Gao W, Ding WX, Stolz DB, Yin XM. Induction of macroautophagy by exogenously introduced calcium. Autophagy 2008; 4:754-61; PMID:18560273; http://dx.doi.org/10.4161/auto.6360
- Chen X, Li M, Chen D, Gao W, Guan JL, Komatsu M, Yin XM. Autophagy induced by calcium phosphate precipitates involves endoplasmic reticulum membranes in autophagosome biogenesis. PloS One 2012; 7:e52347; PMID:23285000; http://dx.doi.org/10.1371/journal.pone.0052347
- Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgó J, Díaz J, Lavandero S, Harper F, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 2007; 14:1029-39; PMID:17256008
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170:1101-11; PMID:16186256; http://dx.doi.org/10.1083/jcb.200504035
- Luciani DS, Gwiazda KS, Yang TL, Kalynyak TB, Bychkivska Y, Frey MH, Jeffrey KD, Sampaio AV, Underhill TM, Johnson JD. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and β-cell death. Diabetes 2009; 58:422-32; PMID:19033399; http://dx.doi.org/10.2337/db07-1762
- Khan MT, Joseph SK. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J Biol Chem 2010; 285:16912-20; PMID:20308071; http://dx.doi.org/10.1074/jbc.M110.114207
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 2005; 24:3470-81; PMID:16148948; http://dx.doi.org/10.1038/sj.emboj.7600777
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell 2006; 21:521-31; PMID:16483933; http://dx.doi.org/10.1016/j.molcel.2006.01.010
- Pfisterer SG, Mauthe M, Codogno P, Proikas-Cezanne T. Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMP-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy. Mol Pharmacol 2011; 80:1066-75; PMID:21896713; http://dx.doi.org/10.1124/mol.111.071761
- Maulik D, Ashraf QM, Mishra OP, Delivoria-Papadopoulos M. Effect of hypoxia on calcium influx and calcium/calmodulin-dependent kinase activity in cortical neuronal nuclei of the guinea pig fetus during development. Am J Obstetr Gynecol 2002; 186:658-62; PMID:11967487; http://dx.doi.org/10.1067/mob.2002.122392
- Sakaki K, Kaufman RJ. Regulation of ER stress-induced macroautophagy by protein kinase C. Autophagy 2008; 4:841-3; PMID:18670192; http://dx.doi.org/10.4161/auto.6607
- Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem 2008; 283:15370-80; PMID:18356160; http://dx.doi.org/10.1074/jbc.M710209200
- Madaro L, Marrocco V, Carnio S, Sandri M, Bouche M. Intracellular signaling in ER stress-induced autophagy in skeletal muscle cells. FASEB J 2013; 27:1990-2000; PMID:23388382; http://dx.doi.org/10.1096/fj.12-215475
- Williams JA, Hou Y, Ni HM, Ding WX. Role of intracellular calcium in proteasome inhibitor-induced endoplasmic reticulum stress, autophagy, and cell death. Pharmaceutical Res 2013; 30:2279-89; PMID:23893020; http://dx.doi.org/10.1007/s11095-013-1139-8
- Appelqvist H, Waster P, Kagedal K, Ollinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 2013; 5:214-26; PMID:23918283; http://dx.doi.org/10.1093/jmcb/mjt022
- Sano R, Hou YC, Hedvat M, Correa RG, Shu CW, Krajewska M, Diaz PW, Tamble CM, Quarato G, Gottlieb RA, et al. Endoplasmic reticulum protein BI-1 regulates Ca(2)(+)-mediated bioenergetics to promote autophagy. Genes Dev 2012; 26:1041-54; PMID:22588718; http://dx.doi.org/10.1101/gad.184325.111
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID:21189453; http://dx.doi.org/10.4161/auto.7.3.14487
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9-14; PMID:21179058; http://dx.doi.org/10.1038/nrm3028
- Kraft C, Reggiori F, Peter M. Selective types of autophagy in yeast. Biochim Biophys Acta 2009; 1793:1404-12; PMID:19264099; http://dx.doi.org/10.1016/j.bbamcr.2009.02.006
- Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci 2013; 126:3237-47; PMID:23908376
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/10.1074/jbc.M702824200
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/10.1016/j.cell.2007.10.035
- Lamark T, Kirkin V, Dikic I, Johansen T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009; 8:1986-90; PMID:19502794; http://dx.doi.org/10.4161/cc.8.13.8892
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015; 522:354-8; PMID:26040720; http://dx.doi.org/10.1038/nature14498
- Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009; 5:732-3; PMID:19398892; http://dx.doi.org/10.4161/auto.5.5.8566
- Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505-16; PMID:19250911; http://dx.doi.org/10.1016/j.molcel.2009.01.020
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 2009; 17:87-97; PMID:19619494; http://dx.doi.org/10.1016/j.devcel.2009.06.013
- Isakson P, Holland P, Simonsen A. The role of ALFY in selective autophagy. Cell Death Differ 2013; 20:12-20; PMID:22653340; http://dx.doi.org/10.1038/cdd.2012.66
- Behl C. BAG3 and friends: co-chaperones in selective autophagy during aging and disease. Autophagy 2011; 7:795-8; PMID:21681022; http://dx.doi.org/10.4161/auto.7.7.15844
- Schuck S, Gallagher CM, Walter P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J Cell Sci 2014; 127:4078-88; PMID:25052096; http://dx.doi.org/10.1242/jcs.154716
- Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 2004; 167:35-41; PMID:15466483; http://dx.doi.org/10.1083/jcb.200406136
- Zhang X, Yuan Y, Jiang L, Zhang J, Gao J, Shen Z, Zheng Y, Deng T, Yan H, Li W, et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014; 10:1801-13; PMID:25126734; http://dx.doi.org/10.4161/auto.32136
- Vannuvel K, Renard P, Raes M, Arnould T. Functional and morphological impact of ER stress on mitochondria. J Cell Physiol 2013; 228:1802-18; PMID:23629871; http://dx.doi.org/10.1002/jcp.24360
- Rubio N, Verrax J, Dewaele M, Verfaillie T, Johansen T, Piette J, Agostinis P. p38(MAPK)-regulated induction of p62 and NBR1 after photodynamic therapy promotes autophagic clearance of ubiquitin aggregates and reduces reactive oxygen species levels by supporting Nrf2-antioxidant signaling. Free Radic Biol Med 2014; 67:292-303; PMID:24269898; http://dx.doi.org/10.1016/j.freeradbiomed.2013.11.010
- Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, Caballero B, Kiffin R, Segura-Aguilar J, Cuervo AM, et al. Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Hum Mol Genet 2012; 21:2245-62; PMID:22337954; http://dx.doi.org/10.1093/hmg/dds040
- Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 2009; 23:2294-306; PMID:19762508; http://dx.doi.org/10.1101/gad.1830709
- Hyrskyluoto A, Bruelle C, Lundh SH, Do HT, Kivinen J, Rappou E, Reijonen S, Waltimo T, Petersén Å, Lindholm D, et al. Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: involvement of the proteasome and ER stress-activated kinase IRE1alpha. Hum Mol Genet 2014; 23:5928-39; PMID:24951540; http://dx.doi.org/10.1093/hmg/ddu317
- Lee H, Noh JY, Oh Y, Kim Y, Chang JW, Chung CW, Lee ST, Kim M, Ryu H, Jung YK. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet 2012; 21:101-14; PMID:21954231; http://dx.doi.org/10.1093/hmg/ddr445
- Jarome TJ, Kwapis JL, Hallengren JJ, Wilson SM, Helmstetter FJ. The ubiquitin-specific protease 14 (USP14) is a critical regulator of long-term memory formation. Learn Mem 2014; 21:9-13
- van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol 2012; 14:829-37; PMID:22820375; http://dx.doi.org/10.1038/ncb2536
- Poulsen RC, Carr AJ, Hulley PA. Cell proliferation is a key determinant of the outcome of FOXO3a activation. Biochem Biophys Res Commun 2015; 462:78-84; PMID:25935481; http://dx.doi.org/10.1016/j.bbrc.2015.04.112
- Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol 2002; 22:7842-52; PMID:12391153; http://dx.doi.org/10.1128/MCB.22.22.7842-7852.2002
- Parmentier F, Lejeune FX, Neri C. Pathways to decoding the clinical potential of stress response FOXO-interaction networks for Huntington's disease: of gene prioritization and context dependence. Frontiers Aging Neurosci 2013; 5:22; PMID:23781200; http://dx.doi.org/10.3389/fnagi.2013.00022
- Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 2008; 20:126-36; PMID:18394876; http://dx.doi.org/10.1016/j.ceb.2008.02.005
- Calnan DR, Brunet A. The FoxO code. Oncogene 2008; 27:2276-88; PMID:18391970; http://dx.doi.org/10.1038/onc.2008.21
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96:857-68; PMID:10102273; http://dx.doi.org/10.1016/S0092-8674(00)80595-4
- Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochemical J 2000; 349:629-34; PMID:10880363; http://dx.doi.org/10.1042/bj3490629
- Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA 1999; 96:7421-6
- Tikhanovich I, Cox J, Weinman SA. Forkhead box class O transcription factors in liver function and disease. J Gastroenterol Hepatol 2013; 28 Suppl 1:125-31; PMID:23855308; http://dx.doi.org/10.1111/jgh.12021
- Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, Dalal SN, DeCaprio JA, Greenberg ME, Yaffe MB. Fourteen-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol 2002; 156:817-28; PMID:11864996; http://dx.doi.org/10.1083/jcb.200112059
- Obsilova V, Vecer J, Herman P, Pabianova A, Sulc M, Teisinger J, Boura E, Obsil T. Fourteen-3-3 Protein interacts with nuclear localization sequence of forkhead transcription factor FoxO4. Biochemistry 2005; 44:11608-17; PMID:16114898; http://dx.doi.org/10.1021/bi050618r
- Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 2011; 25:310-22; PMID:21325132; http://dx.doi.org/10.1101/gad.1984311
- Zhou Y, Lee J, Reno CM, Sun C, Park SW, Chung J, Lee J, Fisher SJ, White MF, Biddinger SB, et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nature Med 2011; 17:356-65; PMID:21317886; http://dx.doi.org/10.1038/nm.2293
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 2002; 16:1345-55; PMID:12050113; http://dx.doi.org/10.1101/gad.992302
- Matus S, Nassif M, Glimcher LH, Hetz C. XBP-1 deficiency in the nervous system reveals a homeostatic switch to activate autophagy. Autophagy 2009; 5:1226-8; PMID:19855189; http://dx.doi.org/10.4161/auto.5.8.10247
- Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillón J, Lo Iacono O, Corazzari M, Fimia GM, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis 2014; 5:e1179; http://dx.doi.org/10.1038/cddis.2014.162
- Noh JY, Lee H, Song S, Kim NS, Im W, Kim M, Seo H, Chung CW, Chang JW, Ferrante RJ, et al. SCAMP5 links endoplasmic reticulum stress to the accumulation of expanded polyglutamine protein aggregates via endocytosis inhibition. J Biol Chem 2009; 284:11318-25; PMID:19240033; http://dx.doi.org/10.1074/jbc.M807620200
- Cavalli V, Vilbois F, Corti M, Marcote MJ, Tamura K, Karin M, Arkinstall S, Gruenberg J. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol Cell 2001; 7:421-32; PMID:11239470; http://dx.doi.org/10.1016/S1097-2765(01)00189-7
- Zhou J, Tan SH, Nicolas V, Bauvy C, Yang ND, Zhang J, Xue Y, Codogno P, Shen HM. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res 2013; 23:508-23; PMID:23337583; http://dx.doi.org/10.1038/cr.2013.11
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4:295-305; PMID:18391949; http://dx.doi.org/10.1038/nchembio.79
- Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell 2011; 42:731-43; PMID:21700220; http://dx.doi.org/10.1016/j.molcel.2011.04.024
- Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, Viollet B, Wesselborg S, Proikas-Cezanne T, Stork B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal 2010; 22:914-25; PMID:20114074; http://dx.doi.org/10.1016/j.cellsig.2010.01.015
- Engedal N, Torgersen ML, Guldvik IJ, Barfeld SJ, Bakula D, Saetre F, Hagen LK, Patterson JB, Proikas-Cezanne T, Seglen PO, et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 2013; 9:1475-90; PMID:23970164; http://dx.doi.org/10.4161/auto.25900
- Yu Z, Wang AM, Adachi H, Katsuno M, Sobue G, Yue Z, Robins DM, Lieberman AP. Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet 2011; 7:e1002321
- Zaouali MA, Boncompagni E, Reiter RJ, Bejaoui M, Freitas I, Pantazi E, Folch-Puy E, Abdennebi HB, Garcia-Gil FA, Roselló-Catafau J. AMPK involvement in endoplasmic reticulum stress and autophagy modulation after fatty liver graft preservation: a role for melatonin and trimetazidine cocktail. J Pineal Res 2013; 55:65-78; PMID:23551302; http://dx.doi.org/10.1111/jpi.12051
- Arsham AM, Neufeld TP. A genetic screen in Drosophila reveals novel cytoprotective functions of the autophagy-lysosome pathway. PloS One 2009; 4:e6068; PMID:19562034; http://dx.doi.org/10.1371/journal.pone.0006068
- Askanas V, Engel WK. Inclusion-body myositis: muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer and Parkinson disease brains. Acta Neuropathol 2008; 116:583-95; PMID:18974994; http://dx.doi.org/10.1007/s00401-008-0449-0
- Ganley IG, Wong PM, Jiang X. Thapsigargin distinguishes membrane fusion in the late stages of endocytosis and autophagy. Autophagy 2011; 7:1397-9; PMID:21921693; http://dx.doi.org/10.4161/auto.7.11.17651
- Yamashita S, Abe K, Tatemichi Y, Fujiki Y. The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy 2014; 10:1549-64; PMID:25007327; http://dx.doi.org/10.4161/auto.29329
- Schink KO, Stenmark H. Cell differentiation: midbody remnants - junk or fate factors? Current Biol: CB 2011; 21:R958-60; PMID:22153165; http://dx.doi.org/10.1016/j.cub.2011.10.035
- Grasso D, Ropolo A, Lo Re A, Boggio V, Molejon MI, Iovanna JL, Gonzalez CD, Urrutia R, Vaccaro MI. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J Biol Chem 2011; 286:8308-24; PMID:21173155; http://dx.doi.org/10.1074/jbc.M110.197301
- Ishimura R, Tanaka K, Komatsu M. Dissection of the role of p62/Sqstm1 in activation of Nrf2 during xenophagy. FEBS Letters 2014; 588:822-8; PMID:24492006; http://dx.doi.org/10.1016/j.febslet.2014.01.045
- Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015; 522:359-62; PMID:26040717; http://dx.doi.org/10.1038/nature14506
- Lipatova Z, Segev N. A Role for Macro-ER-Phagy in ER Quality Control. PLoS Genet 2015; 11:e1005390; PMID:26181331
- Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell 2007; 27:53-66; PMID:17612490; http://dx.doi.org/10.1016/j.molcel.2007.06.011
- Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004; 21:81-93; PMID:15345222; http://dx.doi.org/10.1016/j.immuni.2004.06.010
- Shiraishi H, Okamoto H, Yoshimura A, Yoshida H. ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. J Cell Sci 2006; 119:3958-66; PMID:16954146; http://dx.doi.org/10.1242/jcs.03160
- Digaleh H, Kiaei M, Khodagholi F. Nrf2 and Nrf1 signaling and ER stress crosstalk: implication for proteasomal degradation and autophagy. Cell Mol Life Sci 2013; 70:4681-94; PMID:23800989; http://dx.doi.org/10.1007/s00018-013-1409-y
- Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER Stress to Autophagy: Potential Implications for Cancer Therapy. Int J Cell Biol 2010; 2010:930509; PMID:20145727; http://dx.doi.org/10.1155/2010/930509
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol 2002; 22:7405-16; PMID:12370288; http://dx.doi.org/10.1128/MCB.22.21.7405-7416.2002
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334:1081-6; PMID:22116877; http://dx.doi.org/10.1126/science.1209038
- Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem 2002; 277:13045-52; PMID:11821395; http://dx.doi.org/10.1074/jbc.M110636200