
Abstract
Autophagy is a self-degradative process responsible for the clearance of damaged or unnecessary cellular components. We have previously found that persistence of dysfunctional organelles due to autophagy failure is a key event in the pathogenesis of COL6/collagen VI-related myopathies, and have demonstrated that reactivation of a proper autophagic flux rescues the muscle defects of Col6a1-null (col6a1−/−) mice. Here we show that treatment with spermidine, a naturally occurring nontoxic autophagy inducer, is beneficial for col6a1−/− mice. Systemic administration of spermidine in col6a1−/− mice reactivated autophagy in a dose-dependent manner, leading to a concurrent amelioration of the histological and ultrastructural muscle defects. The beneficial effects of spermidine, together with its being easy to administer and the lack of overt side effects, open the field for the design of novel nutraceutical strategies for the treatment of muscle diseases characterized by autophagy impairment.
Abbreviations
| BM | = | Bethlem myopathy |
| i.p. | = | intraperitoneal |
| Spd | = | spermidine |
| UCMD | = | Ullrich congenital muscular dystrophy |
Introduction
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved self-degradative process allowing cells and tissues to cope with several adverse stress conditions.Citation1-3 Autophagy is constantly active at low rates in the whole organism, but various stress or pharmacological stimuli can induce its flux.Citation1,3 In physiological conditions, autophagy plays a crucial role during development, cell differentiation and tissue remodeling, acts as a cell housekeeper by degrading damaged or unnecessary organelles, and allows the retrieval of metabolites under nutrient starvation.Citation3,4 Age-related degeneration processes and neurodegenerative disorders, as well as liver, heart and muscle diseases, are only some of the pathological conditions where deregulated autophagy contributes to the pathophysiological defects, thus confirming the relevance of the autophagy-lysosome system for the homeostasis of organs and tissues.Citation3,5
Skeletal muscle is an extremely plastic tissue, able to undergo extensive changes in response to physical exercise, dietary restriction and metabolic dysregulation.Citation6 In this tissue, autophagy is essential for providing energy and eliminating harmful exhausted organelles.Citation6,7 However, being a degradation process, autophagy needs to be finely regulated to maintain muscle mass and to ensure a proper removal of damaged organelles or protein aggregates. Indeed, alterations of the autophagic flux affect muscle homeostasis and also impinge on the body metabolic state.Citation7 COL6/collagen VI-related myopathies represent a notable group of inherited muscle disorders where a link between dystrophic phenotype and autophagy flux impairment was first demonstrated. The key contribution of defective autophagy in the pathogenesis of muscle defects for these diseases is provided by studies in Col6a1-null (col6a1−/−) mice, in which abnormal signaling causes persistent AKT (v-akt murine thymoma viral oncogene homolog)-MTOR pathway activation and defective autophagosome formation in muscle fibers.Citation8 Further studies in muscle biopsies of patients affected by Ullrich congenital muscular dystrophy (UCMD) and Bethem myopathy (BM), the 2 major human pathologies linked to mutations of COL6-encoding genes, confirm defective regulation of autophagy.Citation8 Thereafter, defective autophagy induction has been reported in murine models for Duchenne muscular dystrophy and Emery-Dreyfuss muscular dystrophy, whereas increased autophagy flux contributes to the phenotype of dy3K/dy3K mice, the animal model of MDC1A congenital muscular dystrophy.Citation5 Notably, the autophagic defects are reversible and rescue of a proper autophagic flux by either genetic, pharmacological or dietary approaches leads to a strong amelioration of the muscle defects, a finding which led to a pilot clinical trial aimed at restoring autophagy in UCMD and BM patients through a low-protein diet approach (ClinicalTrials.gov Identifier NCT01438788).
Spermidine is a cationic polyamine naturally present in living cells at concentrations that decrease with aging. Spermidine supplementation to cell culture media or in vivo administration to entire animals prolongs life span in different model organisms, an effect which is linked to autophagy induction.Citation9,10 Although the mechanisms by which spermidine acts are not yet fully understood, it has been shown that they involve a complex combination of acetylation and deacetylation events whose occurrence determines a shift in the acetylation pattern of the cell proteome without altering the total amount of acetylated proteins.Citation9 Recently, the histone acetyltransferase EP300 has been described as the major target on which spermidine exerts an inhibitory activity, determining autophagy induction in vitro.Citation11 Since in our previous studies we have found that reactivation of autophagy by either prolonged starvation or a low-protein diet is capable of rescuing the muscle pathology of col6a1−/− mice,Citation8 we evaluated in this myopathic model the efficacy of nutraceutical approaches able to forcibly induce autophagy. Here we show that spermidine administration by either addition to drinking water or intraperitoneal (i.p.) injection leads to a significant amelioration of the phenotype of col6a1−/− mice. The ability to reactivate autophagy in COL6-deficient muscles, coupled with its low level of toxicity, points at spermidine as a nutraceutical with potential benefits for the treatment of these muscle diseases.
Results
Spermidine reactivates autophagy in col6a1−/− muscles
To evaluate the therapeutic potential of spermidine in COL6-related muscle pathologies, we investigated wild-type and col6a1−/− mice subjected to spermidine administration at different doses, durations and routes of administration. Spermidine was administered either by oral administration (per os), by addition to drinking water for 30 d, or i.p., by daily injection for 10 d. In both cases, 2 different doses were given (3 or 30 mM, per os; 5 or 50 mg/kg body weight, i.p.), in order to evaluate dose-dependent effects (Table S1).
Per os administration of spermidine at both 3 mM and 30 mM was effective in stimulating autophagy in skeletal muscles. Fluorescence microscope analysis of tibialis anterior showed several GFP-LC3 puncta in wild-type animals after 30 d spermidine treatment (). Autophagy induction was further confirmed by immunoblotting analysis of cleavage and lipidation of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β), quantified both as LC3B-II/-I and LC3B-II/GAPDH ratios, which was increased at both low- and high-dose spermidine treatment (; Fig. S1A and B). Of note, these treatments did not exert any overt change in the response to 24 h starvation in wild-type mice (; Fig. S1A and B). In agreement with previous work,Citation8 col6a1−/− tibialis anterior displayed a markedly decreased autophagy in the 24-h fasting condition, when compared to the corresponding wild-type muscles. Interestingly, spermidine administration induced autophagy in col6a1−/− muscles. Indeed, fluorescence microscopy showed the presence of GFP-LC3 puncta in tibialis anterior of spermidine-treated col6a1−/− animals, which were clearly evident in mice treated with 30 mM spermidine and subjected to 24-h starvation (). Immunoblotting analysis of tibialis anterior extracts confirmed a significant increase of LC3B-II/-I and LC3B-II/GAPDH ratios in fasted col6a1−/− mice treated with 30 mM spermidine, when compared to fasted col6a1−/− mice without spermidine, whereas no significant change in LC3B-II/-I was detected in col6a1−/− mice maintained under fed conditions or treated with the lower dose of spermidine (; Fig. S2A and B). Interestingly, the LC3B-II/GAPDH ratio was instead significantly increased in fed col6a1−/− mice following a low dose per os treatment (Fig. S1A). In order to further clarify the status of the autophagy machinery in the skeletal muscle of fed wild-type and col6a1−/− mice subjected to per os spermidine treatment at different doses, we analyzed by immunoblotting the protein levels of RAB7, a small Ras-like GTPase with an essential role in lysosomal biogenesis and in the late phases of endosome and autophagosome fusion with lysosomes,Citation12-13 and GABARAP, a yeast Atg8 ortholog playing a key role in the final stages of autophagosome biogenesis.Citation14 Both the lipidated form of GABARAP (GABARAP-II) and RAB7 were increased in a dose-dependent fashion following spermidine treatment not only in wild-type but also in col6a1−/− mice. These data indicate an increased rate of autophagosome and autolysosome formation, respectively, and are consistent with autophagy induction in fed animals of both genotypes (Fig. S2A-C). Altogether, these findings indicate that spermidine supplementation in drinking water for 30 d, promptly induces autophagy in wild-type muscles and also affects the autophagic process of col6a1−/− muscles.
Figure 1 (see previous page). Spermidine induces autophagy in wild-type and COL6-deficient tibialis anterior muscles. (A) Detection of fluorescent puncta in cross sections of tibialis anterior muscles from untreated (Ctrl) and per os spermidine-treated (Spd PO) wild-type and col6a1−/− mice bearing a GFP-LC3 reporter construct. Mice were analyzed under fed conditions or after 24 h fasting. White arrowheads point at some GFP-LC3-positive puncta. Nuclei were stained with Hoechst. Scale bar: 25 μm. (B, C) Western blot analysis of LC3B lipidation in protein extracts from tibialis anterior muscles of untreated (–) and per os spermidine-treated (+) wild-type and col6a1−/− mice. Densitometric quantifications of LC3B-II vs LC3B-I ratio, as determined by at least 3 independent western blot experiments, are shown in the lower panels (n = 3 or 4; *, P< 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant). (D) Detection of fluorescent puncta in cross sections of tibialis anterior muscles from untreated (Ctrl) and i.p. spermidine-treated (Spd IP) wild-type and col6a1−/− mice. Mice were analyzed under fed conditions or after 24 h fasting. White arrowheads point at some GFP-LC3-positive puncta. Nuclei were stained with Hoechst. Scale bar: 25 μm. (E, F) Western blot analysis of LC3B lipidation in protein extracts from tibialis anterior muscles of untreated (–) and i.p. spermidine-treated (+) wild-type and col6a1−/− mice. Densitometric quantifications of LC3B-II vs LC3B-I ratio, as determined by at least 3 independent western blot experiments, are shown in the lower panels (n = 3 or 4; *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant). 24 h, twenty-four hour-long starvation; Ctrl, untreated control condition; Spd IP, spermidine i.p.; Spd PO, spermidine per os; WT, wild-type.
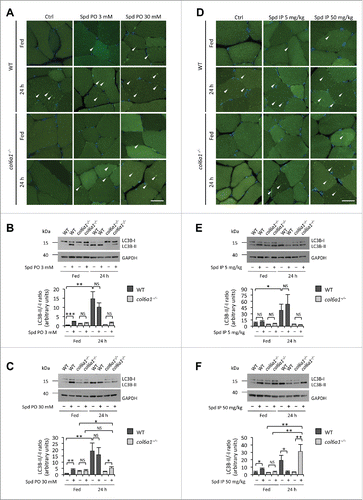
In order to evaluate more precisely the effects of spermidine in inducing autophagy in skeletal muscles, we subjected wild-type and col6a1−/− mice to daily i.p. injection with spermidine for 10 d (Table S1). Spermidine i.p. treatment at 5 mg/kg displayed no significant effects on fluorescent GFP-LC3 puncta and LC3B lipidation, both in wild-type and col6a1−/− tibialis anterior (; Fig. S1C). Slightly increased protein levels of GABARAP-II, but not RAB7, were detected in wild-type and col6a1−/− tibialis anterior (Fig. S2D-F), further confirming that this dosage is unable to elicit a robust upregulation of autophagy. Conversely, spermidine i.p. treatment at 50 mg/kg triggered not only GFP-LC3 puncta formation and LC3B lipidation (; Fig. S1D), but also a very strong increase in RAB7 and GABARAP-II protein levels (Fig. S2D-G and I) in muscles of wild-type mice maintained under fed conditions. Autophagy induction in this condition was further confirmed by the strong accumulation of the autophagosome markers LC3B-II and SQSTM1 (sequestosome 1) in tibialis anterior of wild-type mice treated simultaneously with spermidine i.p. 50 mg/kg and colchicine (Fig. S2G-K), an inhibitor of the late stages of autophagosome maturation and fusion with lysosome that allows investigating the autophagic flux in skeletal muscle.Citation15 Consistently with the role of RAB7 in the intracellular vesicle trafficking and in the fusion of autophagosomes with lysosomes, this protein displayed unaltered levels in spermidine plus colchicine-treated animals with respect to untreated control conditions (Fig. S2G-I). Unexpectedly, i.p. treatment with spermidine at 50 mg/kg combined with 24 h starvation led to decreased LC3B lipidation in wild-type muscles although autophagic vacuoles were clearly detectable (; Fig. S1D). This latter finding may be explained by the intensive and prolonged autophagy induction in this condition, which would lead to the consumption of LC3B protein via autophagosome-lysosome fusion and degradation.
Notably, treatment with spermidine at 50 mg/kg for 10 d led to an overt induction of autophagy in tibialis anterior of 24-h-fasted col6a1−/− mice, as indicated by the presence of a large number of GFP-LC3 puncta and by the conspicuous increase in LC3B lipidation (; Fig. S1C). In fed conditions, autophagy appeared similarly induced in muscles from col6a1−/− mice subjected to i.p. treatment with spermidine at 50 mg/kg, based on the increased presence of GFP-LC3 puncta () and on the higher levels of RAB7 and GABARAP-II (Fig. S2D to F). Moreover, combined treatment with colchicine and i.p. 50 mg/kg spermidine led to a significant accumulation of SQSTM1 and the lipidated form of LC3B in tibialis anterior of col6a1−/− mice. Of note, differently from what we observed for wild-type mice, colchicine treatment alone did not determine an accumulation of any of these autophagic markers in muscles from col6a1−/− mice (Fig. S2G-K). This evidence further confirms the autophagy impairment of COL6-deficient muscles, and represents an unambiguous proof of the concept that spermidine is capable of reactivating autophagy in muscles of col6a1−/− mice.
Altogether, these data indicate that spermidine is able to induce autophagy in skeletal muscle in a dose-dependent manner and is also effective in reactivating autophagy in col6a1−/− muscles.
Spermidine supplementation recovers the histological and ultrastructural defects of col6a1−/− muscles
To assess whether spermidine administration has any effect on the structural defects of COL6-deficient myopathic mice, we performed histological analysis of tibialis anterior muscles from wild-type and col6a1−/− mice subjected to the different protocols of spermidine treatment. In wild-type mice, treatment with spermidine did not cause any sign of histological changes in muscle architecture at the tested doses and routes of administration (). As expected,Citation16 under untreated conditions col6a1−/− tibialis anterior displayed an overt myopathic phenotype, with centrally nucleated myofibers, small atrophic fibers and giant hypertrophic ones. Notably, both i.p. and per os spermidine treatments led to a noticeable amelioration of the myopathic defects. The effects of spermidine seemed to correlate with the dose, as col6a1−/− mice treated with the high doses displayed improved muscle histology (). Indeed, 3 mM per os and 5 mg/kg i.p. spermidine treatments led to a partial rescue of histological alterations in col6a1−/− mice and, albeit less extended, the myopathic defects were still present, with some atrophic and degenerating myofibers (). However, col6a1−/− animals displayed markedly improved muscle morphology both after 30 mM per os and 50 mg/kg i.p. spermidine treatment, with barely detectable signs of myofiber degeneration, more homogeneous myofiber size and rare atrophic fibers (). Some centrally nucleated fibers were present in treated animals, consistent with myofiber regeneration ().
Figure 2 (see previous page). Spermidine treatment ameliorates the myopathic phenotype of col6a1−/− mice. (A, B) Haematoxylin-eosin staining of cross sections of tibialis anterior muscles from untreated (Ctrl) and per os (A) or i.p. (B) spermidine-treated wild-type and col6a1−/− mice. In col6a1−/− mice, centrally nucleated fibers (black arrowheads) and atrophic fibers (white arrowheads) appear reduced after spermidine treatment. Scale bar: 50 μm. (C) Representative electron micrographs of diaphragm thin sections from untreated (a to c) and per os (d to f) or i.p. (g to i) spermidine-treated col6a1−/− mice. Low-power views (upper panels) reveal improvement of muscle ultrastructure following spermidine treatment. High-power magnifications (lower panels) show dilated sarcoplasmic reticulum (SR) and swollen mitochondria (mit) in untreated col6a1−/− mice (b,c), and the presence of autophagic vacuoles (AV) in spermidine-treated col6a1−/− mice (f,i) together with a marked amelioration of organelle morphology (e,h). Scale bar: 1 μm (upper panels) or 500 nm (lower panels). (D) Percentage of morphologically altered mitochondria in the diaphragm of untreated (Ctrl) and spermidine-treated (PO 30, per os 30 mM; IP 50, i.p. 50 mg/kg) col6a1−/− mice (n = 3 or 4; P< 0.05; NS, not significant.) (E) Quantification of apoptosis by TUNEL assay in diaphragm muscles of untreated (Ctrl) and per os (left panel) or i.p. (right panel) spermidine-treated wild-type and col6a1−/− mice (n = 3, each group; **, P < 0.01; ***, P < 0.001). (F) In vivo tetanic force measurements in gastrocnemius muscle of untreated (Ctrl) and per os or i.p. spermidine-treated wild-type and col6a1−/− mice. The histograms report the normalized strength at a stimulation frequency of 100 Hz (n = 5, each group; ***, P<0.001; NS, not significant). Ctrl, untreated control condition; Spd IP, spermidine i.p.; Spd PO, spermidine per os. WT, wild-type.
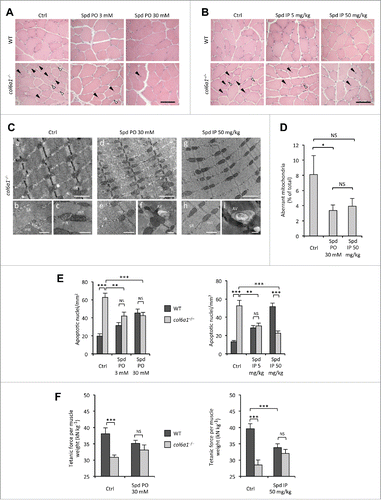
A well-known feature of col6a1−/− muscles is the presence of altered organelles, in particular swollen mitochondria and dilated sarcoplasmic reticulum.Citation8,17,18 Histological staining for SDH/succinate dehydrogenase, a mitochondrial marker, showed a more homogeneous labeling, pointing at an amelioration of the mitochondrial network in tibialis anterior of col6a1−/− animals treated with higher spermidine doses (Fig. S3). Transmission electron microscopy of diaphragm ultrathin sections revealed a marked amelioration of sarcoplasmic reticulum and mitochondria ultrastructure in myofibers of spermidine-treated col6a1−/− mice (). Morphometric analysis confirmed a decreased percentage of aberrant mitochondria in muscles of spermidine-treated col6a1−/− animals, which was significant after 30 mM per os treatment (P = 0.039) whereas it almost reached significance after 50 mg/kg intraperitoneal treatment (P = 0.077) (). Furthermore, electron microscopy confirmed the presence of several autophagic vacuoles in spermidine-treated col6a1−/− animals (), consistently with the elimination of altered mitochondria through spermidine-induced autophagy.
Altogether, these data indicate that spermidine-mediated reactivation of autophagy ameliorates the myopathic defects of col6a1−/− animals, with a significant decrease of swollen mitochondria and a marked improvement of muscle structure.
Spermidine ameliorates the functional deficits of col6a1−/− muscles
Given the noticeable improvement of muscle structural defects, we next investigated whether the tested doses of spermidine were also capable of ameliorating the deterioration in other muscle features. A well-established feature of col6a1−/− mice and of UCMD and BM patients is the increased incidence of apoptotic myofibers in skeletal muscles.Citation8,17 TUNEL analysis for diaphragm revealed that spermidine administration elicited a strongly decreased incidence of apoptotic myofibers in col6a1−/− mice. Indeed, both per os and i.p. treatments led to a significant decrease in the number of apoptotic myonuclei in col6a1−/− diaphragms, and this effect was displayed at all the tested doses of spermidine (). The decrease appeared stronger after i.p. treatment, where the effect positively correlated with the dose. However, per os treatment also displayed a clear effect, with about 35% decrease in the number of apoptotic myonuclei at both low and high doses (). Conversely, wild-type animals displayed an increased number of TUNEL-positive myonuclei directly correlating with the dose both after i.p. and per os treatment (), likely due to the excessive induction of autophagy elicited by spermidine in wild-type muscles.
We have previously demonstrated that under physiological conditions col6a1−/− mice develop significant less strength than wild-type mice.Citation8 Therefore, we performed in vivo tetanic force measurements on gastrocnemius muscles of wild-type and col6a1−/− mice under untreated conditions and following spermidine administration. Both per os and i.p. spermidine treatment led to a positive trend in normalized force in col6a1−/− mice (Fig. S4), although the values obtained at a tetanic stimulation frequency of 100 Hz did not reach a statistically significant difference when compared to untreated animals (P = 0.138, per os treatment; P = 0.163, i.p. treatment). Of note, while the difference in the normalized force developed by wild-type and col6a1−/− mice under standard conditions was highly significant, animals of the 2 genotypes displayed similar normalized force values after spermidine treatment (; Fig. S4). As for apoptosis, high-dose i.p. spermidine administration caused a force decline in wild-type mice, likely due to the excessive autophagy induction elicited in these animals (; Fig. S4B) as we previously observed in wild-type mice subjected to low-protein diet.Citation8
Altogether, these data indicate that spermidine induction of autophagy recovers the apoptotic defects of col6a1−/− animals, with also a positive trend in their muscle strength.
Spermidine effects involve modulation of the AKT signaling pathway
Spermidine induces autophagy through modifications at the level of the acetylproteome, thus modifying the expression levels of several genes.Citation9,10 In previous studies aimed at mechanistic insight into the autophagic defects of COL6-related myopathies, we have demonstrated that col6a1−/− muscles display a hyperactivation of the AKT-MTOR pathway and that this signaling defect plays a key role in the autophagy impairment.Citation8 AKT kinase negatively regulates the activity of FOXO (forkhead box O) transcription factors by phosphorylation, and decreased levels of phosphorylated AKT represent a permissive condition for FOXO activation.Citation19,20 To understand whether the autophagy-inducing effects of spermidine involve the AKT-FOXO axis, we investigated the activation state of AKT and the mRNA levels of several FOXO target genes in tibialis anterior of wild-type and col6a1−/− mice subjected to i.p. treatment with spermidine at 50 mg/kg for 10 d. Chronic spermidine administration led to decreased AKT phosphorylation, an effect which was particularly strong in col6a1−/− mice subjected to 24 h starvation (). Real time PCR for Bnip3 (BCL2/adenovirus E1B 19kDa interacting protein 3), Ctsl (cathepsin L), and Map1lc3b, 3 autophagy-related genes regulated by the AKT-FOXO axis,Citation19 showed significantly increased levels of their transcript in spermidine-treated, 24-h-fasted col6a1−/− mice (). These results indicate that spermidine elicits AKT dephosphorylation, which in turn triggers the translocation of FOXO transcription factors into the nucleus with the consequent increased transcription of FOXO target genes, including autophagy-related genes.
Figure 3. Spermidine modulates the AKT-FOXO axis in skeletal muscle. (A) Western blot analysis of AKT phosphorylation in protein extracts from tibialis anterior muscles of untreated (–) and i.p. spermidine-treated (+) wild-type and col6a1−/− mice. Densitometric quantifications of P-AKT vs AKT, as determined by 3 independent western blot experiments, are shown on the right panel (n = 3; **, P< 0.01; ***, P < 0.001). (B–E) Real-time RT-PCR analysis of mRNA levels for Bnip3 (B), Ctsl (C) and Map1lc3b (E) in tibialis anterior muscles of untreated (–) and i.p. spermidine-treated (+) wild-type and col6a1−/− mice (n = 3 or 4; ***, P < 0.001; ** P < 0.01; *, P < 0.05; NS, not significant). 24 h, twenty-four hour-long starvation; Spd IP, spermidine i.p.; Spd PO, spermidine per os. WT, wild-type.
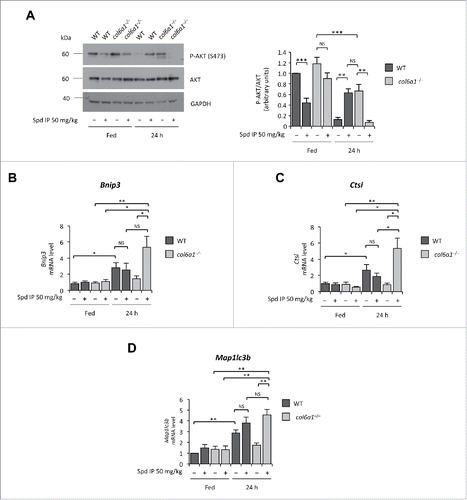
Of note, high-dose spermidine i.p. treatment led to decreased AKT phosphorylation levels in tibialis anterior of wild-type mice even under fed conditions. Yet, AKT phosphorylation was restored when spermidine treatment was followed by 24-h starvation (). This apparently paradoxical finding likely relies upon the stimulatory activity of several FOXO target genes, such as Sesn3 (sestrin 3) and Irs1 (insulin receptor substrate 1), on AKT. In turn, active AKT is known to upregulate protein synthesis and to inhibit FOXO activity, as a part of a feedback mechanism that maintains the balance between protein synthesis and protein degradation inside the cells.Citation21 In agreement with this concept, tibialis anterior of wild-type mice subjected to prolonged (48 h) fasting displayed significantly higher levels of phosphorylated AKT than those of 24-h-fasted wild-type animals (Fig. S5A). At the same time, expression of FOXO target genes was maintained at high levels in these muscles, as found for spermidine-treated and 24-h-fasted wild-type mice (; Fig. S5B-D). Therefore, the excessive and prolonged stimulus for autophagy induction elicited in wild-type muscles by high-dose spermidine administration in combination with fasting induces a feedback mechanism that restores AKT phosphorylation.
Altogether, these data indicate that spermidine affects AKT activation and enhances autophagy by promoting the transcription of FOXO target genes.
Discussion
Spermidine is a nontoxic autophagy-inducing natural compound, with an increasing interest for its applications as a nutraceutical tool for therapies. Spermidine counteracts aging and promotes longevity in different species by increasing the basal autophagy levels.Citation9,10,Citation22 Recent work suggests that this polyamine may be beneficial for counteracting several age-related pathologies, such as arterial aging and memory impairment.Citation23-24 Moreover, spermidine shows a rapamycin-like effect in promoting the removal of brain protein aggregates and ameliorating the phenotype of a mouse model for TDP-43 proteinopathies, suggesting that it may be a useful route for the therapy of neurodegenerative diseases such as frontotemporal lobar dementia.Citation25 Published studies demonstrate the efficacy of spermidine in different models, but no study has ever investigated the effects of this autophagy-inducing compound in muscle diseases.
In this work, we found that spermidine is effective in reactivating autophagy in skeletal muscle and ameliorating the myopathic defects of col6a1−/− mice, the animal model of UCMD and BM, thus showing for the first time the efficacy of this nutraceutical in a genetically determined muscle disease model. The beneficial effects of spermidine administration in col6a1−/− mice are associated with its ability to reactivate the autophagic flux, a process which is overtly impaired in this animal model as well as in the corresponding human myopathies.Citation8 Indeed, we have previously demonstrated that the muscle defects of col6a1−/− mice are strictly linked to a failure to maintain a proper autophagic flux in myofibers.Citation8 Inefficient autophagy in Col6a1-null muscles leads to the progressive accumulation of harmful organelles inside myofibers, which progressively degenerate. However, this autophagy failure is reversible and reactivation of a proper autophagic flux in col6a1−/− mice, through dietary and pharmacological approaches, leads to a substantial rescue of the myopathic pathology.Citation5-8 Here, we investigated the efficacy of spermidine in this well-established myopathic model at different doses and different routes of administration. We observed that high-dose i.p. treatment is very effective in reactivating autophagy, with a significant increase of LC3B lipidation and autophagosome formation. The effects of spermidine on autophagy reactivation in col6a1−/− muscles were reinforced by 24-h starvation. Even under the fed condition, autophagy flux was induced in spermidine-treated col6a1−/− mice, as demonstrated by increased levels of RAB7 and GABARAP-II proteins and by accumulation of LC3-II and SQSTM1 in the presence of colchicine cotreatment, and this was nicely paralleled by a conspicuous amelioration of the histology of tibialis anterior.
BM and UCMD patients suffer from respiratory insufficiency due to a loss of functionality of diaphragm,Citation18 which is also the most affected muscle in col6a1−/− mice where it displays marked ultrastructural defects and high incidence of apoptotic myofibers.Citation16 Both these aspects were rescued by spermidine. High-dose i.p. treatment showed a remarkable effect on the number of TUNEL-positive nuclei in col6a1−/− animals, bringing it to similar levels to wild-type control mice. Moreover, ultrastructural analysis showed a marked decrease in the percentage of aberrant mitochondria and several autophagosomes were observed in the proximity of mitochondria in myofibers of col6a1−/− mice subjected to high-dose i.p. spermidine treatment. Although it is currently not technically feasible to identify the content of these autophagic vacuoles, the major evidence supporting the efficacy of spermidine in inducing autophagy in col6a1−/− muscles is that, at the difference from untreated conditions,Citation8 autophagosomes can be detected in diaphragm of col6a1−/− mice following spermidine treatment. It is plausible that removal of altered mitochondria via spermidine-induced autophagy decreases the levels of proapoptotic factors released by these organelles, as we previously have observed in col6a1−/− mice subjected to prolonged starvation or low-protein diet.Citation8,26 In addition to this, the antiapoptotic effect of spermidine may also depend on a direct influence of this polyamine on the regulation of programmed cell death or on the crosstalk between the regulatory networks of these 2 processes.Citation26,27
Conversely, in wild-type mice, high-dose spermidine administration had some adverse effects, which however were expected. Autophagy induction in spermidine-treated wild-type muscles was promptly detectable even in fed conditions, and it is well known that excessively induced autophagy is detrimental for myofiber homeostasis.Citation8,28 The most detrimental known effect of spermidine is due to its capability to inhibit NOS1 (nitric oxid synthase 1 [neuronal]), but pharmacological studies show that the LD50 of intraperitoneally injected spermidine is 620 mg/kg,Citation29 more than 10 times higher than the highest dose used in the present study. Although we did not observe any overt sign of histological muscle changes in wild-type animals treated with high-dose spermidine, they displayed decreased muscle strength and an increased number of TUNEL-positive myonuclei. These features are typical in skeletal muscle when the autophagy flux is too elevated,Citation8,28 but they may also rely on an action of spermidine on multiple aspects of muscle cell biology. In fact, polyamines are able to induce or inhibit programmed cell death based on the environmental conditions and the cell type examined, through direct or indirect effects on molecules or signaling pathways.Citation26 It would be interesting to investigate in the future the effect of spermidine on muscle contractile functions. It is possible, for example, that this effect is correlated at least in part with the modulation of ion channels activity by polyamines.Citation30
Among the broad range of intracellular targets for spermidine,Citation9-11,Citation30 which may mediate its beneficial effects in col6a1−/− muscles, we focused on the AKT-MTOR axis. We previously demonstrated that the defective autophagy of col6a1−/− muscles is characterized by excessive AKT phosphorylation even after a typical proautophagic stimulus, such as 24-h starvation.Citation8 This persistent AKT activity causes a constitutive activation of the MTOR pathway and prevents the transcription of several autophagy related genes under the control of FOXO transcription factors.Citation8,19 Our data indicate that high-dose i.p. spermidine treatment is capable of normalizing AKT phosphorylation in col6a1−/− tibialis anterior leading to increased expression of FOXO target genes upon starvation, which is sufficient to induce autophagy in skeletal muscle.Citation19 Interestingly, chronically-administered spermidine displayed a robust effect on AKT activity in skeletal muscle. In muscles of wild-type animals, where the AKT pathway is properly working, chronically-administered spermidine led to a significant reduction of AKT phosphorylation under fed conditions, while upon nutrient deprivation AKT phosphorylation was reinstated, an effect that we also observed in wild-type animals subjected to prolonged starvation. This apparently paradoxical effect is likely due to a feedback mechanism, as several transcriptional targets of FOXO are able to upregulate the PI3K-AKT pathway by triggering an increase of the active phosphorylated form of AKT.Citation21 In col6a1−/− animals AKT rephosphorylation is not taking place, and combination of spermidine plus 24 h starvation bring col6a1−/− muscles to the AKT state observed in muscles of wild-type mice subjected to 24 h starvation but without drug treatment. FOXO transcription factors are regulated by different post-translational modifications, and among these inhibitory acetylation by the histone acetyltransferase EP300 has been thoroughly described.Citation31 As spermidine also acts as a histone acetyltransferase inhibitor, and one of its known targets is EP300,Citation11 it is plausible that spermidine controls FOXO activity at multiple levels and determines a permissive condition for its activity by decreasing both its phosphorylation by AKT and its acetylation by EP300.
An interesting finding that emerges from our data is that per os spermidine administration reactivates autophagy and in parallel has beneficial effects on the phenotype of col6a1−/− mice. Even if thus far only proven in the Col6a1-null animal model, these data are of major interest for prospective therapies as they mean that it is possible to deliver a biologically active concentration of this nutraceutical polyamine in myopathic muscles by oral administration. Although beyond the aim of the present work, it will be interesting in the near future to carry out further analysis, including detailed pharmacokinetic studies of spermidine following oral administration, aimed at devising the optimal regimens able to provide the most effective and enduring beneficial outcomes in the different muscles. For example, it would be interesting to treat animals per os with the lowest effective doses of spermidine starting from weaning and throughout their entire life. Furthermore, synergic approaches could be tested using different combinations of spermidine with other nutritional or pharmacological approaches shown to induce autophagy, such as the low-protein diet. Based on our results with spermidine and acute starvation, it is likely that these approaches will show additive effects. A major advantage of spermidine is that it is a natural compound, present at high concentrations in foods such as mushrooms or soybean, and that it can be easily administered.Citation32
In conclusion, our data in col6a1−/− animals point at spermidine as a nutraceutical therapeutic option for COL6-related myopathies and indicate that this approach is valuable for the design of novel treatments, with spermidine alone or in combination with other strategies, and their translation into clinical trials. In perspective, this nutraceutical-based, autophagy-inducing approach could be applied to UCMD and BM therapy, as it has been done for the low-protein diet (ClinicalTrials.gov Identifier NCT01438788), and could also be extended to other inherited muscle diseases involving a defective activity of the autophagy machinery.
Materials and Methods
Animals
C57BL/6N mice were obtained from Charles River. Col6a1+/− mice were backcrossed for at least 8 generations in the inbred C57BL/6N strain. Mice bearing a GFP-LC3 transgene in the C57BL/6J background were obtained from RIKEN BRC (RBRC No. RBRC00806).Citation33 GFP-LC3 mice were backcrossed in the C57BL/6N strain and then crossed with col6a1−/− mice to generate col6a1−/−;GFP-LC3 animals. All mice were housed in a controlled environment with 12 h light/12 h dark cycle and a temperature of 23°C. When maintained in fed conditions, mice were provided with ad libitum standard chow (Laboratorio Dottori Piccioni, #52) and water. For starvation experiments, at 9 a.m. mice were individually moved to clean cages without chow but with free access to the water, for 24 h or 48 h. Experiments were carried out on sex- and age-matched mice, ranging from 18 to 24 wk of age. All the animals were sacrificed by cervical dislocation, and the tissues of interest were immediately dissected and fixed. The adopted procedures were approved by the Ethics Committee of the University of Padova and carried out according to all pertinent Italian laws.
Spermidine treatments
Spermidine (Sigma-Aldrich, S4139) was either dissolved in physiological (NaCl 0.9% w/v) sterile solution or added to drinking water. For the i.p. treatment, each mouse was injected once a d for 10 d with spermidine at 5 mg/kg body weight (low dose) or 50 mg/kg body weight (high dose). Control mice were not injected. Mice were sacrificed within 16 h after the last injection. Per os treatment was performed by adding spermidine to the drinking water at a concentration of 3 mM (low dose) or 30 mM (high dose). Mice had free access to water for 30 d and then they were sacrificed. Water with spermidine was replaced every 2 d, in order to avoid excessive deamination of the active compound.Citation9 Control mice drank water without addition of spermidine. For starvation experiments, mice were deprived of food during the last day of treatment. For colchicine co-treatment, mice received during the last 2 d of treatment daily i.p. injections with 0.4 mg/kg/day of colchicine (Sigma-Aldrich, C9754).
Fluorescence microscopy and histology
For the histological analysis, frozen tibialis anterior muscles were cut in 10-μm-thick sections and stained with haematoxylin-eosin (Sigma-Aldrich, 51275 and HT110116) or with a SDH-specific colorimetric substrate (nitro blue tetrazolium, Sigma-Aldrich, N5514), following standard protocols. Images were captured using a Zeiss Axioplan microscope (Carl Zeiss AG, Oberkochen, Germany) equipped with a Leica DC 500 digital camera (Leica Microsystems GmbH, Wetzlar, Germany). For the fluorescent microscopy analysis of GFP-LC3-positive puncta, tibialis anterior was processed as previously described,Citation33 stained with Hoechst 33258 (Sigma-Aldrich, B1155) and observed with a Leica SP5 confocal microscope (Leica Microsystems GmbH, Wetzlar, Germany).
TUNEL assay
Diaphragm muscles were fixed with paraformaldehyde, embedded in paraffin, and cut into 7-μm-thick transverse sections. Nuclei were stained with Hoechst 33258 (Sigma-Aldrich, B1155) and apoptotic nuclei detected by the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Merck Millipore, S7100). Images of the stained sections were captured randomly using a Zeiss Axioplan fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany) equipped with a Leica DC 500 digital camera (Leica Microsystems GmbH, Wetzlar, Germany), and TUNEL-positive nuclei quantified.
Transmission electron microscopy
Samples were fixed with 1% glutaraldehyde (Sigma-Aldrich, G5882) in 0.2 M sodium cacodylate buffer, pH 7.4. The specimens were washed in distilled water, stained with 2% osmium tetroxide and 0.5% aqueous uranyl acetate, dehydrated in alcohol and embedded in epon resin. Ultrathin (60 nm) sections were cut with a Leica EM UC7 ultramicrotome (Leica Microsystems GmbH, Wetzlar, Germany). Images were acquired from thin sections using a FEI Tecnai 12 transmission electron microscope (FEI Company, Hillsboro, Oregon, USA) equipped with a Veleta CCD digital camera (Olympus Soft Imaging Solutions GmbH, Münster, Germany). For morphometric analysis, mitochondria were classified as “aberrant” when they displayed at least one of the following abnormalities: swollen morphology, faintly stained inner matrix, dilated cristae and/or overtly decreased number of cristae.
Western blotting
Protein extraction from muscles and western blot analysis were performed as previously described.Citation34 Primary antibodies against the following proteins were used: AKT (Cell Signaling Technology, 9272), phospho-AKT (Ser473; Cell Signaling Technology, 4058), LC3B (Thermo Scientific, PA1–16930), GAPDH (Millipore, MAB374), SQSTM1/p62 (Santa Cruz Biotechnology, [H-290], sc-25575), RAB7 (Abcam, [RAB7–117], ab50533), GABARAP (Santa Cruz Biotechnology, [FL-117], sc-28938). Densitometric quantification was performed by ImageJ software (NIH, Bethesda, Maryland, USA).Citation35
Real time RT-PCR
Whole tibialis anterior muscles were blended, and RNA extraction performed using TRIzol reagent (Life Technologies, 15596–018) in accordance with the manufacturer's instructions. 500 ng of total RNA were retrotranscribed using the SuperScript III kit (Life Technologies, 18080–051) following manufacturer's instructions. Quantitative PCR was performed on a RotorGeneQ instrument (Qiagen, Venlo, Netherlands), using specific primers and CyBr green contaning mastermix (Qiagen, 204076). Primer sequences were described elsewhere.Citation8,19
Tetanic force measurements
In vivo determination of force and contraction kinetics of gastrocnemius muscle was carried out as previously described.Citation36 Briefly, mice were anesthetized and contraction elicited by stimulation of the sciatic nerve with multistranded steel wires (Cooner Wire, Chatsworth, CA, USA) implanted on either sides of the nerve. In order to avoid recruitment of the ankle dorsal flexors, and therefore significant reduction of torque, the common peroneal nerve was cut. Torque production of the stimulated plantar flexors was measured using a muscle lever system (Aurora Scientific Corporation, Aurora, Ontario, Canada). The force-frequency curves were determined by stepwise increasing stimulation frequency, pausing at least 30 s between stimuli to avoid effects due to fatigue. Tetanic contraction was reached for stimulations equal to or greater than 100 Hz, and force values obtained in these settings were compared between the different conditions as they represent the maximum contraction muscle can sustain.
Statistical analysis
All data are expressed as mean ± s.e.m. Statistical analysis of data was carried out using the Student t test, except for the electron microscope morphometric analysis where the Wilcoxon rank sum test was used. P < 0.05 was considered as a significant difference.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1108508_Supplemental_Material.zip
Download Zip (9.4 MB)Acknowledgments
We gratefully thank Prof. Noboru Mizushima for providing us with GFP-LC3 reporter mice. Moreover, we are sincerely grateful to Roberto Costa, Andrea Armani, Enrico Moro and Marco Sandri for providing us advice and help.
Funding
This work was supported by Telethon Foundation (Grants GGP10225 and GGP14202) and by the Italian Ministry of University and Research (FIRB Accordo di Programma RBAP11Z3YA_003).
Related Research Data
References
- Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ 2005; 12 Suppl 2:1535-41; PMID:16247501; http://dx.doi.org/10.1038/sj.cdd.4401728
- Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell 2008; 15:344-57; PMID:18804433; http://dx.doi.org/10.1016/j.devcel.2008.08.012
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 2014; 53:167-78; PMID:24462201; http://dx.doi.org/10.1016/j.molcel.2013.12.014
- Sandri M, Coletto L, Grumati P, Bonaldo P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci 2013; 126:5325-33; PMID:24293330; http://dx.doi.org/10.1242/jcs.114041
- Vainshtein A, Grumati P, Sandri M, Bonaldo P. Skeletal muscle, autophagy, and physical activity: the ménage à trois of metabolic regulation in health and disease. J Mol Med (Berl) 2014; 92:127-37; PMID:24271008; http://dx.doi.org/10.1007/s00109-013-1096-z
- Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013; 6:25-39; PMID:23268536; http://dx.doi.org/10.1242/dmm.010389
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010; 16:1313-20; PMID:21037586; http://dx.doi.org/10.1038/nm.2247
- Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 2009; 11:1305-14; PMID:19801973; http://dx.doi.org/10.1038/ncb1975
- Morselli E, Mariño G, Bennetzen M V, Eisenberg T, Megalou E, Schroeder S, Cabrera S, Bénit P, Rustin P, Criollo A, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol 2011; 192:615-29; PMID:21339330; http://dx.doi.org/10.1083/jcb.201008167
- Pietrocola F, Lachkar S, Enot DP, Niso-Santano M, Bravo-San Pedro JM, Sica V, Izzo V, Maiuri MC, Madeo F, Mariño G, et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ 2014; 22:509-16; PMID:25526088
- Hyttinen JMT, Niittykoski M, Salminen A, Kaarniranta K. Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim Biophys Acta 2013; 1833:503-10; PMID:23220125; http://dx.doi.org/10.1016/j.bbamcr.2012.11.018
- Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ 2014; 21:348-58; PMID:24440914; http://dx.doi.org/10.1038/cdd.2013.187
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792-802; PMID:20418806; http://dx.doi.org/10.1038/emboj.2010.74
- Ju JS, Varadhachary AS, Miller SE, Weihl CC. Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy 2010; 6:929-35; PMID:20657169; http://dx.doi.org/10.4161/auto.6.7.12785
- Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D, Bressan GM. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum Mol Genet 1998; 7:2135-40; PMID:9817932; http://dx.doi.org/10.1093/hmg/7.13.2135
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 2003; 35:367-71; PMID:14625552; http://dx.doi.org/10.1038/ng1270
- Bernardi P, Bonaldo P. Dysfunction of mitochondria and sarcoplasmic reticulum in the pathogenesis of collagen VI muscular dystrophies. Ann N Y Acad Sci 2008; 1147:303-11; PMID:19076452; http://dx.doi.org/10.1196/annals.1427.009
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6:458-71; PMID:18054315; http://dx.doi.org/10.1016/j.cmet.2007.11.001
- Van Der Heide LP, Hoekman MFM, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J 2004; 380:297-309; PMID:15005655; http://dx.doi.org/10.1042/bj20040167
- Hay N. Interplay between FOXO, TOR, and AKT. Biochim Biophys Acta 2011; 1813:1965-70; PMID:21440577; http://dx.doi.org/10.1016/j.bbamcr.2011.03.013
- Minois N, Carmona-Gutierrez D, Bauer M a, Rockenfeller P, Eisenberg T, Brandhorst S, Sigrist SJ, Kroemer G, Madeo F. Spermidine promotes stress resistance in Drosophila melanogaster through autophagy-dependent and -independent pathways. Cell Death Dis 2012; 3:e401; PMID:23059820; http://dx.doi.org/10.1038/cddis.2012.139
- LaRocca TJ, Gioscia-Ryan RA, Hearon CM, Seals DR. The autophagy enhancer spermidine reverses arterial aging. Mech Ageing Dev 2013; 134:314-20; PMID:23612189; http://dx.doi.org/10.1016/j.mad.2013.04.004
- Gupta VK, Scheunemann L, Eisenberg T, Mertel S, Bhukel A, Koemans TS, Kramer JM, Liu KSY, Schroeder S, Stunnenberg HG, et al. Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat Neurosci 2013; 16:1453-60; PMID:23995066; http://dx.doi.org/10.1038/nn.3512
- Wang I-F, Guo B-S, Liu Y-C, Wu C-C, Yang C-H, Tsai K-J, Shen C-KJ. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A 2012; 109:15024-9; PMID:22932872; http://dx.doi.org/10.1073/pnas.1206362109
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741-52; PMID:17717517; http://dx.doi.org/10.1038/nrm2239
- Moschou PN, Roubelakis-Angelakis KA. Polyamines and programmed cell death. J Exp Bot 2013; 65:1285-96; PMID:24218329; http://dx.doi.org/10.1093/jxb/ert373
- Carmignac V, Svensson M, Körner Z, Elowsson L, Matsumura C, Gawlik KI, Allamand V, Durbeej M. Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet 2011; 20:4891-902; PMID:21920942; http://dx.doi.org/10.1093/hmg/ddr427
- Sakurada T, Onodera K, Tadano T, Kisara K. Effects of polyamines on the central nervous system. Japan J Pharmacol 1975; 25:653-61; http://dx.doi.org/10.1254/jjp.25.653
- Igarashi K, Kashiwagi K. Modulation of cellular function by polyamines. Int J Biochem Cell Biol 2010; 42:39-51; PMID:19643201; http://dx.doi.org/10.1016/j.biocel.2009.07.009
- Bertaggia E, Coletto L, Sandri M. Posttranslational modifications control FoxO3 activity during denervation. AJP Cell Physiol 2012; 302:C587-96; http://dx.doi.org/10.1152/ajpcell.00142.2011
- Atiya Ali M, Poortvliet E, Strömberg R, Yngve A. Polyamines in foods: development of a food database. Food Nutr Res 2011; 55; PMID:21249160; http://dx.doi.org/10.3402/fnr.v55i0.5455.
- Mizushima N. Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol 2009; 452:13-23; PMID:19200873; http://dx.doi.org/10.1016/S0076-6879(08)03602-1
- Chen P, Cescon M, Megighian A, Bonaldo P. Collagen VI regulates peripheral nerve myelination and function. FASEB J 2014; 28:1145-56; PMID:24277578; http://dx.doi.org/10.1096/fj.13-239533
- Schneider CA, Rasband WS, Eliceiri KW. “NIH Image to ImageJ: 25 years of image analysis.” Nat Methods 2012; 9: 671-675; PMID:22930834; http://dx.doi.org/10.1038/nmeth.2089
- Blaauw B, Canato M, Agatea L, Toniolo L, Mammucari C, Masiero E, Abraham R, Sandri M, Schiaffino S, Reggiani C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J 2009; 23:3896-905; PMID:19661286; http://dx.doi.org/10.1096/fj.09-131870