
abstract
In the context of elevated prevalence of obesity-associated metabolic diseases in the human population wordwide, interest in the autophagy degradation pathway is increasing, due to close links with energy metabolism, nutritional state, and inflammation. Here we highlight recent data focusing on adipose tissue which demonstrate alterations in fat cell autophagic flux in human obesity.
Autophagy is the primary mechanism responsible for the degradation of cell organelles and plays a key role in the maintenance of long-lived cell types. Also, as an inducible process to fuel energy production from intracellular sources upon temporary exhaustion of exogenous nutrients, it has become a focus of interest in the study of common metabolic diseases in which chronic nutritional overload induces obesity, type II diabetes, liver diseases, and cardiovascular complications. Our recent work that focused on adipose tissue autophagy status identified attenuated adipocyte clearance in patients with severe obesity (i.e., body mass index > 35 kg/m2). Comparison of the intensity of autophagic flux in subcutaneous fat cells isolated from normal weight or obese patients eligible for bariatric surgery demonstrated obesity-related adipocyte autophagy impairment, linked to fat cell lipid burden and hypertrophy. Following gastric bypass surgery, this defect can be partially rescued after weight loss, and the improvement is linked to a decrease in fat cell size. In addition, this study associates defective autophagy of obese adipocytes with the loss of expression of a kinase, DAPK2 (death-associated protein kinase 2), which is able to modulate autophagic flux when expressed in cultured fat cells. Thus, disruption of adipocyte autophagy, linked to lipid excess and low-grade chronic inflammation, is likely a contributor in adipose tissue deterioration in obesity.
The above-mentioned data on human adipose tissue samples fit well with a number of studies in mice which initially pointed to autophagy pathways in obesity-related disorders. These studies used mice models with autophagy disruption by tissue-specific inhibition of the Atg7 gene, and described metabolic phenotypes remarkably resembling those seen in obesity and diabetes. For example, deletion of Atg7 in pancreatic β cells compromises insulin production and leads to the development of diabetes. Hepatocyte-specific defective autophagy produces liver steatosis, an important feature in metabolic syndrome, and leads to the discovery of a new mode of mobilization for neutral lipid stores involving lysosomal degradation of lipid droplet organelles by autophagy, a process called lipophagy. Mice with specific macrophage Atg7 deficiency are more prone to develop atherosclerotic plaques with lipid engorged spumous cell infiltration, and, even more surprisingly, autophagy inhibition in a subtype of neurons producing POMC (proopiomelanocortin) also causes obesity in mice by promoting hyperphagia. Consistent with that observed in tissue-specific models, mice with Atg7 haploinsuficiency do not exhibit overt metabolic defects as long as they remain lean, but develop diabetes when crossed with obese ob/ob mice, suggesting that widespread autophagy shortage compromises the adaptive response to metabolic stress. Despite key players in the orchestration of energy maintenance, adipocytes long remained the only metabolic cell type in which the role of autophagy could not be established because it is required for normal fat tissue differentiation, so that adipose-specific Atg7 depletion does not result in an obese, but rather a lean, lipoatrophic phenotype.
Autophagy is a key process for acute response to various stresses. In particular, the adipose tissue microenvironment in obesity is proinflammatory, and because cytokines promote autophagic degradation in the context of host defense against microbial infections a prevailing view is that obese adipose tissue should present with exacerbated autophagy. This was corroborated by studies that indicated elevated expression of adipose tissue autophagic machinery components (such as LC3-II, SQSTM1/p62 or ATG5) in obese patients, and higher expression in the visceral fat depot, which is associated with more pronounced metabolic and cardiovascular risks. However, whereas deletion of autophagy-related genes has profound impacts (see above), it is not established that the opposite situation (i.e., elevated expression of autophagy genes) is sufficient to increase degradative activity and flux. Furthermore, a strong confounding factor in the comparison of autophagy-related gene expression in adipose tissues from lean and obese patients is the huge difference in cell composition of fat, particularly the high proportion of immune infiltrating cells in obese but not in lean adipose tissue. Therefore, increased expression of autophagy-related genes in obese adipose tissue may not reflect activation of autophagic clearance of adipocytes but may simply be brought about by obesity-related changes fat tissue cell composition. According to published autophagy guidelines, monitoring changes in mRNA or protein levels for ATG genes or autophagy regulators may provide some evidence supporting upregulation of a potential to undergo autophagy, but should be used along with other methods to determine autophagic clearance. Our study fills this gap by focusing on adipocytes only, and points to attenuated rather than activated fat cell autophagy in obesity. In line with our finding, others also reported autophagy deterioration in the liver of obese ob/ob mice and in patients developing non-alcoholic fatty liver diseases.
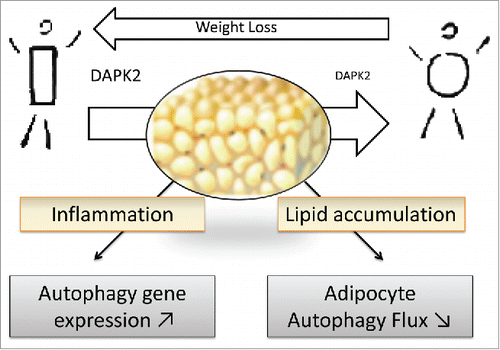
Overall, the picture of adipocyte autophagy status in obesity consists in diminished activity despite elevated expression of autophagy genes, and points to a prominent role of cell engorgement with lipids to constrain autophagic flux (). In the context of medical treatment reserved to severe obesity where sustained caloric restriction can only be obtained by bariatric surgery interventions, still with large variability among patients, the links from autophagic response to weight loss, improved metabolism and inflammation have not been fully investigated. We reported that relative to baseline conditions, fat cell autophagic clearance ameliorates within the first year following bypass surgery, although with large variability among patients. Even if longer follow-up is needed, this may be an encouraging observation to promote trials evaluating the impact of pharmacological autophagy activators as adjuvants to bypass surgery. To what extent adipocyte autophagy reactivation participates in metabolic recovery and can be used to optimize obesity treatments therefore deserves further investigation.
Figure 1. Adipose tissue dysfunction in obesity is characterized by exaggerated lipid engorgement and chronic low grade inflammation with immune cell infiltration. In this context obese adipose tissue expresses high levels of autophagy genes, but attenuated adipocyte autophagic flux. Weight loss by bariatric surgery can partly reverse flux attenuation, likely through normalized expression of an autophagy regulator, DAPK2.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Support from ANR-LIPOCAMD and CORDDIM-Region Ile de France is acknowledged.