
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Autophagy is the primary process for recycling cellular constituents through lysosomal degradation. In addition to nonselective autophagic engulfment of cytoplasm, autophagosomes can recognize specific cargo by interacting with ubiquitin-binding autophagy receptors such as SQSTM1/p62 (sequestosome 1). This selective form of autophagy is important for degrading aggregation-prone proteins prominent in many neurodegenerative diseases. We carried out a high content image-based siRNA screen (4 to 8 siRNA per gene) for modulators of autophagic flux by monitoring fluorescence of GFP-SQSTM1 as well as colocalization of GFP-SQSTM1 with LAMP2 (lysosomal-associated membrane protein 2)-positive lysosomal vesicles. GFP-SQSTM1 and LAMP2 phenotypes of primary screen hits were confirmed in 2 cell types and profiled with image-based viability and MTOR signaling assays. Common seed analysis guided siRNA selection for these assays to reduce bias toward off-target effects. Confirmed hits were further validated in a live-cell assay to monitor fusion of autophagosomes with lysosomes. Knockdown of 10 targets resulted in phenotypic profiles across multiple assays that were consistent with upregulation of autophagic flux. These hits include modulators of transcription, lysine acetylation, and ubiquitination. Two targets, KAT8 (K[lysine] acetyltransferase 8) and CSNK1A1 (casein kinase 1, α 1), have been implicated in autophagic regulatory feedback loops. We confirmed that CSNK1A1 knockout (KO) cell lines have accelerated turnover of long-lived proteins labeled with 14C-leucine in a pulse-chase assay as additional validation of our screening assays. Data from this comprehensive autophagy screen point toward novel regulatory pathways that might yield new therapeutic targets for neurodegeneration.
Introduction
Autophagy is an evolutionarily conserved process that allows cells to meet energetic demands in an ever-changing nutritional environment. When nutrients are scarce, bulk portions of the cytosol are engulfed into double membranous vesicles called autophagosomes and delivered to lysosomes for degradation and recycling. In addition to this nonselective form of autophagy, autophagosomes can selectively isolate damaged organelles, pathogens, and aggregated proteins by binding to a “cargo receptor” such as SQSTM1/p62 (sequestosome 1) that interacts with ubiquitinated substrates.Citation1 This process is particularly important in postmitotic cells, such as neurons, that are unable to reduce the burden of misfolded and aggregation-prone proteins through cell division. In mice, conditional knockout of an essential autophagy gene, Atg7 (autophagy-related 7), in the central nervous system leads to accumulation of inclusion bodies filled with ubiquitinated proteins in neurons, progressive neurodegeneration, and premature death.Citation2
Many aggregation prone proteins that are known to cause neurodegenerative diseases—such as MAPT/tau (microtubule associated protein tau), SNCA/α-synuclein (synuclein α) and HTT (huntingtin)—are substrates for autophagy.Citation3 The decline in autophagic activity and lysosomal proteolytic capacity that occurs with aging may contribute to the prevalence of some proteinopathies late in life.Citation4 Thus, identification of novel regulatory pathways that increase selective forms of autophagy might yield new candidate therapeutic targets for neurodegeneration.
In order to identify modulators of autophagic flux that are involved in selective autophagy, we performed an image-based siRNA screen for genes that alter turnover of SQSTM1 under normal nutritive conditions (; ). Our screen differs from those conducted previously in both the breadth of phenotypes measured via high-content imaging, and in the high count of siRNA sequences used per gene to avoid off-target effects common to RNAi screens. More than 12,000 genes, selected based on likelihood of target druggability, were queried with 4 to 8 individual siRNAs per gene for a total of more than 90,000 wells screened. Reduction in fluorescence of GFP-labeled SQSTM1 in combination with either an increase or decrease in colocalization with lysosomes was interpreted as an increase in autophagic flux. Gene-level hits were confirmed with 6 alternate siRNAs in the same assay in both U2OS and H4 cell lines. In addition to confirming autophagic flux, we assessed whether any of the candidate autophagy modulators were acting independently or downstream of the well-characterized MTOR (mechanistic target of rapamycin [serine/threonine kinase])-signaling pathway. Knockdown of genes that caused a significant reduction in viability as measured by uptake of the cell-impermeable DRAQ7 nuclear dye were excluded from further analysis. As an orthogonal measure for autophagic flux, we measured the number of autophagosomes fused with lysosomes in a high-throughput time-lapse imaging assay. This screening strategy identified known autophagy proteins as well as novel hits that may shed light on previously undescribed regulatory mechanisms.
Figure 1. Screen flow scheme. Twelve thousand thirty seven genes were first screened in a GFP-SQSTM1 and LAMP2 assay in U2OS for phenotypes consistent with upregulation of autophagy. Four to 8 siRNAs were screened per gene in the primary screen; 617 genes were selected for follow-up and screened with 6 new siRNA per gene, with a concerted effort to avoid seeds biased toward off-target effects by applying common seed analysis to primary screen data. Follow-up assays included hit confirmation in the GFP-SQSTM1 and LAMP2 assay, viability assessment, and MTOR profiling, all in both U2OS and H4 cell lines. Based on these data, 96 genes were selected for confirmation assays, which included a live-cell, kinetic autophagic flux assay; 80 of these 96 genes were selected for siRNA knockdown confirmation via qPCR. Ten hits were ultimately identified; knockdown of these targets resulted in phenotypic profiles consistent with upregulation of autophagic flux. Assay details, including primary output measurements, can be found in .
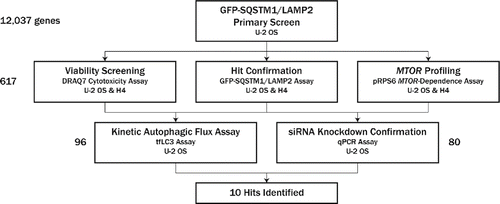
Table 1. Assays used throughout siRNA screening campaign.
Results
To identify novel genes involved in selective forms of autophagic flux, we set up an image-based screening assay to monitor turnover of the autophagy receptor, SQSTM1, fused with GFP. SQSTM1 contains ubiquitin- and LC3-binding domains and acts as a linker to load ubiquitinated protein aggregates into LC3–phosphatidylethanolamine-containing autophagosomes.Citation5 Mature autophagosomes fuse with lysosomes, forming autolysosomes, in which lysosomal hydrolases degrade cargo, including SQSTM1. Previous studies have demonstrated that when autophagic flux is upregulated, GFP-SQSTM1 is degraded, and the intensity of GFP in the cytoplasm decreases.Citation6 Cytoplasmic GFP-SQSTM1 intensity in U2OS fibroblasts decreased ∼50% upon inhibition of MTOR, a negative regulator of autophagy,Citation7 with the selective catalytic inhibitor PP242Citation8 (Fig. S1). U2OS cells stably expressing GFP-SQSTM1 under doxycycline control were reverse-siRNA-transfected for 96 h, formaldehyde-fixed, and stained with Hoechst, anti-TUBB/β-tubulin antibody, and anti-LAMP2 antibody, to identify nuclei, microtubules (for cellular boundary detection), and lysosomes, respectively (). GFP-SQSTM1 is expressed throughout the cytoplasm and aggregates into punctae during autophagosome formation. Upon MTOR knockdown via siRNA transfection, the cytoplasmic intensity of GFP-SQSTM1 decreased by ∼60% (). Immunostaining of LAMP2 allowed us to visualize lysosomes and quantify the extent to which GFP-SQSTM1 and LAMP2 colocalized in the cytoplasm using the Pearson correlation coefficient, r (); a similar approach has been used in primary T cells to quantify autophagic activity.Citation9 Alternative measurements of colocalization were evaluated, including the Manders overlap coefficientCitation10 and the intensity correlation quotient,Citation11 but the Pearson correlation coefficient yielded the highest Z’ scores with LAMP2 and SQSTM1 siRNA controls during assay development; the greater utility of the Pearson correlation coefficient in our assay relative to the Manders overlap coefficient is consistent with previous findings.Citation12 Knockdown of ATP6V0D1 (ATPase, H+ transporting, lysosomal 38kDa, V0 subunit d1), a proton pump subunit required for lysosomal acidification,Citation13 produces a phenotype in which GFP-SQSTM1 is loaded into autolysosomes, but is not fully degraded. This is evident in , where ATP6V0D1 knockdown increases GFP-SQSTM1 and LAMP2 colocalization relative to cells transfected with nontargeting siRNA. Alternatively, knockdown of LAMP2 decreases GFP-SQSTM1 and LAMP2 colocalization as expected ().
Figure 2. Review of primary screen. (A) U2OS cells expressing GFP-SQSTM1 (green, top right) were siRNA-transfected for 96 h and subsequently fixed and stained for nuclear DNA (blue, top left), TUBB (red, bottom left), and LAMP2 (orange, bottom right). Hoechst and TUBB channels were used for segmentation of nuclei and cell boundaries, respectively. (B) GFP-SQSTM1 localizes to the cytoplasm and autophagic puncta. Upon MTOR knockdown (right), autophagy is upregulated, GFP-SQSTM1 is degraded, and cytoplasmic intensity decreases. Degradation of GFP-SQSTM1 was quantified by measuring GFP-SQSTM1 intensity in a 10-pixel wide ring around the nucleus. (C) LAMP2 was stained to identify lysosomes, and cytoplasmic colocalization of GFP-SQSTM1 and LAMP2 was quantified using the Pearson correlation coefficient, r. Knockdown of LAMP2 (right) decreases colocalization relative to nontargeting controls (center), while ATP6V0D1 knockdown (left) attenuates lysosomal acidification and thus increases colocalization. (D) U2OS cells were transfected with 4 to 8 siRNA per gene, and each siRNA's effect on GFP-SQSTM1 mean ring intensity and GFP-SQSTM1 and LAMP2 colocalization was quantified and normalized to nontargeting controls. Individual siRNA results are plotted as percent of control (POC) with positive MTOR control siRNA (red, pink) and neutral control siRNA (blue, light blue) overlaid. Cell count results are reflected in the size of each data point, with higher cell counts represented with larger circles. (E) For each phenotype measured, siRNA POC values were condensed to gene-level P values using a rank-based orthogonal gene averaging (OGA) algorithm. GFP-SQSTM1 and LAMP2 colocalization POC values are plotted on a Kaplan-Meier-like survival plot for LAMP2 (green) and ATP6V0D1 (red) relative to a reference population (black). (F) At the gene level, GFP-SQSTM1 and LAMP2 colocalization uphit P values are plotted against GFP-SQSTM1 mean ring intensity downhit P values. While MTOR and ATP6V0D1 show significant colocalization uphit P values, their autophagy-promoting and autophagy-blocking phenotypes, respectively, are distinguished by their GFP-SQSTM1 mean ring intensity downhit P values. (G) At the gene level, GFP-SQSTM1 and LAMP2 colocalization downhit P values are plotted against GFP-SQSTM1 mean ring intensity downhit P values. While several essential autophagy genes (ATG5, BECN1, PIK3R4, PIK3C3) show significant colocalization downhit P values, these phenotypes are associated with nonsignificant GFP-SQSTM1 mean ring intensity downhit P values.
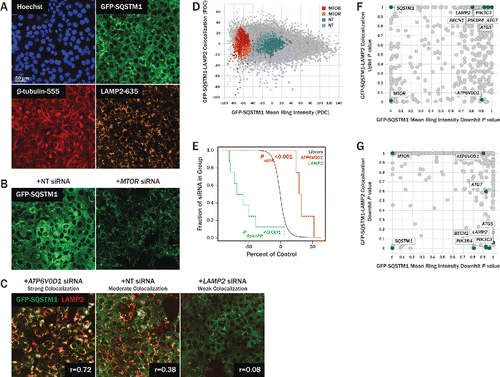
We transfected cells with a siRNA library covering 12,037 genes, with 4–8 siRNAs per gene. Each siRNA was plated into a separate well. Cells were stained, imaged, and GFP-SQSTM1 mean ring intensity, GFP-SQSTM1 and LAMP2 colocalization, and cell count were calculated for each individual well (). Results were normalized to control wells where cells were transfected with nontargeting siRNA; results are reported as percent of control (POC). Roughly 8.3% of the 75,342 siRNAs tested reduced GFP-SQSTM1 mean ring intensity by at least 40% relative to controls (dotted green box in ), and these reductions were associated with a range of GFP-SQSTM1 and LAMP2 colocalization phenotypes. For each phenotype measured, siRNA POC values were condensed to gene-level P values using a rank-based orthogonal gene averaging (OGA) algorithm ().Citation14 In the primary screen, knockdown of 205 genes (1.7%) resulted in significant reductions in GFP-SQSTM1 intensity with OGA P values < 0.05, with MTOR having the 16th lowest P value. OGA P values were used to select primary screen hits with low GFP-SQSTM1 intensity, high cell counts, and both low and high GFP-SQSTM1 and LAMP2 colocalization values. Additional genes were selected for follow-up if 50% or more of their siRNAs resulted in favorable phenotypes. In total, 617 genes were selected for follow-up from the primary screen. OGA P values for GFP-SQSTM1 mean ring intensity, GFP-SQSTM1 and LAMP2 colocalization, and cell count for these genes are tabulated in Table S1, along with P values from several other measurements collected throughout the screening campaign. GFP-SQSTM1 and LAMP2 colocalization uphit and downhit P values are plotted against GFP-SQSTM1 mean ring intensity downhit P values for the 617 genes that were re-evaluated in the confirmation screen (, respectively). Whereas cytoplasmic GFP-SQSTM1 intensity decreased upon stimulation of autophagy via inhibition or knockdown of MTOR, one may expect GFP-SQSTM1 ring intensity to increase when the activity of essential autophagy genes is perturbed. While knockdown of several essential autophagy genes did not significantly increase GFP-SQSTM1 mean ring intensity (Table S1), colocalization between GFP-SQSTM1 and LAMP2 was significantly reduced by RNAi of BECN1 (Beclin 1, autophagy related), PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3), PIK3R4 (phosphoinositide-3-kinase regulatory subunit 4), and ATG5 (autophagy-related 5) (). Furthermore, whereas knockdown of MTOR or ATP6V0D1 significantly increased GFP-SQSTM1 and LAMP2 colocalization, these autophagy-promoting and autophagy-blocking perturbations, respectively, were distinguished by their GFP-SQSTM1 mean ring intensity downhit P values (). Altogether, the combination of these phenotypic measurements, GFP-SQSTM1 mean ring intensity and GFP-SQSTM1 and LAMP2 colocalization, highlighted several autophagy-related genes and provided us with valuable information regarding the direction of autophagy regulation induced by RNAi.
For confirmation and profiling assays, we selected 6 new siRNAs for each gene. To reduce potential off-target effects of siRNA which can contribute to high false-positive rates when screening,Citation15 we carried out common seed analysis using data from the primary screen.Citation16 Briefly, siRNA sequences were grouped by common hexamer and heptamer seeds (nucleotides 2 through 7 or 2 through 8, respectively, of the guide strand), and Kolmogorov-Smirnov (K-S) tests were carried out to assess if the distribution of assay values for a single seed differed from the distribution of assay values for all seeds. The rank order of biased heptamer seeds based on GFP-SQSTM1 intensity measurements was determined and biased seed regions were avoided when selecting siRNAs for confirmation and profiling analyses of the primary screen hits (Fig. S2).
Following the primary screen, 3 assays were carried out in parallel in both U2OS cells and the H4 cell line derived from neuroglioma: i) hit confirmation in the GFP-SQSTM1 and LAMP2 assay, ii) viability screening via a DRAQ7 cytotoxicity assay (), and iii) profiling of MTOR signaling via a phospho-RPS6 (ribosomal protein S6) assay (). Similar to the primary screen in U2OS cells, MTOR knockdown in H4 cells decreased the cytoplasmic intensity of GFP-SQSTM1 by ∼60 to 80% relative to nontargeting controls (Fig. S3), and knockdown of several essential autophagy genes, including BECN1, PIK3C3, and ATG7, strongly reduced colocalization between GFP-SQSTM1 and LAMP2 in H4 cells (Table S1). Gene-level P values were again computed using the OGA algorithm: in U2OS cells, knockdown of 82 genes (13.3%) resulted in significant reductions in GFP-SQSTM1 intensity with OGA P values < 0.05, with MTOR having the 21st lowest P value; in H4 cells, knockdown of 55 genes (8.9%) resulted in significant reductions in GFP-SQSTM1 intensity with OGA P values < 0.05, with MTOR having the 31st lowest P value. 41 genes were common to both GFP-SQSTM1 intensity downhit lists (Fig. S4).
Figure 3. Secondary imaging assay to assess cell viability. (A) DRAQ7, a fluorescent DNA dye that only stains nuclei in dead and permeabilized cells, was used to assess the effect of protein knockdown on cell viability. U2OS cells transfected with nontargeting siRNA (left) show a low percentage of DRAQ7+ cells (red), while cells transfected with siRNA targeting the essential gene EIF4A3 both decrease in count and in viability (right). (B) Gene-level hits from the primary screen were profiled in a DRAQ7 viability assay. Where possible, new sequences were used for each gene to avoid biased seeds identified in common seed analysis. Viability was assessed in both U2OS (shown here) and H4 (not shown) cells. For each siRNA tested, the percentage of live cells (Hoechst+ DRAQ7−) is plotted against the total cell count (Hoechst+). Nontargeting siRNA (blue, light blue) had little effect on cell viability. Upon MTOR knockdown (red, pink), wells maintained a high percentage of live cells, but did, however, contain fewer cells relative to nontargeting siRNA, suggesting a reduction in cell proliferation in this condition. Knockdown of essential genes POLR2A (green) and EIF4A3 (purple) reduced both cell count and the percentage of live cells, as expected.
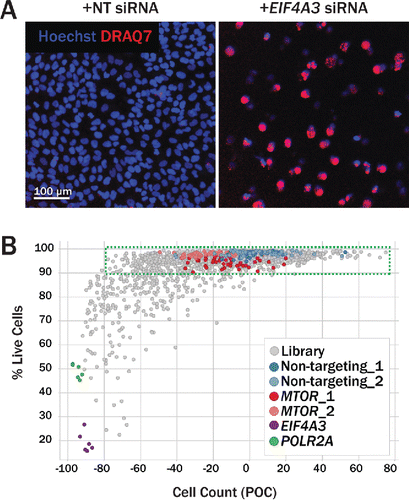
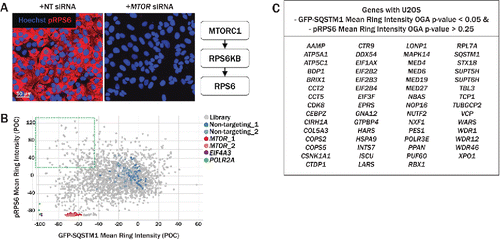
Figure 4. Secondary imaging assay for assessment of MTOR activity. (A) RPS6/S6, is an indirect substrate of MTORC1 and is phosphorylated by RPS6KB. Relative to siRNA transfection with neutral control siRNA (left), MTOR knockdown (right) significantly reduced the intensity of pRPS6, labeled with anti-pRPS6–555 (red). (B) The pRPS6 readout was multiplexed with the primary GFP-SQSTM1 assay, allowing simultaneous measurement of pRPS6 and GFP-SQSTM1 intensities. Upon MTOR knockdown (red, pink), both pRPS6 and GFP-SQSTM1 intensities were dramatically reduced relative to nontargeting controls (blue, light blue). Several siRNAs in the upper left quadrant decreased GFP-SQSTM1 intensity while maintaining high pRPS6 intensity, suggesting an increase in autophagy independent of MTOR inhibition. (C) Genes with U2OS GFP-SQSTM1 mean ring intensity OGA P values < 0.05 and pRPS6 mean ring intensity OGA P values > 0.25 are listed in the table. Knockdown of these genes resulted in an upregulation of autophagy while pRPS6 levels were not significantly reduced.
In order to assess the effect of protein knockdown on cell viability, cells were incubated with DRAQ7, a fluorescent dye that only stains nuclei in dead and permeabilized cells. U2OS cells transfected with nontargeting siRNA remain high in both cell count and in viability (negative for DRAQ7 staining), whereas cells transfected with siRNA targeting the essential gene eukaryotic translation initiation factor 4A3 (EIF4A3) show decreased cell count and viability (positive for DRAQ7 staining) (). The percentage of live DRAQ7-negative cells was calculated for each siRNA treatment, and plotted against normalized cell counts (). While transfection with nontargeting and MTOR siRNA maintained relatively high cell counts with high viability, knockdown of essential genes EIF4A3 and POLR2A (polymerase [RNA] II [DNA directed] polypeptide A, 220kDa) resulted in low cell counts with low viability. Several genes were identified, however, that maintained high viability with low cell counts, suggesting that cell proliferation may be arrested upon knockdown of these targets. U2OS and H4 cell viability downhit P values derived from the DRAQ7 assay are available in Table S1.
Because MTOR is a well-established negative regulator of autophagy, we characterized the extent to which the primary screen hits also impacted MTOR signaling by multiplexing a phospho-RPS6 (pRPS6) readout into the GFP-SQSTM1 imaging assay. RPS6 is an indirect substrate of MTORC1 (), and upon MTOR knockdown, phosphorylation of RPS6 should be reduced. We immunostained cells using an anti-pRPS6 antibody conjugated to Alexa Fluor 555 in both U2OS and H4 cells and measured cytoplasmic intensity. Bright pRPS6–555 intensity was maintained after transfection with nontargeting siRNA while MTOR knockdown greatly diminished the cytoplasmic signal, as expected (). By measuring both pRPS6 and GFP-SQSTM1 cytoplasmic intensities in the same well, we were able to identify a group of genes that when knocked down, reduced GFP-SQSTM1 intensity while maintaining high pRPS6 intensity, suggesting an MTOR-independent mechanism for autophagic clearance of SQSTM1 (); these genes are tabulated in Fig. 4C. Phospho-RPS6 mean ring intensity downhit P values in both U2OS and H4 cells are available in Table S1.
OGA P values were computed for GFP-SQSTM1 intensity, GFP-SQSTM1 and LAMP2 colocalization, cell viability, cell count, and pRPS6 intensity in both cell lines (Table S1). Collectively, these data were used to select 96 genes with autophagy-promoting and viable phenotypes, including necessary controls, to carry forward to the next stage of confirmatory screening, where autophagic flux was measured in an alternate live-cell assay. Additionally, 80 of the 96 genes were prioritized for knockdown confirmation by qPCR; 16 genes with lower viability profiles after knockdown were excluded from this analysis.
In order to confirm autophagic turnover of our hits, we carried out a live-cell assay in which U2OS cells expressed tandem-fluorescent LC3B (tfLC3). The tfLC3 construct consists of the acid-stable tagRFP and acid-quenchable tagGFP2 fused to the N terminus of human MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 β). Upon lipidation, tfLC3 translocates to a budding autophagosome precursor, a phagophore, where the structure fluoresces in both the red and green imaging channels.Citation17 Once an autophagosome fuses with a lysosome, however, the GFP signal is quenched by the acidic environment, while the RFP signal remains stable, and is thus visible only in the red imaging channel (). We initially carried out proof of concept experiments with autophagy promoters and blockers in order to validate the assay system. Treatment with the MTOR inhibitor, PP242, elevated both the number of autophagosomes and autolysosomes per cell relative to untreated cells, while chloroquine increased only autophagosome counts, effectively blocking autophagic flux (Fig. S5). To validate our screen hits, cells were siRNA-transfected and subsequently imaged for 66 h at 6 h intervals, beginning 26 h after transfection. Autophagosomes and autolysosomes were quantified at each time point, and data were normalized by individual cell area in order to account for variations in cell density over the course of the assay. The transfection process caused an initial decrease in both autophagosomes and autolysosomes for the first 45 h after transfection, but the number of vesicles returned to a stable baseline thereafter. MTOR siRNA served as a positive control for autophagic flux and showed a significant increase in the number of autolysosomes relative to nontargeting controls at timepoints >50 h post-transfection (). Conversely, siRNA against essential autophagy genes ATG5, BECN1, and PIK3C3 reduced the number of autolysosomes over time (). Knockdown of 10 genes caused a statistically significant increase in the number of autolysosomes based on OGA P values for time points 56, 74, or 92 h post transfection. Primary and secondary data for these 10 hits along with 7 controls are summarized in . These 10 hits met the following criteria: i) confirmed GFP-SQSTM1 degradation or GFP-SQSTM1 and LAMP2 colocalization phenotype in U2OS and/or H4 cells; ii) autolysosome uphit OGA P value ≤ 0.05 in at least one timepoint in the tfLC3 assay; iii) cell count downhit OGA P value > 0.1 in primary screen, and iv) viability downhit OGA P value > 0.1 in DRAQ7 viability assay.
Figure 5. Tandem-fluorescent LC3 assay to measure autophagic flux over time in live cells. (A) Tandem-fluorescent LC3 (tfLC3) construct consists of tagRFP and tagGFP2 fused to the N terminus of human LC3B. tagRFP pKa = 3.8, while tagGFP2 pKa = 5.0, rendering tagRFP more acid-stable than tagGFP2. Cytosolic RFP-GFP-LC3 is lipidated and translocated to a phagophore (right, GFP+ RFP+ autophagosome). Once an autophagosome has fused with an acidic lysosome, the GFP signal is quenched due to its higher pKa, leaving red-only puncta (left, GFP− RFP+ autolysosome). (B) Merged RFP and GFP channels are shown at 50 h (top) and 80 h (bottom) post-transfection for U2OS cells. Relative to nontargeting controls (left), MTOR knockdown (right), increased the number of GFP− RFP+ puncta, representing autolysosomes, in the cytoplasm, indicating an upregulation of autophagic activity. (C) U2OS cells stably expressing tfLC3 were siRNA-transfected and imaged live for 66 h following a 26 h incubation period post transfection. GFP− RFP+ puncta were identified as autolysosomes and normalized to cell area to quantify autophagic flux over time. siRNA-level results were condensed to gene-level results by aggregating with median values. MTOR knockdown (red) increased autolysosomes/cell area over time relative to nontargeting controls (blue). Several pathway controls, including ATG5 (orange), PIK3C3 (yellow), and BECN1 (green), decreased autophagic flux over time.
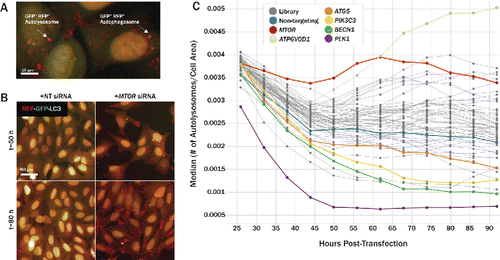
Figure 6. Summary of screening results and confirmation of gene knockdown. (A) Summary heatmap (left) showing P values from various assays carried out in the screening campaign. Measurement values for each column are noted in the table (right). Assay results from several control genes are shown at the top of the heatmap, with the top 10 gene hits, based on various criteria across all assays, shown below. Heatmap is colored such that P values consistent with increased autophagic flux and low cytotoxicity are represented in green and the inverse is represented in red. (B) Gene knockdown was determined in U2OS cells via qPCR. Each data point represents an individual siRNA and the extent to which it reduced expression of that particular target. All data has been normalized to endogenous GAPDH reference levels and is reported relative to cells transfected with nontargeting control siRNA. qPCR was performed in triplicate. Circles mark average values for individual siRNA, with error bars representing SD.
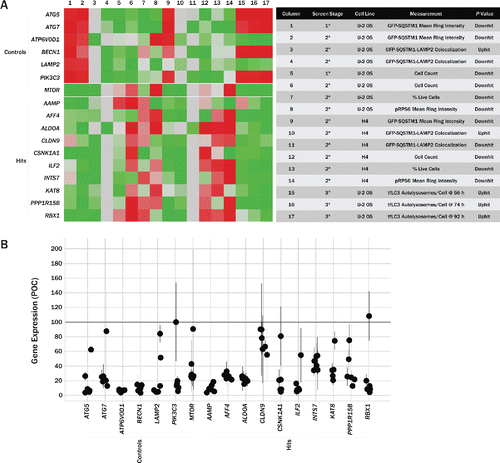
Knockdown of these 10 targets in U2OS cells was also confirmed via qPCR (). While some siRNAs were ineffective at knockdown, the majority of siRNAs used for each gene effectively reduced target levels by at least 40%. GFP-SQSTM1 intensity versus transcript level per siRNA is shown in Fig. S6.
The phenotypes observed for these 10 genes in the primary and secondary screening assays suggest that they may normally suppress autophagy and that inhibition of these targets enhances autophagic flux in the cell types we tested. The cumulative data for CSNK1A1 (casein kinase, α 1) were particularly strong (). siRNA knockdown of CSNK1A1 caused a significant decrease in GFP-SQSTM1 fluorescence in both U2OS and H4 cell lines (OGA P values = 0.001 and 0.006, respectively) and a significant increase in autolysosomes was measured in the tfLC3 assay 96 h after transfection with CSNK1A1 siRNA (OGA P value = 0.03). Interestingly, CSNK1A1 knockdown did not significantly reduce phosphorylation of RPS6 in either the U2OS or H4 lines (OGA P values = 0.749 and 0.999, respectively), indicating that the increase in autophagic flux was independent from or downstream of MTOR signaling.
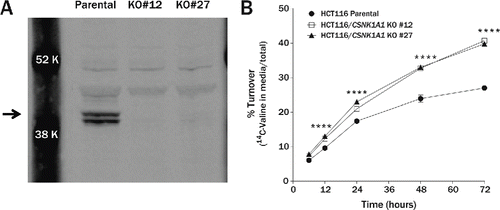
To provide independent confirmation that CSNK1A1 suppresses autophagic flux under normal nutritive conditions, we measured the rate of long-lived protein turnover in TALEN-mediated CSNK1A1 KO cell lines. DNA double-strand breaks were induced by TALENs targeted to exon 1 of CSNK1A1 near the ATG start site in HCT116 colorectal carcinoma cells. Immunoblot analysis showed that the 2 human CSNK1A1 isoforms composed of 365 and 337 amino acids are present in HCT116 cells near the calculated molecular masses of 41.9 and 38.9 kDa but are absent in the 2 CSNK1A1 KO clonal lines (). To measure turnover of long-lived proteins, the parental and KO lines were metabolically labeled with 14C-valine and the fraction released into the media as free amino acid was measured over a period of 3 d. The percent of degraded protein was significantly higher in the KO lines compared to the parental lines (). Total scintillation counts were equivalent in all 3 cell lines indicating that the amount of 14C-valine initially incorporated into protein during the pulse did not differ between the parental and KO lines and the cell counts were equivalent throughout the experiment (data not shown). A second independent experiment yielded similar, statistically significant results. These data suggest that CSNK1A1 may function as a negative regulator of autophagic activity in nutrient-rich conditions.
Figure 7. Turnover of long-lived proteins is accelerated in CSNK1A1 knockout cell lines. (A) Immunoblot of CSNK1A1 protein in homogenates from HCT116 parental and HCT116 CSNK1A1 KO clonal lines. A doublet (arrow) corresponding to human isoforms 1 and 2 are absent in the KO cell lines. (B) In a pulse-chase assay, the percent of degraded long-lived protein was significantly higher in HCT116 CSNK1A1 KO lines compared to HCT116 parental cells at 12 h (parental = 9.6 ± 0.1%, KO#12 = 12.3 ± 0.1%, KO#27 = 13.0 ± 0.3%), 24 h (parental = 17.4 ± 0.1%, KO#12 = 21.0 ± 0.3%, KO#27 = 23.0 ± 0.2%), 48 h (parental = 23.9 ± 0.6%, KO#12 = 32.7 ± 0.2%, KO#27 = 33.0 ± 0.2%), and 72 h (parental = 27.0 ± 0.3%, KO#12 = 40.7 ± 0.2%, KO#27 = 39.8 ± 0.2%) after metabolic labeling with 14C-valine (n = 3 wells per condition). Significance was determined by 2-way analysis of variance followed by the Dunnett multiple comparison test (****P < 0.0001). Error bars represent standard error of the mean.
Discussion
Acceleration of autophagic degradation of misfolded proteins has emerged as a potential therapeutic strategy for several neurodegenerative diseases.Citation3 We screened for suppressors of autophagy that restrain autophagic degradation under normal nutritive conditions, reasoning that inhibition of these targets might represent a viable strategy for enhancing autophagic flux. Because SQSTM1 plays a role in targeting K63-linked ubiquitinated proteins to autophagosomes, and is itself autophagic cargo, we chose to monitor fluorescence of GFP-labeled SQSTM1 as a readout for selective autophagy as previously described.Citation6 Although monitoring SQSTM1 has been used for hit confirmation in other autophagy siRNA screens, GFP-SQSTM1 fluorescence intensity and SQSTM1-lysosomal colocalization have not previously been used to our knowledge as primary screening endpoints. Here, primary screen hits were selected based on GFP-SQSTM1 phenotypes consistent with increased autophagic flux (reduced overall GFP-SQSTM1 fluorescence and/or increased colocalization with the lysosomal protein LAMP2). Because various forms of cellular stress can trigger an increase in autophagy, we also counter-screened for nuclear incorporation of DRAQ7 to exclude targets that may have enhanced autophagy secondary to cytotoxicity. We have also provided a list of hits published in other autophagy RNAi screens and included results for those genes from our primary screen, where applicable (Table S2). For example, SUPT5H (SPT5 homolog, DSIF elongation factor subunit) was a strong hit in the primary screen and has been identified in a previous autophagy screen,Citation18 but it was not included in our follow-up studies because knockdown was also associated with significant cytotoxicity in the cell lines used in our screen (Table S2). Genes with favorable autophagic phenotypes that were excluded from further analysis based on toxicity data have been annotated in Table S1. After retesting the primary screen hits with a second set of 6 siRNAs per gene that were carefully selected using seed region analysis to minimize use of biased sequences, 96 hits were confirmed and prioritized for further analysis. The level of transcript knockdown was also verified by qPCR for 80 of these 96 genes and approximately 80% of the siRNAs reduced target expression by at least 70% (target expression and siRNA vendor information is listed in Table S3). The correlation between transcript knockdown per siRNA and GFP-SQSTM1 fluorescence intensity is not as strong as expected, possibly due to varying thresholds at which knockdown affects autophagy for different targets and a nonlinear relationship between phenotype and transcript level (Fig. S7).
The MTOR signaling complex 1 (MTORC1) is the most extensively studied negative regulator of autophagy and MTOR inhibitors dramatically accelerate autophagic turnover. However, MTOR complexes control a wide variety of signaling pathways related to metabolism and cell cycle progression in addition to autophagy. Although MTOR inhibitors have been successfully developed and are used in cancer treatment and for immunosuppression in organ transplantation, their safety profile limits their use for chronic conditions such as neurodegeneration.Citation19 Thus there is a strong interest in identifying signaling pathways that control autophagy independently of MTOR inhibition. We characterized the phosphorylation state of the downstream MTOR effector RPS6 after knockdown of the confirmed hits to determine if any showed independence from MTORC1 signaling. The weak correlation between GFP-SQSTM1 and pRPS6 intensities suggests that some of the hits might modulate MTOR-independent pathways that control autophagy, and this notion is reinforced by the cluster of phenotypes where both GFP-SQSTM1 degradation and pRPS6 intensity are maximal ().
A potentially confounding factor for the primary screen is that reduction of GFP-SQSTM1 fluorescence can result from mechanisms other than upregulation of autophagy, such as reduced transcription or translation of the reporter protein. In fact, knockdown of several aminoacyl-tRNA synthetases, subunits of RNA polymerase II, and components of the RNA polymerase mediator complex significantly reduced GFP-SQSTM1 intensity in our primary screen. Several of these genes were excluded from follow-up due to their strong ties to protein synthesis. Strong or weak colocalization between GFP-SQSTM1 and LAMP2, however, was associated with RNAi of several autophagy-related genes. By selecting for phenotypes with both outlying colocalization and reduced GFP-SQSTM1 mean ring intensity, we were able to enrich our hit list with genes more likely to be directly involved in autophagy. Furthermore, confirmed primary screen hits were tested in an independent assay for autophagic flux based on quantitation of tfLC3-labeled autolysosomes, which should not be affected by reductions in protein synthesis. We confirmed that knockdown of MTOR caused a significant increase in autolysosomes in the tfLC3 assay, while knockdown of the autophagy essential genes BECN1 and ATG7 caused a significant reduction in autolysosomes compared to nontargeting control siRNAs (). However, knockdown of ATP6V0D1, which encodes a subunit of the vacuolar-type H+-ATPase required for lysosomal acidification, paradoxically caused a significant increase in autolysosomes when loss of lysosomal acidification would be expected to reduce the number of red-only autolysosomal puncta (). One explanation for this unexpected result is that the vacuolar-type H+-ATPase proton pump is a component of the amino acid-sensing complex required for homeostatic activation of MTOR which suppresses autophagy under high nutrient conditions.Citation20 Thus, knockdown of ATP6V0D1 might be expected to deregulate MTOR activity. Consistent with this hypothesis, knockdown of ATP6V0D1 caused a significant reduction in phosphorylation of the downstream MTOR effector RPS6 (P = 0.006), indicating a reduction in MTOR activity. Inhibition of MTOR can also stimulate transcription of autophagy-related and other genes (including ATP6V0D1), which might explain the observed increase in autolysosome formation at the later time points of the tfLC3 assay.Citation21-24 Overall, the control autophagy genes behaved as predicted in the GFP-SQSTM1 and tfLC3 assays indicating that hits that reduced GFP-SQSTM1 and also significantly increased formation of autolysosomes in the tfLC3 assay are likely true modulators of autophagic flux.
We identified 10 primary screen hits that also significantly increased autolysosome formation when knocked down in the tfLC3 assay (). One such gene is the lysine acetyltransferase KAT8, which is the predominant mediator of H4K16 acetylation in mammalian cells.Citation25,26 Acetylation of nonhistone targets, such as forkhead box O (FOXO) transcription factors and essential autophagy proteins, plays a well-described regulatory role in autophagy.Citation27 A role for histone acetylation was also recently demonstrated in a study showing that induction of autophagy by starvation or MTOR inhibition results in degradation of KAT8 and a consequent reduction in H4K16 acetylation that is required for cell survival.Citation28 The authors went on to show that this reduction in H4K16 acetylation is associated with downregulation of autophagy genes. Thus, it is surprising that knockdown of KAT8 appears to induce autophagy in our experiments under normal nutritive conditions. KAT8 knockdown also caused a reduction in pRPS6 in U2OS cells, consistent with inhibition of MTOR signaling. Additional studies are needed to understand the potential interplay among these complex regulatory pathways under different nutritive conditions.
Table 2. Top screen hits and their tfLC3 assay scores.
Among the 10 genes that confirmed in the tfLC3 assay, 3 of these hits (CSNK1A1, AAMP [angio associated migratory cell protein], and INTS7 [integrator complex subunit 7]) increased autophagic flux without apparent reduction in MTOR activity in the pRPS6 assay. While AAMP and INTS7 have not been previously implicated in autophagy regulation, CSNK1A1 has been identified as a modulator of autophagic flux in a siRNA screen of kinases (Table S2).Citation29 Consistent with our finding, Szyniarowski and colleagues also note that phosphorylation of the MTOR substrate RPS6 is not reduced after siRNA knockdown of CSNK1A1. CSNK1A1 encodes the CSNK1A isoform of the CK1 serine/threonine kinase family of which there are 6 distinct human genes: CSNK1A1/α, CSNK1G1/γ1, CSNK1G2/γ2, CSNK1G3/γ3, CSNK1D/δ, and CSNK1E/ε.Citation30 Despite the high sequence homology within the kinase domain region of the CK1 family, we found that only knockdown of CSNK1A1 significantly reduced GFP-SQSTM1 fluorescence in the primary screen (all isoforms were tested with 8 or more siRNAs, and all isoforms are moderately to highly expressed in U2OS cells based on the Cancer Cell Line Encyclopedia and Human Protein Atlas datasets). We chose to more fully characterize CSNK1A1 in a 14C-valine pulse-chase protein turnover assay because the confirmation data for this hit was particularly strong, showing a significant reduction in GFP-SQSTM1 intensity in response to siRNA knockdown in both U2OS osteosarcoma and H4 neuroglioma cell lines (OGA P values = 0.001 and 0.006, respectively).
TALEN-mediated knockout of CSNK1A1 in HCT116 clonal lines leads to a phenotype of accelerated turnover of long-lived proteins. This sustained increase in protein turnover suggests that CSNK1A1 is able to regulate transcription of autophagic and lysosomal proteins to replenish the autophagic machinery that is consumed during the degradation process. This is consistent with a recent study showing that CSNK1A1 phosphorylates the transcription factor FOXO3A and limits its nuclear localization, reducing transcription of autophagy-related genes in RAS-driven cancer cells.Citation31
RNAseq expression profiles reported for human (NextBio Body Atlas, http://www.nextbio.com/b/nextbioCorp.nb)Citation32 and mouseCitation33 brain tissues indicate that at least 5 of the top 10 hits from the screen are likely to be expressed in neurons, with CSNK1A1 showing relatively high levels of expression across multiple brain tissues (Figs. S8 and S9). This is consistent with quantitative immunoblot and immunohistochemical studies showing that CSNK1A1 is widely expressed in human cortex and hippocampus.Citation34,35 Interestingly, Ghoshal et al. report that the CSNK1A1/α, CSNK1D/δ, and CSNK1E/ε isoforms of casein kinase 1 are all elevated in the hippocampus of Alzheimer disease brains. While multiple groups have now reported that CSNK1A1 regulates autophagy in cancer cells, future studies are needed to assess whether inhibition of CSNK1A1 can increase autophagic flux in neurons.
The aim of this study was to provide a comprehensive analysis of cellular phenotypes related to autophagy in an unpooled siRNA-screening format that would support future data mining. We are making this resource available with the hope that the data will shed light on basic cellular pathways as well as initiate new areas of investigation for therapeutics targeting the autophagic process.
Materials and methods
Cell culture
Human osteosarcoma (U2OS; ATCC, HTB-96) and neuroglioma (H4; ATCC, HTB-148) cells were transduced with a lentiviral vector consisting of the human cytomegalovirus promoter, the Tet-On system, an N-terminal Diversa GFP tag (Diversa Corporation), and residues 1 to 441 of the human sequestosome 1 (SQSTM1) core sequence (RefSeq accession number NM_003900); clones were selected for stable expression of GFP-SQSTM1. U2OS cells were cultured in McCoy 5A (Corning, 10–050) supplemented with 10% fetal bovine serum (FBS; Hyclone, SH300070.01), 1% penicillin/streptomycin (Corning, 30–002), 1x L-glutamine (Corning, 25–005), and 1% nonessential amino acids (Corning, 25–025). H4 cells were cultured in high glucose Dulbecco's modified Eagle's medium (Corning, 10–013) supplemented with 10% FBS, 1% penicillin/streptomycin, and 1x L-glutamine. U2OS cells were also transduced with a lentiviral vector consisting of the human cytomegalovirus promoter, N-terminal tagRFP (Evrogen, FP141) and tagGFP2 (Evrogen, FP191) tags, and the human LC3B core sequence (RefSeq accession number NM_022818); a clone was selected for stable expression of tandem-fluorescent LC3B (LC3B), or tagRFP-tagGFP2-HsLC3B. All cells were maintained in a humidified cell-culture incubator (ThermoScientific, Model 370, Waltham, MA) at 37°C and 5% CO2.
Freshly-thawed cryopreserved cells were used in all assays. Cells were thawed at 37°C for 5 min, washed in culture media, and subsequently washed in OptiMEM (Life Technologies, 31985). Cells were then resuspended in OptiMEM with 1% penicillin/streptomycin at appropriate cell concentrations and plated into 384-well plates for assays.
siRNA library and siRNA transfection
siRNA libraries utilized in the primary screen were obtained from Qiagen and Dharmacon. siRNAs were diluted into siRNA dilution buffer (100 mM potassium acetate, 30 mM HEPES-KOH, 2 mM magnesium acetate, pH 7.4) and replated into 384-well storage plates (Life Technologies, AB-0781). Briefly, >70 nontargeting (NT) siRNA were designed and confirmed to have minimal homology to human and mouse transcripts by BLAST.Citation36 Among these sequences, 2 were selected with assay phenotypes similar to that of transfection controls with siRNA buffer only; these 2 nontargeting sequences were included on all plates in all assays. Cells were reverse-transfected with 1 nM siRNA in all assays; siRNAs were diluted into black, clear-bottom, tissue culture-treated 384-well assay plates (BD Biosciences, 353962). Lipofectamine RNAiMAX (Life Technologies, 13778) was diluted in OptiMEM to deliver 0.05 μL RNAiMAX per well, and was added to diluted siRNA. Transfection complexes were incubated 20 min at room temperature, after which freshly thawed cells were seeded into wells at 2000 cells/well. Assay plates were then transferred to incubators at 37°C and 5% CO2. Sixteen h post-transfection, media was refreshed with full growth media. All siRNA transfections were carried out for 96 h prior to cell fixation, except where noted.
GFP-SQSTM1 degradation, as well as GFP-SQSTM1 and LAMP2 colocalization assays
U2OS and H4 cells were reverse-transfected with 1 nM siRNA as described above. The U2OS and H4 lines were selected for screening because they are highly amenable to transfection, they grow in a monolayer that is ideal for imaging assays, and they yielded high Z’ scores in the GFP-SQSTM1 degradation assay using MTOR and nontargeting siRNA as positive and neutral controls. After 72 h of siRNA transfection, doxycycline (Sigma-Aldrich, D9891) diluted in phosphate-buffered saline (PBS)+/+ (PBS with Ca2+ and Mg2+; Corning, 20–030), was added to wells at a final concentration of 0.5 μg/mL to induce expression of GFP-SQSTM1. Twenty-four h later, after 96 h of siRNA transfection, cells were fixed with 3.8% formaldehyde (Sigma-Aldrich, 252549) diluted in PBS−/− (PBS without Ca2+ and Mg2+; Corning, 21–040), for 20 min at room temperature. Cells were then washed 3 times in PBS−/−, permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, T8787), blocked in 10% goat serum (Life Technologies, 16210072) diluted in PBS−/− at room temperature for 30 min, and washed again 3 times in PBS−/−. Cells were then blocked again in 10% goat serum in PBS−/− for 30 min at room temperature. Cells were subsequently incubated with a mouse anti-LAMP2 monoclonal antibody (1 μg/mL; Developmental Studies Hybridoma Bank, H4B4) diluted in blocking buffer and incubated at 4°C overnight. Cells were then washed 3 times with PBS−/− and incubated with a goat anti-mouse Alexa Fluor 635 (1:200; Life Technologies, A31575) secondary antibody, a direct-conjugated rabbit anti-TUBB/β-tubulin-Alexa Fluor 555 antibody (1:200; Cell Signaling Technology, 2116), and Hoechst (1:1000; Life Technologies, H3570) at room temperature for 2 h. Plates were then washed 3 times with PBS−/−, foil-sealed (Agilent PlateLoc, G5402A), and stored at 4°C until imaged.
Plates were imaged on an Opera QEHS (PerkinElmer, Hamburg, Germany) using a 20x air objective (NA 0.45). Eight fields of view were acquired per well with 2-by-2 camera binning. Four channels were acquired in 3 exposures: Hoechst was imaged using 365 nm Xenon lamp excitation; GFP-SQSTM1 and LAMP2 were imaged simultaneously using 488 nm and 635 nm laser excitation, respectively; and TUBB was imaged using 532 nm laser excitation. All channels were acquired with the appropriate emission filters.
Image analysis was carried out offline using Acapella (PerkinElmer) following image acquisition. Briefly, Hoechst and TUBB channels were used for nuclear and cytoplasmic segmentation, respectively. Mean GFP-SQSTM1 intensity was measured in a 10-pixel wide ring surrounding the nucleus, and colocalization between GFP-SQSTM1 and LAMP2 channels was measured in the cytoplasmic region using the Pearson correlation coefficient,where Ri is the intensity of one fluorophore (red, or LAMP2 in this case) in individual pixels and Rav is the arithmetic mean across the region of interest, whereas Gi and Gav are the corresponding intensities for the other fluorophore (green, or GFP-SQSTM1) in the same pixels.
All output measurements were computed at the single-cell level, and well averages, normalized to negative control values, are reported.
DRAQ7 viability assay
U2OS and H4 cells were reverse-transfected with 1 nM siRNA as described above. After 72 h of siRNA transfection, doxycycline (0.5 μg/mL in PBS+/+) and DRAQ7 (1 μM; Cell Signaling Technology, 7406) were added to wells; although GFP-SQSTM1 intensity was not measured in this assay, doxycycline was added to maintain similar conditions to those used in the primary screen. Twenty-four h later, Hoechst was added directly to wells to stain nuclei. After 20 min of incubation at room temperature, live cells were imaged on an Opera QEHS using a 10x air objective (NA 0.4). Four fields of view were acquired per well with 2-by-2 camera binning. Two channels were acquired in a single exposure: Hoechst was imaged using 365 nm Xenon lamp excitation and DRAQ7 was imaged using 635 nm laser excitation. Both channels were acquired with the appropriate emission filters.
Image analysis was carried out offline using Acapella and MATLAB (MathWorks). Nuclei were segmented in the Hoechst channel, and individual nuclear intensities in both the Hoechst and DRAQ7 channels were subsequently calculated and exported to a text file. A MATLAB script was then used to define the DRAQ7 average nuclear intensity threshold below which cells were deemed live, and above which cells were deemed dead, by identifying the trough between peaks in the bimodal distribution of nuclear intensity data; for this step of the analysis, wells in which cells were transfected with nontargeting and essential siRNA were used. This threshold was then used in an Acapella script to determine the number of live, dead, and total cells per well. Total cell count is normalized to negative control values, while the percentage of live cells reported per well is not normalized.
pRPS6 MTOR-dependence assay
U2OS and H4 cells were reverse-transfected with 1 nM siRNA, stimulated with doxycycline to induce GFP-SQSTM1 expression, fixed, Hoechst-labeled, permeabilized, and blocked as described above (GFP-SQSTM1 Degradation and GFP-SQSTM1 and LAMP2 Colocalization Assay). Cells were then incubated with a rabbit anti-phospho-RPS6 (Ser235/236) monoclonal antibody preconjugated to Alexa Fluor 555 (1:50; Cell Signaling Technology, 3985) at 4°C overnight. Plates were then washed 3 times with PBS−/−, foil-sealed, and store at 4°C until imaged.
Plates were imaged on an Opera QEHS with a 20x air objective (NA 0.45). Eight fields of view were acquired per well with 2-by-2 camera binning. Three channels were acquired in 3 separate exposures: Hoechst was imaged using 365 nm Xenon lamp excitation; GFP-SQSTM1 and pRPS6 were imaged using 488 nm and 532 nm laser excitation, respectively. All channels were acquired with the appropriate emission filters.
Image analysis was carried out online using Acapella during image acquisition. Hoechst was used for nuclear segmentation. Mean GFP-SQSTM1 and pRPS6-555 intensities were calculated in a 10-pixel wide ring surrounding the nucleus. All output measurements were computed at the single-cell level, and well averages, normalized to negative control values, are reported.
Tandem-fluorescent LC3 assay
U2OS cells constitutively expressing tagRFP-tagGFP-LC3B were reverse-transfected with 3 nM siRNA as described above. Twenty-six h after transfection, wells were refreshed with full growth media. MicroClime lids (Labcyte, LLS-0310) were injected with 8 mL of water and used during live-cell imaging to maintain proper humidity within the plate for the duration of the experiment.
Live cells were maintained at 37°C, in 5% CO2, and imaged on an Operetta (PerkinElmer) using a 20x air objective (NA 0.45). Nine fields of view were acquired per well. Images were acquired every 6 h for 12 time points, spanning 66 h. At each time point, GFP and RFP signal channels were acquired in separate exposures using Xenon lamp excitation and the appropriate excitation and emission filter sets.
Image analysis was carried out offline using Acapella after image acquisition; the analysis workflow is outlined in Fig. S10. Nuclei and cytoplasms were identified using the RFP channel. Puncta were then identified in the GFP and RFP channels independently, and Boolean logic was applied to determine the number of GFP+ RFP+ structures (representing phagophores and autophagosomes) and GFP− RFP+ structures (representing autolysosomes). Autophagosome and autolysosome counts were then normalized to individual cell area to account for variability in cell density throughout the assay. Absolute counts of autolysosomes per cell area are reported.
Compounds
For validation of the GFP-SQSTM1 assay, U2OS cells were incubated with PP242 (EMD Millipore, 475988) for 18 h. For validation of the tfLC3 assays, U2OS cells were treated with PP242, chloroquine (Sigma, C6628), or both for 8 h.
Quantitative PCR
U2OS cells were reverse-transfected with 1 nM siRNA in 384-well plates as described above. After 48 h, cells were washed with cold PBS−/− (Corning, 21–040) once. PBS was aspirated and 30 μL of lysis buffer from the Ambion Cells-to-CT kit (Life Technologies, AM1728) was added to each well. Lysis and reserve transcription was carried out according to the kit protocol. Four μL of RT samples was used for quantitative PCR (qPCR) with a total well reaction volume of 20 μL. The qPCR procedure is detailed in the Cells-to-Ct kit protocol. qPCR was carried out in a 384-well plate using the ViiA 7 system (Life Technologies, 4453536, Carlsbad, CA), and data was analyzed using the ViiA 7 software. mRNA levels were calculated using the ΔΔCT method; target levels were normalized to an endogenous GAPDH reference and expressed relative to a nontargeting siRNA control calibrator. Six siRNA were assayed in triplicate for each gene.
Gene-level condensing
For each phenotype measured, raw siRNA scores were normalized to nontargeting siRNA controls. These normalized, or percent of control (POC) values, were then condensed to gene-level P values using a rank-based orthogonal gene averaging (OGA) algorithm. Similar to the redundant siRNA activity (RSA) algorithm,Citation37 but different in its implementation, the OGA algorithm reduces the impact of off-target effects of siRNA in large-scale screens and generates a single P value per gene, greatly simplifying downstream analysis. Briefly, the OGA algorithm compares Stouffer-weightedCitation38 siRNA ranks for a particular gene's set of siRNAs to a neutral reference set of siRNA, and computes an up- or down-hit P value for that particular gene using Kaplan-Meier-like survival analysis.
TALEN-mediated knockout of CSNK1A1 in HCT116 cells
CSNK1A1 KO clonal cell lines were generated in HCT116 colorectal carcinoma cells (ATCC) by PNA Bio (Thousand Oaks, CA, USA). DNA double-strand breaks were introduced in exon 1 of the CSNK1A1 gene near the ATG start site with TALENs targeted to the sequences TGGCGAGTAGCAGCGGCTCC and ATTGTCGGAGGGAAATATAA. The TALEN constructs were made with the standard RVD code and linker between TALE and Fok1 domains.Citation39 Clones #12 and #27 had 2 bp and 7,186 bp deletions, respectively. HCT116 parental and KO lines were maintained in growth medium consisting of McCoy 5A Media supplemented with 10% FBS, 1% nonessential amino acids, and 1% penicillin/streptomycin/glutamine. Statistical measures were determined by 2-way analysis of variance followed by the Dunnett multiple comparison test.
Western blot
Cultured HCT116 parental and CSNK1A1 KO cell lines were lysed with cOmplete Lysis-M Buffer (Roche, 4719956001) containing 1% sodium dodecyl sulfate (National Diagnostics, EC-874) and 1x Pierce Protease Inhibitors (Thermo Fisher, 88266). Samples were boiled and benzonase (Sigma-Aldrich, E1014) was added to degrade DNA. Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher, 23225). Ten μg of sample was prepared with NuPage LDS sample loading buffer (Life Technologies, NP0007), boiled, and electrophoresed in 12% Bis-Tris gel (Life Technologies, NP0342). Protein was transferred onto 0.2 μM nitrocellulose membrane and blocked with Odyssey Blocking Buffer (Li-Cor, 927–40000) for 1 h. Rabbit polyclonal anti-CSNK1A1/CK1α antibody (Thermo Fisher, PA5-17536) was diluted 1:1000 in Odyssey Blocking Buffer with 0.2% Tween-20 (Sigma-Alrdich, P7949) and incubated overnight at 4°C. Blots were washed in PBS, incubated in goat anti-rabbit IRDye 800CW secondary antibody (Li-Cor, 926–32211) for 1 h, washed with PBS, and imaged on an Odyssey 9120 imaging system (Li-Cor, Lincoln, NE).
14C-valine pulse-chase turnover assay
The pulse-chase assay was adapted from previously published methods.Citation40 HCT116 parental and CSNK1A1 KO cell lines were plated in 24-well plates at 100,000 cells/well in 1 mL growth medium and cultured overnight. The next day, cells were labeled with 0.2 μCi/mL L-[U-14C] valine (266 mCi/mmol, Amersham Biosciences) in 1 mL growth medium for 23 h. Cells were washed 2 times with 1 mL warm Gibco PBS pH 7.4 (Thermo Fisher, 10010031) and then incubated in 1 mL growth medium supplemented with 1 mM valine (Sigma-Aldrich, 91619) for 1 h. Medium was then replaced with fresh growth medium/1 mM valine and incubated for 6, 12, 24, 48, or 72 h (3 wells per time point). The excess cold valine was added to reduce reincorporation of free 14C-valine into new proteins. At the end of each time point, the medium and cells were collected separately and prepared for scintillation counting to calculate the percent of 14C-valine that had been released into the medium as free amino acid. 900 μL of medium was collected from each well and added to a 1.5 mL tube containing 100 μL of 100% TCA and incubated at 4°C for 22 h to precipitate protein. Tubes were centrifuged at 600xg for 10 min at 4°C and 500 μL of the supernatant was added to a 20 mL counting vial containing 18 mL Ultima Gold AB scintillation fluid (Perkin Elmer, 6013301) and incubated for 48 h before counts were read on a Beckman Coulter LS 6500 Multi-purpose Scintillation Counter (Brea, CA). To prepare cells for scintillation counting, the small volume of residual medium was aspirated and the cells were washed 2 times with PBS. Cells were lysed with 400 μL of EDTA-free Complete Lysis-M, EDTA-free (Roche, 4719964001) plus 500 u/mL benzonase. 200 μL of lysate was then transferred to vials containing 18 mL Ultima Gold AB and counts were read. The percent of protein that was degraded at each time point was estimated by the fraction of 14C-valine that was released into the medium: % degradation = media count/(media count + lysate count).
Abbreviations
| AAMP | = | angio associated, migratory cell protein |
| ATG5 | = | autophagy-related 5 |
| ATG7 | = | autophagy-related 7 |
| ATP6V0D1 | = | ATPase, H+ transporting, lysosomal 38kDa, V0 subunit d1 |
| BECN1 | = | Beclin 1, autophagy related |
| CSNK1A1 | = | casein kinase 1, α 1 |
| DRAQ7 | = | deep red anthraquinone 7 |
| EIF4A3 | = | eukaryotic translation initiation factor 4A3 |
| FBS | = | fetal bovine serum |
| GFP | = | green fluorescent protein |
| H4 | = | human neuroglioma cells |
| H4K16 | = | histone 4 lysine 16 |
| HCT 116 | = | human colorectal carcinoma cell line |
| KAT8 | = | K(lysine) acetyltransferase 8 |
| INTS7 | = | integrator complex subunit 7 |
| KO | = | knockout |
| K-S | = | Kolmogorov-Smirnov |
| LAMP2 | = | lysosomal-associated membrane protein 2 |
| LC3B/MAP1LC3B | = | microtubule associated protein 1 light chain 3 β |
| MTOR | = | mechanistic target of rapamycin |
| OGA | = | orthogonal gene averaging |
| PBS | = | phosphate-buffered saline |
| PIK3C3 | = | phosphatidylinositol 3-kinase, catalytic subunit type 3 |
| PIK3R4 | = | phosphoinositide-3-kinase, regulatory subunit 4 |
| POC | = | percent of control |
| POLR2A | = | polymerase (RNA) II (DNA directed) polypeptide A, 220kDa |
| PP242 | = | pyrazolopyrimidine 242 |
| pRPS6 | = | phosphorylated ribosomal protein S6 |
| qPCR | = | quantitative polymerase chain reaction |
| RFP | = | red fluorescent protein |
| RNAi | = | ribonucleic acid interference |
| siRNA | = | small interfering ribonucleic acid |
| SQSTM1/p62 | = | sequestosome 1 |
| TALEN | = | transcription activator-like effector nuclease |
| tfLC3 | = | tandem fluorescent LC3 |
| U2OS | = | human osteosarcoma cells |
Disclosure of potential conflicts of interest
All authors were employees and shareholders of Amgen Inc. at the time this research was conducted.
Supplementary files
Download Zip (7.3 MB)Acknowledgments
We thank Adam Park, Dan Nebalasca, and Jeanette Villarreal-Kinney for their excellent technical support throughout the screening campaign. We thank Jeff McDowell and Bharath Ramachandran for siRNA informatics support. We also thank Helen McBride and Narender Gavva for their insightful feedback during manuscript preparation.
References
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/10.1083/jcb.200507002
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/10.1038/nature04723
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; http://dx.doi.org/10.1038/nrd3802
- Cuervo AM. Autophagy: Many paths to the same end. Mol Cell Biochem 2004; 263:55-72; PMID:15524167; http://dx.doi.org/10.1023/B:MCBI.0000041848.57020.57
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-A, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:AMBIGUOUS; http://dx.doi.org/10.1074/jbc.M702824200
- Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 2010; 6:784-93; PMID:20574168; http://dx.doi.org/10.4161/auto.6.6.12510
- Yang Z, Klionsky DJ. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/10.1016/j.ceb.2009.11.014
- Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol 2008; 4:691-9; PMID:18849971; http://dx.doi.org/10.1038/nchembio.117
- Phadwal K, Alegre-Abarrategui J, Watson AS, Pike L, Anbalagan S, Hammond EM, Wade-Martins R, McMichael A, Klenerman P, Simon AK. A novel method for autophagy detection in primary cells. Autophagy 2012; 8:677-89; PMID:22302009; http://dx.doi.org/10.4161/auto.18935
- Manders EMM, Verbeek FJ, Ate JA. Measurement of co-localisation of objects in dual-colour confocal images. J Microsc 1993; 169:375-82; http://dx.doi.org/10.1111/j.1365-2818.1993.tb03313.x
- Li Q, Lau A, Morris TJ, Guo L, Fordyce CB, Stanley EF. A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci 2004; 24:4070-81; PMID:15102922; http://dx.doi.org/10.1523/JNEUROSCI.0346-04.2004
- Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry A 2010; 77:733-42; PMID:20653013; http://dx.doi.org/10.1002/cyto.a.20896
- Finbow ME, Harrison MA. The vacuolar H+-ATPase: a universal proton pump of eukaryotes. Biochem J 1997; 324:697-712; PMID:9210392; http://dx.doi.org/10.1042/bj3240697
- Babij C, Zhang Y, Kurzeja RJ, Munzli A, Shehabeldin A, Fernando M, Quon K, Kassner PD, Ruefli-Brasse AA, Watson VJ, et al. STK33 kinase activity is nonessential in KRAS-dependent cancer cells. Cancer Res 2011; 71:5818-26; PMID:21742770; http://dx.doi.org/10.1158/0008-5472.CAN-11-0778
- Ma Y, Creanga A, Lum L, Beachy PA. Prevalence of off-target effects in Drosophila RNA interference screens. Nature 2006; 443:359-63; PMID:16964239; http://dx.doi.org/10.1038/nature05179
- Marine S, Bahl A, Ferrer M, Buehler E. Common seed analysis to identify off-target effects in siRNA screens. J Biomol Screen 2012; 17:370-8; PMID:22086724; http://dx.doi.org/10.1177/1087057111427348
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2014; 3:452-60; PMID:17534139; http://dx.doi.org/10.4161/auto.4451
- McKnight NC, Jefferies HBJ, Alemu E a, Saunders RE, Howell M, Johansen T, Tooze S a. Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J 2012; 31:1931-46; PMID:22354037; http://dx.doi.org/10.1038/emboj.2012.36
- Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf 2013; 12:177-86; PMID:23252795; http://dx.doi.org/10.1517/14740338.2013.752814
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science (80-. ).2011; 334:678-83; PMID:22053050; http://dx.doi.org/10.1126/science.1207056
- Peña-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TAT, Zou L, Xie X-J, Corey DR, Brugarolas J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J 2011; 30:3242-58; PMID:21804531; http://dx.doi.org/10.1038/emboj.2011.257
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31:1095-108; PMID:22343943; http://dx.doi.org/10.1038/emboj.2012.32
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal.2012; 5:ra42-ra42; PMID:22692423; http://dx.doi.org/10.1126/scisignal.2002790
- Martina JA, Diab HI, Lishu L, Jeong-A L, Patange S, Raben N, Puertollano R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal 2014; 7:ra9; PMID:24448649; http://dx.doi.org/10.1126/scisignal.2004754
- Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, Akhtar A. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol 2005; 25:6798-810; PMID:16024812; http://dx.doi.org/10.1128/MCB.25.15.6798-6810.2005
- Smith ER, Cayrou C, Huang R, Lane WS, Côté J, Lucchesi JC. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol Cell Biol 2005; 25:9175-88; PMID:16227571; http://dx.doi.org/10.1128/MCB.25.21.9175-9188.2005
- Bánréti Á, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy 2013; 9:819-29; PMID:23466676; http://dx.doi.org/10.4161/auto.23908
- Füllgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, Joseph B. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 2013; 500:468-71; PMID:23863932; http://dx.doi.org/10.1038/nature12313
- Szyniarowski P, Corcelle-Termeau E, Farkas T, Høyer-Hansen M, Nylandsted J, Kallunki T, Jäättelä M. A comprehensive siRNA screen for kinases that suppress macroautophagy in optimal growth conditions. Autophagy 2011; 7:892-903; PMID:21508686; http://dx.doi.org/10.4161/auto.7.8.15770
- Cheong JK, Virshup DM. Casein kinase 1: Complexity in the family. Int J Biochem Cell Biol 2011; 43:465-9; PMID:21145983; http://dx.doi.org/10.1016/j.biocel.2010.12.004
- Cheong JK, Zhang F, Chua PJ, Bay BH, Thorburn A, Virshup DM. Casein kinase 1α – dependent feedback loop controls autophagy in RAS-driven cancers. J Clin Invest 2015; 125:1401-18; PMID:25798617; http://dx.doi.org/10.1172/JCI78018
- Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, et al. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS One 2010; 5; PMID:20927376; http://dx.doi.org/10.1371/journal.pone.0013066
- Zhang Y, Chen K, Sloan S a, Bennett ML, Scholze AR, Keeffe SO, Phatnani HP, Guarnieri XP, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, Neurons Vasc Cells Cereb Cortex 2014; 34:1-19.
- Kuret J, Johnson GS, Cha D, Christenson ER, DeMaggio AJ, Hoekstra MF. Casein kinase 1 is tightly associated with paired-helical filaments isolated from Alzheimer's disease brain. J Neurochem 1997; 69:2506-15; PMID:9375684; http://dx.doi.org/10.1046/j.1471-4159.1997.69062506.x
- Ghoshal N, Smiley JF, DeMaggio a J, Hoekstra MF, Cochran EJ, Binder LI, Kuret J. A new molecular link between the fibrillar and granulovacuolar lesions of Alzheimer's disease. Am J Pathol 1999; 155:1163-72; PMID:10514399; http://dx.doi.org/10.1016/S0002-9440(10)65219-4
- Swearingen E a, Fajardo F, Wang X, Watson JEV, Quon KC, Kassner PD. Use of cryopreserved cell aliquots in the high-throughput screening of small interfering RNA libraries. J Biomol Screen 2010; 15:469-77; PMID:20371867; http://dx.doi.org/10.1177/1087057110365899
- König R, Chiang C, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y, et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat Methods 2007; 4:847-9; PMID:17828270; http://dx.doi.org/10.1038/nmeth1089
- Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher's approach. J Evol Biol 2005; 18:1368-73; PMID:16135132; http://dx.doi.org/10.1111/j.1420-9101.2005.00917.x
- Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia N V, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 2011; 39:e82; PMID:21493687; http://dx.doi.org/10.1093/nar/gkr218
- Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. 1st ed. Elsevier Inc; 2009.