
Abstract
Regulation of autophagy is required to maintain cellular equilibrium and prevent disease. While extensive study of post-translational mechanisms has yielded important insights into autophagy induction, less is known about post-transcriptional mechanisms that could potentiate homeostatic control. In our study, we showed that the RNA-binding protein, Dhh1 in Saccharomyces cerevisiae and Vad1 in the pathogenic yeast Cryptococcus neoformans is involved in recruitment and degradation of key autophagy mRNAs. In addition, phosphorylation of the decapping protein Dcp2 by the target of rapamycin (TOR), facilitates decapping and degradation of autophagy-related mRNAs, resulting in repression of autophagy under nutrient-replete conditions. The post-transcriptional regulatory process is conserved in both mouse and human cells and plays a role in autophagy-related modulation of the inflammasome product IL1B. These results were then applied to provide mechanistic insight into autoimmunity of a patient with a PIK3CD/p110δ gain-of-function mutation. These results thus identify an important new post-transcriptional mechanism of autophagy regulation that is highly conserved between yeast and mammals.
Abbreviations
| ATG | = | autophagy-related |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| TOR | = | target of rapamycin |
Although macroautophagy (hereafter autophagy) is cytoprotective by recycling cellular material during nutrient stress, it must be tightly regulated to prevent unnecessary cellular degradation. In addition, cell survival mandates a program of rapid autophagy induction when needed, illustrated by yeast cell exposure to starvation. For example, TOR-mediated induction during starvation utilizes constitutive autophagy-related proteins such as Atg13, which upon dephosphorylation, rapidly function to promote autophagy. Although transcription-dependent regulation occurs in yeast and mammalian cells, less is known about post-transcriptional regulation and how it could serve to accelerate this mechanism.
Recent work has suggested that mRNA degradation may be controlled in response to TOR-dependent signals and is facilitated by stabilization factors such as Igo proteins. Control of transcript degradation could complement de novo mRNA synthesis by stabilizing the steady state levels of mRNA that exist under growing conditions. Whereas its regulatory capacity has been poorly understood, the mechanistic steps leading to deadenylation-dependent mRNA degradation in eukaryotes are quite well known. Degradation begins with a reversible deadenylation step, followed by irreversible removal of the mRNA 5’ cap by the decapping enzyme, Dcp2, and subsequent degradation in a 5’- 3’ direction by the exoribonuclease, Xrn1. A family of ancillary proteins includes an RCK member of RNA-binding DExD/H-box proteins—Dhh1 in S. cerevisiae, Vad1 in the fungal pathogen C. neoformans, and DDX6 in mammals.
Two lines of evidence developed independently in our laboratories suggest a role for the post-transcriptional regulation of autophagy.Citation1 From S. cerevisiae, we screened a library of RNA-binding protein mutants for a cell survival phenotype, which identified the RCK member Dhh1 as a potential regulator. In addition, immunoprecipitation of the RCK member Vad1 from C. neoformans identified ATG8 mRNA bound to the decapping complex. Atg8 is a key protein involved in formation of the initial sequestering compartment, the phagophore.Citation2 C. neoformans is a basidiomycetous yeast that is a cause of an estimated half million AIDS-related deaths yearly, and Vad1 had previously been identified as a global regulator of virulence in a mouse model. Autophagy is particularly important for virulence of this fungus, as survival within nutrient-limited host niches such as macrophages and the central nervous system is key to pathogenesis.
Further studies sought to clarify the role of RCK members in the post-transcriptional mRNA regulation of autophagy and to determine if the processes were evolutionarily conserved. We found by mutational studies in S. cerevisiae that Dhh1 suppresses steady-state levels of multiple ATG mRNAs and inhibits autophagic flux measured by turnover of the GFP-Atg8 chimera as well as a Pho8Δ60 enzyme activity assay. The latter assay uses a modified vacuolar alkaline phosphatase precursor that can only be delivered to the vacuole and proteolytically activated via autophagy. Because Dcp2 is also involved in mRNA degradation, we tested a temperature-sensitive dcp2-7Δ mutant that demonstrated a similar, but more penetrant, phenotype associated with elevated levels of 26 of 29 ATG transcripts and increased autophagic flux. The data suggest a role for autophagy regulation by post-transcriptional mechanisms.
In C. neoformans, our initial data led us to ask if recruitment of ATG mRNA to the RCK/Vad1 complex was a dynamic process consistent with true regulation. Previous studies demonstrated binding of yeast RCK/Dhh1 to the Dcp2-containing decapping complex, suggesting a role for RCK members in mRNA recruitment. Immunoprecipitation experiments demonstrated that the Vad1-mRNA complex dissociates upon transfer of cells to either starvation conditions or following treatment with the TOR inhibitor rapamycin, a potent inducer of autophagy. Further studies showed that ATG8 mRNA exhibits time-dependent decapping and degradation under nutrient-rich repressive conditions, which is reversed after starvation, VAD1 deletion or rapamycin treatment. Reduction of levels of multiple ATG mRNAs and autophagic flux were similarly demonstrated in C. neoformans, demonstrating post-transcriptional regulation as a conserved TOR-dependent mechanism of autophagy control in diverse yeast species. In addition, suppression of autophagy by VAD1 overexpression is sufficient to attenuate virulence of the pathogen in a mouse model.
The finding of a role for TOR in this process suggested that phosphorylation could be a key regulatory feature. Previous whole proteome studies had suggested that Dcp2 might be phosphorylated. Western blot analyses demonstrated robust phosphorylation of Dcp2 under nutrient-rich conditions that is markedly reduced in starved cells. In addition, a phosphomimetic S->D mutant of the homologous cryptococcal Dcp2 phosphorylation sites demonstrates constitutive mRNA binding to the degradation complex, resulting in reduced autophagic body formation and autophagic flux. Conversely, a phosphodeficient S->A mutant results in poor mRNA recruitment, and increased autophagy, similar to that induced by starvation or rapamycin.
Given the important role of autophagy in mammalian stress response, cancer and immunity, we tested to see if post-transcriptional regulation occurs in mammals as well.Citation1 Thus, we used 2 independent mouse C57/BL6 embryonic stem cell lines that contain heterozygote gene-trap insertions to reduce expression of the mammalian RCK/DDX6. As in the C. neoformans strains, reductions in RCK/DDX6 result in accumulation of LC3, a mammalian homolog of Atg8, as well as LC3-labeled puncta (corresponding to autophagosomes) and increased autophagic flux. siRNA suppression of DDX6 in human HeLa cells also demonstrates reductions in LC3 mRNA decapping, with resultant accumulation of LC3 mRNA and LC3-labeled puncta. In contrast, overexpression of DDX6 resulted in accelerated decapping of LC3 mRNA with inhibition of autophagy. Since the DCP2 protein is poorly conserved between mammals and mice, mass spectroscopy was used to identify a phosphorylated serine 249 residue (), which undergoes a 10-fold reduction in phosphorylation after rapamycin treatment. Recapitulation of the same phosphomimetic and phosphodeficient homologs of the mammalian DCP2 protein again result in reductions and inductions of autophagy, respectively, similar to that in C. neoformans.
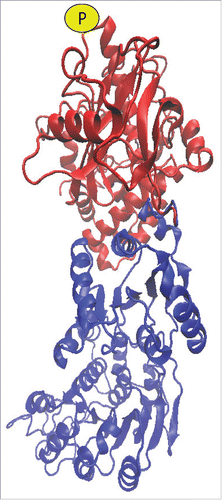
Figure 1. Model of putative mammalian DCP2-RCK interactions with labeling of the S249 phosphorylation site (P). Modeling of the proteins utilized Phyre2,Citation4 interactions were mapped with Zdock,Citation5 and displayed using VMD (www.ks.uiuc.edu/Research/vmd/).Citation6
An important role of autophagy in mammalian immunology is a recently described role in modulating levels of the inflammatory cytokine IL1B, to prevent autoimmune damage of the host. Members of the Uzel laboratory recently described a cohort of patients with a PIK3CD/p110δ gain-of-function mutation with constitutively activated MTOR signaling and autoimmune phenomena;Citation3 thus, we hypothesized that measures of MTOR-dependent post-transcriptional regulation could be used to characterize patients with MTOR-dependent autoimmunity. First, we demonstrated in a macrophage THP-1 cell line that induction of autophagy by the inflammatory inducer lipopolysaccharide leads to reduced decapping and steady state levels of LC3 mRNA. Second, LC3-II and IL1B can be manipulated as predicted, by a set of mammalian DCP2 phosphomimetic and phophodeficient mutants. Interestingly, a patient from the PIK3CD/p110δ gain-of-function cohort demonstrates hyperphosphorylation of DCP2 compared to healthy volunteers based on western blot analyses of peripheral blood mononuclear cells, as well as decreased autophagic vacuole formation and markedly elevated levels of IL1B, suggesting a heretofore unknown regulatory pathway that may constitute an area for further investigation. While additional testing is underway within the cohort, these data suggest the utility of using a measure of post-transcriptional regulation of autophagy as a biomarker of MTOR-dependent autoimmunity.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We would like to acknowledge A. Roy, BCBB/NIAID/NIH for modeling of DDX6-DCP2 interactions.
Funding
This work was funded, in part, by the Intramural Research Program of the NIH, NIAID, NICHD and by National Institutes of Health grant GM053396 (to D.J.K.).
References
- Hu G, McQuiston T, Bernard A, Park YD, Qiu J, Vural A, Zhang N, Waterman SR, Blewett NH, Myers TG, et al. A conserved mechanism of TOR-dependent RCK-mediated mRNA degradation regulates autophagy. Nat Cell Biol 2015; 17:930–42; PMID:26098573; http://dx.doi.org/10.1038/ncb3189
- Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 2008; 19:3290–8; PMID:18508918; http://dx.doi.org/10.1091/mbc.E07-12-1292
- Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, Avery DT, Moens L, Cannons JL, Biancalana M, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol 2014; 15:88–97; PMID:24165795; http://dx.doi.org/10.1038/ni.2771
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015; 10:845–58; PMID:25950237; http://dx.doi.org/10.1038/nprot.2015.053
- Pierce BG, Hourai Y, Weng Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PloS One 2011; 6:e24657; PMID:21949741; http://dx.doi.org/10.1371/journal.pone.0024657
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996; 14:33–8, 27-8; http://dx.doi.org/10.1016/0263-7855(96)00018-5