
ABSTRACT
Perinatal asphyxia induces neuronal cell death and brain injury, and is often associated with irreversible neurological deficits in children. There is an urgent need to elucidate the neuronal death mechanisms occurring after neonatal hypoxia-ischemia (HI). We here investigated the selective neuronal deletion of the Atg7 (autophagy related 7) gene on neuronal cell death and brain injury in a mouse model of severe neonatal hypoxia-ischemia. Neuronal deletion of Atg7 prevented HI-induced autophagy, resulted in 42% decrease of tissue loss compared to wild-type mice after the insult, and reduced cell death in multiple brain regions, including apoptosis, as shown by decreased caspase-dependent and -independent cell death. Moreover, we investigated the lentiform nucleus of human newborns who died after severe perinatal asphyxia and found increased neuronal autophagy after severe hypoxic-ischemic encephalopathy compared to control uninjured brains, as indicated by the numbers of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3)-, LAMP1 (lysosomal-associated membrane protein 1)-, and CTSD (cathepsin D)-positive cells. These findings reveal that selective neuronal deletion of Atg7 is strongly protective against neuronal death and overall brain injury occurring after HI and suggest that inhibition of HI-enhanced autophagy should be considered as a potential therapeutic target for the treatment of human newborns developing severe hypoxic-ischemic encephalopathy.
Introduction
Perinatal asphyxia-induced brain injury, frequently presenting itself as hypoxic-ischemic encephalopathy (HIE), is one of the most common causes of mortality and long-term neurological impairments in term and preterm neonates, such as cerebral palsy, mental retardation, visual and hearing deficiencies, learning disability, and/or epilepsy.Citation1 This is due to lesions that are found predominantly in the gray matter, including the basal ganglia, thalamus and cortex.Citation2
Although human studies are sparse and difficult to perform, the neuronal cell death patterns in response to hypoxia-ischemia (HI) have been reported to consist mostly of necrotic, apoptotic and some hybrid features defined as the “cell death continuum.”Citation3 Many studies have focused on apoptosis as a therapeutic target for preventing neonatal HI brain injury,Citation4,5 but the prevention of neuronal apoptosis has not yet been proven to be clinically relevant. Current therapeutic options for neonatal HIE are predominantly supportive and seek to maintain physiological parameters. Exceptions to this are early post-insult hypothermia and injections of recombinant human erythropoietin.Citation6,7 The efficacy of these treatments, however, is limited, and an improved understanding of neuronal cell death mechanisms after neonatal HI brain injury is needed to develop novel treatments.
In rodents, the patterns of HI-induced neuronal cell death in the immature brain confirm the presence of mixed morphological phenotypes involving necrotic, apoptotic, and autophagic features, each of which corresponding to a unique set of biochemical and morphological criteria.Citation3, 8-11
Autophagy—which is activated by many forms of stressCitation12—is an important physiological mechanism for degrading long-lived cytosolic protein complexes and aggregates and is the only known pathway for degrading organelles.Citation13 Autophagy recycles amino acids and fatty acids to produce energy and removes damaged organelles. Thus, basal autophagy plays an essential role in cell survival.Citation14,15 However inappropriate activation of autophagy can be directly involved in mediating cell death or can trigger the execution of apoptotic or necrotic cell death.Citation16
The immature brain retains its autophagic machinery to a larger extent than the adult brain, as judged by the levels of LC3-II,Citation11 as part of maintaining normal brain development.Citation17,18 Several studies have reported the presence of enhanced neuronal autophagy in rodent models of perinatal cerebral ischemia or HI.Citation11,19-24 Even if a death-mediating role of autophagy has been demonstrated in most of these studies, as indicated by the protective effects of inhibiting autophagy by both pharmacological and genetic methods,Citation11,19,21-23,25 some studies have reported a protective effect of pretreatment with the autophagy inducer rapamycin.Citation20,24
The present study aims to investigate the effect of selective autophagy inhibition in the pathology of rodent neonatal HI brain injury and provides strong evidence for a prodeath role of HI-induced neuronal autophagy in different cerebral regions. We used mice unable to execute autophagy specifically in neurons due to a tissue-specific deletion (or knockout) of the Atg7 gene (atg7 KO), which is involved in autophagy induction and autophagosome formation. A model of severe neonatal HI affecting most of the brain was used to determine the effects of genetically inhibited autophagy on neuronal cell death and brain injury. Moreover, in order to determine whether autophagy could also be involved in human HIE brain damage, we investigated markers of autophagy in autopsied brain tissues of human term newborns who died from severe asphyxia with HIE focusing on the basal ganglia, a part of the brain highly susceptible to HI injury.
Results
HI induces autophagy in neonatal mice
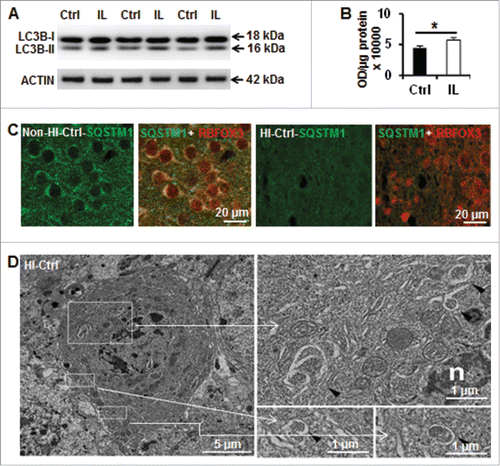
Twenty-four h after HI, immunoblot analysis revealed, as expected, an increase in LC3B-II levels in the ipsilateral hemisphere of Atg7flox/+; Nes-Cre (Ctrl) mice, indicating an increase in autophagosome formation (). Moreover, immunohistochemistry for SQSTM1/p62 (a protein that is selectively degraded during autophagy) shows a reduction of neuronal SQSTM1 expression in the damaged hemisphere (). Ultrastructural studies clearly revealed the formation of autophagosomes in dying neurons () of Ctrl mice after HI. Altogether, these data indicate that autophagy is increased in this mouse model of neonatal HI.
Figure 1. Hypoxia-ischemia induces neuronal autophagy in neonatal Atg7flox/+; Nes-Cre mice. (A) Representative immunoblotting of LC3B in Ctrl (Atg7flox/+; Nes-Cre) mice and (B) its corresponding quantification showed that LC3B-II (16 kDa) was increased in the ipsilateral hemisphere (IL) 24 h after HI compared to an uninjured control brain, (*, P < 0.05, n = 6). (C) Immunostaining of SQSTM1 in the cortex of Ctrl mice suggesting a decrease of its staining in neurons (RBFOX3) 24 h after HI. (D) Dying neurons in Ctrl mice exhibited increased electron density and nuclei with chromatin condensation. They possessed also typical autophagosomes with double membranes in the perikarya (black arrowheads) 24 h after HI. Squared areas are enlarged in the right panels.
Neuronal Atg7 deficiency prevents basal autophagy without increasing stress-related proteins in neonatal mice
In order to investigate the specific role of neuronal enhanced autophagy after HI, we characterized mice bearing a neuron-specific knockout of Atg7 resulting from the inactivation of floxed Atg7 by Cre recombinase expressed under the Nes/nestin promoter (Nes-Cre; Atg7flox/flox). The Nes promoter is commonly used when targeting brain neurons.Citation15 These mice display a nearly complete deficiency of neuronal ATG7 protein, as well as defective autophagy, as shown by the absence of LC3B-II () and pronounced neuronal SQSTM1 accumulation () as early as on postnatal d 9 (P9). Punctate SQSTM1-positive staining was observed in neurons from the cortex, hippocampus, and striatum of atg7 KO mice but not in Ctrl mice, which presented a diffuse labeling ().
Figure 2. Neuronal Atg7 deficiency prevents neuronal basal autophagy and does not alter expression of mitochondria and cell death-related proteins. (A) Representative immunoblotting of ATG7, SQSTM1, and LC3BB in the atg7 KO (Atg7flox/flox; Nes-Cre) and Ctrl (Atg7flox/+; Nes-Cre) mice showing that ATG7 was strongly decreased in KO mice leading to an accumulation of SQSTM1 and absence of LC3BB-II. (B) Representative immunostaining of SQSTM1 in the cortex of Ctrl and atg7 KO mice in the non-HI control condition, confirming the accumulation of SQSTM1 in KO compared to the faint and diffuse expression in Ctrl. (C) Punctate and strong SQSTM1 staining was observed in the cytoplasm of neuronal cells (RBFOX3) in the cortex of atg7 KO mice, but not in the Ctrl as shown by RBFOX3 and SQSTM1. (D) Representative immunoblots from cortical tissue homogenates of P9 Ctrl and atg7 KO mice. Quantification did not show any significant differences between the 2 types of mice for cell death-related proteins AIFM1, CYCS and CASP3, mitochondria-related proteins (SOD2, HSP70, CAT and mitochondrial respiratory chain complexes (COXV, COXIII, COXIV, COXII, COXI). (n = 6/group). KO: atg7 KO (Atg7flox/flox; Nes-Cre) and Ctrl: Atg7flox/+; Nes-Cre.
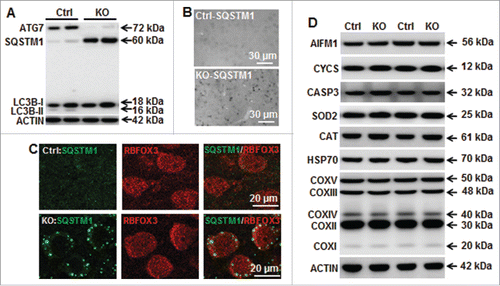
We also verified that the expression of several cell death-related proteins, such as AIFM1/AIF (apoptosis-inducing factor, mitochondria associated), CYCS (cytochrome c, somatic) and CASP3 (caspase 3), was not different between atg7 KO and Ctrl mice (). This indicates that, at the time points investigated, the general apoptotic machinery was transcriptionally intact and unaffected by the ATG7 deficiency. Mitochondria-related proteins, such as SOD2 (superoxide dismutase 2, mitochondrial), HSP70/heat shock protein 70 kDa and CAT (catalase) as well as mitochondria respiratory chain complexes were also similarly abundant in atg7 KO and Ctrl mice (). Thus, neuronal ATG7 deficiency impedes basal autophagy in P9 mice without inducing cellular stress at young developmental stage. This was further confirmed by measuring the transcriptional levels of Keap1-Nfe2l2-related genes (Keap1, Nfe2l2 and Gclc), which are regulators of oxidative stress. Under physiological conditions, the transcription of these genes was not different in atg7 KO mice, indicating that higher levels of Keap1-Nfe2l2 pathway-related genes does not explain the increased resilience to HI (Fig. S1). GSR (glutathione reductase) catalyzes the NADPH-dependent reduction of oxidized glutathione to GSH/reduced glutathione, and thereby maintains adequate levels of GSH. A high GSH/oxidized glutathione ratio is essential for protection against oxidative stress. GSR activity was measured both under control conditions and after HI. GSR activity increased after HI, but the levels were not different between Ctrl mice and atg7 KO mice, neither under control conditions, nor after HI. These data suggest that the protection against HI-induced neuronal death after atg7 KO is not related to enhanced redox capacity (Fig. S2). To further investigate if Atg7 deficiency has effects on mitochondrial function, we examined the transcription of the mitochondrial fission gene Dnm1l/Drp1, and the fusion gene Mfn1 (Fig. S3), as well as 2 key mitochondrial biogenesis-related genes, Ppargc1a/Pgc1a and Nrf1. We also analyzed the mtDNA copy number. In atg7 KO mice (Fig. S4), none of these 4 genes was altered compared with Ctrl mice, and mtDNA copy number was also unaltered. These results suggest that mitochondrial biogenesis, fission and fusion, reflecting the mitochondrial life cycle, were unaffected by atg7 KO.
Neuronal Atg7 deficiency reduced hypoxia-ischemia-induced brain injury
The effects of neuronal Atg7 deficiency on HI-induced autophagy and brain injury were evaluated. In atg7 KO mice, the LC3B-II HI-dependent increase was completely prevented (). Furthermore, at the ultrastructural level, no autophagosomes were observed in atg7 KO mice (data not shown) confirming that ATG7 deficiency efficiently prevented HI-induced neuronal autophagy.
Figure 3. Neuronal Atg7 deficiency reduced hypoxia-ischemia-induced neuronal autophagy and brain injury. (A) Representative immunoblots of LC3BB from non-HI control (Cont) and HI ipsilateral (IL) hemispheres of Ctrl and atg7 KO mice 24 h after HI and the corresponding quantification of LC3BB-II showed that Atg7 deletion completely prevented HI-induced LC3BB-II increase, (*P < 0.05, n = 6/group). (B) Representative MAP2 staining in the ipsilateral hemisphere of the hippocampus (left) and striatum (right) level with the indication of MAP2 negative areas. (C) Representative MAP2 staining of coronal brain sections 8 d after HI at the levels of the dorsal hippocampus (left panels) and striatum (right panels) from Ctrl and atg7 KO mice and (D) measure of total tissue loss volume demonstrated a strong reduction of the lesion in the atg7 KO mice (Ctrl: n = 38, atg7 KO: n = 28), (**, P < 0.01). (E) Pathological scores performed in the cortex (Cx), hippocampus (Hip), striatum (Str), and thalamus (Tha) confirmed the greater resistance of atg7 KO mice compared to Ctrl mice, **, P < 0.01. (F) There was no difference in the neuroprotection provided by Atg7 deletion between males (Ctrl: n = 22, atg7 KO: n = 16) and females (Ctrl: n = 16, atg7 KO: n = 12) (*, P < 0.05). (G) The neuronal cell architecture in the Ctrl and atg7 KO mice in non-HI control and 24 h after HI indicated that dying neurons with condensed chromatin were more frequent in Ctrl than in atg7 KO. Squared areas are enlarged in the right panels. Moreover, the neuronal architecture was relatively well preserved in the atg7 KO mouse compared to that of the Ctrl mice. KO: atg7 KO (Atg7flox/flox; Nes-Cre) and Ctrl: Atg7flox/+; Nes-Cre.
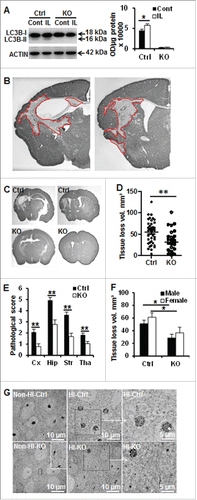
Eight d after neonatal HI, the histologically detectable brain injury encompassed several areas in the cortex, hippocampus, striatum, and thalamus (). The overall brain injury volume—as indicated by tissue loss—was 55.1 ± 4.5 mm3 in the Ctrl mice. Tissue loss was decreased by 42% (32.0 ± 5.0 mm3, p = 0.001) in the atg7 KO mice (). This protective effect was confirmed by calculating the pathological scores of the different affected brain areas (cortex, hippocampus, striatum, and thalamus) (). No significant difference between males and females was observed in tissue loss volume, neither in atg7 KO nor in Ctrl mice (). Ultrastructural studies revealed that the number of dying neurons (per visual field) with condensed chromatin in their nuclei was clearly reduced in atg7 KO mice (86.3 ± 3.5% in Ctrl and 20.9 ± 3.4% in atg7 KO mice P < 0.0001, n = 6/group), indicating that the neuronal architecture was more intact in atg7 KO than in Ctrl mice ().
Neuronal Atg7 deficiency decreased neuronal death in the immature brain after hypoxia-ischemia
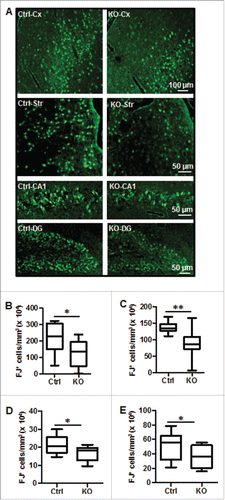
Neuronal cell death in the cortex and striatum as well as in the dentate gyrus (DG) and the cornu ammonis (CA) was investigated by using Fluoro-Jade staining, a nonspecific neuronal marker of cell death, 24 h after HI (). The number of Fluoro-Jade-positive neurons in all 4 cerebral regions was significantly reduced in atg7 KO compared to Ctrl mice after HI ().
Figure 4. Neuronal Atg7 deficiency reduced hypoxia-ischemia-induced neuronal cell death. (A) Representative Fluoro-Jade (FJ) staining 24 h after HI and the corresponding quantifications of the number of FJ-positive cells (B) in the border zone of the cortical infarction (Cx) (2,093,000 ± 287,600/mm3 vs. 1,259,000 ± 230,200/mm3; *, P<0.05), (C) in the striatum (Str) (1,368,000 ± 46,860/mm3 vs. 912,300 ± 134,100/mm3; **, P < 0.01), (D) in the entire CA1 (209,900 cells ± 14,600 cells/mm3 vs. 167,500 cells ± 12,310 cells/mm3; *, P < 0.05) and (E) in the dentate gyrus (DG) (500,700 cells ± 56,880 cells/mm3 vs. 344,000 cells ± 45,150 cells/mm3; *, P < 0.05, n = 11/group). KO: atg7 KO (Atg7flox/flox; Nes-Cre) and Ctrl: Atg7flox/+; Nes-Cre.
Apoptotic cell death pathways, including caspase-dependent and –independent pathways, were investigated by quantifying the activation of CASP3 () and the mitochondrial-nuclear translocation of AIFM (). Immunohistochemical detection of active cleaved CASP3 (but not its inactive precursor, pro-CASP3) revealed a similar pattern as that shown by Fluoro-Jade staining (). Moreover, CASP3 enzymatic activity was 50% lower in the ipsilateral hemispheres of atg7 KO compared to the Ctrl mice 24 h after HI (). CASP3 data were confirmed by immunoblotting against SPTAN1/α-fodrin, revealing a decrease in the production of the caspase-dependent 120-kDa breakdown product (). Caspase-independent apoptotic cell death—as indicated by AIFM1 nuclear translocation, as shown previouslyCitation26—was also reduced in atg7 KO compared to Ctrl mice in all brain regions ().
Figure 5. Neuronal Atg7 deficiency decreased CASP3 activation after hypoxia-ischemia. (A) Representative active CASP3/caspase-3 staining 24 h after HI and the corresponding quantifications of the number of CASP3-positive cells (B) in the cortex (Cx) (81,630 ± 7,671/mm3 vs. 51,210 ± 11,970/mm3; *, P < 0.05), (C) in the striatum (Str) (132,800 ± 5,368/mm3 vs. 47,350 ± 13,110/mm3; ***, P < 0.001), (D) in the CA1 (41,810 cells ± 3,243 cells/mm3 vs. 22,860 cells ± 4,685 cells/mm3; **, P < 0.01) and (E) dentate gyrus (DG) (214,000 cells ± 14,700 cells/mm3 vs. 144,000 cells ± 23,940 cells/mm3; *, P < 0.05, n = 11/group). (F) CASP3 enzymatic activity 24 h after HI was 50% lower in the ipsilateral (IL) hemisphere of atg7 KO mice than in Ctrl (n = 10 for Ctrl, n = 6 for atg7 KO). There was no difference in the nonischemic control brains (Cont) (n = 13 for Ctrl and n = 6 for atg7 KO) or in the contralateral (CL) hemispheres (n = 10 for Ctrl, n = 6 for atg7 KO). (G) Representative SPTAN1/α-fodrin (240 kDa) western blots from control (Cont) and ipsilateral (IL) hemispheres 24 h after HI and (H) the quantification of the caspase-dependent 120 kDa breakdown products confirmed that CASP3 activation was more pronounced in the Ctrl mice. (*, P < 0.05, n = 6/group). KO: atg7 KO (Atg7flox/flox; Nes-Cre) and Ctrl: Atg7flox/+; Nes-Cre.
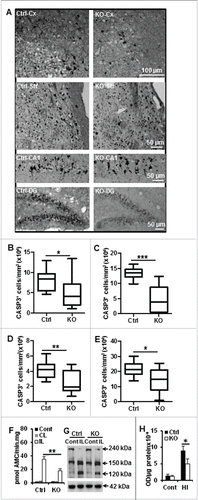
Figure 6. Neuronal Atg7 deficiency reduced hypoxia-ischemia-induced AIFM1 nuclear translocation. (A) Representative AIFM1 staining 24 h after HI and the corresponding quantifications of the number of AIFM1-positive nuclei (B) in the cortex (Cx) (80,610 nuclei ± 4,038 nuclei/mm3 vs. 49,060 nuclei ± 6,115 nuclei/mm3; ***, P < 0.001), (C) in the striatum (Str) (59,610 nuclei ± 2,322 nuclei/mm3 vs. 28,020 nuclei ± 4,287 nuclei/mm3; ***, P < 0.001), (D) in the CA1 (45,620 nuclei ± 3,849 nuclei/mm3 vs. 30,350 nuclei ± 5,123 nuclei/mm3; *, P < 0.05) and (E) dentate gyrus (DG) (48,260 nuclei ± 5,278 nuclei/mm3 vs. 30,730 nuclei ± 4,294 nuclei/mm3; *, P < 0.05). n = 11/group. KO: atg7 KO (Atg7flox/flox; Nes-Cre) and Ctrl: Atg7flox/+; Nes-Cre.
Neuronal Atg7 deficiency decreased hypoxia-ischemia-induced inflammation
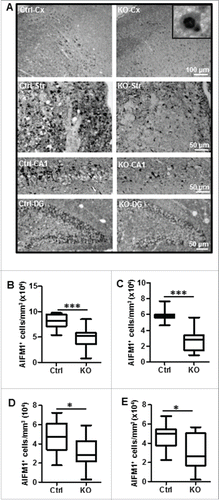
As neuroinflammation is involved in HI-induced cell death, we investigated the effect of neuronal autophagy inhibition on some inflammatory markers. The quantification of selected cytokines and chemokines, such as interleukins (IL1A [interleukin 1 α], IL1B [interleukin 1 β], IL6 [interleukin 6], IL10 [interleukin10], TNF [tumor necrosis factor], CXCL1[chemokine (C-X-C motif) ligand 1] and CCL2 [chemokine (C-C motif) ligand 2]) in brain homogenates revealed that IL1B, IL6, CXCL1 and CCL2 were less expressed in atg7 KO than Ctrl mice 24 h after HI (). After HI, microglial cells positive for AIF1/IBA1 (allograft inflammatory factor 1), were activated and accumulated in the injured area after HI, as indicated by morphological examination (retracted processes and enlarged cell bodies) () and double labeling with LGALS3/galectin-3 (lectin, galactoside-binding, soluble, 3) (). This microglial recruitment was less extensive in atg7 KO mice compared to Ctrl, as shown by quantification of the number of active AIF1-positive cells ().
Figure 7. Neuronal Atg7 deficiency attenuated hypoxia-ischemia-induced cytokine and chemokine expression and microglial activation. (A) Luminex assay of the cytosolic fraction of nonischemic (Cont) (n = 9 for Ctrl and n = 7 for atg7 KO) and for hypoxic-ischemic cortical tissue 24 h after HI (n = 9 for Ctrl and n = 7 for atg7 KO) demonstrated a decreased IL6, CXCL1, CCL2 and IL1B in KO mice. (B) Representative AIF1 immunostaining in the cortex of both Ctrl and atg7 KO mice 24 h after HI. (C) Double labeling of AIF1 and activated microglia marker galectin-3 (LGALS3) in the HI cortex. (D) Quantification of the number of AIF1-positive cells in both the cortex (*, P<0.05) and the striatum (**, P<0.01) confirmed a less pronounced microglia activation in KO mice. (n = 11/group). KO: atg7 KO (Atg7flox/flox; Nes-Cre) and Ctrl: Atg7flox/+; Nes-Cre.
Autophagy is increased in the lentiform nucleus of asphyxiated human term newborns
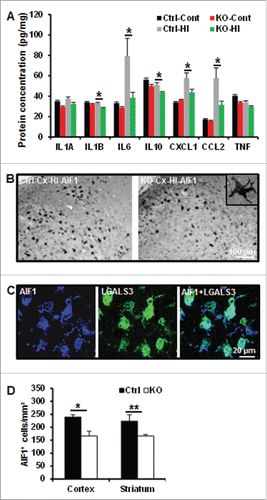
We subsequently investigated whether neuronal autophagy was enhanced in brains from asphyxiated human babies with HIE. We focused our investigations on the lentiform nucleus, which includes the putamen and the globus pallidus within the basal ganglia, a region highly sensitive to HI in neonates. Cerebral MRI diffusion-weighted imaging confirmed that perinatal HI led to induced severe and irreversible lesions in the cerebral cortex and basal ganglia (). Diffusion was severely restricted, confirming the presence of cytotoxic edema. Hematoxylin-eosin staining revealed the presence of pyknotic nuclei and shrunken cells in the lentiform nuclei of all 7 HIE cases, while such signs of cell death were rarely seen in controls. This confirmed the vulnerability of the human neonate basal ganglia to HI ().
Figure 8. Lentiform nucleus of asphyxiated human term newborns is highly vulnerable. (A) Magnetic resonance (MR) diffusion-weighted images with ADC maps (top panel) of hypoxic-ischemic encephalopathy (HIE) case 1 (left) at 36 h of life and HIE case 7 (right) at 23 h of life showed bilateral restricted diffusion in the basal ganglia (mainly in the putamen, arrowheads) and thalamus (long arrows) as well as in the cortex of case 7 (dashed arrows). MR multivoxel proton spectroscopy of HIE case 7 showing a negative double lactate peak at 1.2 ppm indicating an acute energy failure in this region (white arrow) at 23 h of life (lower panel). (B) Representative hematoxylin-eosin staining of the lentiform nucleus region showed the presence of numerous dying neurons (cell shrinkage and pyknotic nuclei, arrow) in newborns subjected to perinatal asphyxia but not in controls (Ctrl).
The presence of autophagosomes was investigated by using LC3B immunohistochemistry and by quantification of the number of LC3B-positive dots in the cytoplasm of individual neurons in the lentiform nuclei. Each of the HIE cases displayed more neuronal LC3B-positive dots (autophagosomes) than the control cases (). On average, the number of LC3B-positive dots was increased by 7-fold compared to controls, indicating that HIE was accompanied by a considerable induction of autophagosome formation in the lentiform nuclei.
Figure 9. Neuronal autophagy is enhanced in the lentiform nucleus of asphyxiated human term newborns. (A) Representative confocal images of LC3B staining (red) in neurons in the lentiform nuclei of human newborns, and quantifications of the numbers of LC3B-positive dots per neuron per μm2 (left histogram) in 6 controls (Ctrl) and 7 hypoxic-ischemic encephalopathy (HIE) cases, showing an increase in autophagosome in all HIE cases. Right histogram showed the average numbers of LC3B-positive dots of control (0.021 ± 0.007) and HIE (0.149 ± 0.018) cases. Representative confocal images of (B) LAMP1 staining (green) and (C) CTSD staining (red) in lentiform nuclei of human newborns and the quantifications of the numbers of positive dots per neuron per μm2 (left histograms) in the different cases individually demonstrated an increase in lysosomes in all HIE cases. Right histograms showed the average numbers of positive dots of control (LAMP1: 0.046 ± 0.005; CTSD: 0.035 ± 0.004) and HIE (LAMP1: 0.172 ± 0.013; CTSD: 0.260 ± 0.014). Quantifications showing the average percentages of (D) LAMP1- and (E) CTSD-positive dots larger than 0.5 μm2 per neuron in control (LAMP1: 2.5 ± 1.9%; CTSD: 2.6 ± 1.8%) and HIE cases (LAMP1: 24.8 ± 6.2%; CTSD: 19.8 ± 3.6%). (F) Representative confocal images of SQSTM1 staining (green) and CTSD (red) in neurons in human newborn lentiform nuclei showed that SQSTM1 expression was not increased (or accumulated) in neurons of HIE cases (right panels) (***, P<0.001).
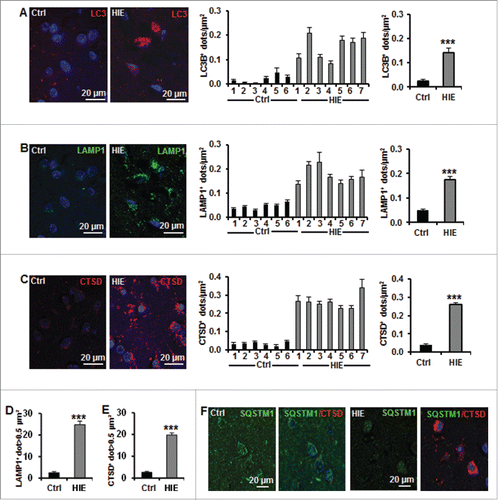
To further confirm that the increased number of autophagosomes was a sign of enhanced autophagic flux, tissues were stained for the lysosomal markers LAMP1 (lysosomal-associated membrane protein 1) and CTSD (cathepsin D). Quantification of the number of LAMP1- () and CTSD-positive dots () revealed an increase in all HIE cases compared to controls and indicated a surge in lysosomal activity. Furthermore, 20% of the positive dots in the HIE cases were very large lysosomes (>0.5 μm2, presumably autolysosomes), while these made up less than 3% of the dots in control cases (). In order to confirm that the increase in autophagosome and lysosomal markers reflected an increase in autophagic flux, we finally investigated the expression of SQSTM1 (). HIE cases did not display an increase (or accumulation) in SQSTM1 staining compared to control cases, suggesting that autophagic flux is not impaired in HIE cases. Altogether, these results strongly suggest that perinatal asphyxia increases neuronal autophagy in the lentiform nuclei of human newborns, as observed in P9 hypoxic-ischemic mice.
Discussion
Previous studies on the role of autophagy in neonatal brain injury have yielded conflicting results, and both inhibition and induction of autophagy by pharmacological agents have been shown independently to decrease the severity of brain injury in rodents.Citation20-22 Since both 3-methyladenine (3-MA) and rapamycin have limited specificity for the regulation of autophagy,Citation27-29 it is important to use more specific methods for inhibiting autophagy,Citation30 and the present approach, genetic KO of an important Atg gene, such as Atg7, is arguably the most specific method available. Here, we show that selective genetic deletion of neuronal Atg7 in mice reduced neuronal cell death, leading to decreased brain injury after severe HI. These results corroborate those of Koike and colleaguesCitation22 obtained in the hippocampus in a milder HI model, and extend the conclusion of a lethal role of HI-enhanced autophagy to other brain regions (cortex, thalamus and striatum), which are highly vulnerable to HI in neonates.
Autophagy is a lysosome-mediated intracellular catabolic mechanism responsible for the bulk degradation and recycling of damaged or dysfunctional cytoplasmic components and is the only pathway for degrading intracellular organelles.Citation14 Inactivation of autophagy results in the accumulation of cytoplasmic protein inclusion bodies leading to cell injury and neurodegeneration. Thus, autophagy plays an essential role in cell survival.Citation31 Incomplete autophagy leads to the accumulation of autophagosomes, and this also contributes to physiological dysfunction.Citation15,32 Nevertheless, the role of autophagy can be different under physiological (basal autophagy) and pathological conditions (induced autophagy). Knockout mouse studies targeting Atg5 or Atg7 have demonstrated that neurons in the brain require at least basal levels of autophagy for the maintenance of their function.Citation14,15 However, the negative effect of Atg7 deficiency only manifests after many weeks of life, meaning that P9 mice do not yet present any signs of neurodegeneration or other abnormalities. This allowed us to use atg7 KO mice for studying the role of induced autophagy in perinatal HI brain injury.
Neuronal cell death after HI in the immature brain displays necrotic, apoptotic, autophagic, and various hybrid morphological features.Citation3 Even though some studies failed to detect neuroprotection with caspase inhibitors in neonatal HI models,Citation9,26 previous studies have shown that apoptosis plays a prominent role in the development of immature brain injuryCitation11,33 and that this involves both caspase-dependent and -independent pathways.Citation4,21,22,34,35 Furthermore, the combined inhibition of both apoptotic pathways provided additive protection against HI injury in this modelCitation34 indicating that these pathways run parallel to each other in neonatal HI. The functional relationship between apoptosis and autophagy is complex, and these 2 processes can act synergistically or suppress each other through their shared regulation by many of the same molecular regulators, such as the BCL2 family of proteins.Citation30 Crosstalk between autophagic and apoptotic cell death pathways has been demonstrated both in vitro and in vivo.Citation21 ATG7 knockdown in a cell line prevents CASP3 activation and cell death, indicating a proapoptotic function of ATG7.Citation36,37 Interestingly, we found that neuron-specific Atg7 deficiency had no influence on the expression of apoptosis-related proteins under physiological condition, contrasting with the reduced activation of both caspase-dependent and -independent apoptotic pathways under HI conditions in P9 mice. This speaks in favor of a proapoptotic function for ATG7 acting upstream of apoptosis under pathological conditionsCitation36 and suggests that HI-enhanced autophagy can partially mediate neuronal death by this pathway. In fact we have previously demonstrated that, in primary cortical neuron cultures, neuronal death induced by classical apoptotic stimuli could be decreased by autophagy inhibition (3-MA, ATG5 and ATG7 downregulation) affecting both CASP3 activation and AIFM1 nuclear translocation.Citation38 The precise mechanisms rendering atg7 KO neurons resistant to HI insults need to be investigated further. Such mechanisms might include effects of atg7 KO on mitochondrial function, redox capacity or other pathways that could be overactivated in response to the autophagy defect. However, it cannot be excluded that atg7 KO might prevent cell death by an autophagy-independent effect, as has been reported for other ATGs.Citation16,39 However, to our knowledge, such a role of Atg7 has not been demonstrated thus far in neurons. Moreover, a recent study targeting BECN1, another autophagic-related protein involved in a different complex along the autophagy machinery, using lentiviral vectors transducing shRNA has also shown that inhibiting autophagy displayed neuroprotection after HI in neonatal rats.Citation25
HI induces microglia activation and local inflammation, exacerbating brain injury through the secretion of proinflammatory cytokines and chemokines. Thus, inhibition of inflammation could provide neuroprotection. Our data show suppressed microglia activation and inflammation after HI in atg7 KO mice. Nonetheless, the precise cause-effect relationship among these observations remains elusive, meaning that reduced microglia activation inflammation might account for tissue protection in Atg7-deficient brains or vice versa. Autophagy plays important roles in the regulation of immune and inflammatory reactions through regulation of cytokine secretion and recruitment of leukocytes to sites of injury.Citation40 Further studies are needed to elucidate if ablation of neuronal autophagy may have direct effects on injury-induced inflammation and microglia activation.
Mitochondria have multiple functions throughout brain development, including mitochondrial biogenesis as part of normal cellular respiration, mitochondrial fusion and fission for maintenance of organelle function, mitophagy to control mitochondrial quality, and regulation of cell death.Citation41 The immature brain is particularly susceptible to HI because of the poorly developed antioxidant defense systems. In this study, we found that Atg7 deficiency neither has any obvious effect on mitochondrial function-related genes, nor on redox capacity in the immature brain. This indicates that increased resilience to HI was not mediated by enhanced mitochondrial function secondary to the autophagy deficiency, and that further investigations are needed to elucidate the underlying mechanism.
Neuronal autophagy is enhanced in the basal ganglia of human severely asphyxiated newborns. Dying neurons in the lentiform nuclei of human HIE cases displayed strong autophagic features (numerous autophagosomes and autolysosomes) compared to control cases who did not display enhanced autophagy and neuronal death. Moreover, the level of SQSTM1, which is inversely correlated with autophagic activity, was reduced in human brains, indicating that autophagic flux (both autophagosome formation and lysosomal degradation) was induced.
In conclusion, the precise mechanisms by which atg7 KO neurons resist HI need to be investigated in further detail. Nonetheless, our findings suggest that transient and specific autophagy inhibition could be a promising strategy that should be developed experimentally in the hope of improving the outcome of perinatal asphyxia.
Materials and methods
Atg7 mice breeding and genotyping
Floxed Atg7 mice were characterized previouslyCitation15 and were crossed with a Nes-Cre-driven line to produce Atg7flox/flox; Nes-Cre knockout (atg7 KO) and Atg7flox/+; Nes-Cre control mice (Ctrl). Genomic DNA was isolated from tail samples according to the manufacturer's instruction (Qiagen, 69506). The following primer sets were used for genotyping: for the Cre transgene (Cre-S:cre sense 55) 5′-TTT GCC TGC ATT ACC GGT CGA TGC AAC-3′ and (Cre-As:Cre AS1000) 5′-TGC CCC TGT TTC ACT ATC CAG GTT ACG GA-3′ and for the Ctrl and Atg7flox alleles (Hind-Fw) 5′-TGG CTG CTA CTT CTG CAA TGA TGT-3′ and (Pst-Rv) 5′-CAG GAC AGA GAC CAT CAG CTC CAC-3′. The reaction mixture for genotyping contained 1 μL of genomic DNA, 0.2 mM dNTP, 2.5 μL 10× PCR buffer (250 mM Tris-HCl, pH 8.3, 375 mM KCl, 15 mM MgCl2, all from Sigma), 1 U of Taq DNA Polymerase (Sigma, D1806), and 0.5 μM of primer. The PCR parameters were 98°C for 20 s, 64°C for 30 s, and 72°C for 90 s for 30 cycles. PCR products were separated on a 1.5% agarose gel containing SYBR Green. A 100-base pair (bp) ladder was used to verify the size of the PCR products. The gels were imaged with an LAS 3000 cooled CCD camera (Fujifilm, Tokyo, Japan). atg7 KO mice were identified by the presence of both 500 bp and 1000 bp DNA bands. Ctrl mice were identified by the presence of 500 bp, 1000 bp, and 1500 bp products.
Hypoxia-ischemia
Postnatal d 9 (P9) atg7 KO and Ctrl mice of either gender were anesthetized with isoflurane (5% for induction, 1.5% to 2.0% for maintenance) in a 1:1 mixture of nitrous oxide and oxygen, and the duration of anesthesia was less than 5 min. The left common carotid artery was cut between double ligatures of Prolene sutures (6.0). After the surgical procedure, the wounds were infiltrated with lidocaine for local analgesia. The pups were returned to their cages for 1 h and then placed in a chamber perfused with a humidified gas mixture (10% oxygen in nitrogen) for 50 min at 36°C. Following hypoxic exposure, the pups were returned to their cages. Control pups were not subjected to HI. Mice were maintained in the Laboratory for Experimental Biomedicine of Gothenburg University. All experiments were performed in accordance with international guidelines and approved by the Gothenburg Committee of the Swedish Animal Welfare Agency (application no. 145-2008).
Human newborn brain specimens
Brain tissue was obtained from 13 autopsied human newborns (Institute of Pathology, University of Lausanne) who died after birth due to severe HIE (n = 7). Control cases (n = 6) were newborns with conditions incompatible with life whose autopsies did not indicate brain injury. Most of the perinatal data from the 2 groups, such as birth weight, gestational age, Apgar score, time of death or resuscitation score, did not show statistical differences. As expected, there was significantly greater metabolic acidosis in the HIE cases as reflected by a lower umbilical artery pH, and all HIE cases experienced postnatal seizures compared to none in the control group. Histological and immunohistological analysis of the control brains revealed very rare dying cells (pyknotic nuclei and shrunken cells, CASP3- and TUNEL-positive cells). Selection was made retrospectively from the death reports of the neonatology unit between 2001 and 2009. Autopsies were performed for medical and legal reasons, and the tissue was fixed within 12 h following death. Informed consent for tissue collection was obtained from the parents, and the local ethical committee approved the use of the anonymized specimens. After fixation in 10% buffered formalin for 3 wk, the samples were embedded in paraffin and 3-μm thick sections were cut. MRI was available for 4 out of 7 HIE cases. Diffusion-weighted images were acquired using spin-echo echo-planar imaging (SE-EPI), with 5-mm thick slices (b values: 0, 500, 1000 mm2/s) and multivoxel proton spectroscopy focused on the basal ganglia and thalamus.
Immunohistochemistry
The animals were deeply anesthetized with phenobarbital and perfusion-fixed with 5% formaldehyde in 0.1 M phosphate-buffered saline (PBS; Gibco, 18912-014) followed by immersion fixation in the same fixative for 24 h at 4°C. After dehydration with graded ethanol and xylene, the brains were paraffin-embedded, serial cut in 5-μm coronal sections, and mounted on glass slides. Every 100th section was stained for MAP2 (microtubule-associated protein 2) (1:1000, Sigma, M4403) for brain injury evaluation (with a distance of 500 μm between slices). The sections were stained for the following cell death-related markers: AIFM1, and active CASP3 as described previously.Citation11 Three adjacent sections with an interval of 250 μm were selected from hippocampal and striatum level from each brain for each cell death-related marker staining. Sections were deparaffinized and rehydrated, and antigen retrieval was performed by heating the sections in 10 mM boiling sodium citrate buffer (pH 6.0) for 10 min. Nonspecific binding was blocked for 30 min with 4% goat (Vector Laboratories, S-1000) or horse (Vector Laboratories, S-2000) or donkey serum (Jackson ImmunoResearch, 118135) (depending on the species used to raise the secondary antibody) in PBS. Rabbit anti-active CASP3 (1:100, BD Pharmingen, 559565), goat anti-AIFM1/AIF (1:100, 2 μg/ml, Santa Cruz Biotechnologies, sc-9416), rabbit anti- SQSTM1 (1:200, Enzo Life Science, BML-PW9860), rabbit anti-AIF1/IBA1 (1:500, Wako, 019-19741), were incubated for 60 min at room temperature followed by the appropriate biotinylated horse anti-goat (1:200, for AIFM1; Vector Laboratories, BA-9500), goat anti-rabbit (1:250, for active CASP3, SQSTM1; Vector Laboratories, BA-1000) secondary antibody for 60 min at room temperature. Endogenous peroxidase activity was blocked with 3% H2O2 for 5 min. Visualization was performed using Vectastain ABC Elite (Vector Laboratories, PK-6200) with 0.5 mg/mL 3,3′-diaminobenzidine (Sigma-Aldrich, D5637-25G) enhanced with 15 mg/mL ammonium nickel sulfate (Sigma-Aldrich, A1827-500G), 2 mg/mL β-D glucose (Chemtronica, G0047), 0.4 mg/mL ammonium chloride, and 0.01 mg/mL β-glucose oxidase (Sigma, G2133-10KU). For the double labeling of SQSTM1-RBFOX3/NeuN (1:200, Millipore, MAB377), AIF1/IBA1-LGALS3 (5 mg/ml; eBioscience, 14–5301), antigen recovery was performed as above followed by incubation with the mixed antibodies in PBS at room temperature for 60 min. After washing, the sections were incubated with secondary antibodies at room temperature for 60 min. All secondary antibodies were from Jackson ImmunoResearch Laboratories and were diluted 1:1000 in PBS. After washing, the sections were mounted using Vectashield mounting medium.
In the human tissue, after deparaffinization and antigen retrieval, the sections were blocked in PBS with 15% donkey serum for 30 min and then incubated with primary antibodies in 1.5% donkey serum overnight at 4°C. The following primary antibodies were used: rabbit anti-LC3B (Abcam, ab48394); goat anti-CTSD (Santa Cruz Biotechnology, sc-6486), mouse anti-LAMP1 (BD Biosciences, 611042) and rabbit anti-SQSTM1 (Sigma-Aldrich, P0067). Sections were then incubated with secondary antibodies, Alexa Fluor 594 donkey-anti-rabbit IgG (H+L) (Invitrogen, A-21207), Alexa Fluor 488 donkey anti-mouse IgG (H+L) (Invitrogen, A-21202), and Alexa Fluor 594 donkey-anti-goat IgG (H+L) (Invitrogen, A-11058). After several washes in PBS and Hoechst staining, the sections were then mounted using FluorSave medium (Calbiochem, 345789).
Fluoro-jade B staining
After deparaffinization, the sections were incubated with freshly prepared 0.06% potassium permanganate for 10 min and rinsed with distilled water. The slides were then incubated with 0.001% Fluoro-Jade B solution (Chemicon, AG310) for 30 min at room temperature. After rinsing with distilled water, the slides were dehydrated with ethanol and placed in xylene for 2 min and covered with mounting medium.
Electron microscopy
The pups were deeply anesthetized with pentobarbital and fixed by cardiac perfusion with 2% paraformaldehyde-2% glutaraldehyde buffered with 0.1 mol/L phosphate buffer for ordinary electron microscopy at 24 h after HI (n = 3/group). Samples were cut into 1 mm thick coronal slices and post-fixed with 2% OsO4 with 0.1 mol/L phosphate buffer, block-stained in 1% aqueous uranyl acetate, dehydrated with a graded series of alcohol, and embedded in Epon 812 (TAAB, T204). Silver sections were cut with an ultramicrotome, stained with uranyl acetate and lead citrate, and observed with an electron microscope (HT7700, Hitachi, Tokyo, Japan). The pyknotic neurons and total neurons were counted per visual field and ratio of pyknotic neurons was calculated.
Immunoblotting
Animals were sacrificed by decapitation at 24 h after HI. Control animals were sacrificed at the same time as HI pups. The brains were rapidly dissected out on a bed of ice. The parietal cortex (including the hippocampus) was dissected out from each hemisphere and immediately frozen in liquid nitrogen and stored at −80°C. The samples were sonicated by adding ice-cold isolation buffer (15 mM Tris-HCl, pH 7.6, 320 mM sucrose, 1 mM DTT, 1 mM MgCl2, 3 mM EDTA-K, 0.5% protease inhibitor cocktail (Sigma, P8340), 1% phosphatase inhibitor cocktail 1 (Sigma, P2850), and 1% phosphatase inhibitor cocktail 2 (Sigma, P5726). The homogenates were centrifuged at 9200 × g for 15 min at 4°C producing a pellet that included nuclear, mitochondrial, and synaptosomal fractions and a crude cytosolic fraction in the supernatant. The pellet fractions were washed in homogenizing buffer and centrifuged at 9200 × g for 15 min. The protein concentrations were determined with the BCA protein assay adapted for microplates. Samples with a volume of 65 μL were mixed with 25 μL NuPAGE LDS 4× sample buffer (ThermoFisher Scientific, NP0007) and 10 μL reducing agent (ThermoFisher Scientific, NP0004) and heated at 70°C for 10 min. Individual samples were run on 4–12% NuPAGE Bis-Tris gels (Novex, NP0323Box) and transferred to reinforced nitrocellulose membranes. After blocking with 30 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.1% Tween 20 (Merck, S4733984) containing 5% fat-free milk powder for 60 min at room temperature, the membranes were incubated with the following primary antibodies: rabbit anti-actin (1:200; Sigma, A2066), mouse anti-SPTAN1/α-FODRIN (0.2 μg/mL; clone AA6, BIOMOL, FG6090), rabbit anti-LC3B (1:1000; Cell Signaling Technology, 2775), rabbit anti-SQSTM1 (1:1000; Enzo Life Science, PW9860), rabbit anti-ATG7 (1:1000; Cell Signaling Technology, 8558), goat anti-AIFM1 (1:1000, Santa Cruz, sc-9416), mouse anti-CYCS (1:500, BD Pharmingen, 556433), rabbit anti-CAYP3 (1:1000, Santa Cruz, sc-7148), mouse anti-CT (1:1000, Frontier Lab Ltd., LF-MA0010); mouse anti-SOD2 (1:2000, Lab Frontier), mouse anti-HSP70 (1:500, Santa Cruz, sc-7298), or mouse total OXPHOS rodent western blotting WB antibody cocktail (COXV, ab14748, COXIII, ab14745, COXIV, ab14705, COXII, ab 14714, COXI, ab110242) (0.5 μg/mL, Abcam, MS604) at room temperature for 60 min. After washing, the membranes were incubated with a peroxidase-labeled secondary antibody for 30 min at room temperature (goat anti-rabbit, 1:2,000, horse anti-goat, 1:2,000, or horse anti-mouse 1:4,000; Vector Laboratories, PI-1000, PtdIns-9500, PI-2000). Immunoreactive species were visualized using the Super Signal West Dura substrate (ThermoFisher Scientific, 34075) and an LAS 3000 cooled CCD camera (Fujifilm, Tokyo, Japan). ACTIN was used as the loading control.
RNA isolation and cDNA synthesis
Total RNA was isolated using the RNeasy mini kit (Qiagen, 74104) according to the manufacturer's instructions. The concentration and purity of all RNA samples were determined using a Nanodrop spectrophotometer (Nanodrop Technologies, Wilmington, USA). The integrity of RNA was measured using Experion™ RNA StdSens analysis kit (Bio-Rad, 7007103) on an Automated Electrophoresis Station machine (Bio-Rad, Hercules, California, USA). One microgram of total RNA was reverse transcribed using Reverse Transcription kit (QuantiTect® Reverse Transcription kit; Qiagen, 205311).
Quantitative real-time PCR
Quantitative real-time PCR was performed using the LightCycler 480 instrument (Roche Diagnostics, Mannheim, Germany) and the SYBR Green (ThermoFisher Scientific, 0253) technique according to the manufacturer's instructions. The primers used in the qPCR reactions were designed by Beacon Designer software (PREMIER Biosoft). These primers are listed in Table S1. The relative expression levels of mRNAs were calculated using the method of geometric averaging of multiple internal control genes.
Mitochondrial DNA copy measurement
Total DNA of cortex was isolated using a genomic DNA isolation kit (Qiagen, 69506). The amount of mitochondrial DNA relative to nuclear genomic DNA was determined by quantitative real-time PCR. Two different genomic genes and mitochondrial genes were used for this purpose, The Ywhaz gene, ‘primers 5′-GAGGAAGAATCGTGAGTTAGTT-3′ (sense) and 5′-TGGTGATGGTTGAGACAGA-3′(anti-sense), and the Rplp0/36B4 gene, primers 5′-GTTGTTAGCCTGTGATAGCA −3′ (sense) and 5′- CCGACCAGCAATGTTCTATT −3′ (anti-sense) from the genome; the mt-Nd4/ND4 gene, primers 5′- CCTCAGTTAGCCACATAGC −3′ (sense) and 5′- GATTCGTTCGTAGTTGGAGTT −3′ (anti-sense), and the D-loop gene, primers 5′- GCCCATGACCAACATAACTG −3′ (sense) and 5′- CCTTGACGGCTATGTTGATG −3′ (anti-sense) from mitochondria. The relative mitochondrial DNA levels were calculated based on the threshold cycle (Ct) as 2−Δ(ΔCt).
Glutathione reductase activity assay
Homogenized tissues were used for this activity assay. GSR activity was measured using the Glutathione Reductase Activity Colorimetric Assay Kit according to the manufacturer's instructions and expressed as mU/ml (BioVison, K761-200).
CASP3 activity assay
The protein concentrations were determined as above. Samples of homogenate (40 μL) were mixed with 60 μL of extraction buffer as described previously.Citation42 Cleavage of Ac-DEVD-AMC (Peptide Institute, 500329) was measured with an excitation wavelength of 380 nm and an emission wavelength of 460 nm and expressed as pmol AMC released per mg protein per minute.
Cytokine and chemokine assay
Cytokines and chemokines were measured in whole-brain homogenate supernatant fractions from P9 Ctrl and atg7 KO mice at 24 h after HI and from P10 control mice. Samples were prepared according to the protocol of the manufacturer. Protein concentration was measured with the BCA protein assay and adjusted to 600 μg/mL with 0.5% bovine serum albumin (Sigma, A2058). Levels of IL1A, IL1B, IL6, CXCL1, CCL2, and TNF were simultaneously measured using the Bio-Plex Multiplex Cytokine Assay (Bio-Rad Laboratories, M60-009RDPD) (Bio-Rad, Hercules, CA, USA). The results were normalized to the amount of protein in the sample.
Injury evaluation
Brain injury was evaluated 8 d after HI based on total tissue loss and neuropathological scoring. The MAP2-positive and -negative areas in each section were measured using Micro Image (Olympus, Japan). The volume was calculated from the MAP2-positive areas according to the Cavalieri principle using the following formula: V = ΣA × P × T, where V = total volume, ΣA = sum of area measurements, P = the inverse of the sampling fraction, and T = the section thickness (5 μm). The MAP2-negative volume was the infarction volume. The total tissue loss was calculated as the MAP2-positive volume in the contralateral hemisphere minus the MAP2-positive volume in the ipsilateral hemisphere. The neuropathological scores for the cortex, hippocampus, striatum, and thalamus were assessed as described previously.
Cell counting in mouse tissue
Area contours were drawn and measured in every 50th section. The active CASP3−, AIFM1+ cells were counted at 400× magnification in the border zone of the injured cortex or striatum within an area of 0.196 mm2 (one visual field) as well as within the whole CA and DG areas by using Micro Image (Olympus, Japan). Fluoro-Jade-positive cells were counted by fluorescence microscopy. Three sections were counted from each brain with an interval of 250 μm. The average was defined as n = 1 when comparing different brains. All of the counting was carried out by investigators blinded to group assignment.
Quantification of autophagic and lysosomal labeling of human tissue
Confocal images of immunocytochemistry against LC3B, CTSD, and LAMP1 were acquired with the same acquisition parameters on the confocal laser-scanning microscope (LSM 710 Meta confocal microscope, Carl Zeiss, Jena, Germany), and images were processed with Adobe Photoshop 10.0. LC3B-, CTSD-, and LAMP1-positive dots were quantified using Image J software and expressed as the number of positive dots per neuron per μm2. For CTSD- and LAMP1-positive dots, the dot area was also quantified.
Statistics
All data were expressed as mean ± s.e.m. The data distribution normality was tested using the Shapiro–Wilk test. The Student t test was used when comparing tissue loss. The Mann–Whitney U-test was used to compare the numbers of immune-positive cells and injury scores between 2 different groups. ANOVA with the Fisher post-hoc or multivariate test was used when comparing more than 2 groups. The significance level was defined as P < 0.05.
Abbreviations
| AIF1/IBA1 | = | allograft inflammatory factor 1 |
| AIFM1/AIF | = | apoptosis-inducing factor, mitochondria-associated 1 |
| Atg7 | = | autophagy-related 7 |
| bp | = | base pair |
| CA | = | cornu ammonis |
| CASP3 | = | caspase 3 |
| CAT | = | catalase |
| CCL2 | = | chemokine (C-C motif) ligand 2 |
| CTSD | = | cathepsin D |
| CXCL1/KC | = | chemokine (C-X-C motif) ligand 1 |
| CYCS/CYC | = | cytochrome c, somatic |
| DG | = | dentate gyrus |
| GSH | = | glutathione |
| HI | = | hypoxia-ischemia |
| HIE | = | hypoxic-ischemic encephalopathy |
| HSP70 | = | heat shock protein 70 |
| IL | = | interleukin |
| IL1A | = | interleukin 1 alpha |
| IL1B | = | interleukin 1 beta |
| IL6 | = | interleukin 6 |
| IL10 | = | interleukin 10 |
| KO | = | knockout |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| LGALS3/galectin-3 | = | lectin, galactoside-binding, soluble, 3 |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 beta |
| MAP2 | = | microtubule-associated protein 2 |
| P9 | = | postnatal d 9 |
| SOD2 | = | superoxide dismutase 2, mitochondrial |
| SQSTM1/p62 | = | sequestosome 1 |
| TNF/TNFα | = | tumor necrosis factor |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary files
Download MS Word (209.6 KB)Acknowledgments
We thank Marie Pascale Pittet and Maria Chiara Osterheld for help in obtaining human tissue.
Funding
This work was supported by the Swedish Research Council, the Swedish Childhood Cancer Foundation, The Swedish Cancer Foundation, The Frimurare Barnhus Foundations in Stockholm and Gothenburg, Swedish governmental grants to scientists working in health care (ALF), the Wilhelm and Martina Lundgren Foundation, Edit Jacobson's Donations fund, the Gothenburg Medical Society, the Swedish Medical Society, the Department of Science and Technology, the Department of Health of Henan Province, the National Nature Science Foundation of China (31271152), the Swiss National Science Foundation (310030-130769 and 310030-163064), the Foundation Emma Muschamp, and the Faculty of Biology and Medicine (University of Lausanne). GK was supported by the LeDucq Foundation.
Related Research Data
References
- Mwaniki MK, Atieno M, Lawn JE, Newton CR. Long-term neurodevelopmental outcomes after intrauterine and neonatal insults: a systematic review. Lancet 2012; 379:445-52; PMID:22244654; http://dx.doi.org/10.1016/S0140-6736(11)61577-8
- Krageloh-Mann I, Helber A, Mader I, Staudt M, Wolff M, Groenendaal F, DeVries L. Bilateral lesions of thalamus and basal ganglia: origin and outcome. Dev Med Child Neurol 2002; 44:477-84; PMID:12162385; http://dx.doi.org/10.1111/j.1469-8749.2002.tb00309.x
- Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol 2011; 69:743-58; PMID:21520238; http://dx.doi.org/10.1002/ana.22419
- Carlsson Y, Schwendimann L, Vontell R, Rousset CI, Wang X, Lebon S, Charriaut-Marlangue C, Supramaniam V, Hagberg H, Gressens P, et al. Genetic inhibition of caspase-2 reduces hypoxic-ischemic and excitotoxic neonatal brain injury. Ann Neurol 2011; 70:781-9; PMID:21674587; http://dx.doi.org/10.1002/ana.22431
- Zhu C, Wang X, Deinum J, Huang Z, Gao J, Modjtahedi N, Neagu MR, Nilsson M, Eriksson PS, Hagberg H, et al. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J Exp Med 2007; 204:1741-8; PMID:17635954; http://dx.doi.org/10.1084/jem.20070193
- Zhu C, Kang W, Xu F, Cheng X, Zhang Z, Jia L, Ji L, Guo X, Xiong H, Simbruner G, et al. Erythropoietin improved neurologic outcomes in newborns with hypoxic-ischemic encephalopathy. Pediatrics 2009; 124:e218-26; PMID:19651565; http://dx.doi.org/10.1542/peds.2008-3553
- Shankaran S, Pappas A, McDonald SA, Vohr BR, Hintz SR, Yolton K, Gustafson KE, Leach TM, Green C, Bara R, et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med 2012; 366:2085-92; PMID:22646631; http://dx.doi.org/10.1056/NEJMoa1112066
- Li Q, Li H, Roughton K, Wang X, Kroemer G, Blomgren K, Zhu C. Lithium reduces apoptosis and autophagy after neonatal hypoxia-ischemia. Cell Death Dis 2010; 1:e56; PMID:21364661; http://dx.doi.org/10.1038/cddis.2010.33
- Puyal J, Clarke PG. Targeting autophagy to prevent neonatal stroke damage. Autophagy 2009; 5:1060-1; PMID:19713756; http://dx.doi.org/10.4161/auto.5.7.9728
- Blomgren K, Leist M, Groc L. Pathological apoptosis in the developing brain. Apoptosis 2007; 12:993-1010; PMID:17453164; http://dx.doi.org/10.1007/s10495-007-0754-4
- Zhu C, Wang X, Xu F, Bahr BA, Shibata M, Uchiyama Y, Hagberg H, Blomgren K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ 2005; 12:162-76; PMID:15592434; http://dx.doi.org/10.1038/sj.cdd.4401545
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383-435; PMID:20959619; http://dx.doi.org/10.1152/physrev.00030.2009
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; http://dx.doi.org/10.1038/nrd3802
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/10.1038/nature04724
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/10.1038/nature04723
- Puyal J, Ginet V, Clarke PG. Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol 2013; 105:24-48; PMID:23567504; http://dx.doi.org/10.1016/j.pneurobio.2013.03.002
- Oppenheim RW, Blomgren K, Ethell DW, Koike M, Komatsu M, Prevette D, Roth KA, Uchiyama Y, Vinsant S, Zhu C. Developing postmitotic mammalian neurons in vivo lacking Apaf-1 undergo programmed cell death by a caspase-independent, nonapoptotic pathway involving autophagy. J Neurosci 2008; 28:1490-7; PMID:18256270; http://dx.doi.org/10.1523/JNEUROSCI.4575-07.2008
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 2013; 106-107:1-16; PMID:23583307; http://dx.doi.org/10.1016/j.pneurobio.2013.04.001
- Ginet V, Puyal J, Clarke PG, Truttmann AC. Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol 2009; 175:1962-74; PMID:19815706; http://dx.doi.org/10.2353/ajpath.2009.090463
- Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 2008; 32:329-39; PMID:18760364; http://dx.doi.org/10.1016/j.nbd.2008.07.022
- Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol 2009; 66:378-89; PMID:19551849; http://dx.doi.org/10.1002/ana.21714
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol 2008; 172:454-69; PMID:18187572; http://dx.doi.org/10.2353/ajpath.2008.070876
- Liu Y, Shoji-Kawata S, Sumpter RM Jr, Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A 2013; 110:20364-71; PMID:24277826; http://dx.doi.org/10.1073/pnas.1319661110
- Carloni S, Girelli S, Scopa C, Buonocore G, Longini M, Balduini W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 2010; 6:366-77; PMID:20168088; http://dx.doi.org/10.4161/auto.6.3.11261
- Ginet V, Spiehlmann A, Rummel C, Rudinskiy N, Grishchuk Y, Luthi-Carter R, Clarke PG, Truttmann AC, Puyal J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy 2014; 10:846-60; PMID:24674959; http://dx.doi.org/10.4161/auto.28264
- Zhu C, Qiu L, Wang X, Hallin U, Cande C, Kroemer G, Hagberg H, Blomgren K. Involvement of apoptosis-inducing factor in neuronal death after hypoxia-ischemia in the neonatal rat brain. J Neurochem 2003; 86:306-17; PMID:12871572; http://dx.doi.org/10.1046/j.1471-4159.2003.01832.x
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol 2005; 17:596-603; PMID:16226444; http://dx.doi.org/10.1016/j.ceb.2005.09.009
- Hughes KJ, Kennedy BK. Cell biology. Rapamycin paradox resolved. Science 2012; 335:1578-9; PMID:22461595; http://dx.doi.org/10.1126/science.1221365
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285:10850-61; PMID:20123989; http://dx.doi.org/10.1074/jbc.M109.080796
- Nassif M, Valenzuela V, Rojas-Rivera D, Vidal R, Matus S, Castillo K, Fuentealba Y, Kroemer G, Levine B, Hetz C. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy 2014; 10:1256-71; PMID:24905722; http://dx.doi.org/10.4161/auto.28784
- Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. J Neurosci 2012; 32:7585-93; PMID:22649237; http://dx.doi.org/10.1523/JNEUROSCI.5809-11.2012
- Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010; 6:482-94; PMID:20400854; http://dx.doi.org/10.4161/auto.6.4.11737
- Wang X, Carlsson Y, Basso E, Zhu C, Rousset CI, Rasola A, Johansson BR, Blomgren K, Mallard C, Bernardi P, et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J Neurosci 2009; 29:2588-96; PMID:19244535; http://dx.doi.org/10.1523/JNEUROSCI.5832-08.2009
- Zhu C, Wang X, Huang Z, Qiu L, Xu F, Vahsen N, Nilsson M, Eriksson PS, Hagberg H, Culmsee C, et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ 2007; 14:775-84; PMID:17039248; http://dx.doi.org/10.1038/sj.cdd.4402053
- Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Holtzman DM. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest 1998; 101:1992-9; PMID:9576764; http://dx.doi.org/10.1172/JCI2169
- Walls KC, Ghosh AP, Franklin AV, Klocke BJ, Ballestas M, Shacka JJ, Zhang J, Roth KA. Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J Biol Chem 2010; 285:10497-507; PMID:20123985; http://dx.doi.org/10.1074/jbc.M110.103747
- Kessel DH, Price M, Reiners JJ Jr. ATG7 deficiency suppresses apoptosis and cell death induced by lysosomal photodamage. Autophagy 2012; 8:1333-41; PMID:22889762; http://dx.doi.org/10.4161/auto.20792
- Grishchuk Y, Ginet V, Truttmann AC, Clarke PG, Puyal J. Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy 2011; 7:1115-31; PMID:21646862; http://dx.doi.org/10.4161/auto.7.10.16608
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8:1124-32; PMID:16998475; http://dx.doi.org/10.1038/ncb1482
- Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol 2009; 10:461-70; PMID:19381141; http://dx.doi.org/10.1038/ni.1726
- Hagberg H, Mallard C, Rousset CI, Thornton C. Mitochondria: hub of injury responses in the developing brain. Lancet Neurol 2014; 13:217-32; PMID:24457191; http://dx.doi.org/10.1016/S1474-4422(13)70261-8
- Wang X, Karlsson JO, Zhu C, Bahr BA, Hagberg H, Blomgren K. Caspase-3 activation after neonatal rat cerebral hypoxia-ischemia. Biol Neonate 2001; 79:172-9; PMID:11275647; http://dx.doi.org/10.1159/000047087