
Abstract
Wear particle-induced osteolysis is the leading cause of aseptic loosening, which is the most common reason for THA (total hip arthroplasty) failure and revision surgery. Although existing studies suggest that osteoblast apoptosis induced by wear debris is involved in aseptic loosening, the underlying mechanism linking wear particles to osteoblast apoptosis remains almost totally unknown. In the present study, we investigated the effect of autophagy on osteoblast apoptosis induced by CoCrMo metal particles (CoPs) in vitro and in a calvarial resorption animal model. Our study demonstrated that CoPs stimulated autophagy in osteoblasts and PIO (particle-induced osteolysis) animal models. Both autophagy inhibitor 3-MA (3-methyladenine) and siRNA of Atg5 could dramatically reduce CoPs-induced apoptosis in osteoblasts. Further, inhibition of autophagy with 3-MA ameliorated the severity of osteolysis in PIO animal models. Moreover, 3-MA also prevented osteoblast apoptosis in an antiautophagic way when tested in PIO model. Collectively, these results suggest that autophagy plays a key role in CoPs-induced osteolysis and that targeting autophagy-related pathways may represent a potential therapeutic approach for treating particle-induced peri-implant osteolysis.
Abbreviations
| 3-MA | = | 3-methyladenine |
| AKT | = | v-akt murine thymoma viral oncogene homolog |
| Alp | = | alkaline phosphatase |
| AP | = | autophagosome |
| ATG | = | autophagy related |
| AVd | = | degradative autophagic vacuole |
| BAX | = | BCL2-associated X protein |
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BGLAP/OCN | = | bone gamma-carboxyglutamate (gla) protein |
| BSA | = | bovine serum albumin |
| BV/TV | = | bone volume/total volume |
| CCK8 | = | cell counting kit-8 |
| CoPs | = | CoCrMo metal particles |
| Col1a2/collagen | = | 1α2, collagen, type I, α 2 |
| CQ | = | chloroquine |
| DAPI | = | 4,6-diamidino-2-phenylindole |
| DRAM1 | = | DNA-damage regulated autophagy modulator 1 |
| EIF2AK3/PERK | = | eukaryotic translation initiation factor 2 α kinase 3 |
| EIF2S1/eIF2α | = | eukaryotic translation initiation factor 2, subunit 1 α |
| E-MAR | = | endosteum mineral apposition rates |
| ER | = | endoplasmic reticulum |
| ERN1/IRE1α | = | endoplasmic reticulum to nucleus signaling 1 |
| HE | = | hematoxylin-eosin |
| IBSP | = | bone sialoprotein |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MAPK8/JNK1 | = | mitogen-activated protein kinase 8 |
| micro-CT | = | microcomputed tomography |
| MMPs | = | matrix metallopeptidases |
| MSCs | = | mesenchymal stem cells |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| PBS | = | phosphate-buffered saline |
| PIO | = | particle-induced osteolysis |
| P-MAR | = | periosteum mineral apposition rates |
| PMMA | = | polymethylmethacrylate |
| Rap | = | rapamycin |
| RIPA | = | radio immunoprecipitation assay |
| SA-GLB1/SA-β-gal | = | senescence-associated β-galactosidase staining |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| THA | = | total hip arthroplasty |
| TRAF2 | = | TNF factor receptor-associated factor 2 |
| UHMWPE | = | ultra-high molecular weight polyethylene |
| XBP1 | = | X-box binding protein 1. |
Introduction
Total hip arthroplasty (THA) is one of the most successful orthopedic procedures performed for the treatment of rheumatoid arthritis, severe trauma and other end-stage joint disease.Citation1 However, periprosthetic osteolysis and consequent aseptic loosening is the most common reason for total hip arthroplasty failure and surgical revision.Citation2,3 Wear particles, originating from the implant components, are the main triggering cause of aseptic loosening.Citation4 Various cell types, including osteoclasts, osteoblasts, macrophages, fibroblasts and lymphocytes, are reportedly involved in the pathogenesis of particle-induced osteolysis.Citation5-7 Wear particles induce a complex adverse local cellular response that disturbs the balance between bone formation and resorption. Numerous studies have focused on the increased osteoclastic bone resorption, which has been mainly related to osteoclasts and macrophages. Although osteoblasts are one of the first cell lines exposed to wear debris, the negative impact of particles on osteoblasts has not been closely investigated.Citation6 Osteoblasts are responsible for bone formation in vivo. The cells secret extracellular matrix (mainly type I collagen) and are responsible for the mineralization of the matrix. Wear particles exhibit adverse effects on osteoblast proliferation, survival and function changes. Various particles (including titanium, PMMA [polymethylmethacrylate] and UHMWPE [ultra-high molecular weight polyethylene]) have been shown to suppress collagen synthesis, inhibit the differentiation of mature osteoblasts from MSCs (mesenchymal stem cells), decrease osteoblast proliferation and increase the apoptosis of osteoblasts, and these changes in osteoblasts can lead to decreased bone formation and improper bone remodeling around the prosthesis site.Citation5,8-11 The disruption of homeostasis between bone formation and degradation results in peri-implant osteolysis and consequent aseptic loosening. However, our knowledge of the mechanism underlying the interaction of wear debris and osteoblasts remains largely unknown.
Recently, countless studies have demonstrated that macroautophagy (hereafter referred to as “autophagy”), an evolutionarily conserved process whereby aggregated proteins, intracellular pathogens, and damaged organelles are degraded and recycled, plays important roles both for normal cellular quality control and in the response to environmental or internal stressors. Furthermore, autophagy engages in complex interplay with apoptosis; various stress pathways sequentially elicit autophagy and apoptosis within the same cell. In general, autophagy blocks the induction of apoptosis, and apoptosis-related caspase activation inhibits the autophagic process. However, in special cases, autophagy or autophagy-associated proteins may help induce apoptosis, and the disruption of the interplay between autophagy and apoptosis will result in serious pathophysiological consequences.Citation12-14 With respect to wear particle-induced osteolysis, studies have reported that the exposure of osteoblasts to several particles can result in apoptosis.Citation15,16 However, the mechanism remains unclear. Recently a growing number of studies have suggested that some nanomaterials can induce autophagy, which may be an attempt to degrade material that is perceived by the cells as foreign or aberrant.Citation15,17-21 In the majority of these studies, autophagosome accumulation induced by nanomaterials was associated with cell death. Additionally, metal wear particles retrieved from patients undergoing aseptic loosening are found to be as small as 50 nm (range 6 to 834 nm).Citation4,22 However, thus far, the relationship between autophagy and wear debris-induced osteolysis remains unclear.
In the present study, we hypothesized that one possible reason for particle-induced osteolysis was wear debris-induced autophagy in osteoblasts. Autophagy induced by wear particles may trigger apoptosis in osteoblasts, decrease bone formation and promote subsequent osteolysis. We tested our hypothesis in osteoblast cell cultures and a particle-induced calvarial osteolysis model.
Results
Activation of autophagy by CoPs-stimulated osteoblasts in murine calvaria resorption models and in vitro
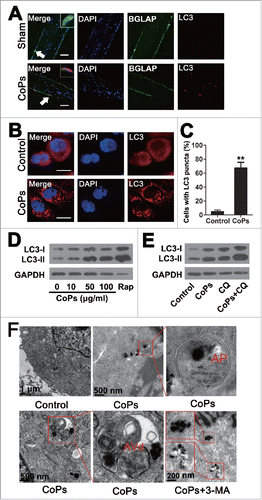
Whether autophagy is involved in wear particle-induced osteolysis is unknown. In the present study, we performed immunofluorescence analysis of MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), a marker of autophagy,Citation23-25 in sections of CoPs-stimulated calvarias obtained from a commonly used murine calvaria resorption model. As shown in , the level of LC3 was significantly increased in osteoblasts compared with that of the control. We further examined the rise of autophagy in osteoblasts in vitro. Confocal microscopy revealed that CoPs can induce the accumulation of LC3-positive puncta (). For western blot analysis, we used LC3-II/GAPDH ratios as indicators of autophagy instead of LC3-II/LC3-I or LC3-II/ (LC3-I+ LC3-II) because LC3-II tends to be more sensitive than LC3-I in immunoblotting.Citation23 We observed a marked increase in LC3-II induced by CoPs when compared with the control (). To further interpret the autophagy induction by CoPs, we treated cells with CQ (chloroquine), a lysosomal degradation inhibitor, and observed that CoPs and chloroquine cotreatment resulted in significantly elevated levels of LC3-II relative to treatment with either CoPs or chloroquine alone (). The accumulation of autophagosomes was also confirmed using electron microscopy. As presented in , under normal conditions, autophagosomes were rarely detected, whereas their levels were elevated upon CoPs treatment. The autophagy inhibitor 3-MA decreased the accumulation of autophagosomes. These results suggested that osteoblasts underwent autophagy in response to CoPs.
Figure 1. Activation of autophagy following exposure to CoPs in murine calvaria resorption models and osteoblast cells. (A) Immunofluorescence was performed to examine the expression of LC3 in osteoblasts. Sections of murine calvaria are presented for animals from each group. Scale bar: 50 µm. Red, LC3; green, osteoblasts (BGLAP); blue, DAPI nuclear staining. (B) Immunofluorescence was performed to examine the expression of LC3 in osteoblast cells. Cells were treated with 50 µg/ml CoPs for 12 h. Scale bar: 10 µm. Red, LC3; blue, DAPI nuclear staining. (C) Quantification of the percentage of cells with LC3 puncta shown in (B). Data are presented as the means ± S.E.M. from 3 independent experiments. **, P < 0.01 vs. control. (D) Western blots performed after osteoblast cells were treated with various concentrations (0, 10, 50, 100 µg/ml) of CoPs for 12 h. Rap, rapamycin. (E) Western blots performed after osteoblast cells were cultured in CQ for 3 h prior to being treated with 50 µg/ml CoPs for 12 h. (F) Transmission electron microscopy of osteoblasts cultured with or without 10 mM 3-MA prior to being treated with 50 µg/ml CoPs for 12 h. The double-membrane autophagosome (AP) containing CoPs clusters and the rough endoplasmic reticulum are shown in the middle and right images of the upper panel. High-magnification view of an autolysosome containing CoPs clusters, partially degraded ribosomes and rough endoplasmic reticulum in the middle image of the lower panel. AVd, degradative autophagic vacuole.
The ERN1-MAPK8 pathway was involved the activation of autophagy induced by CoPs
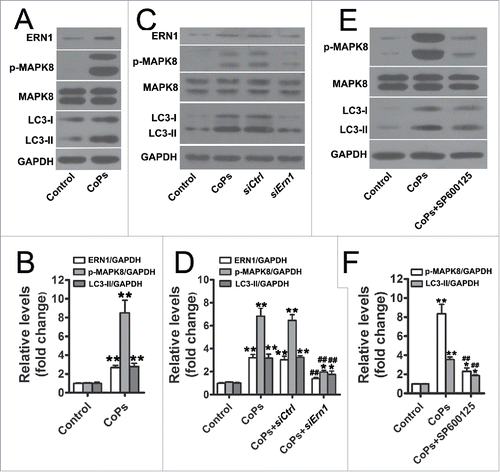
Previous studies have suggested that autophagy can be triggered by members of the endoplasmic reticulum (ER) stress signaling pathway including ERN1/IRE1α (endoplasmic reticulum to nucleus signaling 1) and EIF2AK3/PERK (eukaryotic translation initiation factor 2 α kinase 3), and we have also observed that CoPs can induce ER stress in macrophages.Citation15,26,27 Thus, we sought to determine which ER stress signaling pathway(s) is/are involved in CoPs-stimulated autophagy in osteoblasts. CoPs significantly increased the expression of ERN1 and phosphorylated EIF2AK3 (, and Fig. S1A and B). Small interfering RNA against Ern1 (siErn1) significantly decreased the expression of LC3-II (), but no obvious differences between siEif2ak3 and siControl were observed (Fig. S1C to F). Furthermore, we detected the expression of phosphorylated MAPK8/JNK1 (mitogen-activated protein kinase 8) and XBP1 (X-box binding protein 1), both of which are downstream signaling effectors of ERN1.Citation28-30 Interestingly, we observed significantly increased p-MAPK8 levels in cells treated with CoPs (), but not XBP1 (Fig. S1A and B). Next, we asked whether CoPs-induced autophagy was mediated by MAPK8 after activation by ERN1. Osteoblasts were cotreated with CoPs and SP600125, a MAPK8 inhibitor. LC3-II was significantly reduced in cells treated with MAPK8 inhibitors (). Collectively, these results indicated that CoPs-induced autophagy was mediated by the ERN1-MAPK8 pathway.
Figure 2. The ERN1-MAPK8 pathway mediated the activation of autophagy induced by CoPs. (A) Western blots performed after osteoblast cells were incubated with CoPs (50 µg/ml) for 12 h. (B) The density of the western blots bands shown in (A) was quantified using ImageJ software. (C) Western blots performed after osteoblast cells were incubated with siCtrl or siErn1 before being stimulated with CoPs (50 µg/ml) for 12 h. siCtrl, siControl. (D) The density of the western blots bands shown in (C) was quantified using ImageJ software. (E) Western blots performed after fibroblast cells were incubated with SP600125 (15 µM) before being stimulated with CoPs (50 µg/ml) for 12 h. (F) The density of the western blots bands shown in (E) was quantified using ImageJ software. Data are presented as means ± S.E.M. from 3 independent experiments. *, P < 0.05 and **, P < 0.01 vs. control; ##, P < 0.01 vs. CoPs group.
Autophagy mediated the CoPs-induced upregulation of osteoblast apoptosis in vitro
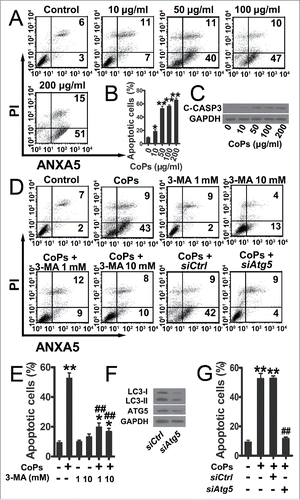
The upregulation of osteoblast death reduces bone formation in the periprosthetic region, which is an etiology for wear particles-induced osteolysis.Citation5,6 To study the role of autophagy in CoPs-treated cells, we examined the cell viability of osteoblasts treated with various concentration of CoPs. CoPs treatment sharply decreased the cell viability (Fig. S2A). The autophagy inhibitor 3-MA could rescue the cell viability in a dose-dependent manner (Fig. S2B). Although 3-MA slightly decreased cell viability, there was no significant effect on cell viability at the dose we used (Fig. S2C). As senescence may be involved in aseptic loosening, we measured cell senescence with an SA-GLB1/SA-β-gal (senescence-associated β-galactosidase staining) staining kit.Citation31 The senescence of the cells was not significantly changed among the various CoPs treatment groups (Fig. S3A to D). To determine whether osteoblast apoptosis is involved in the toxicity of CoPs, we examined the effect of CoPs treatment on osteoblast apoptosis. illustrated that CoPs could increase the apoptotic rate of cells in a dose-dependent manner. The apoptotic rates in cells treated with 100 μg/ml CoPs and control cells were 57% and 9%, respectively. CASP3/caspase 3, a hallmark of apoptosis, was also assessed by western blot, and a dose-dependent increase in cleaved CASP3 was observed after the exposure of cells to increasing concentrations of CoPs (). We then investigated the effect of autophagy on CoPs-induced apoptosis. The elevation of the apoptotic rate was significantly ameliorated by treatment with 3-MA (). To further investigate whether upregulation of particle-induced apoptosis was due to autophagy, siRNA was used to knock down the expression of ATG5 (), a member of the autophagy-related (ATG) protein family that is required for the formation of autophagosomes. The apoptotic rate was significantly decreased in the groups treated with siAtg5 (). Thus, autophagy played a critical role in CoPs-induced osteoblast apoptosis.
Figure 3. Autophagy mediated the increase in osteoblast apoptosis induced by CoPs. (A) Flow cytometry analysis of ANXA5 and propidium iodide staining of osteoblast cells cultured with various concentrations (0, 10, 50, 100, 200 µg/ml) of CoPs for 24 h. (B) Quantification analysis of apoptotic cells in (A) (both upper- and lower-right quadrants in representative dot plots as shown). Data are represented as means ± S.D. from 3 independent experiments. *, P < 0.05 and **, P < 0.01 vs. the control. (C) Western blots performed after osteoblast cells were cultured with various concentrations (0, 10, 50, 100, 200 µg/ml) of CoPs for 24 h. (D) Flow cytometry analysis of ANXA5 and propidium iodide staining of osteoblast cells cultured with various concentrations (0, 1, 10 mM) of 3-MA, siCtrl and siAtg5 prior to being treated with 50 µg/ml of CoPs for 24 h. (E, G) Quantification analysis of apoptotic cells in (D) (both upper- and lower-right quadrants in representative dot plots as shown). Data are presented as means ± S.D. from 3 independent experiments. *, P < 0.05 and **, P < 0.01 vs. control; ##, P < 0.01 vs. CoPs group. (F) Western blots performed after osteoblast cells were cultured in siCtrl or siAtg5 prior to treatment with CoPs (50 µg/ml) for 24 h. c-CASP3, cleaved CASP3; siCtrl, siControl.
Autophagy mediated CoPs-induced upregulation of BAX
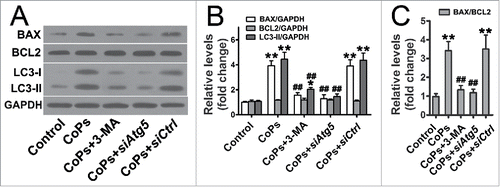
Both the antiapoptotic protein BCL2 (B-cell CLL/lymphoma 2) and the pro-apoptotic protein BAX (BCL2-associated X protein) belong to the BCL2 family. Recently, studies have demonstrated that these BCL2 family proteins are involved in the crosstalk between autophagy and apoptosis.Citation14 In the present study, we first investigated the expression of the BAX and BCL2 proteins in CoPs-stimulated osteoblasts. CoPs treatment increased the level of BAX protein but had no effect on the expression of BCL2, thereby raising the BAX/BCL2 ratio (). To investigate whether autophagy was involved in the upregulation of BAX, we next performed western blot analysis in cells cotreated with CoPs and the autophagy inhibitor 3-MA. As shown in , the upregulation of BAX was significantly decreased when osteoblasts were treated with 3-MA. In addition, 3-MA treatment significantly inhibited the CoPs-induced upregulation of the BAX/BCL2 ratio (). Autophagy is tightly regulated by a limited number of highly evolutionarily conserved molecules called ATGs, including ATG5.Citation32 To further confirm that the upregulation of the BAX/BCL2 ratio was due to autophagy, we used siRNA against Atg5 to inhibit autophagy more specifically at the molecular level ().Citation33,34 SiAtg5 markedly rescued the CoPs-induced upregulation of the BAX/BCL2 ratio ().
Figure 4. Autophagy mediated the upregulation of BAX induced by CoPs. (A) Western blots performed after osteoblast cells were incubated with 3-MA (10 mM), siAtg5 and siCtrl before being stimulated with CoPs (50 µg/ml) for 24 h. (B, C) The density of the western blots bands shown in (A) was quantified using ImageJ software. siCtrl, siControl. Data are presented as means ± S.E.M. from 3 independent experiments. *, P < 0.05 and **, P < 0.01 vs. control; ##, P < 0.01 vs. CoPs group.
Autophagy inhibition rescued the CoPs-induced decreases in osteoblast numbers and functional disturbances that lead to compromised particle-induced osteolysis in animal models
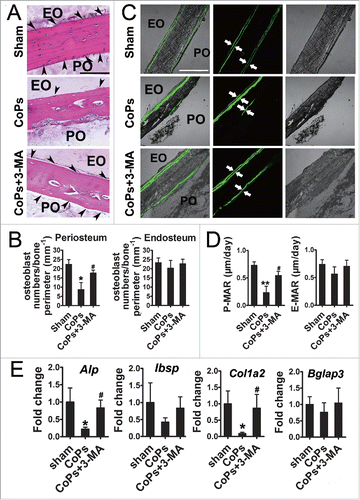
To examine the role of autophagy in vivo, we used a murine calvaria resorption model, which is a commonly used model for studying aseptic loosening. HE (hematoxylin-eosin) staining analysis revealed that treatment with 3-MA, an autophagy inhibitor, markedly ameliorated the reduction in osteoblasts induced by CoPs (). Calcein labeling, which permits dynamic bone histomorphometric analysis in vivo, was performed in sections of calvaria to evaluated new bone formation. CoPs treatment induced a decrease in the P-MAR (periosteum mineral apposition rates) relative to the sham group, but the decrease was significant ameliorated in mice cotreated with 3-MA (). We further examined the expression of osteoblast markers in calvaria samples by real-time PCR analysis. The decreased expression of Alp (alkaline phosphatase) and Col1a2/collagen 1α2 caused by CoPs was largely rescued by 3-MA (). Similar trends were observed in Ibsp (bone sialoprotein) and Bglap3 (bone gamma-carboxyglutamate protein 3), although the increased level in 3-MA-cotreated mice was not statistically significant ().
Figure 5. The autophagy inhibitor 3-MA rescued osteoblast numbers and functional disturbance induced by CoPs. (A) Representative HE staining images of calvaria from each group. Osteoblasts are indicated by black arrowheads. Scale bar: 100 µm. EO, endosteum; PO, periosteum. (B) Osteoblast numbers/bone perimeter (mm−1) of endosteum and periosteum in (A). (C) Representative images of new bone formation of calvaria sections from each group were determined by calcein double labeling. Scale bar: 100 µm. (D) P-MAR and E-MAR in (C). (E) Real-time PCR of Alp, Ibsp, Col1a2 and Bglap3 of calvarial bone extracts from each group. Values are expressed as means ± S.E.M. n = 7 mice per group. *P < 0.05, **P < 0.01 vs. sham; #P < 0.05 vs. CoPs group.
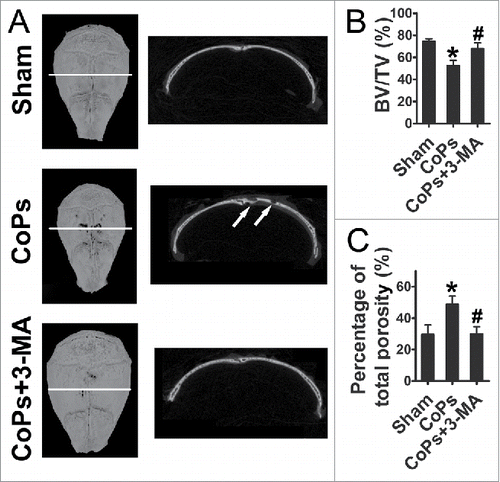
The osteolysis induced by CoPs implantation was examined by micro-CT (micro-CT) with 3-dimensional reconstruction. demonstrated that CoPs-induced osteolysis was suppressed by cotreatment with 3-MA. Quantitative analysis of bone parameters further confirmed that 3-MA markedly ameliorated the BV/TV (bone volume/total volume) and decreased the percentage of total porosity that induced by CoPs ().
Figure 6. The autophagy inhibitor 3-MA ameliorates CoPs-induced mouse calvarial osteolysis assessed by micro-CT. (A) Representative images of micro-CT with 3-dimensional reconstructed images from each group (left). Cross-sectional views of the reconstructed images were indicated by the white horizontal line in the left panel, and the bone osteolysis sites are indicated by white arrows (right). (B) BV/TV and percentage of total porosity of each sample were measured. Values are expressed as means ± S.E.M. n = 7 mice per group. *, P < 0.05, vs. sham; #, P < 0.05 vs. CoPs group.
Autophagy mediated the upregulation of osteoblast apoptosis induced by CoPs in vivo
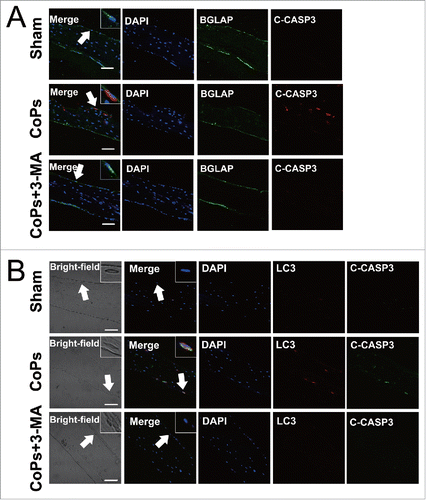
We first confirmed that autophagy was induced by CoPs in osteoblasts in an animal model (Fig. S4). CoPs treatment significantly increased the expression of LC3 in osteoblasts compared with that of the control. Cotreatment with 3-MA resulted in a reduction in LC3 levels in osteoblasts. We then performed immunofluorescence staining of murine calvaria sections to examine the level of cleaved CASP3, a protein marker of apoptosis, in osteoblasts. indicated that the expression of cleaved CASP3 was upregulated by CoPs in osteoblasts in vivo. More importantly, the induction of cleaved CASP3 by particles was markedly inhibited by cotreatment with 3-MA. To further investigate whether the inhibition of autophagy and apoptosis were present simultaneously, we detected the colocalization of LC3 and cleaved CASP3 in osteoblasts. As shown in , CoPs induced the expression of LC3 and cleaved CASP3 in the same osteoblasts, and 3-MA decreased the expression of LC3, as well as cleaved CASP3. Osteoclasts play a crucial role in wear particle-induced osteolysis.Citation1 Thus, we also performed tartrate-resistant acidic phosphatase staining, which detected osteoclasts, to examine the effect of 3-MA on osteoclastogenesis. The data demonstrated that osteoclasts were significantly increased by CoPs (Fig. S5). However, cotreatment with 3-MA resulted in only slightly decreased formation of osteoclasts (Fig. S5). The effects of 3-MA on osteoclasts were not as notable as the effects on osteoblasts. Collectively, these data suggested that autophagy mediated the increased osteoblast apoptosis in this particle-stimulated murine calvaria resorption model.
Figure 7. Autophagy mediated the upregulation of CoPs-induced osteoblast apoptosis in vivo. (A) Immunofluorescence was performed to determine the expression of cleaved CASP3 in osteoblasts. Sections of murine calvaria are presented for the animals from each group. Scale bar: 50 µm. Red, cleaved CASP3 (C-CASP3); green, osteoblasts (BGLAP); blue, DAPI nuclear staining. (B) Immunofluorescence was performed to determine the colocalization of LC3 and cleaved CASP3 in osteoblasts. Sections of murine calvaria are presented for the animals from each group. Scale bar: 50 µm. Red, LC3; green, cleaved CASP3 (C-CASP3); blue, DAPI nuclear staining.
Discussion
Total hip arthroplasty is currently thought to be the most effective surgical procedure for treating severe joint disease and trauma. However, wear debris-induced peri-implant osteolysis and subsequent aseptic loosening remains the most common cause of failed total hip arthroplasty. Wear particles, liberated from the surface of prostheses, interact with multiple cell types in the peri-implant region, resulting in various cell responses that are responsible for the imbalance between bone resorption and formation. Most studies on osteolysis have stressed the stimulation of bone resorption, which is primarily concerned with osteoclasts and macrophages.Citation5,6 Once exposed to wear particles, these cells release various proinflammatory cytokines, including TNF, IL6, IL1B, IL8 and MMPs (matrix metallopeptidases), leading directly to bone resorption and indirectly to the activation of osteoclasts.Citation26,35-37 However, the role of decreased bone formation, which may be related to osteoblasts, has received relatively less research attention.Citation6 Osteoblasts play a key role in periprosthetic osteolysis, as normal bone metabolism relies on the balance between bone formation and degradation, and either increased bone resorption or decreased bone formation can result in the loss of bone stock around the peri-implant area.Citation6 Wear particles have been shown to exert a negative influence on osteoblasts in the osteolytic process. First, wear particles can stimulate osteoblasts to produce proinflammatory mediators, such as IL6, IL8 and TNF.Citation6,38,39 Second, the particles can inhibit the ability of osteoblasts to secret mineralized bone matrix, alkaline phosphatase activity and osteoblast proliferate.Citation6,9,40 Apoptosis has also been demonstrated in primary human osteoblast and osteoblast-like cells following exposure to titanium and PMMA particles.Citation6,41 These changes can influence bone remodeling and ingrowth around the implant area. Although existing studies have demonstrated the important role of osteoblasts in wear debris-induced osteolysis, an obvious limitation of the evidence is the almost exclusive reliance on in vitro studies.Citation6 In our present study, our results indicated that CoPs can affect osteoblasts through the upregulation of osteoblast apoptosis and the downregulation of function.
Several mechanisms of autophagy induction by nanomaterials have been reported, including the induction of endoplasmic reticulum stress or mitochondrial damage, the suppression of AKT (v-akt murine thymoma viral oncogene homolog)-MTOR (mechanistic target of rapamycin [serine/threonine kinase]) signaling, and the alteration of autophagy-related gene and protein expression.Citation15,21 CoPs are classic metal particles that can be taken up by osteoblasts; however, CoPs cannot be digested by cells and thus may induce intracellular stress.Citation15,21,26 This intracellular stress (such as oxidative stress and ER stress) potently further induces autophagy, which is important for organisms to adapt to or overcome harmful conditions.Citation15 The molecular mechanisms linking the stress response to autophagy vary across cell types and organisms and depend on the specific stress conditions.Citation15,21 The involvement of oxidative stress in the induction of autophagy by nanomaterials was evidenced by a study in which fullerene-induced autophagy in HeLa cells is dependent upon photoactivation and is suppressed by the antioxidants N-acetyl-L-cysteine, reduced glutathione, and L-ascorbic acid.Citation15,42 The EIF2AK3 and ERN1 signaling pathways have been implicated as mediators of ER stress-induced autophagy in mammalian cells.Citation27 The EIF2AK3-EIF2S1/eIF2α (eukaryotic translation initiation factor 2, subunit 1 α) signaling pathway is required for the polyglutamine 72 repeat-induced ATG12 upregulation and LC3 conversion in C2C5 cells.Citation43 The ERN1-TRAF2-MAPK8 pathway is essential for the induction of autophagy in mouse embryonic fibroblasts challenged with ER stressors.Citation44 In the present study, we tested both the EIF2AK3 and ERN1 signaling pathways in CoPs-treated osteoblasts and found that ERN1 but not EIF2AK3 mediated the activation of autophagy by CoPs in osteoblasts. These results further confirmed the key role of ERN1 signaling in the crosstalk between autophagy and ER stress.
Recently, autophagy has been recognized as a toxicity mechanism triggered by nanomaterials.Citation15,45,46 Polyamidoamine dendrimer nanoparticles trigger cell death and acute lung injury in mice, both of which can be rescued by autophagy inhibitors.Citation16 Cerium dioxide nanoparticle-induced cell death also can be partially reversed by inhibition of autophagy.Citation47 In our experiment, the CoPs used to stimulate osteoblasts represented a type of metal nanoparticles. Inhibition of autophagy efficiently rescued cell viability and apoptosis in CoPs-treated osteoblasts. The data further confirmed the role of autophagy as a contributor to nanoparticle-induced cell death. Various signaling pathways or proteins are involved in the prodeath mechanisms of autophagy.Citation12,16,47-49 Polyamidoamine dendrimer nanoparticles induce autophagic cell death through the AKT-TSC2-MTOR signaling pathway.Citation16 Autophagosome formation has been implicated in the activation of CASP8/caspase-8.Citation50 In our present study, both the chemical and specific inhibitors of autophagy (3-MA and siAtg5) were able to decrease CoPs-induced BAX expression, which suggested that autophagy mediated the increase in osteoblast apoptosis by BAX. BAX was identified as a BCL2-interacting protein that opposed BCL2 and promoted cell death.Citation51 Furthermore, BAX plays an important role in the crosstalk between autophagy and apoptosis.Citation12,3,52-54 DRAM1 (DNA-damage regulated autophagy modulator 1)-mediated activation of autophagy results in the upregulation of BAX protein expression.Citation52 Autophagy inhibitor was also reported to inhibit the expression of BAX.Citation55 Furthermore, BAX can play opposite roles in the regulation of autophagy under different stimuli.Citation53,54,56,57 Rubinsztein et al. report that BAX reduces autophagy by inducing BECN1/Beclin 1 degradation in Hela cells.Citation53 However, TP53/p53-inducible BH3-only protein-induced autophagy is dependent on BAX-BAK1 and can be reproduced by the overexpression of BAX in U2OS cells.Citation54 Autophagy engages in complex interactions with apoptosis and is able to play both prosurvival and prodeath roles in cells.Citation12 In our present study, autophagy played a prodeath role in CoPs-treated osteoblasts, although studies have reported that autophagy plays a cytoprotective role in hydrogen peroxide- and fluoride-treated osteoblasts.Citation58,59 The different roles that autophagy plays in apoptosis may due to the different inducing factors.Citation12,21,60-62 The stimulus in our research was a type of metal nanoparticle. The nanomaterials exhibit unique physical and chemical properties, which are thought to act as determinants of biologic activity.Citation21 The interactions of nanomaterials with cells and subcellular structures and their biokinetics are likely to be very different from the above-mentioned stimuli (such as hydrogen peroxide).Citation21,58,59,63 Additionally, the intensity of autophagy may be another possible explanation for the different effects of autophagy on apoptosis.Citation12,47,64
To date, strategies for the treatment of wear debris-induced osteolysis have focused on the inhibition of inflammation and target osteoclasts.Citation2,36 However, these strategies have been shown to be ineffective in humans and may cause adverse effects on other organs. Inhibition of TNF/TNFα may lead to blunted reactions to harmful agents, including bacteria and other infective agents. Inhibition of osteoclast-mediated bone resorption with bisphosphonates may result in mandibular lesions, the impairment of fracture healing, pathologic femoral fractures and other adverse events.Citation2 Thus, new strategies and biologics for wear particle-induced osteolysis are urgently needed. Considering the important role of osteoblasts in the pathogenesis of wear debris-induced peri-implant osteolysis, osteoblasts may represent a potential therapeutic target in the treatment of this disease. Because all results discussed in the present study have suggested the key role of autophagy in osteoblast apoptosis and osteolysis induced by CoPs, we aim to further investigate the possibility of alleviating particle-induced osteoblast apoptosis by inhibiting autophagy in osteoblasts and adopting this as a strategy to treat particle-induced osteolysis. In the present study, we demonstrated that inhibition of autophagy could attenuate osteoblast apoptosis, rescue osteoblast function and compromise bone resorption induced by CoPs. Although osteoclasts play a crucial role in wear particle-induced osteolysis, we did not observe an obvious effect of autophagy inhibition on osteoclastogenesis under the in vivo experimental conditions.Citation1 Thus, we attributed the decreased osteolysis in vivo to the inhibition of autophagy in osteoblasts instead of osteoclasts. However, considering that both positive and negative effects of autophagy in modulating osteoclast function and formation have been described previously, systematic dissection of autophagy in wear particle-stimulated osteoclasts in vitro and in vivo is required to fully understand the role of autophagy in aseptic loosening.Citation65-69
In summary, the present study demonstrated that autophagy mediated osteoblast apoptosis and promoted particle-induced osteolysis. Marked upregulation of autophagy was observed in osteoblasts both in vitro and in an animal model of wear debris-induced osteolysis. More importantly, autophagy was involved in the upregulation of apoptosis in osteoblasts in vitro and in vivo. Furthermore, blocking autophagy with 3-MA resulted in significantly amelioration of the osteolysis induced by the implanted particles. Our findings suggested a possible mechanism underlying wear debris-induced osteolysis and identified autophagy inhibition as a potential therapeutic approach for treating aseptic loosening.
Material and Methods
Reagents
BSA (Bovine serum albumin, A4161), 3-MA (M9281), CQ (C6628), rapamycin (R117), SP600125 (S5567) and protease inhibitor cocktail (P8340) were purchased from Sigma-Aldrich. Minimum essential medium α (11900–024) and fetal bovine serum (10099–141) were obtained from Gibco. RIPA lysis buffer (P0013B) was purchased from Beyotime.
Particle preparation
CoCrMo particles, a gift from Dr. Zhenzhong Zhang (The College of Materials Science and Engineering of Nanjing University of Technology), had a mean diameter of 51.7 nm. For decontamination from endotoxins, the particles were autoclaved for 15 min at 121°C and 15 psi. This treatment resulted in negative testing for endotoxin using a quantitative Limulus Amebocyte Lysate (LAL) Assay (Charles River, R13025) at a detection level of <0.25% EU/ml. The particles were suspended in phosphate-buffered saline (PBS; Boster Biological Technology Co., Ltd., AR0030, pH 7.2 to 7.4) at a concentration of 50 mg/ml as stock solutions. For in vitro experiments, the particles were further diluted in cell culture medium to attain concentrations ranging from 10 to 200 µg/ml and ultrasonicated for 20 min before exposing them to cells.
Cell culture
The mouse osteoblastic cell line MC3T3-E1, obtained from the China Center for Type Culture Collection, was cultured in minimum essential medium α containing 10% fetal bovine serum, 1% penicillin and streptomycin and maintained at 37°C and 5% CO2 in a humidified incubator.
Cell viability assay
Cells were seeded in 96-well plates and were cultured with or without various concentrations of 3-MA for 3 h before being stimulated with CoPs for 20 h. Subsequently, cell viability was determined using WST-8 staining with a CCK8 (Cell Counting Kit-8) according to the manufacturer's instructions (Dojindo, CK04). Optical density was determined at 450 nm with a plate reader (Thermo Scientific, Multiskan FC, Waltham, MA, USA).
Flow cytometry analysis
Cells were seeded into 6-well plates at a density of 1 × 106 cells/well and cultured with or without various concentrations of 3-MA for 3 h, or either siAtg5 or siControl for 24 h before being stimulated with CoPs for another 24 h. Subsequently, the cells were collected and stained with the ANXA5 (Annexin V)-FITC apoptosis detection kit (4A Biotech Co. Ltd., FXP018–100) according to the manufacturer's instructions and analyzed by flow cytometry (FACSCalibur, BD. Bioscience, San Jose, CA, USA).
SA-GLB1/SA-β-gal staining
Cells were seeded in 24-well plates and cultured with or without various concentrations of 3-MA for 3 h before being stimulated with CoPs for 24 h. Subsequently, cells were incubated with GLB1/β-galactosidase staining solution overnight at 37°C without CO2 according to the manufacturer's instructions (Beyotime, C0602). The reaction was stopped by the addition of PBS. The cells were then observed and photographed with a light microscope (C2+, Nikon, Tokyo, Japan).
In vivo calvarial resorption model
The wear particle-induced calvarial osteolysis model has been described previously, and all animals received humane care according to the Chinese legal requirements.Citation26 Briefly, 42 6- to 8-wk-old C57BL/J6 mice were assigned randomly into 3 groups: group I, sham-operated PBS mice; group II, CoPs-treated mice; and group III, CoPs cotreatment with 3-MA mice. The mice were anesthetized, and the cranial periosteum was separated from the calvarium by sharp dissection. Forty microliters (30 mg/ml) of the CoPs suspensions was embedded under the periosteum around the middle suture of the calvaria. Group I, which received 40 µl PBS, was used as a sham control group. For group III, 3-MA was administered intraperitoneally at 40 mg/kg/day to inhibit autophagy induction in vivo. 3-MA was injected daily for 2 wk until the animals were sacrificed, and the calvarial caps were removed by dissecting the bone free from the underlying brain tissue. For in vivo fluorescent labeling, 7 mice from each group were injected with calcein (20 mg/kg body weight; Sigma-Aldrich, C0875) intraperitoneally at d 12 and 2 d before sacrifice.
Micro-CT scanning
The mouse calvaria were analyzed using a high-resolution micro-CT (SkyScan1176; SkyScan, Kontich, Belgium) at a resolution of 18 µm and X-ray energy settings of 45 kV and 550 µA. A square region of interest around the midline suture was selected for further qualitative and quantitative analysis after reconstruction. The BV/TV and percentage of total porosity of each sample were measured as reported previously.Citation70
Histologic and histomorphometric analysis
After micro-CT analysis, the calvaria samples were decalcified in 15% EDTA-PBS for 3 wk and embedded in paraffin. The embedded tissues were cut in the coronal plane centered over the area of particle deposition. The sections were stained with HE and tartrate-resistant acidic phosphatase (Sigma-Aldrich, 386A) by standard methods. The specimens were then observed and photographed with a light microscope (C2+, Nikon, Tokyo, Japan). The number of osteoblasts was reported counted in each sample as previously described.Citation71
For Calcein labeling analysis, a histomorphometric measurement of osteoblast activity in vivo, undecalcified sections embedded in plastic were sectioned. The average distance between double-labeled areas was calculated with ImageJ analysis. MAR was calculated by dividing the average double-labeled distance (μm) by the inter-label time of 10 d.
Real-time PCR
Total RNA from calvarial bone was prepared using TRIzol reagent (Invitrogen, 15596–018) according to the manufacturer's instructions. Five micrograms total RNA was reverse transcribed to cDNA with the first-strand cDNA synthesis kit (Invitrogen, 18080–051). Real-time PCR was performed using 2 x SYBR Green qPCR Mix (Zoonbio Biotechnology Co., PC01) according to the manufacturer's protocol. Primers for Actb/β-actin were used as internal controls. The following primers were used: Alp, sense: 5′-GGACAGGACACACACACACA-3′ and antisense: 5′- CAAACAGGAGAGCCACTTCA- 3′; Ibsp, sense: 5′- ACAATCCGTGCCACTCACT-3′ and antisense: 5′- TTTCATCGAGAAAGCACAGG- 3′; Col1a2, sense: 5′- GAGCTGGTGTAATGGGTCCT-3′ and antisense: 5′- GAGACCCAGGAAGACCTCTG- 3′; Bglap3, sense: 5′- CAGACACCATGAGGACCCTC -3′ and antisense: 5′- GGGCGTGGCATCTGTGAGGT- 3′; and Actb, sense: 5′- ATGTGGATCAGCAAGCAGGA-3′ and antisense: 5′- AAGGGTGTAAAACGCAGCTCA-3′ .
Immunofluorescence staining
To detect autophagy in osteoblasts, the cells were stained with an anti-LC3 antibody (Sigma-Aldrich, L7543) at 4°C overnight. For detecting autophagy and apoptosis in calvarias, the sections described above (hematoxylin-eosin staining) were incubated with anti-LC3 (Sigma-Aldrich, L7543), anti-cleaved CASP3 (Cell Signaling Technology, 9664) and anti-BGLAP/OCN (Santa Cruz Biotechnology, sc-390877) antibodies at 4°C overnight as previously reported.Citation26 The secondary antibody, Alexa Fluor 546 donkey anti-rabbit IgG (Molecular Probes, A10040) or Alexa Fluor 488 donkey anti-mouse IgG (Molecular Probes, A21202), was applied at 37°C for 1 h followed by staining of the nuclei with 4, 6-diamidino-2-phenylindole (DAPI, Beyotime, C1005). Cells and sections were photographed using a Nikon confocal microscope (C2+, Nikon, Tokyo, Japan).
Cell interfering experiment
Control siRNA (non-targeting; siControl), siRNA specific for mouse Atg5 (ON-TARGETplus SMARTpool, L-064838–00–0005), Ern1 (ON-TARGETplus SMARTpool, L-041030–00–0005) and siEif2ak3 (ON-TARGETplus SMARTpool, L-044901–00–0005) were obtained from Dharmacon. 20 nM siRNA was transfected into osteoblasts using Lipofectamine 2000 (Invitrogen, 11668027), as reported previously.Citation72 The cells were left to stand for 24 h after transfection before being used for experiments.
Transmission electron microscopy
Cells were seeded in 6-well plates at 1 × 106 cells/well. CoPs (50 μg/ml) were added to the wells on the following day. After 24 h, the cells were collected and fixed with 2.5% glutaraldehyde in 0.1 M sodium dihydrogen phosphate (pH 7.4). The samples were then fixed in 1% OsO4 for 1 h, dehydrated by graded ethanol solutions, and gradually infiltrated with epoxy resin (GENMED, GMS11012). Ultra-thin sections were obtained and stained with uranyl acetate and lead citrate and observed in a transmission electron microscope (JEM-200CX, JEOL, Japan).
Western blotting
The cells were lysed in RIPA lysis buffer with a protein inhibitor cocktail for 30 min on ice, and the lysates were centrifuged at 12,000 g for 10 min at 4°C. The supernatants were collected, and the protein concentrations were measured using a BCA protein assay kit (Biocolor Bioscience and Technology Co., PP1002). Thirty micrograms of each protein was separated by 12% or 15% SDS-PAGE before being transferred to polyvinylidene fluoride membranes (Bio-Rad, 162–0177). Western blotting was performed using the following primary antibodies: anti-LC3 (Sigma-Aldrich, L7543), anti-ERN1 (Cell Signaling Technology, 3294), anti-ATG5 (Cell Signaling Technology, 12994), anti-p-MAPK8 (Cell Signaling Technology, 9255), anti-MAPK8 (Cell Signaling Technology, 9252), anti-BAX (Cell Signaling Technology, 14796), anti-cleaved CASP3 (Cell Signaling Technology, 9664), anti-BCL2 (Cell Signaling Technology, 2876), anti-p-EIF2AK3 (Santa Cruz, sc-32577) and anti-XBP1 (Santa Cruz Biotechnology, sc-7160). The following secondary antibodies were used: horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (Cell Signaling Technology, 7074) and HRP-conjugated anti-mouse IgG antibodies (Santa Cruz Biotechnology, sc-2005). After probing with specific primary antibodies and a HRP-conjugated secondary antibody, the protein bands were detected, and the band density was analyzed using ImageJ 1.41 (National Institutes of Health).
Statistical analysis
Results are expressed as means ± standard error of the means (S.E.M.). Data concerning apoptotic cells analyzed by flow cytometry are expressed as means ± standard deviation (S.D.). The differences between groups were analyzed by the Brown-Forsythe test and, if appropriate, by one-way ANOVA followed by Dunnett's test or Bonferroni test. A P value of less than 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Material.zip
Download Zip (13.5 MB)Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by the National Science Fund for Distinguished Young Scholars (81025019), the National Basic Research Program of China (2012CB517603), the National High Technology Research and Development Program of China (2014AA020707), the Program for New Century Excellent Talents in University (NCET-13–0272), the National Natural Science Foundation of China (31370977, 31400671, 31271013, 31170751, 31200695, 51173076, 91129712 and 81572111), the Department of Orthopedics Clinical Research Center of Jiangsu Province, China (BL2012002), the Key Project of the Chinese Ministry of Education (108059), the Ph.D. Programs Foundation of the Ministry of Education of China (20100091120020, 20130091110037), the Graduate Education Innovation Project of Jiangsu Province (KYZZ15_0045), the Scientific Research Foundation of Graduate School of Nanjing University (2015CL12) and the Nanjing University State Key Laboratory of Pharmaceutical Biotechnology Open Grant (KF-GN-201409).
Related Research Data
References
- Gallo J, Goodman SB, Konttinen YT, Raska M. Particle disease: biologic mechanisms of periprosthetic osteolysis in total hip arthroplasty. Innate Immun 2013; 19:213-24; PMID:22751380; http://dx.doi.org/10.1177/1753425912451779
- Goodman SB, Gibon E, Pajarinen J, Lin TH, Keeney M, Ren PG, Nich C, Yao Z, Egashira K, Yang F, et al. Novel biological strategies for treatment of wear particle-induced periprosthetic osteolysis of orthopaedic implants for joint replacement. J R Soc Interface 2014; 11:20130962; PMID:24478281; http://dx.doi.org/10.1098/rsif.2013.0962
- Wooley PH, Schwarz EM. Aseptic loosening. Gene Ther 2004; 11:402-7; PMID:14724679; http://dx.doi.org/10.1038/sj.gt.3302202
- Nine M, Choudhury D, Hee A, Mootanah R, Osman N. Wear Debris Characterization and Corresponding Biological Response: Artificial Hip and Knee Joints. Materials 2014; 7:980-1016; http://dx.doi.org/10.3390/ma7020980
- Lochner K, Fritsche A, Jonitz A, Hansmann D, Mueller P, Mueller-Hilke B, Bader R. The potential role of human osteoblasts for periprosthetic osteolysis following exposure to wear particles. Int J Mol Med 2011; 28:1055-63; PMID:21850366
- O'Neill SC, Queally JM, Devitt BM, Doran PP, O'Byrne JM. The role of osteoblasts in peri-prosthetic osteolysis. Bone Joint J 2013; 95-B:1022-6
- Purdue PE, Koulouvaris P, Potter HG, Nestor BJ, Sculco TA. The cellular and molecular biology of periprosthetic osteolysis. Clin Orthop Relat Res 2007:251-61; PMID:16980902; http://dx.doi.org/10.1097/01.blo.0000238813.95035.1b
- Fritz EA, Glant TT, Vermes C, Jacobs JJ, Roebuck KA. Chemokine gene activation in human bone marrow-derived osteoblasts following exposure to particulate wear debris. J Biomed Mater Res A 2006; 77A:192-201; http://dx.doi.org/10.1002/jbm.a.30609
- Vermes C, Roebuck KA, Chandrasekaran R, Dobai JG, Jacobs JJ, Glant TT. Particulate wear debris activates protein tyrosine kinases and nuclear factor kappa Beta, which down-regulates type I collagen synthesis in human osteoblasts. J Bone Miner Res 2000; 15:1756-65; PMID:10976995; http://dx.doi.org/10.1359/jbmr.2000.15.9.1756
- Vermes C, Chandrasekaran R, Jacobs JJ, Galante JO, Roebuck KA, Glant TT. The effects of particulate wear debris, cytokines, and growth factors on the functions of MG-63 osteoblasts. J Bone Joint Surg Am 2001; 83A:201-11
- Yao JL, CsSzabo G, Jacobs JJ, Kuettner KE, Glant TT. Suppression of osteoblast function by titanium particles. J Bone Joint Surg Am 1997; 79A:107-12
- Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014; 15:81-94; PMID:24401948; http://dx.doi.org/10.1038/nrm3735
- Booth LA, Tavallai S, Hamed HA, Cruickshanks N, Dent P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal 2014; 26:549-55; PMID:24308968; http://dx.doi.org/10.1016/j.cellsig.2013.11.028
- Mukhopadhyay S, Panda PK, Sinha N, Das DN, Bhutia SK. Autophagy and apoptosis: where do they meet? Apoptosis 2014; 19:555-66; PMID:24415198; http://dx.doi.org/10.1007/s10495-014-0967-2
- Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol 2012; 9:20; PMID:22697169; http://dx.doi.org/10.1186/1743-8977-9-20
- Li C, Liu H, Sun Y, Wang H, Guo F, Rao S, Deng J, Zhang Y, Miao Y, Guo C, et al. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J Mol Cell Biol 2009; 1:37-45; PMID:19516051; http://dx.doi.org/10.1093/jmcb/mjp002
- Li H, Li Y, Jiao J, Hu H-M. Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat Nanotechnol 2011; 6:645-50; PMID:21926980; http://dx.doi.org/10.1038/nnano.2011.153
- Calzolai L, Franchini F, Gilliland D, Rossi F. Protein-Nanoparticle Interaction: Identification of the Ubiquitin-Gold Nanoparticle Interaction Site. Nano Lett 2010; 10:3101-5; PMID:20698623; http://dx.doi.org/10.1021/nl101746v
- Li JJ, Hartono D, Ong C-N, Bay B-H, Yung L-YL. Autophagy and oxidative stress associated with gold nanoparticles. Biomaterials 2010; 31:5996-6003; PMID:20466420; http://dx.doi.org/10.1016/j.biomaterials.2010.04.014
- Yu L, Lu Y, Man N, Yu S-H, Wen L-P. Rare Earth oxide nanocrystals induce autophagy in HeLa cells. Small 2009; 5:2784-7; PMID:19885892; http://dx.doi.org/10.1002/smll.200901714
- Peynshaert K, Manshian BB, Joris F, Braeckmans K, De Smedt SC, Demeester J, Soenen SJ. Exploiting intrinsic nanoparticle toxicity: The pros and cons of nanoparticle-induced autophagy in biomedical research. Chem Rev 2014; 114:7581-609; PMID:24927160; http://dx.doi.org/10.1021/cr400372p
- Doorn PF, Campbell PA, Worrall J, Benya PD, McKellop HA, Amstutz HC. Metal wear particle characterization from metal on metal total hip replacements: Transmission electron microscopy study of periprosthetic tissues and isolated particles. J Biomed Mater Res 1998; 42:103-11; PMID:9740012; http://dx.doi.org/10.1002/(SICI)1097-4636(199810)42:1<103::AID-JBM13>3.0.CO;2-M
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Bejarano E, Yuste A, Patel B, Stout RF, Jr., Spray DC, Cuervo AM. Connexins modulate autophagosome biogenesis. Nat Cell Biol 2014; 16:401-U55; PMID:24705551; http://dx.doi.org/10.1038/ncb2934
- Fukuda T, Ahearn M, Roberts A, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol Ther 2006; 14:831-9; PMID:17008131; http://dx.doi.org/10.1016/j.ymthe.2006.08.009
- Wang R, Wang Z, Ma Y, Liu G, Shi H, Chen J, Dong L, Zhao J, Zhang J. Particle-induced osteolysis mediated by endoplasmic reticulum stress in prosthesis loosening. Biomaterials 2013; 34:2611-23; PMID:23347837; http://dx.doi.org/10.1016/j.biomaterials.2013.01.025
- Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 2007; 14:1576-82; PMID:17612585; http://dx.doi.org/10.1038/sj.cdd.4402200
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304:1500-2; PMID:15131264; http://dx.doi.org/10.1126/science.1096645
- Byun J-Y, Yoon C-H, An S, Park I-C, Kang C-M, Kim M-J, Lee S-J. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis 2009; 30:1880-8; PMID:19783847; http://dx.doi.org/10.1093/carcin/bgp235
- Xu CY, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 2005; 115:2656-64; PMID:16200199; http://dx.doi.org/10.1172/JCI26373
- Landgraeber S, Quint U, Classen T, Totsch M. Senescence in cells in aseptic loosening after total hip replacement. Acta Biomater 2011; 7:1364-8; PMID:21094284; http://dx.doi.org/10.1016/j.actbio.2010.11.016
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931-7; PMID:17712358; http://dx.doi.org/10.1038/nrm2245
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726-34; PMID:16148885; http://dx.doi.org/10.1038/nrc1692
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon H-U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8:1124-32; PMID:16998475; http://dx.doi.org/10.1038/ncb1482
- Kaufman AM, Alabre CI, Rubash HE, Shanbhag AS. Human macrophage response to UHMWPE, TiAlV, CoCr, and alumina particles: Analysis of multiple cytokines using protein arrays. J Biomed Mater Res A 2008; 84A:464-74; http://dx.doi.org/10.1002/jbm.a.31467
- Lin TH, Tamaki Y, Pajarinen J, Waters HA, Woo DK, Yao Z, Goodman SB. Chronic inflammation in biomaterial-induced periprosthetic osteolysis: NF-kappaB as a therapeutic target. Acta Biomater 2014; 10:1-10; PMID:24090989; http://dx.doi.org/10.1016/j.actbio.2013.09.034
- Nich C, Takakubo Y, Pajarinen J, Ainola M, Salem A, Sillat T, Rao AJ, Raska M, Tamaki Y, Takagi M, et al. Macrophages-Key cells in the response to wear debris from joint replacements. J Biomed Mater Res A 2013; 101:3033-45; PMID:23568608; http://dx.doi.org/10.1002/jbm.a.34599
- Takei H, Pioletti DP, Kwon SY, Sung KLP. Combined effect of titanium particles and TNF-alpha on the production of IL-6 by osteoblast-like cells. J Biomed Mater Res 2000; 52:382-7; PMID:10951379; http://dx.doi.org/10.1002/1097-4636(200011)52:2<382::AID-JBM19>3.0.CO;2-V
- Fujii J, Niida S, Yasunaga Y, Yamasaki A, Ochi M. Wear debris stimulates bone-resorbing factor expression in the fibroblasts and osteoblasts. Hip Int 2011; 21:231-7; PMID:21484737; http://dx.doi.org/10.5301/HIP.2011.7977
- Queally JM, Devitt BM, Butler JS, Malizia AP, Murray D, Doran PP, O'Byrne JM. Cobalt ions induce chemokine secretion in primary human osteoblasts. J Orthop Res 2009; 27:855-64; PMID:19132727; http://dx.doi.org/10.1002/jor.20837
- Pioletti DP, Takei H, Kwon SY, Wood D, Sung KLP. The cytotoxic effect of titanium particles phagocytosed by osteoblasts. J Biomed Mater Res 1999; 46:399-407; PMID:10397998; http://dx.doi.org/10.1002/(SICI)1097-4636(19990905)46:3<399::AID-JBM13>3.0.CO;2-B
- Zhang Q, Yang W, Man N, Zheng F, Shen Y, Sun K, Li Y, Wen L-P. Autophagy-mediated chemosensitization in cancer cells by fullerene C60 nanocrystal. Autophagy 2009; 5:1107-17; PMID:19786831; http://dx.doi.org/10.4161/auto.5.8.9842
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2 alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230-9; PMID:16794605; http://dx.doi.org/10.1038/sj.cdd.4401984
- Ogata M, Hino S-i, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophapy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220-31; PMID:17030611; http://dx.doi.org/10.1128/MCB.01453-06
- Yu Y, Duan J, Yu Y, Li Y, Liu X, Zhou X, Ho K-f, Tian L, Sun Z. Silica nanoparticles induce autophagy and autophagic cell death in HepG2 cells triggered by reactive oxygen species. J Hazard Mater 2014; 270:176-86; PMID:24583672; http://dx.doi.org/10.1016/j.jhazmat.2014.01.028
- Zabirnyk O, Yezhelyev M, Seleverstov O. Nanoparticles as a novel class of autophagy activators. Autophagy 2007; 3:278-81; PMID:17351332; http://dx.doi.org/10.4161/auto.3916
- Hussain S, Al-Nsour F, Rice AB, Marshburn J, Yingling B, Ji Z, Zink JI, Walker NJ, Garantziotis S. Cerium dioxide nanoparticles induce apoptosis and autophagy in human peripheral blood monocytes. Acs Nano 2012; 6:5820-9; PMID:22717232; http://dx.doi.org/10.1021/nn302235u
- Afeseh Ngwa H, Kanthasamy A, Gu Y, Fang N, Anantharam V, Kanthasamy AG. Manganese nanoparticle activates mitochondrial dependent apoptotic signaling and autophagy in dopaminergic neuronal cells. Toxicol Appl Pharmacol 2011; 256:227-40; PMID:21856324; http://dx.doi.org/10.1016/j.taap.2011.07.018
- Liu HL, Zhang YL, Yang N, Zhang YX, Liu XQ, Li CG, Zhao Y, Wang YG, Zhang GG, Yang P, et al. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt-TSC2-mTOR signaling. Cell Death Dis 2011; 2:e159; PMID:21593791; http://dx.doi.org/10.1038/cddis.2011.27
- Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang JK, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 ctivation and apoptosis. J Biol Chem 2012; 287:12455-68; PMID:22362782; http://dx.doi.org/10.1074/jbc.M111.309104
- Oltvai ZN, Milliman CL, Korsmeyer SJ. BCL-2 Heterodimerizes in-vivo with a conserved homolog, bax, that accelerates programmed cell-death. Cell 1993; 74:609-19; PMID:8358790; http://dx.doi.org/10.1016/0092-8674(93)90509-O
- Rui S, Qin Z-H. The multiple roles of autophagy in neural function and disease in pathways to cures: Neurodegenerative diseases in China, S. Sanders, Z. Zhang, B. Tang, Eds. Science/AAAS 2013; 342:10.
- Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ 2010; 17:268-77; PMID:19713971; http://dx.doi.org/10.1038/cdd.2009.121
- Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ 2009; 16:1135-45; PMID:19300452; http://dx.doi.org/10.1038/cdd.2009.28
- Chaanine AH, Jeong D, Liang L, Chemaly ER, Fish K, Gordon RE, Hajjar RJ. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis 2012; 3:265; PMID:22297293; http://dx.doi.org/10.1038/cddis.2012.5
- Maiuri MC, Le Toumelin G, Criollo A, Rain J-C, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-X-L and a BH3-like domain in Beclin-1. EMBO J 2007; 26:2527-39; PMID:17446862; http://dx.doi.org/10.1038/sj.emboj.7601689
- Zhou H, Chen J, Lu X, Shen C, Zeng J, Chen L, Pei Z. Melatonin protects against rotenone-induced cell injury via inhibition of Omi and Bax-mediated autophagy in Hela cells. J Pineal Res 2012; 52:120-7; PMID:21883444; http://dx.doi.org/10.1111/j.1600-079X.2011.00926.x
- Yang Y-H, Li B, Zheng X-F, Chen J-W, Chen K, Jiang S-D, Jiang L-S. Oxidative damage to osteoblasts can be alleviated by early autophagy through the endoplasmic reticulum stress pathway-Implications for the treatment of osteoporosis. Free Radic Biol Med 2014; 77:10-20; PMID:25224042; http://dx.doi.org/10.1016/j.freeradbiomed.2014.08.028
- Wei M, Duan D, Liu Y, Wang Z, Li Z. Autophagy may protect MC3T3-E1 cells from fluoride-induced apoptosis. Mol Med Rep 2014; 9:2309-15; PMID:24682525
- Nollet M, Santucci-Darmanin S, Breuil V, Al-Sahlanee R, Cros C, Topi M, Momier D, Samson M, Pagnotta S, Cailleteau L, et al. Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 2014; 10:1965-77; PMID:25484092; http://dx.doi.org/10.4161/auto.36182
- Onal M, Piemontese M, Xiong J, Wang Y, Han L, Ye S, Komatsu M, Selig M, Weinstein RS, Zhao H, et al. Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem 2013; 288:17432-40; PMID:23645674; http://dx.doi.org/10.1074/jbc.M112.444190
- DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, Klumperman J, Tooze SA, Teitelbaum SL, Virgin HW. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell 2011; 21:966-74; PMID:22055344; http://dx.doi.org/10.1016/j.devcel.2011.08.016
- Oberdorster G, Oberdorster E, Oberdorster J. Nanotoxicology: An emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect 2005; 113:823-39; PMID:16002369; http://dx.doi.org/10.1289/ehp.7339
- Balgi AD, Fonseca BD, Donohue E, Tsang TCF, Lajoie P, Proud CG, Nabi IR, Roberge M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. Plos One 2009; 4; PMID:19771169; http://dx.doi.org/10.1371/journal.pone.0007124
- Hocking LJ, Whitehouse C, Helfrich MH. Autophagy: a new player in skeletal maintenance? J Bone Miner Res 2012; 27:1439-47; PMID:22706899; http://dx.doi.org/10.1002/jbmr.1668
- Sambandam Y, Townsend MT, Pierce JJ, Lipman CM, Haque A, Bateman TA, Reddy SV. Microgravity control of autophagy modulates osteoclastogenesis. Bone 2014; 61:125-31; PMID:24463210; http://dx.doi.org/10.1016/j.bone.2014.01.004
- Zhao Y, Chen G, Zhang W, Xu N, Zhu J-Y, Jia J, Sun Z-J, Wang Y-N, Zhao Y-F. Autophagy regulates hypoxia-induced osteoclastogenesis through the HIF-1 alpha/BNIP3 signaling pathway. J Cell Physiol 2012; 227:639-48; PMID:21465467; http://dx.doi.org/10.1002/jcp.22768
- Sanchez CP, He Y-Z. Bone growth during rapamycin therapy in young rats. BMC Pediatr 2009; 9; PMID:19144108; http://dx.doi.org/10.1186/1471-2431-9-3
- Cejka D, Hayer S, Niederreiter B, Sieghart W, Fuereder T, Zwerina J, Schett G. Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis Rheum 2010; 62:2294-302; PMID:20506288; http://dx.doi.org/10.1002/art.27504
- Liu X, Qu X, Wu C, Zhai Z, Tian B, Li H, Ouyang Z, Xu X, Wang W, Fan Q, et al. The effect of enoxacin on osteoclastogenesis and reduction of titanium particle-induced osteolysis via suppression of JNK signaling pathway. Biomaterials 2014; 35:5721-30; PMID:24767789; http://dx.doi.org/10.1016/j.biomaterials.2014.04.006
- Xue Y, Xiao Y, Liu J, Karaplis AC, Pollak MR, Brown EM, Miao D, Goltzman D. The calcium-sensing receptor complements parathyroid hormone-induced bone turnover in discrete skeletal compartments in mice. Am J Physiol Endocrinol Metab 2012; 302:E841-E51; PMID:22275754; http://dx.doi.org/10.1152/ajpendo.00599.2011
- Yang SY, Kim NH, Cho YS, Lee H, Kwon HJ. Convallatoxin, a dual inducer of autophagy and apoptosis, inhibits angiogenesis in vitro and in vivo. Plos One 2014; 9