
Abstract
Autophagosome biogenesis requires efficient mobilization and delivery of membranes from intracellular sources. How these membranes are mobilized remains poorly understood. Our recent work reported an autophagic signal-induced membrane mobilization event from the ER-Golgi intermediate compartment (ERGIC) to generate an early autophagosomal membrane precursor. We found that starvation activates the autophagic phosphatidylinositol 3-kinase, which promotes a relocation of COPII proteins from the ER-exit sites to the ERGIC. The relocation of COPII generates ERGIC-derived COPII vesicles as a membrane template for LC3 lipidation, a key step for autophagosome biogenesis.
Autophagosome biogenesis is initiated by intricate protein signaling cascades to mobilize membranes from the endomembrane system. The membrane precursors derived from the mobilization process are assembled into membrane cisternae, also known as phagophores, which elongate to form cup-shaped membranes that seal at their extremity to become autophagosomes. Although the step from phagophore elongation to autophagosome formation has been morphologically characterized by imaging techniques, less is known about a very early step, the conversion of the endomembrane to an early autophagosomal precursor, due to the lack of distinct membrane morphology to trace this event. A variety of autophagosomal precursors have been proposed to arise from multiple locations in the cellular membrane. In the current study, our attention focused on the generation of membrane precursors responsible for LC3 lipidation. Precursors are produced in early stages of autophagosome biogenesis and are required for completion of the autophagosome. LC3 lipidation depends on a ubiquitin-like conjugation process during which the protein complex ATG12–ATG5-ATG16L1, and the proteins ATG3 and ATG7 function to covalently link LC3 to phosphatidylethanolamine on autophagosomal precursor membranes. The process is modulated by the class III phosphatidylinositol 3-kinase (PtdIns3K) complex consisting of ATG14, BECN1/Beclin 1, PIK3C3/VPS34 and PIK3R4/VPS15/p150. It is proposed that activation of the PtdIns3K complex produces phosphatidylinositol-3-phosphate (PtdIns3P) which, together with PtdIns3P effectors and downstream factors, triggers the membrane conversion toward the phagophore.
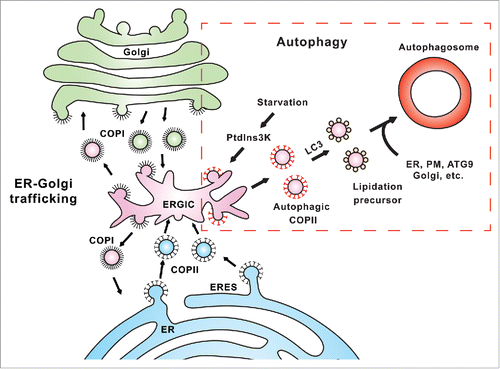
Previously, we established a cell-free LC3 lipidation assay that recapitulates the PtdIns3K-dependent lipidation of LC3. Here, we employed this assay to trace the membrane mobilization event induced by the PtdIns3K complex. We monitored the generation of precursors for LC3 lipidation in Atg5 knockout (KO) mouse embryonic fibroblasts (MEFs) where phagophore formation is blocked at the LC3 lipidation step downstream of the PtdIns3K. We detected a PtdIns3K-dependent accumulation of membrane precursors active for LC3 lipidation in starved Atg5 KO MEFs. These precursors are small vesicles likely produced by budding from a donor membrane. Based on this observation, we developed a cell-free small vesicle formation assay based on our well-established in vitro COPII budding system. We combined this assay with cell-free LC3 lipidation to reconstitute a process of starvation-induced and PtdIns3K-dependent generation of small LC3 lipidation-active vesicles from a donor membrane. Through a membrane fractionation experiment, we identified the endoplasmic reticulum (ER)-Golgi intermediate compartment (ERGIC) as the donor that produces small vesicles active for LC3 lipidation. In addition, components of the PtdIns3K complex, ATG14 and BECN1, as well as upstream regulators such as RB1CC1/FIP200, are required for the generation of small vesicles active for LC3 lipidation. These data are consistent with our previous study demonstrating a critical role of the ERGIC in triggering LC3 lipidation by providing a platform for the docking of ATG14, the key targeting factor of the PtdIns3K. Therefore, together with our previous study, the current data indicate that the ERGIC generates the small membrane precursors for LC3 lipidation in response to an activated PtdIns3K.
The ERGIC is a sorting station undergoing dynamic membrane exchange with COPI and COPII vesicles. Furthermore, both COPI and COPII machineries function to bud small vesicles from donor membranes. We found that inhibition of COPII but not COPI blocks the generation of small membranes active for LC3 lipidation, indicating that the COPII machinery is involved in budding the precursors from the ERGIC. This observation was surprising because it is thought that in vesicular transport, the COPII machinery buds vesicles exclusively from the ER (). We proposed that generation of COPII vesicles from the ERGIC could thus be a special event for autophagy-related membrane mobilization induced by starvation. To test this hypothesis, we performed membrane fractionation and found relocation of COPII machinery to the ERGIC after starvation, which did not occur under nutrient-rich conditions and was largely compromised in the presence of PtdIns3K inhibitors. The starvation-induced and PtdIns3K-dependent recruitment of COPII to the ERGIC was further confirmed by colocalization analysis performed with both confocal and deconvolution microscopy. Notably, COPII vesicles generated from the ERGIC fraction may have a different composition than those derived from the ER because they are enriched in the activity responsible for triggering LC3 lipidation (about 5-fold compared with those from the ER). These data indicate a mechanism for membrane mobilization induced by an autophagy signal, which generates early autophagosomal precursors. In this process, PtdIns3K activation induces translocation of the COPII machinery from the ER to the ERGIC. This then generates special COPII vesicles as precursors for LC3 lipidation, an essential step for development of the autophagosome ().
Figure 1. A proposed model for the diversion of COPII machinery from ER-Golgi trafficking to autophagosome biogenesis. In ER-Golgi trafficking, COPII vesicles are generated from special subdomains of the ER called ERES to deliver cargo proteins in the secretory pathway. The ERGIC is a recycling station between the ER and Golgi that receives COPII vesicles from the ER, and COPI vesicles from the Golgi. In the ERGIC, cargoes are sorted and either move forward to the Golgi or backward to the ER through COPI vesicles. In autophagy (boxed region), stress signals, such as starvation, activate the PtdIns3K which subsequently acts on the ERGIC to promote relocation of COPII proteins to the ERGIC. The process leads to the generation of ERGIC-derived COPII vesicles as templates for LC3 lipidation, an essential step of autophagosome biogenesis. These LC3-lipidated vesicles are subsequently transported to the PAS where they fuse with other membrane precursors derived from the ER, ATG9 compartment, and possibly also from the plasma membrane (PM) and Golgi, etc. to complete the formation of the autophagosome.
The involvement of COPII in autophagy was first indicated by the Ohsumi group over a decade ago. However, it has been difficult to pinpoint if COPII vesicles directly provide membrane to the forming autophagosome or indirectly affect autophagy via their role in membrane trafficking. Recently, significant progress has been made to indicate a direct contribution of COPII vesicles to autophagosome biogenesis. In mammalian cells, the Yoshimori and Yamamoto labs, using electron microscopy, found clusters of COPII-positive vesicles adjacent to a developing phagophore. In yeast, the Ohsumi, Suzuki and Nunnari labs used fluorescence microscopy and genetics to demonstrate an association of the ER exit site (ERES, the site of COPII assembly) with the phagophore assembly site (PAS). The Klionsky and Ferro-Novick labs identified Trs85 as a protein factor that determines the autophagic function of the protein tethering complex TRAPPIII. The TRAPPIII tethering complex was then shown to associate with COPII components by the Ferro-Novick, Reinisch and Walz labs. Our study provides direct evidence for the contribution of COPII vesicles in autophagosome biogenesis by showing that COPII vesicles are a membrane template for LC3 lipidation. In addition, we found that autophagic COPII vesicles can be distinguished from those for ER-Golgi trafficking by the different locations of generation and composition i.e. autophagic COPII vesicles are generated from the ERGIC instead of the ER, and they are more active for LC3 lipidation. Finally, we demonstrated that autophagic COPII vesicles are induced by the activation of PtdIns3K during starvation.
Our study indicates a switch of COPII location from the ERES to the ERGIC for autophagosome biogenesis in mammalian cells. However, there has been no observation of a distinct ERGIC structure in yeast. How could yeast distinguish autophagic COPII vesicles from those for ER-Golgi trafficking? It is possible that the ER may allocate a specific site for generation of autophagic COPII vesicles. This notion is supported by studies from the Ohsumi and Nunnari labs, which indicated a spatial distribution of a COPII formation site adjacent to the PAS. This COPII formation site could be specifically dedicated for autophagy. The delivery of these autophagic COPII vesicles to the PAS requires similar protein machineries as those for ER-Golgi trafficking, including a vesicle tethering complex (TRAPPIII), a small GTPase (Ypt1) and a protein kinase (Hrr25), as shown by the Ferro-Novick lab. It would be interesting to know if these autophagic COPII vesicles in yeast are functionally customized for autophagy purposes, similar to those derived from the ERGIC in mammalian cells.
In summary, our study identifies a pathway for the generation of autophagosomal precursors from the ERGIC. The process includes an autophagic PtdIns3K-induced relocation of COPII machineries to produce ERGIC-derived COPII vesicles as a membrane template for LC3 lipidation. Future questions include how and what components of the COPII machinery are relocated from the ERES to the ERGIC during starvation; what the molecular requirements are for the delivery of LC3-lipidated COPII vesicles to the PAS; what the difference is between the 2 kinds of COPII vesicles; and how these LC3-lipidated COPII vesicles contribute to the growth of the phagophore.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledegments
L.G. was a fellow of the Jane Coffin Childs Fund and is now a fellow of NIH Pathway to Independence Award (Parent K99/R00) supported by National Institute of General Medical Sciences (Grant Number: 1K99GM114397–01). R.S. is an Investigator of the HHMI and a Senior Fellow of the UC Berkeley Miller Institute.