
Abstract
The highly conserved cellular degradation pathway, macroautophagy, regulates the homeostasis of organelles and promotes the survival of T lymphocytes. Previous results indicate that Atg3-, Atg5-, or Pik3c3/Vps34-deficient T cells cannot proliferate efficiently. Here we demonstrate that the proliferation of Atg7-deficient T cells is defective. By using an adoptive transfer and Listeria monocytogenes (LM) mouse infection model, we found that the primary immune response against LM is intrinsically impaired in autophagy-deficient CD8+ T cells because the cell population cannot expand after infection. Autophagy-deficient T cells fail to enter into S-phase after TCR stimulation. The major negative regulator of the cell cycle in T lymphocytes, CDKN1B, is accumulated in autophagy-deficient naïve T cells and CDKN1B cannot be degraded after TCR stimulation. Furthermore, our results indicate that genetic deletion of one allele of CDKN1B in autophagy-deficient T cells restores proliferative capability and the cells can enter into S-phase after TCR stimulation. Finally, we found that natural CDKN1B forms polymers and is physiologically associated with the autophagy receptor protein SQSTM1/p62 (sequestosome 1). Collectively, autophagy is required for maintaining the expression level of CDKN1B in naïve T cells and selectively degrades CDKN1B after TCR stimulation.
Abbreviations
| Atg | = | autophagy related |
| CDK | = | cyclin-dependent kinase |
| CDKN1B | = | cyclin-dependent kinase inhibitor 1B (p27 Kip1) |
| CFSE | = | carboxyfluorescein succinimidyl ester |
| cfu | = | colony-forming units |
| CKI | = | cyclin-dependent kinase inhibitor |
| LM | = | L. monocytogenes |
| OVA | = | ovalbumin |
| RT | = | room temperature |
| SQSTM1/p62 | = | sequestosome 1 |
| TCR | = | T cell receptor |
Introduction
The highly conserved cellular homeostasis pathway, macroautophagy (referred to as autophagy thereafter), degrades large protein aggregates, removes damaged or extraneous organelles, recycles nutrients, and promotes cell survival during stressful conditions.Citation1-3 In the immune system, autophagy is a mechanism to eliminate intracellular pathogensCitation4 and deliver endogenous antigens to MHC II-bearing compartments.Citation5,6 Autophagy plays an essential role in the development and function of T lymphocytes. Autophagy regulates the calcium mobilization and energy metabolism in T cells,Citation7–9 is critical for effector CD8+ T cell survival and memory formation,Citation10,11 promotes the proliferation and survival of invariant NKT cellsCitation12,13 and tunes down TCR signaling by degrading BCL10.Citation14 Autophagy starts with a membrane precursor called the phagophore. The phagophore elongates and engulfs cytosolic materials to form a typical double-membrane structure, called an autophagosome. The final maturation step involves the fusion of the autophagosomes with lysosomes to form autolysosomes, with the sequestered macromolecules degraded by lysosomal proteases and lipases. A series of molecules, termed autophagy-related (Atg) genes, regulate the initiation, elongation and maturation of autophagosomes. The formation of autophagosome requires 2 ubiquitination-like systems to generate membrane inserting ATG12–ATG5 conjugates and LC3. During this process, ATG7 functions as an E1-like activating enzyme, and ATG10 and ATG3 function as E2-like conjugating enzymes.Citation15 Both ATG7 and ATG3 are required for autophagy in T cells.Citation16-18 Previous studies from other groups and our lab indicated that autophagy-deficient conventional T and invariant NKT lymphocytes fail to proliferate efficiently,Citation12,18-21 but the mechanism of the proliferation defect is not clear.
TCR activation-induced proliferation is triggered by the interaction between the T cell receptor-CD3 complex (TCR-CD3 complex) and the MHC-peptide complex on antigen presenting cells. This process expands antigen-specific T cell clones and ultimately generates the adaptive immune response. On the cellular level, T cell expansion is regulated by the progression of the cell cycle. In mammalian cells, the cell cycle consists of the transition from G0 to G1 then to S-phase, and is controlled by positive regulators, the cyclins and cyclin-dependent kinases (CDKs), as well as negative regulators, the CDK inhibitors (CKIs).Citation22,23 CCND (cyclin D) forms a complex with CDK4 or CDK6, while CCNE (cyclin E) or CCNA (cyclin A) associates with CDK2. The expression levels of cyclins and CDKs are very low in resting T cells, whereas they are significantly increased in G1 phase after upon TCR stimulation.Citation24 The negative regulators, CDKIs, include 2 families: the INK4 inhibitors and the Cip/Kip inhibitors. The INK4 family contains CDKN2A/p16/INK4a, CDKN2B/p15/INK4b, CDKN2C/p18/INK4c, and CDKN2D/p19/INK4d, which bind to CDK4 or CDK6, and inhibit kinase activity. The Cip/Kip (CDK-interacting protein/kinase inhibitor protein) family includes CDKN1A/p21/Cip1, CDKN1B/p27/Kip1, and CDKN1C/p57/Kip2, which inhibit all CDKs.Citation25
CDKN1B is abundantly expressed in T lymphocytes and is the major negative regulator of the cell cycle in T lymphocytes.Citation26 CDKN1B is accumulated in quiescent cells, and the degradation of CDKN1B facilitates the transition from G1- to S-phase in T lymphocytes after T cell activation. Previous data have shown that CDKN1B is degraded through the ubiquitin-proteasome pathway.Citation27 The increased expression of cyclin D and CDK4 or CDK6 results in the decrease of CDKN1B in G1 phase, and the activation of CCNE-CDK2. The activated CCNE-CDK2 phosphorylates threonine 187 (T187) of CDKN1B. Phosphorylated CDKN1B interacts with the F-box protein SKP2 (S-phase kinase-associated protein 2, E3 ubiquitin protein ligase).Citation28 SKP2 associates with SKP1, CUL1, RBX1/ROC1 and CKS1B/CKS1 to form the SKP1–CAND1/CDC53-CUL1–F-box protein (SCF) ubiquitin-protein ligase (E3) complex.Citation29-32 Finally CDKN1B is degraded by the 26S proteasome.Citation27 A SKP2-independent pathway, which is mediated by the ubiquitin ligase KIP1 ubiquitination-promoting complex (KPC), also regulates the degradation of CDKN1B during the G1 phase.Citation33 Besides regulating cell cycle, CDKN1B also plays roles in mediating other functions of T cells. For example, CDKN1B negatively regulates the CD4 memory response by promoting the apoptosis and inhibiting proliferation in both effector and memory CD4+ T cells.Citation34 CDKN1B also regulates the homeostasis of CD8+ T cells and limits the proliferation of effector memory cells.Citation35 Finally, CDKN1B is required for tolerance induction in T cells.Citation36
Here we show that ATG7-deficient T cells cannot proliferate in vitro, similar to what is observed in Atg5- and Atg3-deficient T cells.Citation18,19 We identify that the impairment of proliferation causes the intrinsic defect in the primary immune response in autophagy-deficient CD8+ T cells to infection by L. monocytogenes. Furthermore, we show that autophagy-deficient T cells fail to enter into S-phase after TCR stimulation and correspondingly, the cell-cycle inhibitor, CDKN1B, is abnormally accumulated in autophagy-deficient naïve T lymphocytes and cannot be degraded after activation. The genetic deletion of Cdkn1b restored the proliferative capacity in autophagy-deficient T cells. Interestingly, natural CDKN1B forms polymers that are physiologically associated with the autophagy receptor protein, SQSTM1/p62. Taken together, our data indicates that autophagy regulates the proliferation of T lymphocyte through selectively degradation of the cell-cycle inhibitor, CDKN1B.
Results
The primary immune response is defective in autophagy-deficient T cells
In previous studies, our group and others have found that Atg5- or Atg3-deficient T cells cannot proliferate efficiently.Citation18-21 To further confirm that autophagy is essential for the proliferation of T lymphocytes, the proliferation of Atg7-deficient (Atg7f/f Lck-Cre) T cells was analyzed. Proliferation in both CD4+ and CD8+ Atg7-deficient T cells was severely impaired. The percentage of proliferating T cells in Atg7-deficient T cells was significantly reduced compared to that in floxed control (Atg7f/f) T cells under all TCR stimulation conditions (). Compared to anti-CD3 antibody stimulation alone, costimulation with anti-CD28 could only partially improve the proliferation in Atg7-deficient T cells. Interestingly, the use of PMA and ionomycin, which bypasses the TCR proximal signaling, did not rescue the proliferation defect in Atg7-deficient T cells (). These data suggest that the defective proliferation in autophagy-deficient T cells is downstream of proximal TCR signaling. These results are consistent with previous data, which indicated that the proximal TCR signaling remained intact in autophagy-deficient T cells.Citation17,18
Figure 1. Defective proliferation of Atg7-deficient T cells. Splenocytes from Atg7f/f and Atg7f/fLck-Cre mice were loaded with CFSE and stimulated with coated anti-CD3 mAb (2C11), soluble anti-CD3 plus anti-CD28 (sCD3+CD28) or PMA together with ionomycin for 72 h. The CFSE-diluted cell populations were analyzed by flow cytometry and all cells were gated on 7-AAD negative cells. These experiments were repeated 3 times. (A) Representative flow cytometry profiles of CD4+ or CD8+ T cell proliferation from Atg7-deficient T cells. (B) The percentages of CFSE-diluted CD4+ or CD8+ T cells from Atg7f/f and Atg7f/fLck-Cre mice. Each symbol represents one mouse.
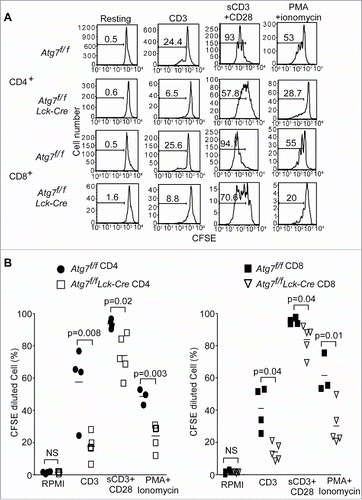
The survival of autophagy-deficient T cells is impaired.Citation10,18-20,37 To exclude the possibility that the proliferation defect is caused by cell death, all cells in the carboxyfluorescein succinimidyl ester (CFSE) dilution assay were gated on 7-AAD negative live cells (). The death of autophagy-deficient T cells after anti-CD3 stimulation was determined. The survival of autophagy-deficient T cells was improved after TCR stimulation (Fig. S1).
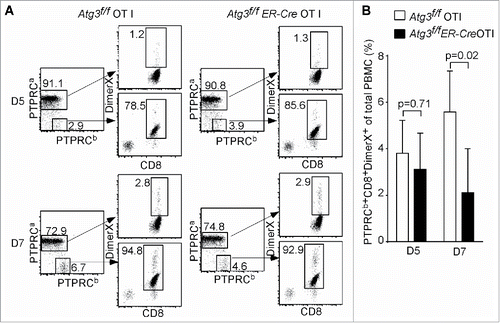
To further analyze the physiological function of autophagy in T cells, primary immune responses of autophagy-deficient T cells were analyzed using adoptive transfer and L. monocytogenes infection. We utilized a recombinant strain of L. monocytogenes expressing chicken OVA (LM-OVA).Citation38 The use of an inducible deletion system, Atg3f/f ER-Cre, provides an ideal tool since the numbers of mature T cells are comparable between the floxed control and Atg3f/f ER-Cre after the deletion of Atg3.Citation18 As shown in , the percentage of Atg3-deficient PTPRCb/CD45.2+ donor cells at d 5 after infection was comparable to that of floxed control donor cells. This indicates comparable short-term engraftment between Atg3-deficient and floxed control T cells. However, the percentage of Atg3-deficient donor cells was significantly lower than that of floxed control donor cells at d 7 after infection, when the immune response against L. monocytogenes infection reaches its peak ().Citation39 Therefore, both in vitro proliferation assays and in vivo adoptive transfer infection experiments indicate that the autophagy-deficient T cells cannot proliferate efficiently and the primary immune response against L. monocytogenes infection may be defective.
Figure 2. Impaired primary T cell immune response in autophagy-deficient T cells. (A) Analysis of autophagy-deficient T cells in primary response against the infection of L. monocytogens through adoptive transfer assay. One pair of Atg3f/f OT-I and Atg3f/fER-Cre OT-I mice were used to prepare the donor cells. Purified CD8+ cells were transferred to 3-5 PTPRCa/CD45.1 host mice. The blood was withdrawn at d 5 and d 7 after infection, and the frequency of antigen-specific CD8+ T cells was analyzed by gating on the PTPRCb/CD45.2+ CD8+ cells. The experiments were repeated 3 times. (B) The frequencies of antigen-specific CD8+ T cells (PTPRCb/CD45.2+ Dimer X+ CD8+) pooled from 3 mice that received Atg3f/f OT-I and 5 mice that received Atg3f/fER-Cre OT-I cells.
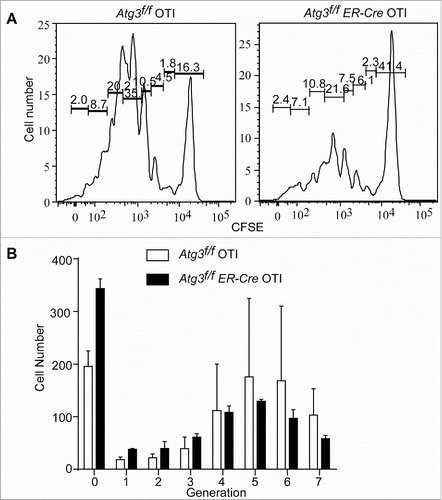
To directly test whether an impaired primary immune response was due to the incapability of autophagy-deficient T cells to proliferate, the division of antigen-specific CD8+ T cells responding to LM-OVA was analyzed in vivo. CFSE-labeled OT-I CD8+ T cells or Atg3-deficient OT-I T cells were transferred to normal host mice. Recipient mice were infected with LM-OVA at 24 h post transfer. The division of antigen-specific T cells was analyzed at 48 h after infection. While control OT-I T cells divided very well as expected, autophagy-deficient T cells failed to proliferate normally (). The cell numbers in each generation after generation 4 were reduced in autophagy-deficient OT-I cells comparing to floxed control T cells (), indicating that the proliferation of autophagy-deficient T cells is impaired.
Figure 3. Defective in vivo proliferation of autophagy-deficient T cells. Atg3f/f OT-I and Atg3f/fER-Cre OT-I mice were injected with tamoxifen to induce the deletion of Atg3. Purified CD8+ T cells were loaded with CFSE and then transferred to 3 to 5 normal host mice for each donor cell population intravenously. The next day after transfer, host mice were infected with L. monocytogenes-OVA (1×104cfu/mouse). At 48 h after infection, the splenocytes were harvested. Live, CFSE+ CD8+cells were analyzed for CFSE dilution. This experiment was repeated twice. (A) Representative data of flow cytometry profiles from a pair of mice that received CFSE-labeled Atg3f/fOT-I or Atg3f/fER-CreOT-I T cells are shown. (B) The cell numbers of each generation from 3 pairs of mice that received Atg3f/fOT-I or Atg3f/fER-CreOT-I were analyzed and calculated by Flowjo.
Autophagy-deficient T cells fail to enter into S phase after TCR stimulation
Since autophagy-deficient T cells have obvious deficiencies in proliferation after TCR stimulation, cell-cycle analysis of autophagy-deficient T cells was performed after TCR stimulation in both Atg3- () and Atg7- (Fig. S2) deficient models (Atg3f/f Lck-Cre or Atg7f/f Lck-Cre). As shown in , 20% of floxed control CD4+ and more than 40% floxed control CD8+ T cells entered into S-phase after stimulation with soluble anti-CD3 in the presence of B cells. However, there were reduced percentages of autophagy-deficient CD4+ and CD8+ T cells entering into S-phase.
Figure 4. Autophagy-deficient T cells fail to enter into S-phase after TCR stimulation. (A) Splenocytes from Atg3f/f and Atg3f/fLck-Cre mice were stimulated with soluble anti-CD3 plus anti-CD28 antibodies overnight. Cell cycle was analyzed by flow cytometry. The statistical analysis in the lower panel was derived from 3 pairs of Atg3f/f and Atg3f/fLck-Cre mice (mean±SD). The experiment was repeated twice. (B) CDKN1B is accumulated in autophagy-deficient T cells. Atg3f/fOT-I and Atg3f/fER-Cre OT-I mice were inducibly deleted the Atg3 through tamoxifen injection. At d 6 or d 35 after the first injection, the CD8+ T cells were purified and cell lysates were prepared. The expression level of CDKN1B and CDKN1 were analyzed by western blot. The numbers are the ratios of the intensity of target molecule to the loading control ACTB/actin. The normalized intensities from 3 pairs of Atg3f/f OT-I and Atg3f/fER-Cre OT-I mice are shown in the right panel (mean±SD). (C) Impaired degradation of CDKN1B in autophagy-deficient T cells after TCR-mediated activation. Splenocytes were stimulated with anti-CD3 plus anti-CD28 antibodies or without any stimulation overnight. Total T cells were purified and cell lysates were prepared. The expression levels of CDKN1B and CDKN1 were analyzed by western blot. The normalized intensities from 3 pairs of Atg7f/f and Atg7f/f Lck-Cre mice are shown in the lower panel (mean±SD).
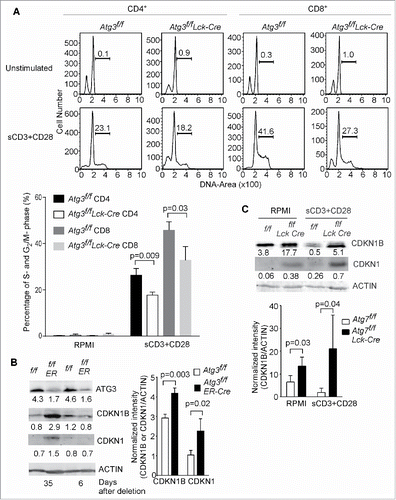
In T lymphocytes, the cell cycle is primarily regulated by the cyclin-dependent kinase inhibitor CDKN1B, which is degraded after TCR stimulation and IL-2 production. The levels of CDKN1B and other Kip/Cip family members were quantified in naïve T cells, in either an inducible deletion system of autophagy related gene or a Lck-Cre system. As shown in , the protein level of CDKN1B was comparable between autophagy-competent and autophagy-deficient T cells within 1 wk of Atg3 deletion. The protein expression level of CDKN1B in naïve floxed control and autophagy-deficient T cells was relatively low, while the CDKN1 expression level was extremely low (). However, the expression level of both CDKN1B and CDKN1 increased significantly in autophagy-deficient T cells if the deletion of autophagy related genes was induced for a longer time period (, right panel). Similarly, the expression levels of CDKN1B and CDKN1 were also increased in autophagy-deficient Atg7f/fLck-Cre T cells compared to floxed control T cells (), in which Atg7 was deleted at DN3 stage of thymocyte development. These results suggest that autophagy constitutively regulates the expression level of CDKN1B and CDKN1. It has been shown that the degradation of CDKN1B after TCR-mediated T cell activation facilitates T cell entry into the S phase and proliferation.Citation26 As shown in , the protein level of CDKN1B was decreased in floxed control T cells after TCR stimulation, while the protein level of CDKN1B in autophagy-deficient T cells remained unchanged. This suggests that autophagy is required for the degradation of CDKN1B after TCR stimulation. It has been reported that concanavalin A and IL-2 stimulation transcriptionally decrease the abundance of CDKN1B.Citation40 However, there was no difference in the expression of Cdkn1b at the mRNA level between floxed control and autophagy-deficient T cells (data not shown). IL-7 promotes T cell proliferation through the regulation of stability of CDKN1B at the protein level, which is dependent on PRKCQ/PKC-θ (protein kinase C, theta).Citation41 But IL-7 fails to rescue the proliferation defect in autophagy-deficient T cells (our unpublished data). Interestingly, the protein level of CDKN1 was slightly increased in floxed control T cells after TCR stimulation, but it was strongly elevated in resting and activated autophagy-deficient T cells (). CDKN1 may be regulated at the level of both expression and degradation in activated T cells. Taken together, the proliferation defect in autophagy-deficient T cells may be due to a degradation defect of CDKN1B and CDKN1, resulting in the incapacity of autophagy-deficient T cells to enter into S-phase.
The defect in proliferation is caused by the abnormal accumulation of CDKN1B in autophagy-deficient T cells
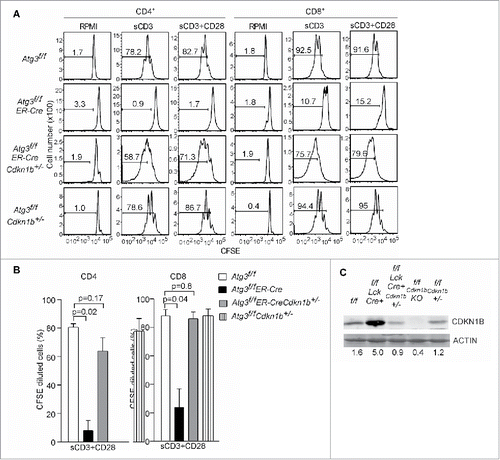
The increased protein levels of CDKN1B and CDKN1, along with the impaired proliferation in autophagy-deficient T cells after TCR activation, suggest that CDKN1B and CDKN1 may control the cell cycle and proliferation in T lymphocytes after activation. To test whether the decreased level of CDKN1B can restore proliferation, Atg3f/fER-Cre mice were bred to Cdkn1b-/- mice to generate atg3-/-Cdkn1b+/- T cells. Consistent with our hypothesis, deletion of just one allele of Cdkn1b in autophagy-deficient T cells restored the capability to enter into S-phase and permitted proliferation (). The percentage of proliferating T cells from Atg3f/fER-CreCdkn1b+/- mice was comparable to floxed control cells in both CD4+ and CD8+ compartments (). As expected, no obvious accumulation of CDKN1B was observed in autophagy-deficient Cdkn1b+/- T cells (). These results suggest that the abnormal accumulation of CDKN1B in autophagy-deficient T cells blocked their proliferation.
Figure 5. Deletion of CDKN1B restores proliferative capacity in autophagy-deficient T cells. (A) Atg3f/f, Atg3f/fER-Cre, Cdkn1b+/-, Atg3f/fER-Cre Cdkn1b+/- and Atg3f/fCdkn1b+/- mice were injected with tamoxifen to inducibly delete Atg3. Splenocytes from Atg3f/f, Atg3f/fER-Cre, Atg3f/fER-Cre Cdkn1b+/- and Atg3f/fCdkn1b+/- mice were isolated, loaded with CFSE, and stimulated with soluble anti-CD3 mAb, or soluble anti-CD3 plus anti-CD28 antibodies for 72 h. Live cells were analyzed for CFSE dilution. All cells were gated on CD4+7-AAD- or CD8+7-AAD- populations. There were 3 or 4 mice in each group in 2 independent experiments. (B) The percentages of CFSE-diluted CD4+ or CD8+ T cells after stimulation with soluble anti-CD3 plus anti-CD28 were analyzed (n=3 or 4). (C) The expression levels of CDKN1B in purified T cells from the above mice by western blot. ACTB/actin serves as loading control. The numbers are normalized expression levels for CDKN1B.
Autophagy selectively degrades CDKN1B
Previously published data has demonstrated that CDKN1B is degraded through the proteasomal pathway.Citation27 The accumulation of CDKN1B in autophagy-deficient T cells suggests that autophagy may be involved in maintaining homeostatic levels of CDKN1B. Interestingly, CDKN1B formed polymers in nonreducing conditions as analyzed by western blot (). Under reducing conditions, the CDKN1B bands showed up at 50 kDa and 27 kDa. Under nonreducing conditions, in which the disulfide bonds remained intact, CDKN1B migrated at larger molecular masses in addition to the other 2 relative weak bands ().
Figure 6. Autophagy selectively degrades CDKN1B in T lymphocytes. (A) CDKN1B forms polymers naturally. Cell lysates of purified T cell from wild-type mice were prepared. The cell lysates were mixed with reducing sample buffer, which contains 2-ME or nonreducing sample buffer without any 2-ME. The western blot was probed with anti-CDKN1B antibody. (B) CDKN1B interacts with SQSTM1. 293T cells were cotransfected with Sqstm1-HA and Cdkn1b-FLAG constructs. Then tagged proteins were immunoprecipitated with an anti-FLAG antibody and probed with an anti-HA antibody (upper panel). The interaction between endogenous of CDKN1B and SQSTM1 is shown in the lower panel. Atg3f/f and Atg3f/fER-Cre mice were injected with tamoxifen to delete Atg3. Splenocytes from Atg3f/f and Atg3f/fER-Cre mice were isolated and cell lysates were prepared. CDKN1B was immunoprecipitated with anti-CDKN1B antibody and probed for SQSTM1 interaction. (C) Ubiquitination of CDKN1B in splenic T cells. Splenocytes from normal mice were stimulated with soluble anti-CD3 plus anti-CD28 antibodies for 6 h. Then CDKN1B was immunoprecipitated with anti-CDKN1B mAb and the western blot membrane was probed with an anti-ubiquitin antibody. (D) CDKN1B localized in the same autophagy-related membrane structures as that of LC3-II and SQSTM1. Splenocytes from 3 normal mice were stimulated with anti-CD3 antibody in RPMI with choloroquine (50 µg/ml) for 6 h. The homogenates were prepared with protease inhibitors and choloroquine (50 µg/ml) using Dounce homogenizers. Postnuclear subcellular compartments were added into ultracentrifuge tubes on the top of prepared gradients. The distributions of target molecules in each fraction were detected by western blot in reducing condition. The arrow represents the concentration of the gradients from lower to higher densities. P, cell pellet; L, cell lysate. (E) The degradation of CDKN1B was analyzed in the presence of lactacystin or bafilomycin A1 treatment. Splenocytes from normal mice were stimulated with soluble anti-CD3 plus anti-CD28 antibodies with or without lactacystin or bafilomycin A1 treatment. The cells were collected at different time points and the T cells were purified. The expression levels of CDKN1B in T cells were analyzed by western blot.
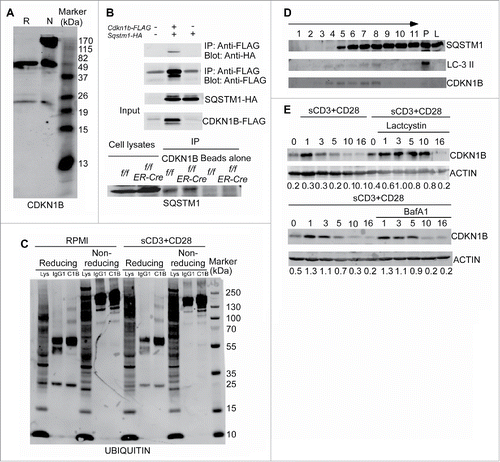
Selective autophagy can degrade specific targets. Several autophagy receptor proteins facilitate the loading of targets into phagophores.Citation42 The receptor molecule SQSTM1 is one of these autophagy receptors. SQSTM1 can associate with LC3 during the induction of autophagy and at the same time binds ubiquitinated proteins.Citation43 To test the interaction between SQSTM1 and CDKN1B, recombinant HA-tagged SQSTM1 and FLAG-tagged CDKN1B were coexpressed in 293T cells. CoIP indicated that SQSTM1-HA could interact with CDKN1B-FLAG ( upper panel). The endogenous association between CDKN1B and SQSTM1 was analyzed as well. As shown in the lower panels of , the endogenous SQSTM1 interacts with CDKN1B in both floxed control and ATG3-deficient splenocytes. Autophagy-deficient CD4+ and CD8+ T cells have relatively higher ubiquitination levels compared to floxed control T cells (Fig. S3). To directly determine its ubiquitination status, CDKN1B was isolated from normal T cells by immunoprecipitation in the presence or absence of TCR activation, followed by western blot under both reducing and nonreducing conditions. Under reducing condition, ubiquitinated CDKN1B shown as multiple bands with higher molecular weight was detected (). Under nonreducing conditions, some ubiquitinated CDKN1B could also be detected (). TCR stimulation did not obviously enhance ubiquitinated CDKN1B (). To further confirm that CDKN1B can translocate to phagophores, a subcellular fractionation assay was performed. CDKN1B was localized within the same fractions (fraction: 4 to 8) in which the autophagosome marker LC3-II was fractionated (). Therefore, our results indicate that autophagy selectively maintains the expression level of CDKN1B and degrades CDKN1B when T cells are activated.
Our data do not exclude the functions of ubiquitin/proteasome pathway in the degradation of CDKN1B. The kinetic degradation of CDKN1B in T cells after TCR stimulation was analyzed. After stimulation, CDKN1B started being degraded at 3 h, further decreased at 5 to 10 h, and was mostly degraded at 10 h or more post stimulation (). The proteasome inhibitor lactacystin inhibited and delayed the degradation of CDKN1B (). Bafilomycin A1, which is an endosomal and lysosomal acidification inhibitor that can inhibit the fusion of autophagosomes and lysosomes, also delayed the degradation of CDKN1B (). Another autophagy inhibitor, 3-MA, could more clearly inhibit the degradation of CDKN1B in activated T cells (Fig. S4). 3-MA did not affect the activation of T cells as detected by the expression of murine IL2RA/CD25 and CD69 (data not shown). Considering the published data that the proliferation of T cells from CDKN1BT187A knockin mice is decreased, but can still be promoted by IL-2,Citation44 there may be a proteasome-independent pathway for the degradation of CDKN1B. Our data shown here from mouse genetic models demonstrate that autophagy is required for CDKN1B degradation in both naïve and activated T cells.
Discussion
We have previously reported that autophagy developmentally regulates the homeostasis of organelles, such as the ER and mitochondria, and promotes T cell survival.Citation18,19 Here we demonstrate that Atg7-deficient T cells behave like Atg5- or Atg3-deficient T cells and cannot proliferate efficiently in vivo and in vitro. Interestingly, a recent report shows that autophagy-deficient invariant NKT cells also exhibit defective proliferation.Citation12 However, the molecular mechanism underlying the defective proliferation in both types of T cells is not clear. The primary response of autophagy-deficient CD8+ T cells against the LM infection is severely impaired due to proliferative effects both in vitro and in vivo. Consistently, autophagy-deficient T cells fail to enter into S phase after TCR stimulation. The main negative cell-cycle regulator, CDKN1B, is accumulated in naïve autophagy-deficient T cells and CDKN1B cannot be degraded after T cell activation in the absence of autophagy. Furthermore, genetic deletion of a single CDKN1B allele restores the proliferative capacity in autophagy-deficient T cells. Although both CDKN1B and CDKN1 are accumulated in autophagy-deficient T cells, the genetic restoration by deleting a single CDKN1B allele suggests that CDKN1 is not the primary inhibitor of proliferation of autophagy-deficient T cells. Moreover, we found that CDKN1B is ubiquitinated and physiologically associated with an autophagy receptor molecule, SQSTM1. Most importantly, natural CDKN1B forms large polymers. This is the first evidence to indicate that autophagy regulates the cell-cycle negative regulator, CDKN1B, in naïve T cell and reduces its abundance when T cells are activated to enter into the cell cycle.
The level of CDKN1B remains higher during the early to middle of the G1 phase to prevent cell-cycle progression by inhibiting the kinase activity of CCNE- or cyclin A-CDK2 complex. Intensive studies have been published about the degradation of CDKN1B during cell-cycle regulation. Previous reports showed that CDKN1B is degraded by the ubiquitin/proteasome system upon cell-cycle entry. This proteasome degradation of CDKN1B is initiated by CCNE-CDK2-dependent CDKN1B phosphorylation at T187 and then recognized by the SCF-SKP2 ubiquitin ligase complex.Citation27,45 CDKN1B is also phosphorylated at a highly conserved tyrosine 88 (Y88) by the Src-family kinase LYN or the oncogenic product, BCR-ABL. The phosphorylation of Y88 facilitates the phosphorylation of T187 and is followed by SKP2-dependent degradation.Citation46 CDKN1B localizes to the nucleus to inhibit cell-cycle progression and can be redistributed to the cytosol for degradation upon cell-cycle entry. This cytoplasmic relocation is dependent upon the phosphorylation of serine 10. Although CDKN1B is generally degraded in the cytosol, it can also be degraded in the nucleus.Citation47 The degradation of CDKN1B can also be accomplished through a SKP2-independent ubiquitination pathway demonstrated in SCF (SKP2)-deficient cells.Citation48,49 Another study indicates that CDKN1B is downregulated through endo-lysosomal degradation, mediated by an interaction with endosomal protein SNX6 (sorting nexin 6).Citation50
Although it has been demonstrated that CDKN1B is degraded by the proteasome, our genetic mouse model provides evidence that the intracellular homeostasis of CDKN1B is maintained by autophagy in both naïve and activated T lymphocytes as CDKN1B is not degraded efficiently in the absence of autophagy. Why does CDKN1B require autophagy to maintain its basal levels in naïve T cells and to be further degraded upon activation to enter into S phase? The reason may be that native CDKN1B forms large polymers which require autophagy for breaking down. Our data suggest that autophagy is a downstream step for degradation of CDKN1B.
The proteasome and autophagosome are the main organelles to degrade misfolded proteins or protein aggregates.Citation42 Ubiquitination of target proteins designates them as a substrate for degradation. Indeed, CDKN1B is ubiquitinated. K48-linked ubiquitin chains target the protein to the proteasome, while the K63-linked ubiquitin chain proteins are shuttled into the autophagy pathway.Citation51 Whether the ubiquitination on CDKN1B is K48- or K63-linked remains to be determined. We examined the role of proteasome-mediated degradation of CDKN1B by testing CDKN1B levels in the presence of proteasome inhibitors in normal T cells. Interestingly, proteasome inhibitors delayed but not completely prevented CDKN1B degradation. This result is consistent with a role of autophagy in CDKN1B degradation. Knockdown of the CDKN1B E3 ligase SCFSkp2 caused cell-cycle arrest and increased apoptosis in which the cells displayed characteristics of both activation and autophagy.Citation52 The energy-sensing pathway regulates the phosphorylation of threonine 198 (T198) of CDKN1B and increases its stability. Ectopic expression of the phosphomimetic T198 to aspartic acid mutation (T198D) induces the activation of autophagy.Citation53 This suggests that autophagy and the cell cycle may regulate each other, especially during stressful conditions, including proliferation.
Our finding that autophagy selectively degrades CDKN1B may provide new insights for regulating immune responses through autophagy inhibition or induction. Previous studies have provided examples of selective targeting of ubiquitinated BCL10 in T cells by autophagy.Citation14 A recent study also demonstrates that chaperone-mediated autophagy regulates T cell response by degrading RCAN1.Citation54 Two recently published findings indicate that autophagy is critical in the regulation of memory CD8+ T cells using virus infection models.Citation10,11 Autophagy is reduced in T cells from aging mice and enhancing autophagy could restore CD8 memory response.Citation11 Our data provides new clues and explanations for these published findings. Also, selective degradation of CDKN1B by autophagy may be a mechanism of action of chloroquine in the treatment of autoimmune diseases. Considering the function of CDKN1B in the regulation of cancer cell growth, inhibition of autophagy may be used as a strategy to treat cancer. Our preliminary experiments indicated that the autophagy inhibitor, 3-MA, could inhibit the proliferation of a leukemia cell line Jurkat (data not shown). Above all, our data suggests that autophagy regulates the abundance of the cell-cycle inhibitor, CDKN1B, and selectively degrades CDKN1B upon cell activation and entry into the cell cycle.
Materials and Methods
Mice
Atg3 floxed mice (Atg3f/f) were generated in our lab and described in detail.Citation18 Atg7 floxed (Atg7f/f) mice have been previously described.Citation55 The Lck-Cre (012837), ER-Cre (004847), Cdkn1b+/- (B6.129S4-Cdkn1btm1Mlf/J; 002781), OT-I (003831) and B6.SJL-Ptprca Pepcb/BoyJ(CD45.1, 002014) mice were purchased from The Jackson Laboratory. All mice were housed in a specific pathogen-free facility at Duke University and used at 6 to 8 wk of age. All animals were used according to protocols approved by the Duke University Institutional Animal Care and Use Committee.
Proliferation assay
Splenocytes from Atg7f/f, Atg7f/fLck-Cre, Atg3f/f, Atg3f/f ER-Cre or Atg3f/fER-Cre Cdkn1b+/- mice were incubated with CFSE (5 μM; Life Technologies, C34554) for 5 min at room temperature (RT) in phosphate-buffered saline (PBS; Life Technologies, 10010–023) containing 5% fetal bovine serum. Then the CFSE loaded cells were washed 3 times with 5% fetal bovine serum-PBS. After washing, the splenocytes were stimulated with plate bound anti-CD3 (5 μg/ml in PBS, 2C11; 100302), soluble anti-CD3 (5 μg/ml) plus anti-CD28 (2 μg/ml; 122003), or PMA (10 ng/ml; Sigma, P8139) plus ionomycin (300 ng/ml; Sigma, I9657) for 72 h. CFSE diluted cells were analyzed by flow cytometry (FACSCanto II, BD, San Jose, CA). All antibodies are from Biolegend. ANXA5/Annexin V-FITC is from BD Bioscience (556420).
Adoptive transfer and Listeria monocytogenes infection
Atg3f/f OT-I or Atg3f/fER-Cre OT-I mice were injected with 0.4 mg per mouse tamoxifen (Sigma, T5648) intraperitoneally (i.p.) once a day for 3 d. Three wk after the first injection, the splenocytes were harvested. Using an EasySep mouse CD8 T cell negative-selection kit (StemCell Technologies, 19753), the purified CD8+ OT-I T cells (2×104 per mouse) from Atg3f/f OT-I or Atg3f/fER-Cre OT-I mice were transferred to PTPRCa/CD45.1 through i.v. injection. The following day, recombinant L. monocytogenes which express chicken OVA (LM-OVA 1×104 colony-forming units [cfu] per mouse) was injected i.v.. Peripheral blood was drawn at d 5 and d 7 after infection. Antigen-specific CD8+ T cells recognizing OVA were stained with Dimer X (BD Bioscience, 550750) loaded with OVA peptide 257–264 (American Peptide Company, 74-1-16A). The donor cells (PTPRCb/CD45.2+) and host cells (PTPRCa/CD45.1+) were distinguished by anti-PTPRCa/CD45.1-APC (BioLegend, 110713) and anti-PTPRCb/CD45.2-APC/Cy7 antibodies (BioLegend, 109823).
In vivo proliferation assay
CD8+ T cells were purified from splenocytes of tamoxifen inducibly deleted Atg3f/fER-Cre OT I mice or Atg3f/f OT I control mice. The purified CD8+ T cells were loaded with CFSE and injected into normal host mice intravenously. Twenty-four h later, the adoptively transferred mice were infected with LM-OVA (1×104 cfu/mouse in 0.1ml) intravenously. The splenocytes were harvested 48 h after infection. CFSE dilution was analyzed in CD8+Dimer X+CFSE+ cells. The cell number of each generation was analyzed and calculated by Software Flowjo (TreeStar Inc., Ashland, OR).
Cell-cycle analysis
Splenocytes from Atg3f/f, Atg3f/f Lck-Cre or Atg7f/f, Atg7f/fLck-Cre mice were stimulated with anti-CD3 (2C11, 5 µg/ml) plus anti-CD28 (2 µg/ml) overnight. Stimulated and control cells were harvested and fixed with 70% ethanol, incubated on ice for 15 min, washed, and stained with propidium iodide (PI) solution (50 µg/ml PI [Sigma, P4170], 0.1 mg/ml RNAse A [Sigma, R6513], 0.05% Triton X-100 [Sigma, X100] in PBS) for 30 min at 37˚C. After washing, the cell cycle was analyzed by flow cytometry (FACScan or Canton II, BD Bioscience, San Jose, CA).
Western blot
Total T cells, CD4, or CD8 T cells were purified by EasySep mouse CD4 or CD8 T cell negative-selection kits (StemCell Technologies, 19752, 19753). The purity was more than 95%. Cell lysates were prepared. Samples were separated by SDS-PAGE and transferred to Immobilon-FL polyvinylidene difluoride (PVDF) membranes (Millipore, IPVH00010). The PVDF membranes were blocked with 5% nonfat milk for 1 h at RT. The primary antibodies were incubated with working concentration in 3% BSA (Sigma, A2153)-PBS (Life Technologies, 10010023) solution with 0.02% Tween 20 (Sigma, P1379) at 4˚C overnight. After washing 3 times, the membranes were incubated with antibodies specific for the pertinent species and labeled with Alexa Fluor 680- (Life Technologies, A-21059 or A-21096) or IRDye 800- (Rockland Immunochemicals, 605–732–002) secondary antibodies at RT for 1 h. After washing, the blots were visualized using an Odyssey Infrared Imaging System and analyzed using Odyssey software (LI-COR Bioscience, Lincoln, NE). Anti-BCTB/Actin antibody (sc-1616) and anti-CDKN1B mAb (sc-1641) are from Santa Cruz Biotechnology, Inc. Anti-ubiquitin mAb (646301) is from BioLegend.
Subcellular fractionation
Subcellular fractionation was performed with OptiPrep (Axis-Shield PoC, 1114542), according to the literature and protocols recommended by the manufacturer with some modifications.Citation56 The working solution of 50% (w/v) iodixanol was prepared by diluting the OptiPrep with buffer (130 mM KCl, 25 mM NaCl, 6 mM EGTA, 150 mM Tris-Cl, pH 7.4). Splenocytes from 3 normal unmanipulated mice were suspended in RPMI with chloroquine (50 µg/ml; Sigma, C6628) and stimulated with anti-CD3 antibody (2C11) at 37oC for 6 h to activate autophagy. The activation of T cells was confirmed by CD69 and IL2RA/CD25 expression. The cells were harvested and washed with precooled homogenization buffer (130 mM KCl, 25 mM NaCl, 1 mM EGTA, 25 mM Tris-Cl, pH 7.4), and suspended in homogenization buffer with chloroquine and protease inhibitors (Sigma, P1860). The homogenate was prepared manually by dounce tissue grinder with glass pestle (1 ml capacity; Wheaton, 357538) on ice. The homogenates were centrifuged at 1000 g for 5 min to remove the nuclei. The supernatant fraction (containing all subcellular membrane structures) was transferred to another centrifuge tube (Beckman, Brea, CA) and adjusted to 30% (w/v) iodixanol with the working solution of OptiPrep. Different concentrations of OptiPrep gradients were layered in the following amount and order: 1 ml 30%, 1.5 ml 25%, 2 ml 20%, 2 ml 15%, 1.5 ml 10% and 1 ml 5%. The adjusted supernatant fraction (2 ml) was layered at the top of centrifuge tube and the swing-bucket 40Ti rotor (Beckman) was used to centrifuge at 48,000 g for 18 h at 4˚C. One ml of each fraction was collected. The expression levels of target proteins in each fraction were analyzed by SDS-PAGE and western blot.
Affinity isolation assay
The Cdkn1b and Sqstm1 were cloned by RT-PCR with the cDNA from mouse T cells and inserted into a pMI expression vector. The cloning primer sequences are: Cdkn1b forward (F), GCGGATCCATGTCAAACGTGAGAGTGTCTAACGGGAGC, Cdkn1b reverse (R), GCGAATTCCTTGTCGTCATCGTCTTTGTAGTC CGTCTGGCGTCGAAGGCCGGGCTTC, Sqstm1 F, GCGGATCCATGGCGTCGTTCACGGTGAAGGCCT, Sqstm1 R, GC GTCGACAGCGTAATCTGGAACATCGTATGGGTACAATGGTGGAGGGTGCTTCGAATACTGGAT. The reverse primer contains the FLAG or HA sequences. CDKN1B was tagged with FLAG and SQSTM1 was tagged with HA. After 293T cells were cotransfected with CDKN1B-FLAG and SQSTM1-HA, the cell lysates were prepared in lysis buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1 mM PMSF, 1 mM sodium vanadate (Sigma, 72060), 2 mM sodium pyrophosphate (Sigma, P8010),10 mM β-glycerol phosphate (Sigma, 50020), 10 µg/ml leupeptin (Sigma, 62070), 10 µg/ml aprotinin (Sigma, A1153), and 1% Triton X-100). The immunoprecipitation was performed with anti- FLAG antibody (Biolegend, 923601) using Dynabeads Protein G beads (Life Technologies, 10003D). After SDS-PAGE and western blot, the membranes were probed with anti-HA antibody (Sigma, H3663). The endogenous association was analyzed with the cell lysates from splenocytes from Atg3f/f, Atg3f/fER-Cre mice and inducibly-deleted Atg3 by tamoxifen injection. Anti-SQSTM1 antibody is from MBL (PM045).
Statistics analysis
The 2-tail distribution Student t test was used to analyze the significance through Excel. A p value < 0.05 was considered statistical significance.
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest.
Supplemental.docx
Download MS Word (1 MB)Acknowledgments
We thank Dr. Qi-Jing Li for critical discussion and suggestions. We thank Dr. Michael Cook, Lynn Martinek, Nancy Martin and Dr. Bin Li for helping with the flow cytometry.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work is supported by NIH grants AI074754 and AI074944 to YWH.
Related Research Data
References
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/10.1038/ncb0910-814
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/10.1016/j.cell.2011.10.026
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722-37; PMID:24064518; http://dx.doi.org/10.1038/nri3532
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005; 307:593-6; PMID:15591165; http://dx.doi.org/10.1126/science.1104904
- Munz C. Antigen processing via autophagy–not only for MHC class II presentation anymore? Curr Opin Immunol 2010; 22:89-93; PMID:20149615; http://dx.doi.org/10.1016/j.coi.2010.01.016
- McLeod IX, Jia W, He YW. The contribution of autophagy to lymphocyte survival and homeostasis. Immunol Rev 2012; 249:195-204; PMID:22889223; http://dx.doi.org/10.1111/j.1600-065X.2012.01143.x
- He MX, McLeod IX, Jia W, He YW. Macroautophagy in T lymphocyte development and function. Front Immunol 2012; 3:22; PMID:22566906
- Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol 2010; 185:7349-57; PMID:21059894; http://dx.doi.org/10.4049/jimmunol.1000576
- Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol 2014; 15:1152-61; PMID:25362489; http://dx.doi.org/10.1038/ni.3025
- Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, Watson AS, Cerundolo V, Townsend AR, Klenerman P, et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife 2014; 3:e03706 (1-21); PMID:25385531
- Pei B, Zhao M, Miller BC, Vela JL, Bruinsma MW, Virgin HW, Kronenberg M. Invariant NKT Cells Require Autophagy To Coordinate Proliferation and Survival Signals during Differentiation. J Immunol 2015; 194:5872-84; PMID:25926673; http://dx.doi.org/10.4049/jimmunol.1402154
- Salio M, Puleston DJ, Mathan TS, Shepherd D, Stranks AJ, Adamopoulou E, Veerapen N, Besra GS, Hollander GA, Simon AK, et al. Essential role for autophagy during invariant NKT cell development. Proc Natl Acad Sci U S A 2014; 111:E5678-87; PMID:25512546; http://dx.doi.org/10.1073/pnas.1413935112
- Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity 2012; 36:947-58; PMID:22658522; http://dx.doi.org/10.1016/j.immuni.2012.04.008
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 2007; 7:767-77; PMID:17767194; http://dx.doi.org/10.1038/nri2161
- Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol 2009; 182:4046-55; PMID:19299702; http://dx.doi.org/10.4049/jimmunol.0801143
- Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol 2011; 186:1564-74; PMID:21191072; http://dx.doi.org/10.4049/jimmunol.1001822
- Jia W, He YW. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol 2011; 186:5313-22; PMID:21421856; http://dx.doi.org/10.4049/jimmunol.1002404
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 2007; 204:25-31; PMID:17190837; http://dx.doi.org/10.1084/jem.20061303
- Parekh VV, Wu L, Boyd KL, Williams JA, Gaddy JA, Olivares-Villagomez D, Cover TL, Zong WX, Zhang J, Van Kaer L. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of vps34. J Immunol 2013; 190:5086-101; PMID:23596309; http://dx.doi.org/10.4049/jimmunol.1202071
- Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, Graham DB, Mizushima NN, Xavier R, Virgin HW, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy 2009; 5:625-35; PMID:19276668; http://dx.doi.org/10.4161/auto.5.5.8133
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501-12; PMID:10385618; http://dx.doi.org/10.1101/gad.13.12.1501
- Murray AW. Recycling the cell cycle: cyclins revisited. Cell 2004; 116:221-34; PMID:14744433; http://dx.doi.org/10.1016/S0092-8674(03)01080-8
- Lupino E, Buccinna B, Ramondetti C, Lomartire A, De Marco G, Ricotti E, Tovo PA, Rinaudo MT, Piccinini M. In CD28-costimulated human naive CD4+ T cells, I-kappaB kinase controls the expression of cell cycle regulatory proteins via interleukin-2-independent mechanisms. Immunology 2010; 131:231-41; PMID:20465575; http://dx.doi.org/10.1111/j.1365-2567.2010.03297.x
- Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell 2008; 14:159-69; PMID:18267085; http://dx.doi.org/10.1016/j.devcel.2008.01.013
- Mohapatra S, Agrawal D, Pledger WJ. CDKN1B regulates T cell proliferation. J Biol Chem 2001; 276:21976-83; PMID:11297537; http://dx.doi.org/10.1074/jbc.M009788200
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995; 269:682-5; PMID:7624798; http://dx.doi.org/10.1126/science.7624798
- Kipreos ET, Pagano M. The F-box protein family. Genome Biol 2000; 1:REVIEWS3002; PMID:11178263; http://dx.doi.org/10.1186/gb-2000-1-5-reviews3002
- Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999; 1:193-9; PMID:10559916; http://dx.doi.org/10.1038/12013
- Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W. p45SKP2 promotes CDKN1B degradation and induces S phase in quiescent cells. Nat Cell Biol 1999; 1:207-14; PMID:10559918; http://dx.doi.org/10.1038/12027
- Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol 2001; 3:321-4; PMID:11231585; http://dx.doi.org/10.1038/35060126
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 2004; 6:661-72; PMID:15130491; http://dx.doi.org/10.1016/S1534-5807(04)00131-5
- Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol 2004; 6:1229-35; PMID:15531880; http://dx.doi.org/10.1038/ncb1194
- Jatzek A, Tejera MM, Singh A, Sullivan JA, Plisch EH, Suresh M. p27(Kip1) negatively regulates the magnitude and persistence of CD4 T cell memory. J Immunol 2012; 189:5119-28; PMID:23071285; http://dx.doi.org/10.4049/jimmunol.1201482
- Singh A, Jatzek A, Plisch EH, Srinivasan R, Svaren J, Suresh M. Regulation of memory CD8 T-cell differentiation by cyclin-dependent kinase inhibitor CDKN1B. Mol Cell Biol 2010; 30:5145-59; PMID:20805358; http://dx.doi.org/10.1128/MCB.01045-09
- Li L, Iwamoto Y, Berezovskaya A, Boussiotis VA. A pathway regulated by cell cycle inhibitor CDKN1B and checkpoint inhibitor Smad3 is involved in the induction of T cell tolerance. Nat Immunol 2006; 7:1157-65; PMID:17013388; http://dx.doi.org/10.1038/ni1398
- Pua HH, He YW. Autophagy and lymphocyte homeostasis. Curr Top Microbiol Immunol 2009; 335:85-105; PMID:19802561
- Dunkle A, Dzhagalov I, Gordy C, He YW. Transfer of CD8+ T cell memory using Bcl-2 as a marker. J Immunol 2013; 190:940-7; PMID:23269245; http://dx.doi.org/10.4049/jimmunol.1103481
- Zhang N, He YW. The antiapoptotic protein Bcl-xL is dispensable for the development of effector and memory T lymphocytes. J Immunol 2005; 174:6967-73; PMID:15905539; http://dx.doi.org/10.4049/jimmunol.174.11.6967
- Bagui TK, Cui D, Roy S, Mohapatra S, Shor AC, Ma L, Pledger WJ. Inhibition of CDKN1B gene transcription by mitogens. Cell Cycle 2009; 8:115-24; PMID:19158484; http://dx.doi.org/10.4161/cc.8.1.7527
- Li WQ, Jiang Q, Aleem E, Kaldis P, Khaled AR, Durum SK. IL-7 promotes T cell proliferation through destabilization of CDKN1B. J Exp Med 2006; 203:573-82; PMID:16492801; http://dx.doi.org/10.1084/jem.20051520
- Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ 2013; 20:21-30; PMID:22722335; http://dx.doi.org/10.1038/cdd.2012.72
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259-69; PMID:19450525; http://dx.doi.org/10.1016/j.molcel.2009.04.026
- Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakides TR, Roberts JM. A mouse knock-in model exposes sequential proteolytic pathways that regulate CDKN1B in G1 and S phase. Nature 2001; 413:323-7; PMID:11565035; http://dx.doi.org/10.1038/35095083
- Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev 1999; 13:1181-9; PMID:10323868; http://dx.doi.org/10.1101/gad.13.9.1181
- Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jäkel H, Kullmann M, Kriwacki RW, Hengst L. Cdk-inhibitory activity and stability of CDKN1B are directly regulated by oncogenic tyrosine kinases. Cell 2007; 128:269-80; PMID:17254966; http://dx.doi.org/10.1016/j.cell.2006.11.047
- Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J 2001; 20:6672-82; PMID:11726503; http://dx.doi.org/10.1093/emboj/20.23.6672
- Hara T, Kamura T, Nakayama K, Oshikawa K, Hatakeyama S. Degradation of p27(Kip1) at the G(0)-G(1) transition mediated by a Skp2-independent ubiquitination pathway. J Biol Chem 2001; 276:48937-43; PMID:11682478; http://dx.doi.org/10.1074/jbc.M107274200
- Hara T, Kamura T, Kotoshiba S, Takahashi H, Fujiwara K, Onoyama I, Shirakawa M, Mizushima N, Nakayama KI. Role of the UBL-UBA protein KPC2 in degradation of p27 at G1 phase of the cell cycle. Mol Cell Biol 2005; 25:9292-303; PMID:16227581; http://dx.doi.org/10.1128/MCB.25.21.9292-9303.2005
- Fuster JJ, Gonzalez JM, Edo MD, Viana R, Boya P, Cervera J, Verges M, Rivera J, Andrés V. Tumor suppressor p27(Kip1) undergoes endolysosomal degradation through its interaction with sorting nexin 6. FASEB J 2010; 24:2998-3009; PMID:20228253; http://dx.doi.org/10.1096/fj.09-138255
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 2008; 17:431-9; PMID:17981811; http://dx.doi.org/10.1093/hmg/ddm320
- Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, Corral LG, Krenitsky VP, Xu W, Moutouh-de Parseval L, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008; 111:4690-9; PMID:18305219; http://dx.doi.org/10.1182/blood-2007-09-112904
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 2007; 9:218-24; PMID:17237771; http://dx.doi.org/10.1038/ncb1537
- Valdor R, Mocholi E, Botbol Y, Guerrero-Ros I, Chandra D, Koga H, Gravekamp C, Cuervo AM, Macian F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol 2014; 15:1046-54; PMID:25263126; http://dx.doi.org/10.1038/ni.3003
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/10.1083/jcb.200412022
- Velikkakath AK, Nishimura T, Oita E, Ishihara N, Mizushima N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell 2012; 23:896-909; PMID:22219374; http://dx.doi.org/10.1091/mbc.E11-09-0785