
Abstract
An attractive strategy for cancer therapy is to stop cell proliferation by means of agents that directly arrest the cell cycle. Microtubule poisons such as taxanes block mitosis, eventually leading to cell death in a process frequently known as mitotic catastrophe. However, some cells are able to bypass this mitotic arrest and survive, thus contributing to chemoresistance to those therapies. We have recently observed that mitotic arrest induces an early autophagic flux response that results in autophagy-dependent mitochondrial degradation and a dramatic energetic deficit. The subsequent increase in the AMP/ATP ratio results in the activation of the metabolic sensor AMPK followed by phosphorylation and activation of PFKFB3, an enzyme required for glycolysis. Thus, mitophagy can be considered as a critical effector of the therapeutic effect of mitotic therapies, while both AMPK and PFKFB3 are critical for survival. The manipulation of these molecular routes may therefore have therapeutic benefits in the presence of microtubule poisons.
The special structural requirements that accompany chromosome segregation during mitosis make cells very sensitive to cell death during this process. Multiple lines of evidence suggest that mitotic cell death is caspase-dependent although the presence of undefined caspase-independent processes has also been suggested. Whether autophagy was or not present in mitosis has been a matter of debate in the past few years. Using a combination of genetic and chemical models, as well as time-lapse microscopy to monitor progression throughout the cell cycle, we recently found that autophagy also plays critical roles in cell survival during prolonged mitotic arrest (). This process is induced a few hours after mitotic arrest and has a strong impact on mitotic cell death. Knockdown of the autophagy regulators ULK1, PIK3C3/Vps34 and BECN1/Beclin1, as well as treatment with the class III PtdIns3K inhibitor 3-methyladenine, blocks mitotic cell death to a similar extent as caspase inhibition. In fact, autophagy is likely upstream of apoptosis as caspase inhibitors do not prevent autophagosome formation, whereas autophagy inhibitors delay caspase activation during mitotic cell death. Bax Bak1 double-knockout cells also die during prolonged mitotic arrest suggesting the presence of both caspase-dependent and -independent pathways in this process. Interestingly, induction of autophagy by knocking down RPTOR or treating cells with AKT-MTOR inhibitors increases cell death in mitotic arrested cells, indicating a pro-death role in this particular setting. Autophagy is mainly a prosurvival mechanism, and cell death is frequently prevented by a safety mechanism that leads to the activation of MTOR after prolonged stimuli, thus resulting in autophagy attenuation. However, protein synthesis is limited during mitosis due to the lack of general transcription and translation so it could be speculated that the normal controls that limit the autophagy-dependent degradation of intracellular components are absent during mitotic arrest.
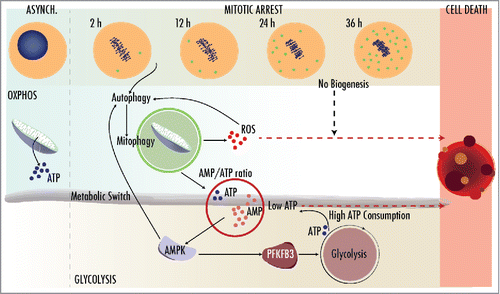
Figure 1. Mitotic arrest, such as the one imposed by microtubule poisons, induces a series of cellular changes to prolong cell survival. The induction of autophagy in the absence of organelle biogenesis results in mitophagy (green dots in the upper panel) and the depletion of functional mitochondria, leading to the presence of reactive oxygen species (ROS) and low ATP levels. This stimulates the activity of AMPK resulting in PFKFB3 phosphorylation and the induction of glycolysis, in order to fulfill the high energetic requirements of mitotic cells. The balance between ROS and ATP levels determines whether cells eventually die using caspase-dependent or -independent pathways before cells can escape from the mitotic arrest. (Art designs by [email protected].)
The productive autophagic flux observed as early as 3 h after mitotic arrest results in a marked decrease in mitochondrial numbers. Using a quantitative method that we have recently developed based on the determination of MitoTracker Deep Red levels by flow cytometry, we found that inhibiting lysosomal activity as well as preventing autophagy or mitophagy restores mitochondrial levels. Mitochondrial fission occurs during mitosis to allow the proper distribution of mitochondria into daughter cells. Mitochondrial fragmentation is also a prerequisite for mitophagy and, in fact, knockdown of DNM1L/Drp1 reduces mitochondria fragmentation and the colocalization of autophagy markers with mitochondria, thus preventing the loss in mitochondrial levels and delaying cell death. Therefore, our data place mitochondrial fission and the subsequent mitophagy likely upstream of all catastrophic events observed during prolonged mitotic arrest.
The decrease in functional mitochondria, together with the high energetic requirements of mitotic cells, provokes an energetic crisis after 8-12 h of mitotic arrest. Interestingly, this is accompanied by an increase in lactate production and a reduction in oxygen consumption rate indicative of a metabolic switch from oxidative phosphorylation towards glycolysis. This is likely mediated by AMPK, an essential metabolic regulator that when activated blocks anabolic pathways while simultaneously inducing cellular catabolism to sustain energy homeostasis. In tumor cell lines, AMPK phosphorylates PFKFB3, a critical mediator of the Warburg effect, thus inducing glycolysis and cell survival in mitotically arrested cells. Accordingly, knockdown of PFKFB3 or its pharmacological inhibition reduces survival of cells in mitosis, whereas its overexpression rescues the defects observed after the downregulation of AMPK.
It is interesting to note that, during mitosis, mitophagy lies upstream of AMPK activation. The opposite has been previously observed, as AMPK can directly induce autophagy via phosphorylation of ULK1. We also found that effect as AMPK downregulation results in decreased mitophagy, suggesting an amplification loop between autophagy and AMPK activation during prolonged mitotic arrest (). Yet, the exact molecular mechanism by which mitophagy is activated during mitotic arrest remains elusive. In addition to the consequences imposed by mitochondrial fission, we could speculate a role for PARK2/Parkin during prolonged mitotic arrest. The reduction in mitochondrial membrane potential leads to PARK2 translocation to the outer mitochondrial membrane, where it ubiquitinates mitochondrial outer membrane proteins that recruit the autophagosome machinery. We indeed observed an increase in PARK2 levels as early as 2 h after mitotic arrest, although how PARK2 is regulated during mitosis and its requirements for mitotic mitophagy remain to be investigated.
The fine balance between degradation and biogenesis seems to be critical for mitochondrial function. The coordination of these processes requires transcription factors that increase both mitophagy and mitochondrial biogenesis. Intriguingly, AMPK also phosphorylates PPARGC1A/PGC1alpha, one of the master regulators of mitochondrial biogenesis. However, all these transcriptional responses are not active in mitosis due to the lack of transcription in condensed chromosomes. It is therefore possible that lack of transcription—and limited translation—during mitosis stands out as the major culprit for the loss of mitochondria. In this special setting, mitophagy is not coupled to mitochondrial biogenesis, triggering rapid mitochondrial loss and ATP depletion. Cells then entirely depend on glycolysis, and blocking this adaptation program results in cell death. This effect can be therapeutically exploited and the combination of taxol (to induce mitotic arrest) with either autophagy inducers, or AMPK and glycolysis inhibitors cooperates in reducing survival. These data may open new avenues in therapeutic strategies against cancer based on the special energetic requirements of proliferating cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Work in the authors' laboratories is funded by grants from MINECO (SAF2012-36079 to PB and SAF2012-38215, SAF2014-57791-REDC, and BFU2014-52125-REDT to MM), and Comunidad de Madrid (S2010/BMD-2470).