
abstract
Regeneration of skeletal muscle relies on its resident stem cells, also known as satellite cells, which are normally quiescent. With aging, satellite cell quiescence is lost concomitant with a muscle regenerative decline. Here we demonstrate that autophagy sustains quiescence over time and that its failure with age drives senescence, which accounts for stem cell loss of function. Pharmacological and genetic reestablishment of autophagy restores homeostasis and regenerative functions in geriatric satellite cells, which has relevance for the elderly population.
The formation of skeletal myofibers (myogenesis) takes place during embryonic and fetal development, and during postnatal and adult life as a regenerative response for replacing damaged muscle tissue. Because of the post-mitotic nature of myofibers, their regeneration after injury requires a unique population of tissue-resident stem cells, named satellite cells. Under normal conditions, satellite cells are in quiescence (a G0 reversible arrest state), but in response to damage, they are activated, proliferate and differentiate to form new myofibers. A small satellite cell subpopulation, however, returns to quiescence (self-renewal) to reestablish the stem cell pool. Because adult skeletal muscle has little turnover in resting conditions, quiescence appears as an ideal state to maintain the stem cell population at a low activity rate, yet functionally ready for future regenerative demands.
Our previous studies demonstrated that the number and regenerative capacity of muscle stem cells decline with aging, with the decline being maximal at advanced geriatric age. At this late stage of life, skeletal muscles show a dramatic impairment of their regenerative capacity, due to the entrance of satellite cells into senescence (a G0 irreversible arrest state), at the expense of their normal quiescence status.
Disruption of autophagic activity with aging occurs in many organs and tissues, being one of the major contributors to the global aging process. The mechanisms driving this autophagic decline are still poorly understood, but likely involve alteration of multiple age-related pathways.
The goal of our study is to understand how satellite cells are able to maintain quiescence throughout most of their life, avoiding senescence, and to reveal the age-associated mechanisms underlying the eventual decline in their regenerative capacity. In particular, we have investigated whether autophagy plays a role in this process.
We have demonstrated for the first time that quiescent satellite cells have active basal autophagy that is responsible for protein and organelle turnover. Our results indicate that in very old (geriatric) mice, this basal autophagic flux is impaired in muscle stem cells, resulting in accumulation of damaged proteins and subcellular organelles, including mitochondria. Toxic waste accumulation in satellite cells during aging affects their fitness and causes loss of the bona fide quiescent state. Thus, constitutive autophagy is a quality-control process that is essential to maintain cellular homeostasis and stemness: The capacity to regenerate damaged myofibers and self-renew to replenish the satellite cell pool. Consistent with these conclusions, autophagy disruption in post-mitotic cells, such as neurons, results in accumulation of abnormal proteins and organelles that lead to neurodegeneration.
Of importance, we have found that in vivo pharmacological treatment (rapamycin or spermidine), as well as genetic manipulation (LV-Atg7) ex vivo, are able to restore autophagy in geriatric satellite cells, which allows their proliferative expansion while preventing senescence entry, thus enabling new muscle-fiber formation. In contrast, constitutive deletion of Atg7 in the satellite cell lineage (Atg7ΔPax7 mice) leads to reduction of the number of satellite cells in postnatal muscle. Interestingly, the remaining cells show signs of premature aging including reduced regenerative capacity, reminiscent of satellite cells in physiologically aged mice. Furthermore, when Atg7 was deleted specifically in satellite cells of adult mice (Atg7ΔPax7ER mice), the satellite cell population suffers also a major numerical and functional drop, confirming that basal autophagy is required for both the postnatal establishment and the life maintenance of the quiescent stem cell population. In contrast to these results, basal autophagy in hematopoietic stem cells (HSCs) is induced with aging, in agreement with preservation of the HSC total population over time. Of interest, recent studies also showed that induction of autophagy is required for the activation from quiescence of young satellite cells, to provide the nutrients necessary to meet bioenergetics demands during this critical cellular transition stage.
How could loss of autophagy in young quiescent satellite cells induce premature aging? We found that the age-related autophagy impairment involves failure of mitophagy. In geriatric satellite cells, mitochondrial turnover is affected, and, consequently, higher levels of reactive-oxygen species (ROS) are generated. We have provided evidence that constitutive autophagy/mitophagy can be pharmacologically restored in vivo, not only by rapamycin or spermidine, but also by treatment with trolox, a vitamin E analog that neutralizes excessive ROS (). These treatments restore autophagy, avoid the accumulation of intracellular damage and improve cellular fitness of satellite cells from geriatric mice, as shown by their increased proliferative and regenerative activities.
Figure 1. Age-impaired autophagy in muscle stem cells leads to the accumulation of intracellular components and damaged organelles such as mitochondria, thereby promoting ROS generation. This increase in ROS levels induces cellular transition from quiescence into senescence through the activation of CDKN2A. Pharmacological or genetic manipulation of autophagy is able to modulate this transition in muscle stem cells.
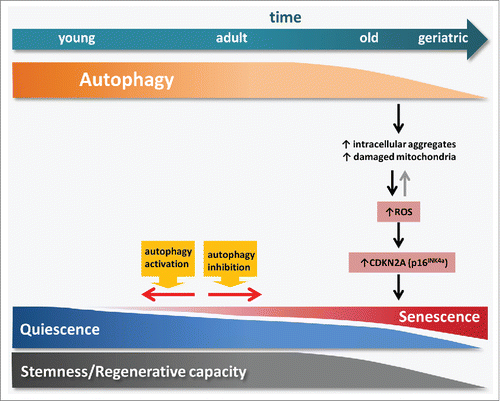
Our previous studies implicated induction of the senescence-associated gene Cdkn2a/p16INK4a in the loss of reversible quiescence in satellite cells at geriatric age. However, what caused induction of Cdkn2a in aged satellite cells remained unknown. We have now shown that high levels of ROS, such as those generated in geriatric satellite cells by dysfunctional mitochondria, are sufficient to trigger the epigenetic-mediated derepression of the Cdkn2a locus, and this contributes to the acquisition of the senescence fate (). Remarkably, as observed in mice, human geriatric satellite cells also exhibit autophagy defects and senescence traits that preclude their normal myogenic functions. Importantly, the rapamycin-mediated activation of autophagy is able to restore cell fitness and myogenesis in cultured human geriatric cells.
In conclusion, we have provided evidence that autophagy is essential to maintain quiescence in satellite cells and guarantee their “stemness.” Age-associated deficits in autophagy and mitophagy increase ROS levels and drive senescence in aged satellite cells. These findings highlight the possibility of pharmacologically restoring the homeostatic quiescence state in old satellite cells through the modulation of autophagy.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.