
Abstract
Autophagy dysfunction is implicated in the pathogenesis of Parkinson disease (PD). BECN1/Beclin 1 acts as a critical regulator of autophagy and other cellular processes; yet, little is known about the function and regulation of BECN1 in PD. In this study, we report that dopamine D2 and D3 receptor (DRD2 and DRD3) activation by pramipexole and quinpirole could enhance BECN1 transcription and promote autophagy activation in several cell lines, including PC12, MES23.5 and differentiated SH-SY5Y cells, and also in tyrosine hydroxylase positive primary midbrain neurons. Moreover, we identified a novel FOS (FBJ murine osteosarcoma viral oncogene homolog) binding sequence (5′-TGCCTCA-3′) in the rat and human Becn1/BECN1 promoter and uncovered an essential role of FOS binding in the enhancement of Becn1 transcription in PC12 cells in response to the dopamine agonist(s). In addition, we demonstrated a critical role of intracellular Ca2+ elevation, followed by the enhanced phosphorylation of CAMK4 (calcium/calmodulin-dependent protein kinase IV) and CREB (cAMP responsive element binding protein) in the increases of FOS expression and autophagy activity. More importantly, pramipexole treatment ameliorated the SNCA/α-synuclein accumulation in rotenone-treated PC12 cells that overexpress wild-type or A53T mutant SNCA by promoting autophagy flux. This effect was also demonstrated in the substantia nigra and the striatum of SNCAA53T transgenic mice. The inhibition of SNCA accumulation by pramipexole was attenuated by cotreatment with the DRD2 and DRD3 antagonists and Becn1 siRNAs. Thus, our findings suggest that DRD2 and DRD3 agonist(s) may induce autophagy activation via a BECN1-dependent pathway and have the potential to reduce SNCA accumulation in PD.
Abbreviations
| ACTB | = | actin, β; AP-1, activating protein-1 |
| BafA1 | = | bafilomycinA1 |
| BECN1 | = | Beclin 1, autophagy related |
| [Ca2+]i | = | intracellular calcium |
| CAMK4 | = | calcium/calmodulin-dependent protein kinase IV |
| ChIP | = | chromatin immunoprecipitation |
| CQ | = | chloroquine |
| CREB | = | cAMP responsive element binding protein |
| DRD2 | = | dopamine D2 receptor |
| DRD3 | = | dopamine D3 receptor |
| EBSS | = | Earle's balanced salt solution |
| EGFP | = | enhanced green fluorescent protein |
| FOS | = | FBJ murine osteosarcoma viral oncogene homolog |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| HTT | = | huntingtin |
| JUN | = | jun proto-oncogene |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| LAMP2 | = | lysosomal-associated membrane protein 2 |
| MAPK8/JNK1 | = | mitogen-activated protein kinase 8 |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 β |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| PD | = | Parkinson disease |
| RPS6KB1 | = | ribosomal protein S6 kinase, 70 kDa, polypeptide 1 |
| PCR | = | polymerase chain reaction |
| PLCB1 | = | phospholipase C, β 1 (phosphoinositide-specific) |
| PPX | = | pramipexole |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| RA | = | retinoic acid |
| SN | = | substantia nigra |
| SNCA | = | synuclein, α (non A4 component of amyloid precursor) |
| SQSTM1/p62 | = | sequestosome 1 |
| Str | = | striatum |
| Tf-LC3B | = | tandem fluorescent mRFP-GFP-LC3B |
| tg | = | transgenic |
| TH | = | tyrosine hydroxylase |
| TPA | = | phorbol 12-myristate 13-acetate |
| TUBB | = | tubulin, β class I |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| WT | = | wild type |
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disorder, affecting 1% to 2% of the population over 65 y old. Its etiology is not fully understood. Pathologically, it is characterized by dopaminergic neuron degeneration in the substantia nigra (SN) pars compacta and other affected brain regions, and the presence of Lewy bodies, the cytoplasmic inclusion largely comprised of SNCA/α-synuclein. Growing evidence indicates that the aggregation of SNCA due to its multiplication or point mutations (A30P, A53T, E46K, H50Q, G51D, A53E) contributes to PD pathogenesis,Citation1-7 although the physiological function of SNCA remains elusive.
Autophagy is a catabolic process that removes aggregated proteins and damaged organelles via lysosomal degradation. In recent years, many studies demonstrate that autophagy dysregulation may be implicated in the development of PD,Citation8-10 and that SNCA aggregate is mainly degraded via autophagy.Citation11,12 BECN1/Beclin 1 is a mammalian homolog of yeast Vps30/Atg6. It interacts with the class III phophatidylinositol 3-kinase (PtdIns3K) and forms a core complex to recruit other autophagy-related proteins that orchestrate autophagosome formation, thus playing a central role in autophagy induction.Citation13 The changes in BECN1 levels in the brains of PD patients remain unknown, albeit the downregulation of BECN1 has been reported and is associated with the accumulation of amyloid β in Alzheimer disease.Citation14 Yet, BECN1 overexpression activates autophagy, reduces SNCA accumulation, and ameliorates the neuritic alterations in vitro and in vivo.Citation15 Therefore, BECN1 may represent a potential therapeutic target for neurodegenerative disorders. The function and regulatory machinery of BECN1 deserves to be explored intensively. Thus far, several transcription factors like HIF1A (hypoxia inducible factor1, α subunit), FOXO3 (forkhead box O3), NFKB1 (nuclear factor of kappa light polypeptide gene enhancer in B-cells 1), JUN (jun proto-oncogene) and E2F1 (E2F transcription factor 1) have been reported to regulate BECN1 transcription and expression under certain conditions.Citation16-20 It remains to be determined whether other factors may exist in regulation of BECN1 transcription.
The dopamine D2-like receptors (DRD2, DRD3, DRD4) agonist pramipexole (PPX) alleviates both motor and nonmotor symptoms of PD patients, via mechanisms dependent or independent of the receptors.Citation21-24 Moreover, Li et al. report an accumulation of autophagic vacuoles in the brains of PPX-treated mice,Citation25 implying PPX may increase autophagy induction or inhibit autophagy flux. However, the molecular mechanism(s) is poorly studied. It is also unknown whether this is a common effect of D2-like receptor agonists. More importantly, it remains to be clarified whether the PPX-modulated autophagy activity affects the accumulation of SNCA.
In the present study, we revealed an essential role of BECN1 in the autophagy induced by the DRD2 and DRD3 agonists PPX and quinpirole. Moreover, we identified a novel FOS (FBJ murine osteosarcoma viral oncogene homolog) binding sequence in the rat and human Becn1/BECN1 gene promoter, and verified that FOS was critical for Becn1/BECN1 transcription increase, thus showing autophagy activation in response to PPX treatment. Functionally, we demonstrated that PPX treatment could ameliorate the SNCA accumulation in rotenone-treated PC12 cells that overexpress wild-type (WT) and mutant SNCA, and also in SNCAA53T transgenic (tg) mice, via promoting BECN1-dependent autophagy activity.
Results
PPX and quinpirole induce autophagy activation
To study the effect of D2-like receptor agonists on autophagy, PC12 cells were treated with increasing concentrations (10, 50, and 100 μM) of PPX for the indicated time periods. The protein levels of autophagy-related markers including LC3B-II/MAP1LC3B-II, BECN1 and SQSTM1/P62 were assessed by western blot. The results showed that LC3B-II and BECN1 levels increased while SQSTM1 decreased in response to PPX treatment (). Moreover, the expression of BECN1 and LC3B-II was still higher at 24 h after PPX treatment, and did not decline to the basal level until 48 h later (Fig. S1A). By contrast, dexpramipexole (DexPPX), which is the R (+) enantiomer of PPX and is virtually devoid of the agonist activity, had little effect on these autophagic markers (). Another selective DRD2 and DRD3 agonist quinpirole (10 μM) also enhanced the LC3B-II and BECN1 levels but reduced SQSTM1 protein levels in PC12 cells (). In order to verify the LC3B-II increase detected by western blotting, we also examined the changes of EGFP-LC3B distribution in PC12 cells transfected with EGFP-LC3B plasmid. As shown in , treatment with PPX for 12 h elevated the EGFP-LC3B puncta number in PC12 cells overexpressing EGFP-LC3B. The autophagosome formation was further validated by an electron microscopy study. and Figure S1B show that in responses to PPX and quinpirole treatment, numerous double-membrane structured vacuoles (green arrows), which reflect the autophagic characteristics, appeared in the cytoplasm of PC12 cells. In contrast, such vacuoles were rarely seen in untreated cells. These results suggest that DRD2 and DRD3 agonists have the ability to induce autophagy activation in PC12 cells.
Figure 1. For figure legend, see page 2074.Figure 1 (See previous page). Pramipexole (PPX) and quinpirole induce autophagy activation in PC12 cells. (A to D) PC12 cells were treated with PPX (A and B), dexpramipexole (DexPPX, C), or quinpirole (D) at various concentrations for the indicated time. The changes of LC3B-II, BECN1, and SQSTM1 proteins levels in response to the treatments were analyzed by western blotting. ACTB/actin served as loading controls for this and other figures. N=3 or 4. One-sample t test. (E) Effect of PPX on EGFP-LC3B dots formation. PC12 cells were transfected with EGFP-LC3B plasmid for 48 h and subjected to 100 μM PPX treatment for 12 h, followed by confocal microscopy study for EGFP-LC3B dots. At least 30 cells per group were included for the counting and quantification. Scale bar: 20 μm. Student t test. (F) Transmission electron microscopy (TEM) images for autophagic vacuoles (green arrows pointed) in normal PC12 cells following 100 μM PPX treatment for 12 h. (ii) and (iii) represents the insets in black square in (i) and (ii), while (v) and (vi) indicates the insets in (iv) and (v), respectively. (G) PC12 cells were pretreated with or without BafA1 (100 nM) or CQ (30 μM) for 30 min, followed by PPX treatment for 12 h. One-sample t test and unpaired t test. N=3. (H) PC12 cells were transfected with the tandem fluorescent mRFP-GFP-LC3B (Tf-LC3B) plasmid, and then treated with 100 μM PPX for 12 h at 48 h post-transfection. The RFP- and GFP-LC3B puncta per cell were counted as described in (E). Student t test. Scale bar: 20 μm. *, P<0 .05; **, P<0 .01; ***, P<0 .001; n.s., not significant.
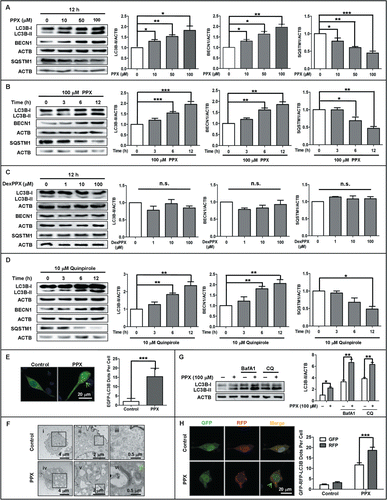
Next, we assessed the LC3B-II turnover with or without the blockage of lysosomal degradation in PC12 cells. The lysosome inhibitors bafilomycin A1 (BafA1) and chloroquine (CQ) were individually added into the culture medium at 30 min before and during the agonist treatment. Western blot analysis showed that the endogenous LC3B-II level induced by PPX () and quinpirole (Fig. S1C) further increased in the presence of BafA1 or CQ, implying the enhancement of autophagic flux by the agonist. The autophagic flux was further monitored with the tf-LC3B assay, which relies on the different nature of GFP and RFP fluorescence under acidic conditions. GFP fluorescence is sensitive to the acidic condition of the lysosome lumen while RFP is relatively stable under acidic conditions. Thus, the colocalization of GFP and RFP signals (yellow dots) indicate the phagophores or autophagosomes that have not fused with lysosomes, whereas RFP-only signals (red puncta) stain the autolysosomes. In our study, the confocal microscopic scanning demonstrated that 100 μM PPX treatment for 12 h increased the number of yellow dots per cell, with more elevations in RFP-only LC3B dots in PC12 cells that were transfected with the tandem fluorescent mRFP-GFP-LC3B (tf-LC3B) plasmid.()
Similar observations were obtained in MES23.5 cells, another commonly used cell culture model for PD. Figure S1D showed that treatment with PPX (10, 50, and 100 μM) for 12 h resulted in the elevations of LC3B-II and BECN1, accompanied by a reduction in SQSTM1 in MES23.5 cells. Then, we asked if PPX treatment could also induce endogenous autophagosome formation in dopaminergic neurons. To this end, the rat primary midbrain neurons were treated with 100 μM PPX for 12 h and then assessed for LC3B puncta formation using the antibody specific for LC3B and the antibody against TH (tyrosine hydroxylase) to label dopaminergic neurons. Figure S1E demonstrated that the puncta formation of endogenous LC3B was profoundly increased in TH positive midbrain neurons following PPX treatment, implying autophagy activation in dopaminergic neurons. Taken together, all the data suggest the increases in autophagy induction and flux by PPX and quinpirole.
The cell viability was also studied since autophagic cell death was frequently reported in different types of cells. It was observed that treatment with ≤100 μM PPX or ≤ 10 μM quinpirole for up to 24 h did not affect the cell survival of PC12 cells, as determined by CCK-8 assay (Fig. S2). These 2 agonists decreased the cell viability of PC12 cells only at higher concentrations (PPX ≥ 500 μM, quinpirole ≥50 μM). In support of this, cleaved CASP3/caspase-3 was almost undetected by western blot in PC12 cells treated with PPX (10 and 100 μM) for 24 h; however, an obvious induction and elevation of cleaved CASP3 was observed in etoposide (a classic apoptosis inducer as a positive control) -treated cells as compared to untreated cells. Therefore, it excludes a possibility of increased cell death or apoptosis associated with enhanced autophagy activity in PC12 cells in response to the agonist(s) treatment within certain concentration ranges.
BECN1 is required for PPX-induced autophagy activation
Next, we studied the signaling pathways that may be involved. Since MTOR (mechanistic target of rapamycin [serine/threonine kinase]) is a major negative regulator of autophagy, we focused on MTOR signaling first. Consistent with previous reports,Citation26 our data revealed that starvation of PC12 cells by culturing in Earle's balanced salt solution (EBSS) immediately reduced the phosphorylation of MTOR and its substrate RPS6KB1 (ribosomal protein S6 kinase, 70kDa, polypeptide 1), accompanied by the increases of ULK1 (unc-51 like autophagy activating kinase 1) dephosphorylation at the S757 site and phosphorylation at S555 (Fig. S3A), implying that nutrient limitation by EBSS inhibits MTOR and enhances ULK1 activity in PC12 cells. To our surprise, PPX treatment enhanced the phosphorylation of MTOR, RPS6KB1 and ULK1 at S757, without any significant effect on ULK1 phosphorylation at the S555 site in PC12 cells cultured in complete medium (Fig. S3B). Interestingly, quinpirole failed to affect either of the above-mentioned signaling molecules in nutrient-rich conditions (Fig. S3C). The results indicate that an MTOR-independent pathway may be involved in the induction of autophagy by these agonists.
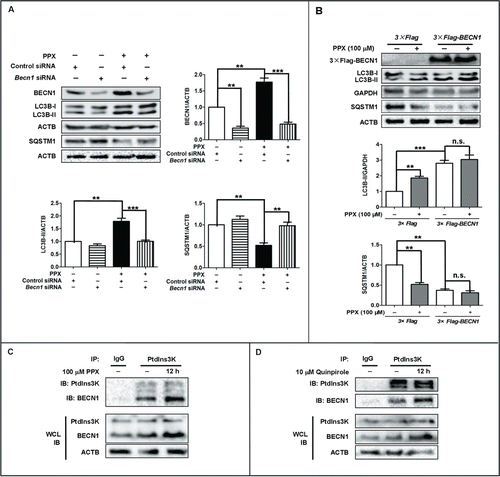
We then examined BECN1, another critical regulator of autophagy, since it was upregulated by these 2 agonists (). Western blot analysis revealed that knockdown of Becn1 with siRNA abolished the changes in LC3B-II and SQSTM1 protein levels induced by PPX in PC12 cells (). Moreover, PPX failed to increase LC3B-II and reduce SQSTM1 levels in BECN1-deficient cells. On the contrary, BECN1 overexpression was sufficient to enhance the autophagy level, as evidenced by LC3B-II elevation and SQSTM1 reduction in PC12 cells (). Coimmunoprecipitation analysis revealed that both PPX and quinpirole enhanced endogenous BECN1 expression and the binding with PtdIns3K, without significant alterations in PtdIns3K levels at 12 h after treatment in PC12 cells (). Neither the LAMP1 nor the LAMP2 level was altered in PPX- or quinpirole- treated PC12 cells (Fig. S4). These results suggest that BECN1 is required for the autophagy induction by PPX and quinpirole.
Figure 2. BECN1 is required for PPX-induced autophagy activation. (A) PC12 cells were transfected with Becn1 siRNA or control siRNA for 48 h, followed by 100 μM PPX treatment for 12 h. The efficiency of BECN1 knockdown, as well as LC3B-II and SQSTM1 protein levels, was assessed by western blotting. ACTB and GAPDH served as loading controls. (B) BECN1 overexpression resulted in an enhanced level of autophagy. PC12 cells were transfected with 3×Flag-tagged BECN1 or vector, followed by PPX treatment for 12 h. **, P<0 .01; ***, P<0 .001; n.s., not significant. One-sample t test and Student t test. (C and D) PPX and quinpirole enhanced BECN1 expression and binding to PtdIns3K, without affecting PtdIns3K protein levels. PC12 cells were treated with PPX for 12 h. Whole cell lysates (WCL) and PtdIns3K or IgG immunoprecipitated lysates were analyzed by western blotting with anti-BECN1 or anti-PtdIns3K antibody. The results were independently repeated 3 times.
DRD2 and DRD3 mediate the autophagy induction by PPX
To determine whether the autophagy induction was mediated by the D2-like receptors, several dopamine receptor expressing cell lines were applied and subjected to PPX treatment in the presence or absence of a D2-like receptor antagonist. Western blot analysis and reverse transcription PCR () showed that PC12 and MES23.5 cells express DRD2 at a low abundance while undifferentiated SH-SY5Y cells express DRD2 at an almost undetectable level. The DRD3 protein abundance in PC12 and undifferentiated SH-SY5Y cells is very low; however, its abundance in MES23.5 is quite high, almost comparable to that in retinoic acid (RA) and phorbol 12-myristate 13-acetate (TPA)-differentiated SH-SY5Y cells. This indicates that these cell lines differentially express dopamine receptor(s) in various situations. Thus, the receptor-specific effect on autophagy was validated in different cell lines. As shown in , pretreatment with the DRD2 antagonist L741,626 (50 nM) dampened the alterations in LC3B-II, BECN1 and SQSTM1 protein levels induced by PPX in PC12 cells, which predominantly express the DRD2 subtype. In MES23.5 cells, which express both DRD2 and DRD3, inhibition of either the DRD2 by 50 nM L741,626 or DRD3 by 20 nM GR103,691 suppressed the increases of LC3B-II and BECN1 levels in PPX-treated cells (). GR103,691 seemed to be more effective than L741,626. The combination of L741,626 and GR103,691 almost abolished the increase of LC3B-II and BECN1 expression induced by PPX.
Figure 3. For figure legend, see page 2077.Figure 3 (See previous page). DRD2 and DRD3 mediate the autophagy activation by PPX. The protein (A) and mRNA (B) expression of DRD2 and DRD3 in PC12, MES23.5, undifferentiated (UN) and, RA and TPA differentiated SH-SY5Y cells were analyzed by western blotting and reverse transcription PCR, respectively. PC12 (C) or MES23.5 cells (D) were treated with 100 μM PPX for 12 h, with (+) or without (−) 50 nM L741,626 or 20 nM GR103691 pretreatment 30 min before and during PPX treatment. N=4. One-sample t test and Student t test. (E) PPX treatment resulted in significant increases in LC3B-II and BECN1 protein levels in PC12 and RA and TPA differentiated SH-SY5Y cells, but not in undifferentiated cells. One-sample t test. N=4. (F to H) SH-SY5Y cells were transfected with GFP-DRD2 plasmid or GFP-DRD3 or both. The changes in DRD2 and DRD3 mRNA levels (F) were analyzed by reverse transcription PCR at 24 h after transfection. (G) The DRD2 and DRD3 receptors (GFP fluorescence) predominantly localized to the plasma membrane of cells, visualized by the confocal microscopy. Scale bar: 10 μm. (H) SH-SY5Y cells were treated with PPX at 24 h post-transfection. The protein levels of GFP-DRD2/3, LC3B, and BECN1 were analyzed by western blotting. One-sample t test and Student t test. N=5 to 7. *, P<0.05; **, P<0.01; ***, P<0.001. UN, undifferentiated; RA/TPA, RA and TPA differentiated; n.s., not significant.
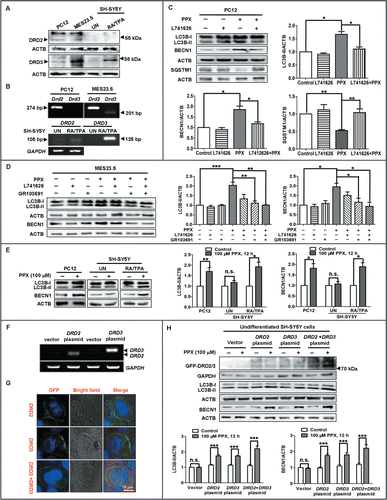
In addition to pharmacological inhibition, we also used genetic approaches to assess the role of DRD2 and DRD3 in autophagy induction by PPX. In accordance with previous observations,Citation27 undifferentiated SH-SY5Y cells expressed few DRD2 or DRD3, while RA and TPA-differentiated SH-SY5Y expressed these 2 subtypes at a relatively higher abundance, especially the DRD3 subtype. We found that PPX treatment caused a dramatic increase in LC3B-II and BECN1 levels in RA and TPA-differentiated SH-SY5Y cells but not in undifferentiated cells (). In support of this, PPX treatment for 12 h obviously upregulated the LC3B-II and BECN1 levels in undifferentiated SH-SY5Y cells that were transfected with GFP-conjugated DRD2 or DRD3 plasmid, or a combination of both, compared to vector-transfected cells ( to H). DRD2 and DRD3 predominantly localized to the plasma membrane of the transfected cells. These results, in combination with the data that PPX rather than its enantiomer DexPPX induced autophagy activation, indicate that the autophagy induction by PPX may be mediated by DRD2 and DRD3 signaling.
FOS is essential for rat and human Becn1/BECN1 transcription upregulation and autophagy induction by PPX
We then moved on to study the molecular regulation of BECN1 expression. The quantitative PCR analysis revealed that PPX enhanced Becn1 mRNA levels in PC12 cells in a dose-dependent manner (). Gene sequence analysis identified a putative AP-1 binding site (5′-TGCCTCA-3′) within the rat Becn1 promoter (−278 to −272 from the transcription start site) (). Transcription factors JUN and FOS often form heterodimers to bind AP-1 site and regulate the target gene transcription.Citation28 JUN activation is reported to regulate BECN1 transcription and mediate autophagic cell death in cancer cells.Citation17 However, we observed that PPX had little impact on the phosphorylation of MAPK8/JNK1 or JUN in PC12 cells (Fig. S5), which is in line with the previous report.Citation29 Therefore, we focused on FOS signaling in the following study. As presented in , FOS expression in PC12 cells increased following PPX treatment, which rose early at 1 h, peaked at 12 h and remained to be elevated at 24 h after treatment. Moreover, the chromatin immunoprecipitation (ChIP) assay revealed that FOS bound to this AP-1 site in the rat Becn1 promoter region, and this binding could be enhanced by PPX treatment (). A similar AP-1 sequence was identified in the human BECN1 promoter (−431 to −206), and the FOS binding to human BECN1 promoter was also verified by ChIP assay (Fig. S6). More importantly, the reporter luciferase assay displayed that PPX resulted in an elevation of BECN1 promoter activity, and this was abolished in cells transfected with the AP-1-deleted mutants of the BECN1 reporter (), indicating that FOS is critical for the increase of BECN1 transcription through binding to the AP-1 site in its promoter.
Figure 4. FOS is essential for PPX-induced BECN1 upregulation and autophagy increases. (A) PC12 cells were treated with PPX at the indicated concentration, and Becn1 mRNA levels were determined by quantitative PCR at 6 h after treatment. One-sample t test. N=5. (B) The schematic model for the localization of the putative AP-1 site (−278 to −272) in the rat Becn1 promoter. (C) PC12 cells were treated with 100 μM PPX for the indicated time, followed by western blot analysis for FOS protein levels. One-sample t test, n=4. (D) PC12 cells were treated without (−) or with (+) 100 μM PPX for 12 h. ChIP assays were then performed using 10 μg anti-FOS or normal IgG, and the mRNA levels of immunoprecipitated rat Becn1 promoter region spanning the putative AP-1 site (−319 to −98) were measured by reverse transcription PCR. Input DNA was shown as controls. (E) PPX resulted in an increase in the promoter activity of WT but not mutant (MUT) Becn1 in PC12 cells. One-sample t test. N=3. (F) PC12 cells were transfected with control or Fos siRNA for 36 h, followed by PPX treatment for 12 h. One-sample t test and Student t test. N=4. (G) Effect of FOS overexpression on the levels of autophagy markers. PC12 cells were transfected with FOS construct or its vector for 48 h and subjected to 100 μM PPX for 12 h. One-sample t test. N=3 or 4. *, P<0 .05; **, P<0 .01; ***, P<0 .001; n.s., not significant.
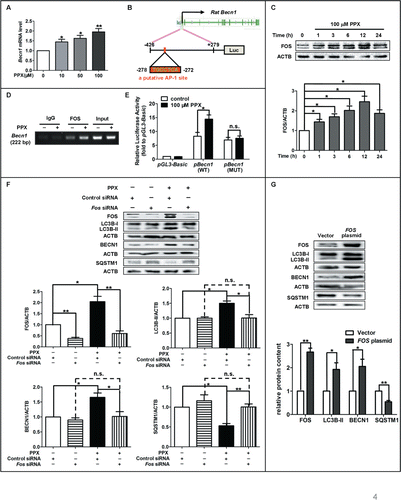
Next, we studied whether FOS increase was essential for PPX-induced autophagy. To achieve this, cells were transfected with Fos-specific or control siRNA for 48 h, followed by 100 μM PPX treatment for 12 h. As expected, the siRNA transfection resulted in at least a 50% reduction of FOS protein level (). The PPX-induced autophagy increase, as evidenced by the elevations of LC3B-II and BECN1 and reductions of SQSTM1 protein level, was diminished by FOS knockdown. PPX failed to alter the protein levels of LC3B-II, BECN1 and SQSTM1 in FOS-deficient cells. However, FOS overexpression alone caused about 2-fold increases in both LC3B-II and BECN1 protein levels and about 50% reduction in SQSTM1 proteins compared to vector-transfected PC12 cells (), confirming that FOS upregulation may be relevant in the BECN1 transcription and autophagy increases induced by PPX.
The role of Ca2+ and CAMK4 (calcium and calmodulin-dependent protein kinase IV) signaling in FOS increases and autophagy activation induced by PPX
We further examined the signaling events that mediate FOS upregulation. Previous studies report that D2-like receptor activation triggers Ca2+-calmodulin signaling.Citation30 Here we found that 100 μM PPX caused an elevation of intracellular Ca2+ ([Ca2+]i), which rose and peaked at about 30 min after treatment in PC12 cells. This [Ca2+]i increase was resistant to EDTA cotreatment (), implying the elevated [Ca2+]i mainly comes from intracellular Ca2+ stores. In support of this, [Ca2+]i chelation by BAPTA-AM or Quin-2-AM consistently reversed the increases of FOS expression and autophagy activity caused by PPX treatment ( and C). Moreover, PPX induced a time-dependent increase in CAMK4 (T196) phosphorylation (). Given that Ca2+ and CAMK4 are implicated in the regulation of cAMP response element (CRE)-dependent gene transcription and that FOS is highly regulated by CREB (cAMP responsive element binding protein), we further studied and found that the phosphorylation of CREB (S133) exhibited similar changes to that of CAMK4 (T196) following PPX treatment in PC12 cells (). Herein, these data indicate that Ca2+ and CAMK4 signaling may play a key role in PPX-triggered FOS upregulation and thereby autophagy induction, and that CREB phosphorylation may be involved in this signaling cascade.
Figure 5. A critical role of intracellular Ca2+ in PPX-induced autophagy activation. (A) Representative Ca2+ tracings and group data showed that PPX elevated [Ca2+]i in both Krebs bicarbonate buffer and EDTA-treated Ca2+-free buffer. Student t test. (B and C) Intracellular Ca2+ depletion with BAPTA-AM (B, 10 μM) or Quin-2-AM (C, 10 μM) abolished the increases of FOS expression and autophagy induced by PPX. PC12 cells were pretreated with Ca2+ chelator for 30 min, followed by 100 μM PPX treatment for 6 h. One-sample t test and Student t test. (D) PC12 cells were treated with 100 μM PPX for the indicated time periods. The levels of p-CAMK4(T196) and p-CREB(S133) were analyzed by western blotting. One-sample t test. N=3 or 4. *, P<0 .05; **, P<0 .01; ***, P<0 .001.
![Figure 5. A critical role of intracellular Ca2+ in PPX-induced autophagy activation. (A) Representative Ca2+ tracings and group data showed that PPX elevated [Ca2+]i in both Krebs bicarbonate buffer and EDTA-treated Ca2+-free buffer. Student t test. (B and C) Intracellular Ca2+ depletion with BAPTA-AM (B, 10 μM) or Quin-2-AM (C, 10 μM) abolished the increases of FOS expression and autophagy induced by PPX. PC12 cells were pretreated with Ca2+ chelator for 30 min, followed by 100 μM PPX treatment for 6 h. One-sample t test and Student t test. (D) PC12 cells were treated with 100 μM PPX for the indicated time periods. The levels of p-CAMK4(T196) and p-CREB(S133) were analyzed by western blotting. One-sample t test. N=3 or 4. *, P<0 .05; **, P<0 .01; ***, P<0 .001.](/cms/asset/c9e5aa80-6c3b-47d1-ae26-c24c9345c4e1/kaup_a_1100930_f0005_oc.gif)
PPX promotes SNCA degradation via BECN1-dependent autophagy in rotenone-treated cells
We also sought to examine whether PPX could affect SNCA accumulation since it enhanced autophagy activity. We found that 50 nM rotenone treatment for 24 h suppressed autophagy activity, as indicated by LC3B-II reduction, and SQSTM1 accumulation, and caused endogenous SNCA accumulation in PC12 cells (). This was reversed by PPX cotreatment. The mRNA level of endogenous Snca was not affected by rotenone or PPX treatment alone, or in combination (), implying that the rotenone-induced SNCA accumulation mainly results from post-transcriptional increase. Strikingly, BECN1 protein level remained unaltered following rotenone exposure. Lentivirus infection with WT and mutant variants (A53T, A30P) of SNCA into PC12 cells had little effect on BECN1 expression either (Fig. S7A). Remarkably, overexpression of WT or mutant SNCA differentially affected the LC3B-II level in PC12 cells. Specifically, LC3B-II level was decreased in WT-SNCA overexpressing cells but not in SNCAA53T- or SNCAA30P-transduced PC12 cells as compared to vector-infected cells.
Figure 6. PPX reduces the accumulation of SNCA in vitro. (A to C) Normal PC12 cells (A and B) or PC12 cells that stably overexpress mutant SNCAA53T (A53T-PC12, C) were pretreated with 100 μM PPX for 30 min, followed by 50 nM rotenone cotreatment for 24 h. The protein levels of SNCA and autophagy markers were evaluated by western blotting while Snca mRNA levels were examined by reverse transcription PCR. ACTB and GAPDH served as loading controls. (D) A53T-PC12 cells were transfected with control siRNA or Becn1 siRNA for 24 h and then subjected to 100 μM PPX or DexPPX pretreatment for 30 min, followed by rotenone (Rot) cotreatment for another 24 h. For D2(L)R antagonist cotreatment group, 50 nM L741,626 and 20 nM GR103691 were mixed together and added at 30 min before PPX treatment. Whole lysates were analyzed by western blotting. One-sample t test and Student t test. N=4. *, P<0 .05; **, P<0 .01; ***, P<0 .001; n.s., not significant.
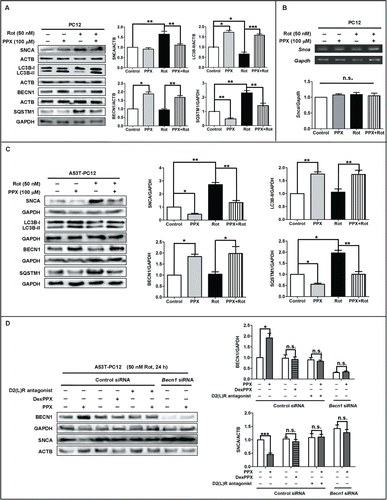
In addition to endogenous SNCA, we also examined the accumulation of exogenously transduced WT SNCA and SNCAA53T since multiplication and point mutations of SNCA is pathologically relevant for PD development. Our data showed that rotenone exposure for 24 h resulted in about 2- and one-fold increases of SNCA level in PC12 cells that overexpress SNCAA53T () and WT SNCA(Fig. S7B). PPX cotreatment efficiently decreased the SNCA accumulation induced by rotenone in both cells, along with the elevations of LC3B-II and BECN1 and reductions in SQSTM1 level. PPX also resulted in a decrease of SNCA accumulation in SNCAA53T overexpressing PC12 cells (A53T-PC12) without rotenone exposure, with less effect in WT-SNCA overexpressing cells (WT-PC12). More importantly, PPX, but not DexPPX, enhanced BECN1 expression and reduced SNCA accumulation in A53T-PC12 cells following rotenone exposure for 24 h, and this effect was abolished by DRD2 and DRD3 antagonists cotreatment (). Further, PPX failed to decrease SNCA accumulation in BECN1-deficient cells compared to control siRNA-transfected cells. These results indicate that the beneficial effect of PPX on SNCA clearance may be secondary to DRD2 and DRD3 activation and BECN1 upregulation.
To verify the effect of PPX on SNCA clearance, we further separated SNCA into NP40-soluble and -insoluble fractions, followed by western blotting under denaturing conditions. The results showed that both NP40-soluble and -insoluble SNCA levels increased in WT-PC12 cells after rotenone exposure for 24 h (Fig. S7C), with more increase in the NP40-insoluble fraction, thereby implying a possibility that more SNCA aggregates may form following rotenone treatment. Nevertheless, PPX cotreatment ameliorated the accumulation of both NP40-soluble and -insoluble SNCA caused by rotenone. ACTB/β-actin was detected in both fractions whereas GAPDH was only detected in the soluble fraction, indicating the purity of insoluble fraction was sufficient.
To confirm the effect of autophagy induction by PPX on the clearance of protein aggregates, we tested the effect of PPX on another aggregate-prone protein huntingtin (HTT) in a parallel study. To achieve this, 2 fragments HTT-552–18Q and HTT-552–100Q, which represent for WT and mutant variant of HTT proteins that express amino acids 1 to 552 of the full length, were infected into PC12 cells via adenovirus delivery. Western blot analysis showed that the levels of both HTT-552–18Q and HTT-552–100Q fragments were exceptionally high compared to that in noninfected (Control) and vector-infected (Null) cells, implying the successful infection of these 2 fragments (Fig. S7D). PPX treatment dramatically reduced the levels of both fragments, supporting a beneficial role of PPX in attenuating protein accumulation.
PPX decreases SNCA accumulation in SNCAA53T tg mice
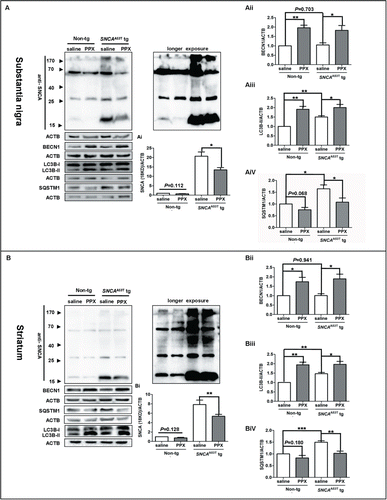
Finally, the in vitro observations were validated in SNCAA53T tg mice. SNCAA53T tg mice and age-matched Non-tg mice were intraperitoneally injected with PPX at 0.5 mg/kg or saline twice daily for 3 consecutive wk. As shown in , PPX administration decreased the SNCA level (18 kDa) in both the SN (I) and the Str (I) of SNCAA53T tg mice. Moreover, PPX injection resulted in significant increases of BECN1 and LC3B-II levels in the SN () and the Str () of both non-tg and SNCAA53T tg mice. PPX injection also reduced the SQSTM1 levels in both brain tissues of SNCAA53T tg mice, with less effect in non-tg mice (AiV, P=0.068, SN; BiV, P=0.180, Str). These results indicate that PPX may reduce SNCA accumulation in the brains of SNCAA53T tg mice, probably via promoting autophagy activity. This is consistent with the previous report that PPX administration led to the accumulation of autophagic vacuoles in the SN of adult C57BL/6 mice.Citation25 Notably, as observed in vitro (Fig. S7A), the BECN1 levels in the brain tissues of SNCAA53T tg mice did not differ significantly from that in non-tg mice (P=0.703 for SN, and P=0.941 for Str).\raster="rgFigKAUP_A_1100930_F0007_B"
Figure 7. PPX administration increases autophagy and ameliorates SNCA accumulation in the nigrostriatal pathway of SNCAA53T tg mice. SNCAA53T tg (12 to 15 mo old) and age-matched non-tg mice were intraperitoneally injected with PPX or saline at 0.5 mg/kg twice daily for 3 wk. The protein levels of SNCA, LC3B, BECN1 and SQSTM1 in the SN (A) and the Str (B) were subjected to western blot analysis for shorter or longer exposure time as specified. Molecular mass markers are indicated on the left of the blots. One-sample t test and Student t test. N=5 mice for each group. *, P<0 .05; **, P<0 .01; ***, P<0 .001.
Discussion
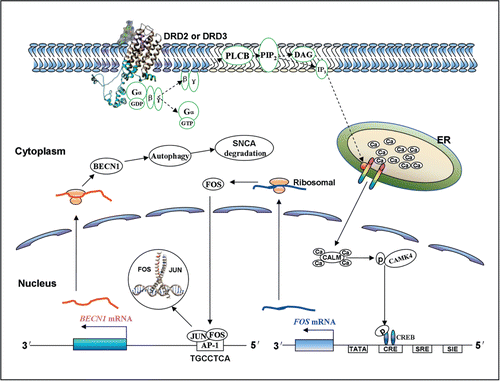
In this study, we demonstrated that the DRD2 and DRD3 agonists PPX and quinpirole had the ability to induce autophagy activation via an MTOR-independent but BECN1-dependent pathway, and reduce SNCA accumulation in vitro and in vivo (summarized in ). Second, we identified a novel FOS binding site (5′-TGCCTCA-3′) within the rat and human Becn1/BECN1 promoter and revealed that FOS was critical for BECN1 transcription increase and autophagy induction by the agonists. The endogenous LC3B-II level and the number of EGFP-LC3B and mRFP-LC3B puncta increased, along with a decrease in SQSTM1 protein level, in various cell lines and TH-positive midbrain neurons in response to the 2 agonists treatment. Moreover, the agonist-induced LC3B-II level was further increased in the presence of the lysosome inhibitor BafA1 or CQ. The transmission electron microscopy assessment also revealed an increase in the number of autophagic vacuoles in agonist-treated cells. Hence, these results clearly point to autophagy activation by DRD2 and DRD3 agonist treatment.
Figure 8. Schematic summary of the signaling cascades that underlie the induction of autophagy and SNCA degradation by DRD2 and DRD3 agonists. Once binding to DRD2 and DRD3, the agonist(s) may mobilize Ca2+ release from endoplasmic reticulum via PLCB1 and IP3 signaling. Ca2+ may then bind to CALM/calmodulin and promote the activation of CAMK4, which further phosphorylates CREB and enhances FOS transcription. Next, FOS may bind to the AP-1 sequence in the rat and human Becn1/BECN1 gene promoter, and thereby induces BECN1 upregulation, which plays an important role in promoting autophagy and SNCA degradation via autophagy in dopaminergic cells. DAG, diacylglycerol; Gα,β,γ, α,β, and γ subunits of G protein; PIP2, phosphatidylinositol 4,5-bisphosphate, also known as PtdIns(4,5)P2.
A growing body of literature implicates autophagy dysfunction in the pathogenesis of PD.Citation9,10,31-35 For example, earlier studies report that autophagic vacuoles accumulate in the brains of PD patients.Citation36 SNCA aggregates are mainly degraded via the autophagy-lysosome pathway.Citation37,38 Conditional disruption of autophagy leads to protein aggregates formation and progressive losses of dopaminergic neurons in the SN.Citation39 More importantly, overexpression of BECN1 activates autophagy, reduces SNCA accumulation and ameliorates the neurodegenerative pathology in SNCA tg mice.Citation15 In line with this, our present study showed that PPX induced BECN1-dependent autophagy activation and attenuated SNCA accumulation in rotenone-treated cells and also in SNCAA53T tg mice, supporting an important role of autophagy in clearing SNCA aggregates. Notably, the failure of autophagy may also promote the exocytosis and intercellular transfer of SNCA to neighboring cells and thus lead to the spread of SNCA pathogenesis.Citation40,41 SNCA can be secreted by exosomes in a Ca2+-dependent manner.Citation42 Therefore, this adds to the complexity with regard to the role of Ca2+ in autophagic regulation of SNCA. So far, there remains controversy over the role of Ca2+ in modulating autophagy activity.Citation43 Some studies reported that enhancing [Ca2+]i could induce autophagy, while others held the opposite opinion. Our results suggest that the [Ca2+]i elevation may be involved in autophagy activation because [Ca2+]i depletion with BAPTA-AM or Quin-2-AM abolished the autophagy induction by PPX. The relative [Ca2+]i levels and the duration of [Ca2+]i elevation as well as the types of cells or stimuli are important factors that determine the autophagy status, and may thus account for the controversies. However, whether the Ca2+-dependent secretion of SNCA contributes to the reduction of intracellular SNCA level induced by PPX was not examined in our study, which deserves further investigation in future. Regardless of this, the PPX-induced decreases in SNCA accumulation was abolished in BECN1-deficient cells. Therefore, the autophagy flux induced by PPX may be responsible for the decrease in SNCA accumulation.
In this study, we found that WT SNCA overexpression in PC12 cells via lentivirus transduction reduced LC3B-II levels whereas the mutant forms of SNCA (A53T and A30P) had no effect on it, similar to a previous report in human neuroblastoma cells (SKNSH).Citation44 It is likely that either the A53T or A30P mutant SNCA has little effect on autophagy compared with WT SNCA. Alternatively, these mutants may also suppress autophagy in a similar way to WT SNCA; however, it may be masked by the compensatorily activated macroautophagy due to the inhibition of chaperone-mediated autophagy by the mutants.Citation45 Conflicting data also exist regarding the effect of mutant SNCA on autophagy status.Citation44,46-49 Several factors such as the level of SNCA overexpression, the vector as well as culture condition may be involved.
We also demonstrate that the DRD2 and DRD3 agonist PPX could promote protein aggregate degradation via autophagy based on several findings. First, PPX not only decreased the endogenous SNCA level, but also reduced the accumulation of exogenously overexpressed WT and mutant SNCAA53T in rotenone-treated PC12 cells along with the increases in autophagy activity, without altering the Snca mRNA level. Moreover, PPX was able to reduce the protein levels of NP-40 insoluble SNCA, in addition to the soluble fraction. More importantly, PPX administration for 3 wk decreased the levels of SNCA protein at both 18 kDa and higher molecular mass in the nigrostriatal pathway of SNCAA53T tg mice. Apart from SNCA, PPX ameliorated the levels of another aggregate-prone protein HTT in PC12 cells that overexpress the HTT-552–18Q or HTT-552–100Q fragment.
BECN1 is an adaptor protein that assembles a core complex with PtdIns3K and thus functions in autophagy initiation. The transcriptional and post-translational regulation of the BECN1-PtdIns3K complex crucially controls the autophagy activity.Citation17,50,51 Understanding of BECN1 transcription and translation regulation is thus relevant for autophagy and its related disorders. Our data suggest that PPX and quinpirole induced autophagy activation in an MTOR-independent but BECN1-dependent manner. This is different from the machinery for nutrient deprivation-triggered autophagy activation. These 2 DRD2 and DRD3 agonists upregulated Becn1 transcription and expression and enhanced its binding with PtdIns3K, without affecting PtdIns3K expression levels. Further, a FOS binding site (5′-TGCCTCA-3′), slightly different from the consensus AP-1 sequence (5′-TGACTCA-3′), was identified in the rat and human Becn1/BECN1 promoter. In fact, a recent study also defined the sequence 5′-TGCCTCA-3′ as a putative AP-1 site in cancer cells.Citation52 FOS often interacts with JUN and regulates the transcription of AP-1 responsive genes.Citation28 However, PPX did not modulate JUN phosphorylation or expression in our study. Instead, PPX enhanced FOS expression. Furthermore, PPX failed to enhance the promoter activity of BECN1 that was deleted with the 5′-TGCCTCA-3′ sequence. This deletion did not alter its basal activity. These data indicate that FOS may serve as a transcription factor that regulates Becn1/BECN1 transcription, at least in the rat and human species since we did not find the putative AP-1 site in the mouse Becn1 promoter. Rather, a classic CREB site (5′-TGACGTCA-3′) exists in the mouse Becn1 promoter. Thus, it is likely that in mice, PPX may upregulate BECN1 expression, as we observed in SNCAA53T tg mice, through the phosphorylation of CREB, bypassing the FOS signaling.
The role of BECN1 in autophagy and neurodegenerative disorders emerges as a hot topic in recent years. The alterations of BECN1 expression and activity in PD brains remain unknown. Our study, consistent with previous studies,Citation15,53 showed that BECN1 expression levels were not altered in SNCA overexpressing cells or in the brains of SNCAA53T tg models of PD. Nevertheless, BECN1 overexpression via lentivirus delivery is reported to be beneficial for PD.Citation15 In agreement with this, our study showed that the elevation of BECN1 expression by PPX was sufficient to enhance autophagy and reduce SNCA accumulation in vitro and in vivo. In addition, no obvious toxicity of PPX or quinpirole was observed at the tested concentrations although they induced BECN1 upregulation. These results highlight a potential role of BECN1 as a therapeutic target for PD. It should be cautioned that the subcellular localization or function of BECN1 may be compromised in PD although its total expression level remains unchanged. Hence, the molecular machinery of BECN1 function in PD deserves further investigation in future.
D2-like receptors-mediated signaling is complex. Our data showed that PPX elevated the [Ca2+]i level even in the presence of EDTA and that [Ca2+]i chelator abolished the autophagy induction by PPX, indicating a role of [Ca2+]i in the processes of autophagy activation. This [Ca2+]i elevation may result from the liberation of Gβγ subunits, due to DRD2 and DRD3 activation and subsequent stimulation of PLCB1 (phospholipase C, β 1 [phosphoinositide-specific]), which mobilizes the Ca2+ release from endoplasmic reticulum stores.Citation30,54-58 Ca2+ could bind with calmodulin and promote the phosphorylation of CAMK4, which further phosphorylates CREB and induces FOS expression.Citation59 We also tested this possibility and found that PPX-induced [Ca2+]i elevation was associated with the increases of CAMK4 (T196) and CREB (S133) phosphorylation. Therefore, these 2 signaling molecules may be involved in the cascade that linked [Ca2+]i elevation to FOS upregulation.
Both receptor -dependent and -independent neuroprotection exerted by PPX have been reported.Citation27,60,61 Our data suggest that the BECN1 upregulation and autophagy induction by PPX may be mediated by DRD2 and DRD3. First, quinpirole and PPX, but not the R (+) enantiomer DexPPX, increased BECN1 expression and autophagy level in various cells including midbrain neurons. Second, the effect of PPX on autophagy could be blocked by the DRD2 and DRD3 antagonist. PPX only elicited these effects in RA and TPA-differentiated or DRD2- and DRD3-overexpressing SH-SY5Y cells, not in undifferentiated SH-SY5Y cells that express DRD2 and DRD3 at a very low abundance. More importantly, we provided the evidence that PPX reduced SNCA accumulation in rotenone-treated PC12 cells and in SNCAA53T tg mice, uncovering a novel neuroprotective effect of PPX. However, a recent clinical trial named “Pramipexole On Underlying Disease (PROUD)” claims that PPX did not have disease-modifying effects.Citation62 Notably, the PROUD study recruited the patients that suffered from severe motor symptoms, in which the dopaminergic neuron losses in the SN probably reached beyond 60%. It is conceivable that it would be difficult to “rescue” or “restore” the neurons at this stage. Yet, it may induce autophagy, ameliorate SNCA accumulation, and even prevent the spread of SNCA pathogenesis if given earlier.
Taken together, our findings identify a new AP-1 sequence (5′-TGCCTCA-3′) in the rat and human Becn1/BECN1 promoter, and demonstrate that in response to the DRD2 and DRD3 agonist treatment, FOS may bind to the AP-1 site and trigger BECN1-dependent autophagy activation, which subsequently ameliorates the accumulation of SNCA in rotenone-challenged PC12 cells and SNCAA53T tg mice. This reveals a novel endogenous venue for autophagy regulation that comes from G protein-coupled DRD2 and DRD3 signaling, and may also provide some clues for the understanding of autophagy dysregulation in PD.
Materials and Methods
Reagents and antibodies
Pramipexole dihydrochloride (4174), (-)-quinpirole hydrochloride (1061), L-741,626 (1003) and GR103,691 (1109) were purchased from Tocris Bioscience. Retinoic acid (R2625), TPA (P8139), dexpramipexole dihydrochloride (SML0392), EBSS, and all other reagents unless specified were purchased from Sigma-Aldrich. BAPTA-AM (B-1205) and Fura-2-AM (F1221) were from Invitrogen. Quin-2-AM (SC-215769) was from Santa Cruz Biotechnology. The primary antibodies for immunoblot analysis were listed as follows: LC3B (Abcam, ab62721), BECN1 (Santa Cruz Biotechnology, sc11427), SQSTM1 (Sigma, P0067), FOS (Abcam, ab7963), p-MTOR (S2448)/MTOR (Cell Signaling Technology [CST], 5536/2938), p-RPS6KB1 (T389)/RPS6KB1 (CST, 9208/2708), p-ULK1 (S757; CST, 6888), p-ULK1 (S555; Chemicon, 2449230), ULK1 (CST, 4773), PtdIns3K (CST, 4263s), DRD2 (Chemicon, AB1558O), DRD3 (Chemicon, AB1785P), p-CAMK4 (T196)/CAMK4 (Santa Cruz Biotechnology, sc-28443/sc-166156), p-CREB (S133)/CREB (CST, 9198S/9104S), p-MAPK8 (Thr183/Tyr185)/MAPK8 (CST, 4671/3708), p-JUN (S63)/JUN (Santa Cruz Biotechnology, sc822/sc1694), SNCA (CST, 2642), HTT (Chemicon, MAB2166), ACTB (Sigma, A3584), TUBB/β-tubulin (Sigma, T0198), and GAPDH (Beyotime, AG019).
Cell lines and cell culture
Rat pheochromocytoma PC12 cells were purchased from the Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China) and cultured in RPMI1640 medium. MES23.5 cells (a gift from professor Wei-dong Le, Institute of Health Science, Shanghai Institutes For Biological Sciences, CAS) were cultured in Dulbecco's Modified Eagle's Medium (DMEM)/F12 with 2 mM glutamine and Sato's chemically defined medium to a final concentration of 5 mg/ml insulin, 48.6 mg/ml pyruvic acid, 5 mg/ml transferrin, 6.3 ng/ml progesterone, 5 ng/ml sodium selenite, 4 mg/ml putrescine and 5% fetal bovine serum.Citation63 Undifferentiated human neuroblastoma (SH-SY5Y) cells were purchased from ATCC (CRL®−2266TM) and cultured in DMEM. For differentiation, SH-SY5Y cells were treated with 10 µM RA for 3 d, followed by 80 nM TPA treatment for another 3 d.Citation27 All culture medium except that for MES23.5 was supplemented with 10% fetal bovine serum and 100 U/ml penicillin and streptomycin, and cultured in a 5% CO2 atmosphere at 37°C.
Animals
M83 transgenic (tg) mice expressing the mutant human SNCAA53T under the direction of the mouse prion protein promoter were obtained from Model Animal Research Center of Nanjing University (Nanjing, China). Mice were housed in an SPF-grade animal room (12/12 h light/dark cycle at 24 ± 1°C and 70% ± 4% relative humidity) with food and water ad libitum. All experimental procedures were performed according to the guidelines of the Institutional Animal Care and Use Committee of Soochow University. A total of 20 SNCAA53T tg mice and age-matched Non-tg mice at 12 to 15 mo old were used in our study (N=5 for each group), and were intraperitoneally injected with PPX at 0.5 mg/kg twice daily for 3 wk. An equal volume of saline was given as a control. The SN and Str tissues were dissected and lysed on ice at the end of treatment, and the lysates were subjected to western blotting under denaturing conditions.
Western blotting
Cells lysates were prepared by homogenization in lysis buffer (25 mM Tris, 150 mM NaCl, 5 mM EDTA, 1% NP-40 [AppliChem, A1694], pH 7.5) with protease inhibitor cocktail tablets (Roche Diagnostics, Penzberg, Germany), and phosphatase inhibitor cocktail tablets (Thermo Scientific, 88667) for phosphorylation analysis. Protein lysates were separated by 10–12% sodium dodecyl sulfate-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Millipore, ISEQ00010). Next, blots were blocked with 5% milk in Tris buffered saline/Tween20 buffer (TBST: 10 mM Tris, 150 mM NaCl, 0.1% Tween-20 [Sigma, P1379], pH 8.0) for 1 h and incubated with primary antibodies at 4°C overnight. After that, blots were briefly washed and incubated with secondary antibodies for another 1 h. After that, blots were visualized using a chemiluminescence kit (Bio-Rad, 170–5061). The densitometric analysis was performed using ImageJ software (National Institute of Health, Bethesda, MD, USA). ACTB, GAPDH and TUBB served as loading controls for different protein analyses.
To monitor the levels of soluble and insoluble SNCA fractions, cells were lysed using NP-40 buffer (10 mM HEPES, 142.5 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1% NP-40, pH 7.5) with protease inhibitor cocktail and rotated at 4°C for 1 h. Next, the lysates were centrifuged at 12,000 g for 10 min at 4°C. The supernatant fractions were then transferred to a new tube and mixed with 2*SDS buffer (12.5% 1 M Tris-HCl pH 6.8, 20% glycerine, 4% SDS [Genshare, 77–86–1]) to obtain the NP-40-soluble fraction. The sediments were subsequently washed with NP-40 buffer 3 times and mixed with 1*SDS buffer (6.25% 1 M Tris-HCl, pH 6.8, 10% glycerine, 2% SDS), followed by sonication to make sure that the sediment was completely lysed. After centrifugation, the supernatant fraction was then harvested and referred to as the NP-40-insoluble fraction.
Midbrain neuron cultures
The ventral midbrains of rat embryos (E13–14) were dissected, with meninges carefully removed. Tissues were then minced and digested in 0.125% trypsin at 37°C for 15 min and cleared by centrifugation at 100 g for 5 min. Cells were resuspended in neurobasal medium (Invitrogen, 21103049) supplemented with B27 and glutamine, and seeded on poly-D-lysine precoated culture dishes. One d later, the medium was replaced by fresh medium with 5 mM Ara-C (Sigma, C6645). After that, about half of the medium was replaced every 3 d and maintained for at least one wk for neuron maturation.
Immunoprecipitation
The coimmunoprecipitation kit (Thermo Scientific, 26149) was used according to the manufacturer's instructions. In brief, 10 μg monoclonal rat anti-PtdIns3K antibody was immobilized onto AminLink Plus Coupling Resin, and precleared protein extracts (containing protease and phosphatase inhibitor) were added to the column overnight at 4°C. The next day, the columns were washed and the samples were eluted. The PtdIns3K-immnoprecipitated proteins were subjected to immunoblot analysis.
Reverse transcription and quantitative PCR
Reverse transcription and quantitative PCR was performed as previously described,Citation64,65 with primers as listed in . The PCR products were separated in 1.5–2% agarose gels and the band densities were analyzed with ImageJ software. The quantitative PCR reactions were performed on the 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) and the results were normalized to 18S RNA.
Table 1. The primers listed for reverse transcription PCR and quantatative PCR.
Immunostaining
Immunostaining was performed as we previously described.Citation66 Briefly, cells seeded in coverslips were fixed with 4% paraformaldehyde and probed with antibodies against LC3B and TH, followed by incubation with appropriate fluorophore-conjugated secondary antibodies (Thermo Fisher Scientific, A-21206 and A-31570). Coverslips were observed and photographed under a confocal microscope (LSM 700, Zeiss, Oberkochen, Germany).
Cell imaging and LC3B puncta counting
Cells were fixed with 4% paraformaldehyde for 10 min. Coverslips were then mounted with mounting solution with DAPI (Vector Laboratories, H-1200). Finally, cells were observed and photographed under a Zeiss fluorescence microscope (Axio Scope A1, Goettingen, Germany) or confocal microscope. The LC3B puncta in cells were manually counted, and at least 30 cells were randomly selected for counting in each group.
Transmission electron microscopy
Cells were pre-fixed with ice-cold 2.5% glutaraldehyde in 0.1 Mphosphate-buffered saline (pH 7.4; HyClone SH30256.01B) and postfixed with 1% osmium tetroxide buffer. After dehydration in a gradient series of ethyl alcohol, cells were embedded in epoxy resin. Ultrathin sections (60-nm thick) were stained with uranyl acetate and lead citrate, and examined using a transmission electron microscope (JEM 1230, JOEL, Tokyo, Japan).
Cell viability measurement
Cell viability assay was carried out using the cell counting kit-8 (CCK-8) (Dojindo Laboratories, CK04). Briefly, PC12 cells were seeded in a 96-well microplate and then pretreated with PPX or quinpirole for 24 h at a series of concentrations. Subsequently, 10 µl CCK-8 solution was added to 100 µl medium per well and then the plate was incubated at 37°C for 4 h. The optical densities were read on a microplate reader at a wavelength of 450 nm. The percentage of cell viability was calculated by the ratio of optical density of the experimental wells in relation to the control wells.
Transient transfection
The small-interfering RNAs (siRNA) targeting rat Becn1 (5′-UGA GGA UGA CAG UGA ACA GTT-3′ and 5′-CUG UUC ACU GUC AUC CUC ATT-3′), rat Fos (5′-CGU CUC UAG UGC CAA CUU UTT-3′ and 5′-AAA GUU GGC ACU AGA GAC GTT-3′), and scrambled siRNA duplexes were synthesized by GenePharma (Shanghai, China). The EGFP-LC3B (21073), GFP-DRD2 (24099), GFP-DRD3 (24098), FOS (8966) plasmids were purchased from Addgene. The siRNA duplexes and plasmids were transfected using lipofectamine 2000. Lentiviral vector driving the recombinant eukaryotic expression of pLentiVENUS-YFP-SNCA that contains WT SNCA or its mutant variants (A53T, A30P) were gifts from Wei-ling Dong, Department of Neurobiology, Soochow University. N-terminal fragments of WT HTT (HTT-552–18Q) and mutant HTT (HTT-552–100Q) that cloned into pDC316 adenovirus shuttle were gifts from Zheng-Hong Qin, Department of Pharmacology, Soochow University.
Chromatin immunoprecipitation
ChIP assay was carried out using a commercially available kit (CST, 9004). In brief, cells were cross-linked by 1% formaldehyde for 10 min and stopped by glycine. The extracted chromatin was digested and fragmented into 150 to 900 bp, which was then immunoprecipitated by ChIP grade antibodies against FOS (CST, 2250s) or a normal IgG using protein G agarose beads. After that, the DNA and transcriptional factor complexes were uncross-linked to obtain the pure DNA fragment. PCR was then performed using the primers spanning the putative FOS binding site in rat Becn1 (forward 5′-AGG GCG AGT TTC AGG AGA CA-3′; reverse 5′-GAG GGT TCA CTC ACG ATT TG-3′) or human BECN1 promoter (forward 5′-CAC TGC AAC CTC CAC CTC C-3′; reverse 5′-TGG CTC ACG CCT GTA ATC TC-3′).
Reporter plasmid construction and activity assays
The fragments relative to the transcription start site of rat Becn1 genomic sequence (spanning from −426 to +279) were synthesized by GenScript (Nanjing, China). The fragments were cloned into the pGL3-basic vector (Promega, E1751) to generate a rat Becn1 (−426 to +279)-luc. The mutant plasmid was constructed by deleting the putative activating protein-1 (AP-1) binding site 5′-TGCCTCA-3′ (−278 to −272) from the Becn1 reporter plasmid as described above. The construct was verified by DNA sequencing. The luciferase activity was assessed using a luciferase assay kit (Promega, E2000). Briefly, cells were transfected with various reporter plasmids or pGL3-basic vector, and pSV-β-galactosidase control vector as well (Promega, E1081). At 36 h posttransfection, cells were treated with or without PPX for 12 h. The Becn1 reporter activity was normalized to that of pSV-β-galactosidase.
Intracellular calcium level ([Ca2+]i) measurement
To measure [Ca2+]i, PC12 cells were incubated with 4 μM Fura-2-AM for 30 min. The unincorporated dye was removed. Thereafter, the Fura-2-AM loaded cells were maintained at room temperature for another 30 min and then transferred to an inverted microscope, which was coupled with a dual-wavelength excitation spectrofluorometer. Cells were then perfused with Krebs bicarbonate buffer (118 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1.25 mM CaCl2, 25 mM NaHCO3, 11 mM glucose, pH 7.4) or EGTA-treated Ca2+-free Krebs bicarbonate buffer (2 mM EGTA, 118 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM NaHCO3, 11 mM glucose, pH 7.4). 100 μM PPX was carefully added into the incubation buffer, and Ca2+ tracings were recorded. The ratio of fluorescent signals obtained at 340 nm (F340) and 380 nm (F380) excitation wavelengths were calculated and represented as the [Ca2+]i changes.
Statistical analyses
All results are presented as mean ± SEM and represent data from a minimum of 3 independent experiments unless otherwise stated. For cell viability assay, an ANOVA was used for the determination of significance followed by Tukey post-hoc analysis. For all other results, the statistical significance of difference between groups was determined by unpaired Student t test or one-sample t test if values were compared with normalized control values using GraphPad Prism and PASW Statistics software. Differences with P value less than 0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1100930_Supplemental_Material.zip
Download Zip (16.9 MB)Acknowledgments
The authors would like to thank Prof. Guang-Yin Xu and Hong-Yan Zhu for the technical support for Ca2+ measurements and Wei-ling Dong and Li-Dong Shan for providing pLentiVENUS-YFP-SNCA.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by grants from National Basic Research Program of China (973 Program, 2011CB510003), National Natural Science Foundation of China (81171213, 81301091 and 81171212), Jiangsu Provincial Special Program of Medical Science (BL2014042), and Suzhou Medical Key Discipline Project. This was also partly supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Related Research Data
References
- Bradbury J. Alpha-synuclein gene triplication discovered in Parkinson disease. Lancet Neurol 2003; 2:715; PMID:14649238; http://dx.doi.org/10.1016/S1474-4422(03)00601-X
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson disease. Lancet 2004; 364:1167-9; PMID:15451224; http://dx.doi.org/10.1016/S0140-6736(04)17103-1
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schöls L, Riess O, et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson disease. Nat Genet 1998; 18:106-8; PMID:9462735; http://dx.doi.org/10.1038/ng0298-106
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human α-synuclein-harboring familial Parkinson disease-linked Ala-53 –> Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA 2002; 99:8968-73; PMID:12084935; http://dx.doi.org/10.1073/pnas.132197599
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004; 55:164-73; PMID:14755719; http://dx.doi.org/10.1002/ana.10795
- Rutherford NJ, Moore BD, Golde TE, Giasson BI. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of α-synuclein. J Neurochem 2014; 131:859-67; PMID:24984882; http://dx.doi.org/10.1111/jnc.12806
- Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M, Paetau A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson disease-type pathology. Neurobiol Aging 2014; 35:2180 e1-5; PMID:24746362; http://dx.doi.org/10.1016/j.neurobiolaging.2014.03.024
- Xilouri M, Stefanis L. Autophagic pathways in Parkinson disease and related disorders. Expert Rev Mol Med 2011; 13:e8; PMID:21418705; http://dx.doi.org/10.1017/S1462399411001803
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson disease. Brain 2008; 131:1969-78; PMID:18187492; http://dx.doi.org/10.1093/brain/awm318
- Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci 2010; 33:541-9; PMID:20947179; http://dx.doi.org/10.1016/j.tins.2010.09.001
- Xilouri M, Vogiatzi T, Vekrellis K, Stefanis L. α-synuclein degradation by autophagic pathways: a potential key to Parkinson disease pathogenesis. Autophagy 2008; 4:917-9; PMID:18708765; http://dx.doi.org/10.4161/auto.6685
- Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 2010; 11:777-88; PMID:20944667; http://dx.doi.org/10.1038/nrm2993
- Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M, Vandenabeele P. Beclin1: a role in membrane dynamics and beyond. Autophagy 2012; 8:6-17; PMID:22170155; http://dx.doi.org/10.4161/auto.8.1.16645
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest 2008; 118:2190-9; PMID:18497889
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson and Lewy body diseases. The J Neurosci 2009; 29:13578-88; PMID:19864570; http://dx.doi.org/10.1523/JNEUROSCI.4390-09.2009
- Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol Cell Biol 2001; 21:6820-32; PMID:11564866; http://dx.doi.org/10.1128/MCB.21.20.6820-6832.2001
- Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009; 28:886-98; PMID:19060920; http://dx.doi.org/10.1038/onc.2008.441
- Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol 2009; 29:2594-608; PMID:19289499; http://dx.doi.org/10.1128/MCB.01396-08
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472-83; PMID:18054316; http://dx.doi.org/10.1016/j.cmet.2007.11.004
- Bohensky J, Shapiro IM, Leshinsky S, Terkhorn SP, Adams CS, Srinivas V. HIF-1 regulation of chondrocyte apoptosis: induction of the autophagic pathway. Autophagy 2007; 3:207-14; PMID:17224629; http://dx.doi.org/10.4161/auto.3708
- Herrero MT, Pagonabarraga J, Linazasoro G. Neuroprotective role of dopamine agonists: evidence from animal models and clinical studies. Neurologist 2011; 17:S54-66; PMID:22045327; http://dx.doi.org/10.1097/NRL.0b013e31823968fc
- Joyce JN, Millan MJ. Dopamine D3 receptor agonists for protection and repair in Parkinson disease. Curr Opin Pharmacol 2007; 7:100-5; PMID:17174156; http://dx.doi.org/10.1016/j.coph.2006.11.004
- Iida M, Miyazaki I, Tanaka K, Kabuto H, Iwata-Ichikawa E, Ogawa N. Dopamine D2 receptor-mediated antioxidant and neuroprotective effects of ropinirole, a dopamine agonist. Brain Res 1999; 838:51-9; PMID:10446316; http://dx.doi.org/10.1016/S0006-8993(99)01688-1
- Joyce JN. Dopamine D3 receptor as a therapeutic target for antipsychotic and antiparkinsonian drugs. Pharmacol Ther 2001; 90:231-59; PMID:11578658; http://dx.doi.org/10.1016/S0163-7258(01)00139-5
- Li C, Guo Y, Xie W, Li X, Janokovic J, Le W. Neuroprotection of pramipexole in UPS impairment induced animal model of Parkinson disease. Neurochem Res 2010; 35:1546-56; PMID:20635141; http://dx.doi.org/10.1007/s11064-010-0214-3
- Ghislat G, Patron M, Rizzuto R, Knecht E. Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-β (CaMKK-β). J Biol Chem 2012; 287:38625-36; PMID:23027865; http://dx.doi.org/10.1074/jbc.M112.365767
- Presgraves SP, Ahmed T, Borwege S, Joyce JN. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotoxicity Res 2004; 5:579-98; http://dx.doi.org/10.1007/BF03033178
- Chiu R, Boyle WJ, Meek J, Smeal T, Hunter T, Karin M. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell 1988; 54:541-52; PMID:3135940; http://dx.doi.org/10.1016/0092-8674(88)90076-1
- Fujita Y, Izawa Y, Ali N, Kanematsu Y, Tsuchiya K, Hamano S, Tamaki T, Yoshizumi M. Pramipexole protects against H2O2-induced PC12 cell death. Naunyn-Schmiedeberg's Arch Pharmacol 2006; 372:257-66; http://dx.doi.org/10.1007/s00210-005-0025-2
- Mizuno K, Kurokawa K, Ohkuma S. Regulation of type 1 IP(3) receptor expression by dopamine D2-like receptors via AP-1 and NFATc4 activation. Neuropharmacol 2013; 71:264-72; http://dx.doi.org/10.1016/j.neuropharm.2013.03.036
- Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson disease. Cold Spring Harbor Perspectives Med 2012; 2:a009357; http://dx.doi.org/10.1101/cshperspect.a009357
- Tan CC, Yu JT, Tan MS, Jiang T, Zhu XC, Tan L. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging 2014; 35:941-57; PMID:24360503; http://dx.doi.org/10.1016/j.neurobiolaging.2013.11.019
- Cheung ZH, Ip NY. The emerging role of autophagy in Parkinson disease. Mol Brain 2009; 2:29; PMID:19754977; http://dx.doi.org/10.1186/1756-6606-2-29
- Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci 2006; 29:528-35; PMID:16859759; http://dx.doi.org/10.1016/j.tins.2006.07.003
- Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol 2007; 6:352-61; PMID:17362839; http://dx.doi.org/10.1016/S1474-4422(07)70076-5
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson disease. Histol Histopathol 1997; 12:25-31; PMID:9046040
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 2006; 76:89-101; PMID:17118264; http://dx.doi.org/10.1016/S0070-2153(06)76003-3
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 2004; 24:1888-96; PMID:14985429; http://dx.doi.org/10.1523/JNEUROSCI.3809-03.2004
- Ahmed I, Liang Y, Schools S, Dawson VL, Dawson TM, Savitt JM. Development and characterization of a new Parkinson disease model resulting from impaired autophagy. J Neurosci 2012; 32:16503-9; PMID:23152632; http://dx.doi.org/10.1523/JNEUROSCI.0209-12.2012
- Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med 2013; 45:e22; PMID:23661100; http://dx.doi.org/10.1038/emm.2013.45
- Poehler AM, Xiang W, Spitzer P, May VE, Meixner H, Rockenstein E, Chutna O, Outeiro TF, Winkler J, Masliah E, et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014; 10:2171-92; PMID:25484190; http://dx.doi.org/10.4161/auto.36436
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 2010; 30:6838-51; PMID:20484626; http://dx.doi.org/10.1523/JNEUROSCI.5699-09.2010
- Decuypere JP, Bultynck G, Parys JB. A dual role for Ca(2+) in autophagy regulation. Cell Calcium 2011; 50:242-50; PMID:21571367; http://dx.doi.org/10.1016/j.ceca.2011.04.001
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, et al. α-Synuclein impairs macroautophagy: implications for Parkinson disease. J Cell Biol 2010; 190:1023-37; PMID:20855506; http://dx.doi.org/10.1083/jcb.201003122
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004; 305:1292-5; PMID:15333840; http://dx.doi.org/10.1126/science.1101738
- Song JX, Lu JH, Liu LF, Chen LL, Durairajan SS, Yue Z, Zhang HQ, Li M. HMGB1 is involved in autophagy inhibition caused by SNCA/α-synuclein overexpression: a process modulated by the natural autophagy inducer corynoxine B. Autophagy 2014; 10:144-54; PMID:24178442; http://dx.doi.org/10.4161/auto.26751
- Yan JQ, Yuan YH, Gao YN, Huang JY, Ma KL, Gao Y, Zhang WQ, Guo XF, Chen NH. Overexpression of human E46K mutant α-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol Neurobiol 2014; 50:685-701; PMID:24833599; http://dx.doi.org/10.1007/s12035-014-8738-1
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 2001; 21:9549-60; PMID:11739566
- Jiang TF, Zhang YJ, Zhou HY, Wang HM, Tian LP, Liu J, Ding JQ, Chen SD. Curcumin ameliorates the neurodegenerative pathology in A53T α-synuclein cell model of Parkinson disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J Neuroimmune Pharmacol 2013; 8:356-69; PMID:23325107; http://dx.doi.org/10.1007/s11481-012-9431-7
- Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 2010; 3:ra42; PMID:20501938
- Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl−XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/10.1038/embor.2008.246
- Gong C, Qu S, Liu B, Pan S, Jiao Y, Nie Y, Su F, Liu Q, Song E. MiR-106b expression determines the proliferation paradox of TGF-β in breast cancer cells. Oncogene 2013; 34(1):84-93; PMID:24292682; http://dx.doi.org/10.1038/onc.2013.525
- Smith WW, Liu Z, Liang Y, Masuda N, Swing DA, Jenkins NA, Copeland NG, Troncoso JC, Pletnikov M, Dawson TM, et al. Synphilin-1 attenuates neuronal degeneration in the A53T α-synuclein transgenic mouse model. Hum Mol Genet 2010; 19:2087-98; PMID:20185556; http://dx.doi.org/10.1093/hmg/ddq086
- Berridge MJ. Neuronal calcium signaling. Neuron 1998; 21:13-26; PMID:9697848; http://dx.doi.org/10.1016/S0896-6273(00)80510-3
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res 2004; 24:165-205; PMID:15521361; http://dx.doi.org/10.1081/RRS-200029981
- Mikoshiba K. The Ins(1,4,5)P3 receptor/Ca2+ channel and its cellular function. Biochem Soc Symp 2007; 74:9-22; PMID:17233576; http://dx.doi.org/10.1042/BSS2007c02
- Stewart AJ, Morgan K, Farquharson C, Millar RP. Phospholipase C-eta enzymes as putative protein kinase C and Ca2+ signalling components in neuronal and neuroendocrine tissues. Neuroendocrinol 2007; 86:243-8; http://dx.doi.org/10.1159/000107795
- Clapham DE, Neer EJ. G protein β gamma subunits. Ann Rev Pharmacol Toxicol 1997; 37:167-203; http://dx.doi.org/10.1146/annurev.pharmtox.37.1.167
- Sato K, Suematsu A, Nakashima T, Takemoto-Kimura S, Aoki K, Morishita Y, Asahara H, Ohya K, Yamaguchi A, Takai T, et al. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat Med 2006; 12:1410-6; PMID:17128269; http://dx.doi.org/10.1038/nm1515
- Ling ZD, Robie HC, Tong CW, Carvey PM. Both the antioxidant and D3 agonist actions of pramipexole mediate its neuroprotective actions in mesencephalic cultures. J Pharmacol Exp Therapeutics 1999; 289:202-10
- Ferrari-Toninelli G, Maccarinelli G, Uberti D, Buerger E, Memo M. Mitochondria-targeted antioxidant effects of S(−) and R(+) pramipexole. BMC Pharmacol 2010; 10:2; PMID:20137065; http://dx.doi.org/10.1186/1471-2210-10-2
- Schapira AH, McDermott MP, Barone P, Comella CL, Albrecht S, Hsu HH, Massey DH, Mizuno Y, Poewe W, Rascol O, et al. Pramipexole in patients with early Parkinson disease (PROUD): a randomised delayed-start trial. Lancet Neurol 2013; 12:747-55; PMID:23726851; http://dx.doi.org/10.1016/S1474-4422(13)70117-0
- Crawford GD, Jr., Le WD, Smith RG, Xie WJ, Stefani E, Appel SH. A novel N18TG2 x mesencephalon cell hybrid expresses properties that suggest a dopaminergic cell line of substantia nigra origin. J Neurosci 1992; 12:3392-8; PMID:1356145
- Wang Y, Jia J, Ao G, Hu L, Liu H, Xiao Y, Du H, Alkayed NJ, Liu CF, Cheng J. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J Neurochem 2014; 129:827-38; PMID:24673410; http://dx.doi.org/10.1111/jnc.12695
- Wang XH, Wang F, You SJ, Cao YJ, Cao LD, Han Q, Liu CF, Hu LF. Dysregulation of cystathionine gamma-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage. Cell Signal 2013; 25:2255-62; PMID:23872072; http://dx.doi.org/10.1016/j.cellsig.2013.07.010
- Li D, Shi JJ, Mao CJ, Liu S, Wang JD, Chen J, Wang F, Yang YP, Hu WD, Hu LF, et al. Alteration of dynein function affects α-synuclein degradation via the autophagosome-lysosome pathway. Int J Mol Sci 2013; 14:24242-54; PMID:24351814; http://dx.doi.org/10.3390/ijms141224242