
Abstract
HPSE (heparanase) is the predominant enzyme in mammals capable of cleaving heparan sulfate, an activity highly implicated in cellular invasion and tumor metastasis. HPSE expression is induced in many types of cancer and increased HPSE levels are most often associated with increased tumor metastasis and reduced patient survival post operation. In addition, HPSE induction is associated with progression of the primary tumors but the mechanism(s) underlying tumor expansion by HPSE have not been sufficiently resolved. Our results establish a role for heparanase in modulating autophagy in normal and malignant cells, thereby conferring growth advantages as well as resistance to chemotherapy.
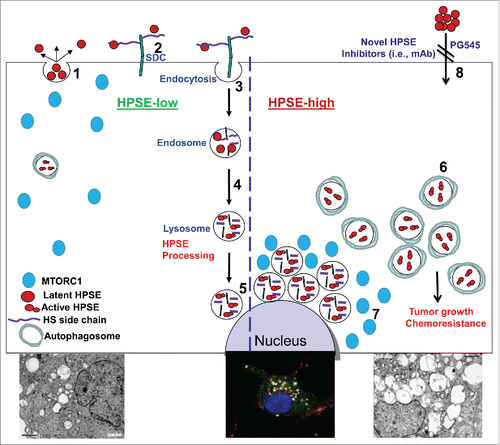
We have shown previously that following secretion as a latent 65-kDa enzyme (; 1), HPSE rapidly interacts with cell membrane heparan sulfate proteoglycans (HSPG; e.g., SDC/syndecans; ; 2), followed by internalization (; 3) and processing into a highly active enzyme composed of 50- and 8-kDa subunits (; 4). Notably, active HPSE resides primarily within endocytic vesicles, assuming a polar, peri-nuclear localization and colocalizing with lysosomal markers (; 5 and lower middle panel). In spite of its localization in a highly active protein degradation environment such as the lysosome, HPSE appears stable and exhibits a half-life of about 30 h, leading us to hypothesize that HPSE plays a role in lysosomal function. More specifically, we examined the possible involvement of HPSE in autophagy in the context of tumor growth and chemoresistance.
Figure 1. A schematic model of HPSE trafficking and function in autophagy. Once secreted (1), HPSE rapidly interacts with cell membrane HSPGs such as SDC/syndecans (2), followed by rapid endocytosis of the HPSE-HSPG complex (3). Conversion of endosomes to lysosomes (4) results in HPSE processing and activation (5). Typically, HPSE appears at perinuclear lysosomal vesicles (5 and middle lower image). Lysosomal HPSE drives fusion with autophagosomes and controls the basal levels of autophagy. Cancer cells that exhibit a high content of HPSE (HPSE-high) are endowed with increased levels of autophagy (6 and left vs. right lower electron micrographs) that promote tumor growth and chemoresistance. Enhanced autophagy by HPSE is associated with reduced RPS6KB phosphorylation levels and accumulation of MTORC1 at perinuclear areas (7) vs. a more diffuse distribution in control (HPSE-low) cells. This function of HPSE within the cell encourages the development of a new class of inhibitors that will prevent HPSE uptake and lysosomal accumulation (8). HS, heparan sulfate; mAb, monoclonal antibody.
We found that HPSE localizes to autophagosomes, colocalizing with LC3. Moreover, autophagy (i.e., LC3-II levels) is decreased in mouse embryonic fibroblasts (MEFs) and tissues originating from HPSE-deficient mice compared with control mice, whereas increased LC3-II levels are noted in tissues of transgenic mice overexpressing HPSE. An even higher increase of autophagy was observed following overexpression of HPSE in tumor-derived cells. Autophagy is markedly increased under resting conditions, and a further increase in autophagy levels is noted following chloroquine treatment, evident also by electron microscopy (; 6 and left vs. right lower panels). We next examined whether induction of autophagy underlies the pro-tumorigenic function of HPSE applying inhibitors of the lysosome (chloroquine), HPSE (PG545) or both. Utilizing the MTT assay we found that chloroquine treatment reduces cell viability compared with control untreated cells, a decrease that was most pronounced when combined with pharmacologically relevant concentrations of the HPSE inhibitor PG545. Moreover, colony formation in soft agar is markedly reduced by chloroquine treatment, synergizing with PG545 in term of colony number and size. The in vitro results were closely recapitulated in a tumor model in vivo, where tumor growth is attenuated by chloroquine and PG545 as single agents and is most evident when the 2 compounds are combined. These results strongly suggest that the pro-tumorigenic properties of HPSE are mediated, at least in part, by promoting autophagy.
Mechanistically, autophagy induction by HPSE appears to involve the mechanistic target of rapamycin (serine/threonine kinase) complex 1 (MTORC1). This nutrient-sensing kinase acts as a master negative regulator of autophagy because during starvation, MTORC1 is inhibited and this induces autophagy. In cells overexpressing HPSE we observed reduced phospho-RPS6KB/p70 S6-kinase (an MTORC1 substrate) levels, in agreement with increased autophagy. Moreover, a substantial increase in RPS6KB phosphorylation level is observed in HPSE-deficient MEF vs. control cells, in agreement with reduced autophagy in these cells. In accordance with these results we observed increased phosphorylation of RPS6KB in tumor xenografts following treatment with inhibitors of HPSE (PG545) or the lysosome (chloroquine). The alterations in RPS6KB phosphorylation in relation to HPSE levels are associated with changes in the cellular localization of MTORC1. In control MEFs, MTORC1 is localized within scattered vesicles but appears diffusely distributed in HPSE-deficient MEFs. We also found that MTORC1 assumes a perinuclear localization in cells with a high content of HPSE (; 7) compared with a more diffuse distribution in control cells, and noted that MTORC1 colocalizes with HPSE and LysoTracker that labels acidic lysosomal vesicles.
We further found that HPSE endows cancer cells with stress resistance, and chemoresistance. For example, amino acid starvation reduces the viability of control cells to half compared with untreated cells but does not reduce the viability of HPSE overexpressing cells. Notably, the resistance of HPSE-overexpressing cells is reversed by combining amino acid starvation with chloroquine, suggesting that resistance to starvation stress is mediated by induction of autophagy in these cells and can be prevented by autophagy inhibition. More importantly, HPSE overexpressing carcinoma cells are more resistant to cisplatin treatment, and combining cisplatin and chloroquine similarly prevents this tolerance. This implies that HPSE provides cells with chemoresistance that is mediated, in part, by increased autophagy.
In addition to providing a novel mechanistic insight for the pro-tumorigenic function of HPSE, these results are important in at least 2 other aspects. First, while understanding the role of HPSE in pathological disorders (tumorigenesis, inflammation, diabetes) is advancing, its role under normal cell physiology is still unclear. Decreased autophagy levels observed in MEFs and tissues derived from HPSE-deficient mice suggest that HPSE functions to control autophagy levels thereby contributing to maintain homeostasis. From a translational point of view, the results strongly imply that HPSE functions inside the cell to promote autophagy and tumor growth. Thus, while the traditional thinking envisions HPSE as an enzyme that functions extracellularly to cleave heparan sulfate and facilitate remodeling of the extracellular matrix and release of extracellular matrix-bound growth-promoting factors, our results suggest that HPSE may also function from inside the cells. Targeting HPSE in the lysosome may therefore become as important as its inhibition extracellularly, but the ability of currently available HPSE inhibitors, some of which are being evaluated in Phase I clinical trials (i.e., PG545 and Roneparstat), to cross the plasma membrane and enter the cell is unclear. Alternatively, the pro-autophagy function of HPSE can be inhibited by decreasing its lysosomal content. For example, autophagy is decreased by the HPSE inhibitor PG545 and this is associated with accumulation of HPSE in the cell culture medium and, concomitantly, reduced HPSE levels in the lysosomes. This opens the way for the development of a new class of highly specific inhibitors (i.e., monoclonal antibodies) that prevent HPSE uptake by targeting its heparin-binding domain (; 8).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by research grants awarded to I.V. by the Israel Science Foundation (grant 601/14); National Cancer Institute, NIH (grant CA106456); the United States-Israel Binational Science Foundation (BSF); the Israel Cancer Research Fund (ICRF); and the Rappaport Family Institute Fund. I. Vlodavsky is a Research Professor of the ICRF.