
ABSTRACT
Autophagy dysfunction is a common feature in neurodegenerative disorders characterized by accumulation of toxic protein aggregates. Increasing evidence has demonstrated that activation of TFEB (transcription factor EB), a master regulator of autophagy and lysosomal biogenesis, can ameliorate neurotoxicity and rescue neurodegeneration in animal models. Currently known TFEB activators are mainly inhibitors of MTOR (mechanistic target of rapamycin [serine/threonine kinase]), which, as a master regulator of cell growth and metabolism, is involved in a wide range of biological functions. Thus, the identification of TFEB modulators acting without inhibiting the MTOR pathway would be preferred and probably less deleterious to cells. In this study, a synthesized curcumin derivative termed C1 is identified as a novel MTOR-independent activator of TFEB. Compound C1 specifically binds to TFEB at the N terminus and promotes TFEB nuclear translocation without inhibiting MTOR activity. By activating TFEB, C1 enhances autophagy and lysosome biogenesis in vitro and in vivo. Collectively, compound C1 is an orally effective activator of TFEB and is a potential therapeutic agent for the treatment of neurodegenerative diseases.
Introduction
Macroautophagy (herein referred to as autophagy) is a highly conserved cellular process for the bulk degradation of long-lived proteins and organelles mediated by lysosomes. Defects in the autophagy-lysosome pathway (ALP) have been linked to a variety of human diseasesCitation1,2 including neurodegenerative disorders caused by toxic, aggregate-prone proteins.Citation3,4 Recently, TFEB (transcription factor EB) was identified as a master regulator of autophagy and lysosomal biogenesis.Citation5-7 Starvation, lysosomal stress or inhibition of the mechanistic target of rapamycin (serine/threonine kinase) complex 1 (MTORC1) activates TFEB by promoting its translocation to the nucleus,Citation8-10 where it binds to the CLEAR (coordinated lysosomal expression and regulation) elements and activates genes involved in autophagy and lysosomal biogenesis.Citation5,6 TFEB overexpression or small molecules capable of stimulating the expression and/or nuclear translocation of endogenous TFEB, has been shown to promote clearance of pathologic lysosomal substrates in lysosomal storage disorders (LSDs)Citation11-13 and to be neuroprotective by promoting the clearance of toxic protein aggregates in cell and animal models of neurodegenerative disorders such as Parkinson disease (PD),Citation14 Alzheimer disease (AD)Citation15-17 and Huntington disease (HD).Citation18
Curcumin is a natural polyphenolic compound derived from the herbal medicine turmeric (Curcuma longa Linn.), which is nontoxic and possesses diverse pharmacologic effects.Citation19 It is well documented that curcumin enhances autophagy via inhibiting the phosphoinositide 3-kinase-AKT-MTOR signaling pathway.Citation20,21 However, the poor absorption and low bioavailability of curcumin curtails its clinical application.Citation19,22 To improve the bioavailability and potency, a number of derivatives of curcumin have been chemically synthesized.Citation23,24 Among these derivatives, monocarbonyl analogs of curcumin without the β-diketone moiety have exhibited enhanced stability, improved pharmacokinetic profiles and better in vitro and in vivo activities.Citation25-28
By screening a series of synthetic monocarbonyl analogs of curcumin, an analog termed C1 was identified as a potent TFEB activator. Unlike currently known TFEB activators, C1 activates TFEB by directly binding to TFEB and promotes its entry into the nucleus, without affecting TFEB phosphorylation or inhibiting the activities of MTOR and MAPK1/ERK2 (mitogen-activated protein kinase 1)-MAPK3/ERK1. C1 is orally effective in enhancing autophagy and lysosome biogenesis in the brain.
Results
New MTOR-dependent and -independent autophagy enhancers identified from monocarbonyl analogs of curcumin
A series of monocarbonyl analogs of curcumin () were tested for their autophagy-enhancing activities in the mouse neuroblastoma neuro-2a (N2a) cells. First, the cytotoxicity of the tested compounds was determined by LDH (lactate dehydrogenase) release assay (Fig. S1). The compounds were nontoxic at the concentration of 1 µM and were used in subsequent autophagy assays. Curcumin at 1 µM showed no effects on autophagy (data not shown). Curcumin (10 µM) and its analogs A2, B1, B3, C1, E2, E3 and E4 (1 µM) significantly increased the levels of LC3B-II, the lipidated and phagophore- or autophagosome-associated form of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β) in N2a cells compared to the vehicle control (0.1% DMSO) (). In the presence of the lysosomal inhibitor chloroquine (CQ), these analogs further increased LC3B-II levels (). The results indicate that curcumin analogs enhance autophagy rather than blocking lysosomal degradation. Among the compounds tested, C1 shows the best autophagy-enhancing effect.
Figure 1. New MTOR dependent- and independent- autophagy enhancers identified from monocarbonyl analogs of curcumin. (A) Chemical structure of curcumin and its monocarbonyl analogs. (B) N2a cells were treated with curcumin (Cur, 10 μM) and its analogs (1 μM) for 12 h. The expression of LC3B-II was determined by western blot. (C) Relative intensity is normalized to that of ACTB/β-actin. Data are presented as the mean ± SD from 3 independent experiments. *, P< 0.05 vs. the control (0.1% DMSO). (D) N2a cells were treated with Cur (10 μM) and its analogs (1 μM) in the presence of chloroquine (CQ, 20 μM) for 12 h. The expression of LC3B-II was determined by western blot. (E) Relative intensity is normalized to that of ACTB/β-actin. Data are presented as the mean ± SD from 3 independent experiments. *, P < 0.05 vs. the control (0.1% DMSO); #, P < 0.05 vs. CQ treatment alone. (F) Effects of curcumin analogs on the MTOR pathway. N2a cells were treated with curcumin (Cur, 10 μM) and its analogs (1 μM) for 12 h. Torin1 (1 μM) treatment for 2 h was used as a positive control. Representative blots show the expression of phosphorylated (p-) and total RPS6KB1/p70S6K and MTOR. (G) Data are presented as the mean ± SD from 3 independent experiments. *, P < 0.05 vs. the control (0.1% DMSO).
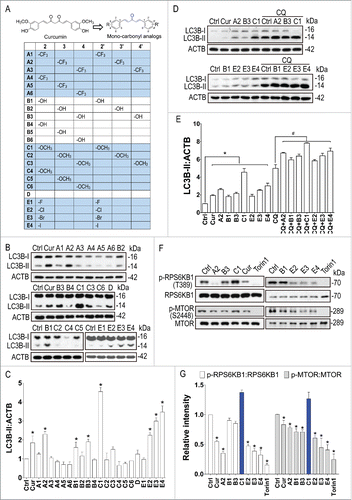
Since curcumin enhances autophagy through inhibiting the MTOR pathway,Citation20,21,29 we next determined the effects of these newly identified autophagy enhancers on the MTOR pathway. Torin1, a potent MTOR inhibitorCitation30 was used as a positive control. Similar to curcumin, most of these analogs inhibited phosphorylation of RPS6KB1/p70S6K (ribosomal protein S6 kinase, polypeptide 1) and MTOR (). Compound E4 showed the best inhibition of the MTOR pathway. Unexpectedly, compound C1 significantly promoted phosphorylation of RPS6KB1 and MTOR, indicating that C1 enhanced autophagy without inhibiting the MTOR pathway. Meanwhile, we found that C1 treatment had no significant effects on the activity of the MTOR-related kinases, including AMP activated protein kinase (AMPK) and ULK1 (unc-51 like kinase 1), which play important roles in autophagy regulation (Fig. S2).Citation31 Together we identified a potent MTOR-independent (C1) and a MTOR-dependent (E4) autophagy enhancer from monocarbonyl analogs of curcumin.
Monocarbonyl analogs of curcumin activate TFEB
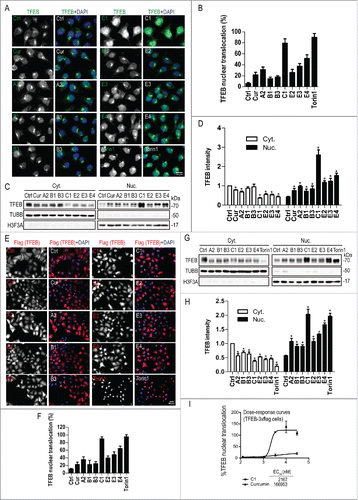
Pharmacological inhibition of MTORC1 activates TFEB by promoting its nuclear translocation.Citation8-10 We therefore tested whether curcumin and its analogs could activate TFEB. First, we determined the distribution of endogenous TFEB in N2a cells treated with curcumin and its analogs with autophagy-enhancing effect. Curcumin (10 µM) treatment showed a mild effect on TFEB nuclear translocation (∼20% of cells). Curcumin analogs A2, B1, B3, E2, E3 and E4 triggered different levels of TFEB nuclear translocation (), which is correlated with their MTOR-inhibiting activity (). Compounds E4, which showed the best MTOR-inhibiting activity, induced ∼50% TFEB nuclear translocation. Interestingly, compound C1, which did not inhibit MTOR, showed the best effect on the TFEB nuclear translocation (more than 80% cells) (). Quantification of TFEB levels in the cytosolic and nuclear fractions by western blots further confirmed that C1 potently induced nuclear translocation of endogenous TFEB in N2a cells (). Next we tested the effects of curcumin and its analogs on the translocation of TFEB in HeLa cells stably expressing 3xFlag-TFEBCitation5,6 by western blot and by a cell-based quantitative high-content assay.Citation9 Similar to torin1 treatment, compound C1 significantly promoted the nuclear translocation of Flag-TFEB with an EC50 value of 2167 nM (). Therefore, compound C1 may represent a novel MTOR-independent TFEB activator.
Figure 2. Effects of curcumin and its analogs on the nuclear translocation of TFEB. (Ato D) Effects of curcumin and its analogs on the nuclear translocation of endogenous TFEB. N2a cells were treated with curcumin (Cur, 10 μM) and its analogs (1 μM) for 12 h. (A) Cells were fixed and stained with TFEB antibody (green) and DAPI (blue). Representative images are shown. (B) Quantification of the number of cells with nuclear TFEB localization. Data are presented as mean ± SD of 3 replicates in a representative experiment. At least 500 cells were analyzed in each treatment group. (C) The expression of endogenous TFEB in the cytosolic (Cyt.) and nuclear (Nuc.) fractions were determined by western blotting. TUBB/β-tubulin and H3F3A (H3 histone, family 3A) were used as loading controls of the cytoplasmic and nuclear fraction, respectively. (D) Data are presented as the mean ± SD from 3 independent experiments. *, P< 0.05 vs. the control (0.1% DMSO). (E to I) Effects of curcumin and its analogs on the nuclear translocation of 3xFlag-TFEB. (E) HeLa cells stably expressing 3xFlag-TFEB were treated with Cur (10 μM) and its analogs (1 μM) for 12 h. Torin1 (1 μM) treatment for 2 h was used as a positive control. Cells were fixed and stained with anti-Flag antibody (red) and DAPI (blue). (F) Quantification of the number of cells with nuclear TFEB localization. Data are presented as mean ± SD of 3 replicates in a representative experiment. At least 500 cells were analyzed in each treatment group. (G) The expression of 3xFlag-TFEB in the cytosolic (Cyt.) and nuclear (Nuc.) fractions were determined by western blotting. TUBB/β-tubulin and H3F3A were used as loading controls of the cytoplasmic and nuclear fraction, respectively. (H) Data are presented as the mean ± SD from 3 independent experiments. *, P < 0.05 vs. the control (0.1% DMSO). (I) Dose–response curves of TFEB nuclear translocation induced by C1 and curcumin. Stable HeLa cells overexpressing 3xFlag-TFEB were seeded in 96-well plates, cultured for 12 h, and treated with 6 different concentrations of curcumin and C1, ranging from 123 nM to 30 μM. The graph shows the percentage of nuclear translocation at the different concentrations of each compound (in log of the concentration). The EC50 for each compound was calculated using Prism software.
Curcumin analog C1 does not inhibit serine-phosphorylation of TFEB or MAPK1/3 activity
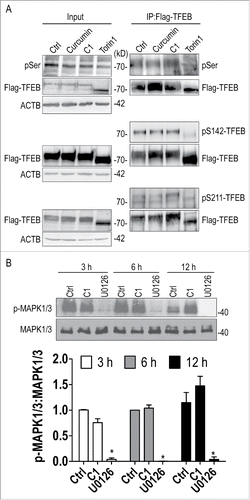
MTORC1 controls TFEB subcellular localization by phosphorylating key serine residues such as S142 and S211.Citation5,9 To further confirm that curcumin analog C1 activates TFEB in a MTOR-independent manner, we determined the effects of C1 on the serine-phosphorylation of TFEB. First, we found that C1 treatment did not affect the total levels of phosphoserine on TFEB (). Second, by using site-specific phosphoserine antibodies, we found that C1 treatment did not affect phosphorylation of S142 and S211 on TFEB (), 2 key target sites phosphorylated by MTOR. Similarly, C1 treatment did not trigger a molecular weight downshift of TFEB bands, indicating that C1 had no effects on serine-phosphorylation of TFEB (). As a positive control, torin1 treatment caused a significant downshift of TFEB bands and dephosphorylation of total phosphoserine, S142 and S211. Since MAPK1/3 inhibition also partially contributes to TFEB nuclear translocation,Citation5,9 we tested the effects of C1 treatment on MAPK1/3 phosphorylation at several time points and found that C1 did not inhibit MAPK1/3 activity (). These results further confirmed that C1 activates TFEB independent of MTOR inhibition.
Figure 3. Curcumin analog C1 does not inhibit serine-phosphorylation of TFEB and MAPK1/3 activity. (A) HeLa cells stably overexpressing 3xFlag-TFEB were treated with curcumin (10 μM) and C1 (1 μM) for 12 h, and torin1 (1 μM) for 2 h. Protein extracts were subjected to immunoprecipitation using anti-FLAG antibody and then blotted with anti-phosphoserine (p-Ser) and anti-phospho-S142-TFEB (pS142-TFEB) antibodies. Phosphorylation of TFEB on serine 211 (pS211-TFEB) was detected using an antibody against the specific YWHA/14-3-3 motif that recognizes p-S211-TFEB. Blots are representative of at least 2 independent experiments. (B) Stable HeLa cells overexpressing 3xFlag-TFEB were treated with C1 (1 μM) and the MAPK1/3 inhibitor U0126 (50 μM) for the indicated time periods. The ratio of phospho-MAPK1/3 (p-MAPK1/3) to total MAPK1/3 was quantified. Data are presented as the mean ± SD from 3 independent experiments. *, P< 0.05 vs. the control (0.1% DMSO).
Curcumin analog C1 directly binds to TFEB
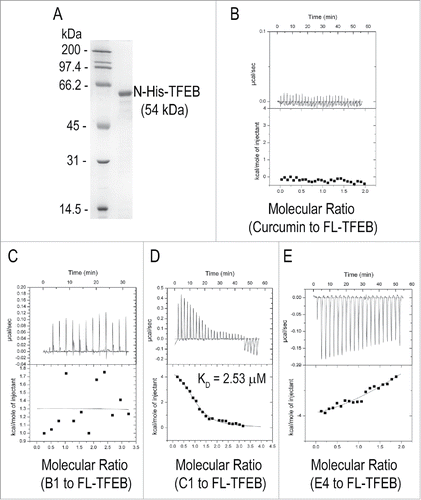
Since curcumin analog C1 activates TFEB in an MTOR-independent manner, we hypothesized that C1 directly binds to and activates TFEB. First, a solid-phase binding assayCitation32 was performed to determine the direct binding of C1 to recombinant TFEB. A specific His-TFEB band at ∼55 kDa was observed only in the pellet of C1, but not curcumin, indicating a direct binding of C1 to TFEB (Fig. S3). To further confirm the binding, we prepared high-purity recombinant full-length TFEB with a N-terminal histidine tag () and determined the binding affinities of C1 to TFEB by isothermal titration calorimetry (ITC). The dissociation constant (KD) of C1 to TFEB was determined to be 2.53 μM (). To confirm the specific binding of C1 to TFEB, we used curcumin (), B1 () and E4 () as parallel controls. In the titration of TFEB with curcumin, which differs significantly in structure from C1 (), the heat release of each injection was too low with respect to the background reading at the highest available concentration of the protein and ligand, for successful KD determination (). This suggests a significantly weaker binding affinity of curcumin to TFEB.
Figure 4. Curcumin analog C1 directly binds to recombinant TFEB. (A) SDS-PAGE gel of the purified recombinant His-TFEB. (B) ITC thermogram for curcumin (200 μM) and FL-TFEB (20 μM). (C) ITC thermogram for B1 (200 μM) and FL-TFEB (20 μM). (D) ITC thermogram for C1 (200 μM) and FL-TFEB (10 μM). Data were fitted with one site binding model, n = 0.99 ± 0.03, KD = 2.53 ± 0.33 μM, ΔH = 5.09 ± 0.24 kcal/mol−1, ΔS = 42.7 cal/mol−1 K−1. (E) ITC thermogram for E4 (200 μM) and FL-TFEB (20 μM).
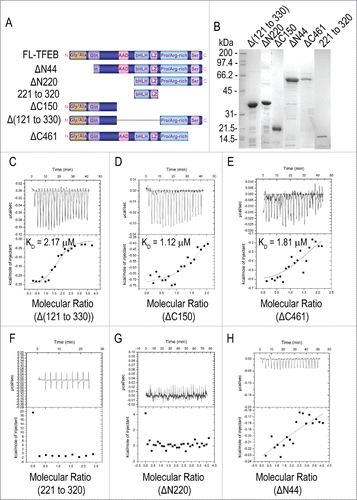
Isothermal titration of B1 (with di-ortho-hydroxyl groups) and E4 (with di-ortho-iodide groups) to the full-length (FL) TFEB was performed under similar conditions. The di-hydroxyl substitution in B1 reduced the ligand-protein binding to an undetectable level (), and the ITC signals of E4 binding to TFEB were reduced greatly but not completely abolished (). These results indicate that curcumin and its MTOR-inhibiting analogs have no interaction with TFEB while C1 with the di-ortho-methoxyl groups is a specific and direct binding small molecule of TFEB. To identify the exact C1 binding sites on TFEB, we expressed and purified various truncated forms of TFEB () and determined their binding affinity with C1 using ITC (). The titration curves for C1 binding to Δ(121 to 330), ΔC150 and ΔC461, all with the intact glycine and alanine (Gly and Ala)-rich region, fit a one-site model of binding, with dissociation constants, KD, of 2.17 μM, 1.12 μM and 1.81 μM, respectively (). Other truncated mutants with N-terminal deletions failed to interact with C1 (). The results indicate that the binding site of C1 is likely located at the N-terminal Gly and Ala-rich domain of TFEB.
Figure 5. Curcumin analog C1 binds to TFEB at the N-terminal Gly and Ala-rich domain. (A) Map of TFEB protein domains. N-terminal deletions (ΔN) and carboxyl-terminal (ΔC) deletions are indicated with the corresponding amino acid positions. (B) SDS-PAGE of the purified recombinant TFEB deletion mutants. (C to E) Binding of C1 to TFEB truncations Δ(121 to 330) (C), ΔC150 (D) and ΔC461 (E). The concentration of each TFEB truncation was 10 μM and the concentration of C1 was 200 μM (for Δ(121 to 330)) and 100 μM (for ΔC150 and ΔC461). ((F)to H) ITC data showing TFEB truncation 221 to 320 (F), ΔN220 (G) and ΔN44 (H) has no binding with C1. Titration curves are shown with the baseline-corrected raw data with background subtraction.
Curcumin analog C1 weakens TFEB-YWHA interaction
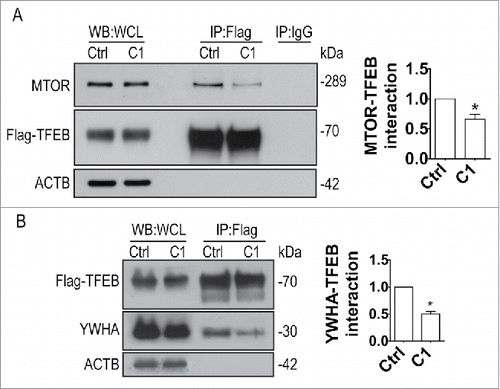
MTORC1-dependent phosphorylation of TFEB results in YWHA/14-3-3 interactions that promote the cytoplasmic retention of TFEB.Citation8-10 Conversely, pharmacological inhibition of MTORC1 causes dissociation of the TFEB-YWHA complex and rapid translocation of TFEB to the nucleus.Citation8 In HeLa cells stably expressing Flag-TFEB, C1 treatment did not affect the levels of endogenous MTOR and YWHA (). However, the levels of YWHA coimmunoprecipitated with Flag-TFEB significantly decreased after C1 treatment compared with the control (), suggesting that C1 could reduce TFEB-YWHA interaction independently of MTOR activity. At the same time, we observed an elevation of TFEB nuclear translocation, suggesting that C1 modulates TFEB subcellular localization by the weakening of the TFEB-YWHA interaction. Accordingly, the level of MTOR coimmunoprecipitated with FLAG-TFEB was also reduced (), due to the increased nuclear localization of TFEB promoted by C1 treatment.
Figure 6. Curcumin analog C1 inhibits the MTOR-TFEB-YWHA interaction. HeLa cells stably expressing 3xFlag-TFEB were treated with C1 (1 μM) for 12 h. Endogenous MTOR (A) and YWHA (B) were coimmunoprecipitated with Flag-TFEB. The levels of immunoprecipitated MTOR and YWHA were normalized to their corresponding levels in whole cell lysates (WCL). Data are presented as the mean ± SD from 3 independent experiments. *, P< 0.05 vs. the control.
Curcumin analog C1 promotes autophagy flux and lysosomal biogenesis in cell cultures
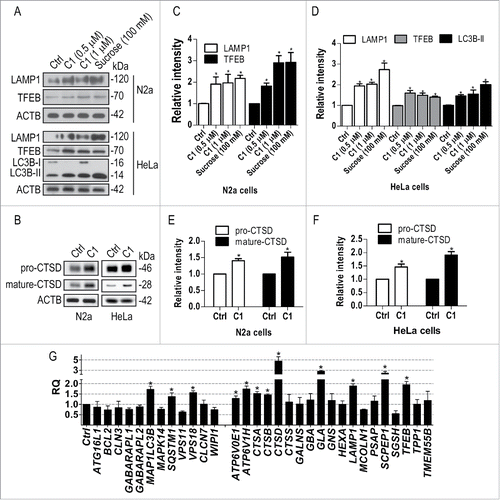
After translocation into the nuclei, TFEB triggers a transcriptional program activating multiple genes involved in autophagy and lysosomal function.Citation5 Since curcumin analog C1 increases the nuclear translocation of TFEB, we investigated whether C1 was able to promote TFEB-mediated autophagy and lysosome biogenesis. First, we confirmed that C1 treatment dose-dependently increases the levels of LC3-II and SQSTM1/p62 (sequestosome 1) in N2a cells (), and the effects require at least 9 h of treatment (). However, 6 h of C1 treatment was sufficient to significantly promote TFEB nuclear translocation (data not shown), indicating a delayed autophagy response via TFEB activation.
Figure 7. Curcumin analog C1 promotes autophagy flux and lysosomal degradation. (A) N2a cells were treated with increasing concentration of C1 for 12 h. (B) N2a cells were incubated with C1 (1 μM) for the indicated times. The protein levels of LC3B-II and SQSTM1/p62 were determined by western blotting. Data are presented as the mean ± SD from 3 independent experiments. *, P< 0.05 vs. the control (0.1% DMSO). (C) N2a cells were cotreated with cycloheximide (CHX, 10 μg/ml) and C1 (1 μM) for 12 h. The level of SQSTM1 was quantified as mean ± SD from 3 independent experiments. *, P < 0.05 vs. CHX treatment alone (N.S., not significant). (D) N2a cells were transfected with Flag-tagged SQSTM1 for 24 h and then treated with the indicated compounds for 12 h (C1, 1 μM; CQ, 20 μM; rapamycin [Rap], 5 µM). The level of Flag-SQSTM1 was quantified as mean ± SD from 3 independent experiments. *, P < 0.05 vs. the control (0.1% DMSO); #, P < 0.05 vs. C1 treatment alone (NT, nontransfected). (E) N2a cells were transiently transfected with mRFP-GFP-LC3 (tfLC3) plasmid and then treated with the indicated compounds (C1, 1 μM for 12 h; CQ, 50 μM for 12 h; torin1, 0.5 μM for 3 h). Representative images are shown (F) The numbers of red-only puncta per cell were quantified. n–20 randomly selected cells from 3 independent experiments. Data are presented as the mean ± SD. *, P < 0.05 vs. the control (0.1% DMSO); #, P < 0.05 vs. C1 treatment alone. (G) N2a cells were preincubated with 10 μg/ml of DQ-BSA-Red for 1 h, washed and then treated with the indicated compounds (C1, 1 μM for 12 h; CQ, 50 μM for 12 h; torin1, 0.5 μM for 3 h). Cells were fixed and then stained with LC3B antibody. (H) The number of DQ-BSA puncta (red) per cell was quantified. (I) The colocalization of DQ-BSA puncta (red) and LC3B puncta (green) were quantified. n–20 randomly selected cells from 3 independent experiments. Data are presented as the mean ± SD. *, P < 0.05 vs. the control (0.1% DMSO); #, P < 0.05 vs. the control (0.1% DMSO).
![Figure 7. Curcumin analog C1 promotes autophagy flux and lysosomal degradation. (A) N2a cells were treated with increasing concentration of C1 for 12 h. (B) N2a cells were incubated with C1 (1 μM) for the indicated times. The protein levels of LC3B-II and SQSTM1/p62 were determined by western blotting. Data are presented as the mean ± SD from 3 independent experiments. *, P< 0.05 vs. the control (0.1% DMSO). (C) N2a cells were cotreated with cycloheximide (CHX, 10 μg/ml) and C1 (1 μM) for 12 h. The level of SQSTM1 was quantified as mean ± SD from 3 independent experiments. *, P < 0.05 vs. CHX treatment alone (N.S., not significant). (D) N2a cells were transfected with Flag-tagged SQSTM1 for 24 h and then treated with the indicated compounds for 12 h (C1, 1 μM; CQ, 20 μM; rapamycin [Rap], 5 µM). The level of Flag-SQSTM1 was quantified as mean ± SD from 3 independent experiments. *, P < 0.05 vs. the control (0.1% DMSO); #, P < 0.05 vs. C1 treatment alone (NT, nontransfected). (E) N2a cells were transiently transfected with mRFP-GFP-LC3 (tfLC3) plasmid and then treated with the indicated compounds (C1, 1 μM for 12 h; CQ, 50 μM for 12 h; torin1, 0.5 μM for 3 h). Representative images are shown (F) The numbers of red-only puncta per cell were quantified. n–20 randomly selected cells from 3 independent experiments. Data are presented as the mean ± SD. *, P < 0.05 vs. the control (0.1% DMSO); #, P < 0.05 vs. C1 treatment alone. (G) N2a cells were preincubated with 10 μg/ml of DQ-BSA-Red for 1 h, washed and then treated with the indicated compounds (C1, 1 μM for 12 h; CQ, 50 μM for 12 h; torin1, 0.5 μM for 3 h). Cells were fixed and then stained with LC3B antibody. (H) The number of DQ-BSA puncta (red) per cell was quantified. (I) The colocalization of DQ-BSA puncta (red) and LC3B puncta (green) were quantified. n–20 randomly selected cells from 3 independent experiments. Data are presented as the mean ± SD. *, P < 0.05 vs. the control (0.1% DMSO); #, P < 0.05 vs. the control (0.1% DMSO).](/cms/asset/926d836b-a921-426c-903c-3f0c522fe3d0/kaup_a_1179404_f0007_oc.gif)
SQSTM1 is a selective substrate of autophagy and its degradation is considered as an indicator of autophagic flux.Citation33 However, SQSTM1 is also a target gene of TFEB.Citation5 Overexpression of TFEB upregulates both LC3B-II and SQSTM1,Citation5,8 which is consistent with the results of C1 treatment. To determine whether C1 promotes the degradation of SQSTM1, cells were cotreated with C1 and the protein synthesis inhibitor cycloheximide (CHX) for 12 h. The protein level of SQSTM1 was significantly decreased by C1, but not CQ in the presence of CHX (), indicating that C1 enhances autophagic degradation of SQSTM1. Furthermore, C1 significantly degraded the exogenously expressed Flag-tagged SQSTM1, and CQ blocked this effect (). Rapamycin was used as a positive control. TFEB is also reportedly activated by lysosomal stress such as CQ.Citation9,10 To further confirm that C1 indeed promotes autophagy flux rather than causing lysosomal stress, N2a cells were transfected with a tandem fluorescent mRFP-GFP-LC3 (tfLC3)Citation34 construct and then treated with the compounds indicated. CQ treatment induced accumulation and colocalization of both GFP and mRFP fluorescence, indicating the blockade of autophagosome-lysosome fusion (). In contrast, C1 or torin1 significantly increased the number of red-only puncta, indicating the formation of autolysosomes (). CQ inhibited the effect of C1 on the formation of autolysosomes.
Meanwhile, the bovine serum albumin (BSA) derivative dequenched-BSA (DQ-BSA) combined with endogenous LC3B staining were used to determine the effect of C1 on the proteolytic activity of functional lysosomes.Citation26,33 Proteolysis of DQ-BSA-Red resulted in dequenching and the release of intense red fluorescence. CQ treatment significantly inhibited the degradation of DQ-BSA-Red indicated by the decreased number of red puncta compared with the control. In contrast, C1 and torin1 promoted formation of DQ-BSA red puncta, which colocalized well with endogenous LC3B puncta (green) (). The results indicate that C1 enhances autophagy flux and increases lysosomal degradation capacity.
Next, the effects of C1 on lysosomal biogenesis were determined. We found that C1 treatment for 12 h increased the levels of TFEB and the lysosome marker LAMP1 (lysosomal-associated membrane protein 1) in N2a and HeLa cells (). Sucrose, a TFEB activatorCitation5 was used as a positive control. Meanwhile, C1 (1 μM) treatment increased the levels of both the precursor (46 kDa) and mature (28 kDa) forms of CTSD (cathepsin D) (). To extend our findings, we confirmed that C1 also induced TFEB nuclear translocation (Fig. S4A), and increased the protein levels of TFEB, LAMP1, LC3B-II and CTSD in the human neuroblastoma cell line SH-SY5Y (Fig. S4B to D). At the gene expression level, C1 increased the transcription of a series of TFEB target genes in both HeLa () and SH-SY5Y cells (Fig. S4E).
Figure 8. Curcumin analog C1 promotes lysosomal biogenesis. (A to F) N2a and HeLa cells were treated with C1 (0.5, 1 μM) or sucrose (100 mM) for 12 h. The protein levels of the autophagy marker (LC3B-II), TFEB and lysosome markers (LAMP1, CTSD) were determined by western blotting. (A, B) Representative blots. (C to F) Relative intensities are normalized to that of ACTB/β-actin. Data are presented as the mean ± SD from 3 independent experiments. *, P< 0.05 vs. the control (0.1% DMSO). (G) HeLa cells were treated with C1 (1 μM) for 12 h. mRNA transcript abundance was assessed by real-time PCR using specific primers for the indicated genes. Relative quantification (RQ) is presented as means ± SD of 3 independent experiments. *, P < 0.05 vs. the control (0.1% DMSO).
TFEB is required for curcumin analog C1 to enhance autophagy
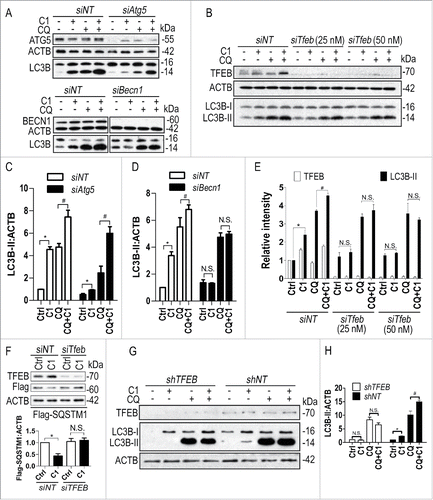
To determine whether TFEB is specifically required for C1 to enhance autophagy, we knocked down the key autophagy genes Atg5 (autophagy-related 5), Becn1 (Beclin 1, autophagy related) and Tfeb in N2a cells and then treated the cells with C1 in the presence or absence of CQ. The LC3B-II level significantly decreased in Atg5 knockdown (KD) cells treated with C1 and C1+CQ, compared to that in cells transfected with nontarget siRNA and treated with C1 and C1+CQ. However, C1 treatment could still significantly increase LC3B-II compared with the control and C1+CQ could further increase LC3B-II compared with CQ treatment alone in Atg5 KD cells (). In contrast, in Becn1 KD and Tfeb KD cells, C1 could not increase LC3B-II in the presence or absence of CQ (), indicating that Becn1 and Tfeb were required for C1 to enhance autophagy. Meanwhile, Tfeb KD blocked the effects of C1 on the degradation of the exogenously expressed SQSTM1 in N2a cells (). In HeLa cells with stable knockdown of TFEB by shRNA, we further confirmed that C1 enhanced autophagy depending on TFEB ().
Figure 9. Curcumin analog C1-induced autophagy requires TFEB and BECN1. (A-E) N2a cells were transfected with nontarget siRNA (siNT, 25 nM), Atg5 siRNA (siAtg5, 25 nM), Becn1 siRNA (siBecn1, 25 nM) and Tfeb siRNA (siTfeb, 25 and 50 nM) for 48 h and then treated with C1 (1 μM) in the presence or absence of CQ (25 μM) for an additional 12 h. (A, B) Representative blots are shown. (C to E) Relative intensity of LC3B-II is normalized to that of ACTB/β-actin. Data are presented as the mean ± SD from 3 independent experiments. *, P< 0.05 vs. the control; #, P < 0.05 vs. CQ treatment alone. (F) N2a cells were transfected with Flag-tagged SQSTM1 for 24 h and then transfected with nontarget siRNA (siNT, 25 nM) or Tfeb siRNA (siTfeb, 50 nM) for another 48 h. Cells were treated with C1 (1 μM) for 12 h and the level of Flag-SQSTM1 was quantified as mean ± SD from 3 independent experiments. *, P < 0.05 vs. the control (0.1% DMSO). (G) HeLa cells were infected with lentivirus expressing nontarget shRNA (shNT) or TFEB shRNA (shTFEB) for 48 h and then treated with C1 (1 μM) in the presence or absence of CQ (25 μM) for an additional 12 h. Representative blots are shown. (H) Relative intensity of LC3B-II is normalized to that of ACTB/β-actin. Data are presented as the mean ± SD from 3 independent experiments. *, P < 0.05 vs. the control; #, P < 0.05 vs. CQ treatment alone. N.S. (not significant).
Curcumin analog C1 activates TFEB and enhances autophagy and lysosome biogenesis in rat brain
The TFEB transgene is neuroprotective by promoting the clearance of neurodegenerative protein aggregates.Citation14,16-18 Small molecule activators of TFEB with satisfactory brain penetration and low toxicity have great potential for the treatment of neurodegenerative disorders. In neuronal and non-neuronal cell cultures, we have identified curcumin analog C1 as a TFEB activator, which enhances autophagy-lysosome biogenesis. Before performing in vivo activity tests, we determined the acute toxicity of C1 in rats by single-dose intravenous (IV) tail vein injection, and we determined that the medium lethal dose (LD50) value of C1 was 175 mg/kg (data not shown). Short-term oral administration of C1 (10 mg/kg and 25 mg/kg) dose-dependently increased the expression of LC3B-II and TFEB in the liver (Fig. S5), frontal cortex () and striatum (Fig. S5) of the brain. However, short-term administration of C1 did not affect the levels of endogenous SQSTM1/p62 in the brain (). Meanwhile, C1 treatment dose-dependently increased the expression of LAMP1 in the frontal cortex. Real-time PCR analysis of brain lysates showed that C1 upregulated TFEB and several autophagy and lysosomal genes (). C1 could pass the blood-brain barrier (BBB). The average concentration of C1 in brain tissues was 0.26 ± 0.063 μg/g (equivalent to 0.885 ± 0.213 μM) as determined by HPLC after oral administration of C1 (10 mg/kg) for 6 h (data not shown). Based on these data, we concluded that short-term treatment of C1 was sufficient to activate TFEB and autophagy in rat brains.
Figure 10. Oral administration of C1 activates TFEB and enhances autophagy and lysosome biogenesis in rat brain. SD rats (n = 6 per group) were orally administered with C1 (L, low dosage, 10 mg/kg; H, high dosage, 25 mg/kg) or vehicle (Veh., 1% CMC-Na) for 24 h. An additional dosage of C1 was given 6 h prior to sacrifice. (A) Representative blots show the levels of TFEB, autophagy and lysosome markers, p-MTOR and MTOR, as well as p-RPS6KB1 and RPS6KB1 in the frontal cortex. (B, C) Data are presented as the mean ± SD (n = 6). *, P< 0.05 vs. the vehicle treatment. (D) Representative blots shows that C1 treatment promotes TFEB nuclear translocation in the frontal cortex (n = 4). ACTB/β-actin and H3F3A (H3 histone, family 3A) were used as loading control of cytoplasmic and nuclear fraction respectively. (E) Data are presented as the mean ± SD (n = 4). *, P < 0.05 vs. vehicle treatment. (F) Levels of autophagy and lysosome genes in the frontal cortex were analyzed by real- time PCR. Relative quantification (RQ) is presented as means ± SD (n = 4). *P < 0.05 vs. the vehicle treatment. (G) Immunoprecipitation shows that C1 treatment inhibits MTOR-TFEB interaction in the frontal cortex (n = 4). (H) The levels of immunoprecipitated MTOR were normalized to its corresponding levels in cell lysates (Input). Data are presented as the mean ± SD (n = 4). *, P < 0.05 vs. the vehicle treatment.
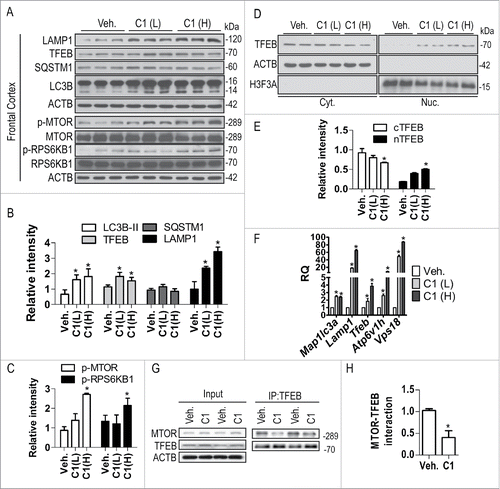
Next, we examined the MTOR pathway and TFEB translocation in the frontal cortex of rats orally treated with C1. Consistent with the in vitro observation, C1 treatment (25 mg/kg) significantly increased the phosphorylation of MTOR and RPS6KB1 (), confirming that C1 promoted MTOR activity. C1 administration dose-dependently promoted the nuclear translocation of TFEB in the brain (). The interaction between endogenous TFEB and MTOR was significantly inhibited in the frontal cortex of rats treated with C1 (25 mg/kg) ().
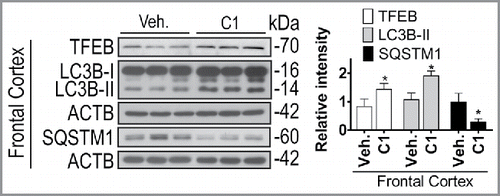
Finally, we examined the effects of chronic C1 treatment on TFEB and autophagy. Rats received oral administration of C1 (10 mg/kg per day) for 21 d. Another dose of C1 was given 6 h prior to sacrifice and the average concentration of C1 in brain tissues was found to be 0.849 ± 0.302 μg/g (equivalent to 2.884 ± 1.028 μM) (data not shown). Then the autophagy markers in the livers and brains were analyzed. Similar to the results of short-term treatment, long-term C1 treatment increased the levels of LC3B-II in the livers (Fig. S6). In the frontal cortex () and striatum (Fig. S6) of brain, C1 treatment significantly increased the levels of TFEB and LC3B-II. Notably, the levels of endogenous SQSTM1 significantly decreased in rat brains treated with C1 for 21 d (; Fig. S6), indicating that long-term administration of C1 was sufficient to promote autophagy-mediated degradation of protein aggregates in the brain. During the treatment, no changes in body weight and no behavior abnormalities were observed. Histological evaluation revealed no morphological abnormalities in major organs such as liver, lung, kidneys and pancreas (data not shown).
Figure 11. Chronic administration of C1 activates TFEB and enhances autophagy in rat brains. SD rats (n = 6 per group) were orally administered by gavage with C1 (10 mg/kg per day) or vehicle (1% CMC-Na) for 21 d. An additional dosage of C1 was given 6 h prior to sacrifice. Representative blots show the protein levels as indicated in the frontal cortex. Data are presented as the mean ± SD (n = 6). *, P< 0.05 vs. the vehicle treatment.
Discussion
Autophagy dysfunction is a common feature in neurodegenerative proteinopathies.Citation35 The essential role of TFEB in the regulation of autophagy and lysosomal biogenesis makes it a promising therapeutic target for human diseases including neurodegenerative disorders.Citation36 TFEB deregulation is involved in polyglutamine (polyQ) diseaseCitation37 and PD.Citation14 Increasing evidence demonstrates that virus-mediated gene transfer of TFEB reduces protein aggregates, including SNCA,Citation14 APP (amyloid β [A4] precursor protein),Citation38 amyloid-β (Aβ),Citation17 MAPT/TAUCitation16 and HTT (huntingtin),Citation18 ameliorates neurotoxicity, and rescues neurodegenerative phenotypes. Therefore, small molecules that can increase TFEB expression and/or stimulate its nuclear translocation hold promise as disease-modifying therapies to block the progression of neurodegeneration. Currently known TFEB activators are mainly MTOR inhibitors. The catalytic inhibitors of MTOR such as PP242 and torin1 activate TFEB by triggering its nuclear translocation while the allosteric inhibitor rapamycin has minimal effects.Citation10,39 MTOR kinase is centrally involved in the regulation of cell growth and metabolism, and the unpredictable side effectsCitation40 of MTOR inhibitors make them less likely to be useful for long-term use.Citation14 Therefore, discovery of novel MTOR-independent activators of TFEB may be more clinically important.
In this study, we found that the natural MTOR inhibitor curcuminCitation20,21,29 mildly promotes TFEB nuclear translocation and that several monocarbonyl analogs of curcumin potently inhibit MTOR activity, promote TFEB nuclear translocation and enhance autophagy. Interestingly, a curcumin analog termed C1 was found to potently activate TFEB and promote TFEB-mediated autophagy and lysosome biogenesis without inhibiting MTOR activity. Meanwhile, compared to torin1, which causes TFEB dephosphorylation on serine residues, C1 has no effects on serine-phosphorylation of TFEB. Furthermore, we excluded the possibility that C1 activates TFEB via MAPK1/3 inhibition (). These findings led us to hypothesize that C1 may directly bind to and activate TFEB.
By using a series of binding assays, we have confirmed a direct and specific binding of C1 to recombinant TFEB. ITC titration results for C1 showed endothermic binding with apparently positive ΔS and ΔH values, indicating the binding is a typical entropy-driven interaction with favorable entropy change and unfavourable enthalpy change, as commonly observed in other proteins associating with hydrophobic membrane or detergent micelles.Citation41,42 Increase of entropy is mainly contributed by ligand-protein hydrophobic reaction and release of loosely bound water from the binding cavity of C1 in TFEB. The disfavored enthalpy change in this binding process probably indicates that formation of hydrogen bond(s) is absent and we speculate that the TFEB contains a hydrophobic binding pocket to accommodate C1. Notably, deletion of just the 44 amino acids at the N terminus completely blocked the binding of C1 to TFEB (), indicating the binding site of C1 localizes within the N-terminal Gly and Ala-rich domain of TFEB. It has been reported that several residues (S3 and R4, as well as Q10 and L11) within the first 30 amino acids regulate the cytosolic localization of TFEB.Citation43 A possible mechanism of C1 action on TFEB could be that its binding with TFEB at the N terminus changes TFEB conformation to expose the nuclear localization signal as TFEB dephosphorylation in S211 does.Citation10 Alternatively, or in addition to the former hypothesis, C1 binding to TFEB may block TFEB-YWHA interaction facilitating its nuclear translocation. Future work on resolving the structure of TFEB will definitely help to delineate how C1 binds to and promotes TFEB activation, and will facilitate design of new specific TFEB activators using C1 as a lead compound. Interestingly, the osteoclast differentiation factor TNFSF11/RANKL promotes lysosomal biogenesis once osteoclasts are differentiated through the selective activation of TFEB through phosphorylation modification that stabilizes and increases TFEB protein levels.Citation44 We also showed that C1 increases TFEB protein levels; most likely through its positive effect on its own transcription after nuclear translocation,Citation7 although further studies are required to investigate whether C1 treatment stabilizes TFEB.
In several cell lines, C1 upregulated a series of autophagy-lysosomal genes and proteins controlled by TFEB and its ability to enhance autophagy was completely blocked by knocking down Tfeb and its target gene Becn1Citation45 These facts support our conclusion that C1 promotes TFEB-mediated autophagy and lysosome biogenesis.
Finally, we addressed the critical issue of whether curcumin analog C1 activates TFEB and enhances autophagy in vivo, especially in the mammalian brain. Autophagy is induced in most organs including the liver in response to nutrient starvation. However, enhancing autophagy is relatively difficult in the central nervous system by food withdrawal or exercise.Citation46,47 BBB permeability, stability, potency and duration of action are important factors that determine the efficacy of a drug in the brain. In a previous study, the same structure of C1 (term B63) showed much better cellular uptake, delayed degradation and had better antitumor effects than curcumin.Citation48 After determining the acute toxicity of C1, 2 relatively safe doses (10 mg/kg and 25 mg/kg) of C1 were selected for animal studies. Six h after the last oral administration, the brain content of C1 was approximately 1 μM, which is an effective concentration for TFEB activation and autophagy enhancement in cell cultures. Consistent with our in vitro findings, C1 treatment activated TFEB, inhibited MTOR-TFEB interaction, and promoted autophagy and lysosome biogenesis without inhibiting MTOR activity in the brain (). Notably, chronic treatment of C1 significantly promoted the degradation of the autophagy substrates SQSTM1 ().
Overall, the curcumin analog C1 is identified as a direct activator of TFEB in the present study. Structural stability and good BBB permeability make it a good drug candidate deserving further studies in animal models of neurodegenerative diseases.
Materials and methods
Reagents and antibodies
A to D series of mono-carbonyl analogs of curcumin were synthesized according to our previous study.Citation49 The procedure for the synthesis of E series is described in Supplementary Methods. Curcumin (08511), chloroquine (C6628), doxycycline (D9891), anti-Flag M2 (F1804) and anti-SQSTM1/p62 (P0067) were purchased from Sigma-Aldrich. Torin1 (2273–5) was purchased from BioVision Inc. Anti-TFEB (4240), anti-SQSTM1/p62 (5114), anti-phospho-MTOR (Ser2448) (2971), anti-MTOR (2983), anti-phospho-RPS6KB1/P70S6K (Thr389) (9234), anti- RPS6KB1/p70S6K (9202), anti-phospho-ULK1 (Ser757) (14202), anti-ULK1 (D8H5; 8054), anti-phospho-PRKAA/AMPKα (Thr172; 2535), anti-PRKAA/AMPKα (D5A2; 5831), phospho-(Ser) YWHA/14-3-3 binding motif (9601), pan-YWHA/14-3-3 (8312) and anti-H3F3A/histone H3 (D1H2; 4499) antibodies were purchased from Cell Signaling Technology. Anti-phosphoserine (ab9332), anti-LAMP1 (ab24170) and anti-CTSD/cathepsin D (ab75852) antibodies were purchased from Abcam. HRP-conjugated goat anti-mouse (115-035-003) and goat anti-rabbit (111-035-003) secondary antibodies were purchased from Jackson ImmunoResearch. Anti-CTSD/cathepsin D (H-75; sc-10725), anti-TUBB/β-tubulin (H-235; sc-9104) and anti-ACTB/β-actin (sc-47778) was purchased from Santa Cruz Biotechnology. Anti-ATG5 (NB110-53818), anti-BECN1/Beclin 1 (NB110-87318) and anti-LC3B (NB100-2220) antibodies were purchased from Novus Biologicals. Anti-TFEB (13372-1-AP) was purchased from Proteintech. Anti-TFEB (A303-673A) was purchased from Bethyl Laboratories, Inc. Mouse Atg5 siRNA (L-064838-00-0005), Becn1 siRNA (L-055895-00-0005), Tfeb siRNA (L-050607-02-0005) and nontarget siRNA were purchased from Dharmacon. DMEM (11965–126), fetal bovine serum (FBS; 10270–106), Opti-MEM I (31985–070), horse serum (16050–122), G418 (10131–035), DQ-BSA-Red (D12051), Alexa Fluor® 488 goat anti-mouse IgG (A-11001) and Alexa Fluor® 594 goat anti-rabbit IgG (A-11012) were purchased from Life Technologies. SQSTM1 plasmid (MYC-DDK-tagged) (RC203214) and recombinant human TFEB protein (TP760282) were purchased from OriGene Technologies, Inc.
Cell culture and drug treatment
N2a and HeLa cells were cultured in DMEM supplemented with 10% FBS. SH-SY5Y cells were cultured in DMEM/F12 (Life Technologies, 10565042) supplemented with 10% FBS. HeLa cells stably expressing 3x-Flag-TFEBCitation5,6 were maintained in DMEM supplemented with 10% FBS and 500 μg/ml G418. For drug treatment, the full medium was replaced by fresh Opti-MEM I containing the compounds (in 0.1% DMSO) and incubated for the indicated time periods.
LDH assay
The cytotoxicity was determined by measurement of LDH release from damaged cells using the LDH Kit (Roche, 11644793001) according to the manufacturer's protocol.
High-content TFEB nuclear translocation assay
To quantify TFEB subcellular localization, a high-content assay was performed using stable HeLa cells overexpressing 3xFlag-TFEB according to our previous protocols.Citation9
Protein preparation
Full-length human TFEB (FL-TFEB) was subcloned into the pET28a plasmid (Novagen, 69864) from the TFEB-3×FLAG vectorCitation5 to generate the N-terminal 6xHis-tagged form for E. coli overexpression. Deletions of the TFEB N and C termini were performed by PCR amplification of the truncated fragments and ligation into pET28a vector or ppSUMO (a generous gift from Prof. Jiahai Zhou, Shanghai Institute of Organic Chemistry) vector yielding N-terminal 6xHis-tagged or N-terminal 6xHis-SUMO-fusion protein. TFEB Δ(121 to 330) was generated via a one-step site-directed deletion of the 121st to 330th amino acids from the full length TFEB-pET28a plasmid using the Quickchange II site-directed mutagenesis kit (Agilent, 200524).Citation50 The subcloned genes were confirmed by full-length DNA sequencing. The oligodeoxynucleotides used in the gene amplification are listed in Table S1.
The N-terminal 6xHis-tagged human TFEB was overexpressed in E. coli BL21 (DE3; ThermoFisher, C600003) cells. The E. coli culture was grown in LB medium at 37°C and shaken at 220 rpm until the OD600 reached 0.8, at which point IPTG (Sigma-Aldrich, I6758) was added to a final concentration of 0.1 mM. The cells were then grown for an additional 12 h for protein overexpression shaken at 30°C and 180 rpm. Cells were harvested by centrifugation at 3,000 g, 4°C and resuspended in the lysis buffer containing 20 mM Tris, pH 8.0, 10 mM imidazole, 500 mM NaCl, 0.6% BriJ-58 (Sigma-Aldrich, P5884) and 10% glycerol (ThermoFisher, 15514–029) for sonication. The cell lysate was treated with DNase I (Roche, 10104159001) for removal of bound DNA fragments in the presence of 5 mM Mg2+ and 2 mM Ca2+. After DNase I digestion, the cell debris was removed by centrifugation at 14,000 g, 4°C. The soluble His-tagged TFEB was purified by Ni2+ affinity chromatography using HisTrap Ni-NTA column (GE Healthcare Life Sciences, 11-0004-58) in the presence of 0.6% Brij-58. Extra detergent was removed by an anion exchange column (HiPrep DEAE FF, GE Healthcare Life Sciences, 28-9365-41). The purified TFEB was aliquoted and stored in 25 mM Tris, pH 8.0, 150 mM NaCl, 2 mM DTT, 20% glycerol, 0.1% Brij-58.
All TFEB variants were overexpressed in E. coli BL21-Codonplus (DE3). The cells were grown in ZYB-5052 complex auto-inducing mediaCitation51 containing 1% N-Z-amine A (Amresco Inc., J853), 0.5% yeast extract, 25 mM Na2HPO4, 25 mM KH2PO4, pH 7.5, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.5% glycerol, 0.05 % glucose, 0.1% galactose (Sigma-Aldrich, G0750) supplemented with 50 mg/L kanamycin and 38 mg/L chloramphenicol. The cultures were shaken at 37°C, 270 rpm until lightly turbid (OD600 < 1) and then transferred to a 25 to 30°C environment for protein overexpression and incubated for an additional 12- to 24-h period until saturated (OD600 ∼7). The cells were lysed in the lysis buffer containing 20 mM Tris, pH 8.0, 10 mM imidazole (Sigma-Aldrich, 792527), 1 M NaCl, 1% sarkosyl (Sigma-Aldrich, L5777) and 10% glycerol. The detergent sarkosyl was excluded in all other buffers and gradually removed via dilution in the following steps of Ni2+ affinity chromatography purification. The ppSUMO plasmid-encoded deletions (ΔC461, ΔC150 and 221 to 330) were treated by Ulp1 SUMO-specific protease (a generous gift from Prof. Jiahai Zhou, Shanghai Institute of Organic Chemistry) on ice for ∼1 h after purification to remove the SUMO tag. After the protease and SUMO tag were removed by the Ni-NTA column, the proteins were concentrated, aliquoted and stored in 25 mM Tris, pH 8.0, 400 mM NaCl, 2 mM β-mecaptoethanol, 10% glycerol. Protein qualities were verified on an SDS-PAGE gel to be more than 90% pure. Protein concentrations of 221 to 330 and ΔC150 were determined using Pierce™ Coomassie (Bradford) Protein Assay Kit (Thermo Scientific, No.23200). Concentrations of other truncations and the full-length protein were determined based on extinction coefficient ϵ280 calculated via ProteinCalculator v3.4 developed by Scripps Institute (http://www.scripps.edu/cgi-bin/cdputnam/protcalc3).
Isothermal titration calorimetry (ITC) binding assay
ITC experiments including C1 to FL-TFEB, C1 to ΔC150, C1 to ΔC461, C1 to Δ(121 to 330), curcumin to the FL-TFEB, B1 to FL-TFEB and E4 to FL-TFEB were carried out in a VP-ITC microcalorimeter (Malvern Instruments Ltd., Worcestershire, UK). Protein and ligand solutions were prepared by dilution with the corresponding protein storage buffer containing 6% DMSO, to the required concentration. All solutions were thoroughly degased under vacuum for 5 min with gentle stirring before use. Typically, ligand solutions at 100 to 200 μM in a 250-μl syringe were titrated into 1.4 ml solution of the proteins at 10 to 20 μM in the sample cell, respectively. Titration trials were performed at 25°C with a reference cell power of 9. Injections of 10 to 14 μl of ligand solutions were performed at an interval of 120 to 150 s in to the protein solution with stirring at 307 rpm. Titration experiments of C1 to other truncations were performed using MicroCal-ITC 200 (Malvern Instruments Ltd. Worcestershire, UK). The 40-μl syringe and 200-μl cell were filled with 200 μM ligand solution and 10 to 20 μM protein solution, respectively. The experiments were carried out at 25°C. An initial injection of 0.1 μl was excluded for data analysis, followed by 16 injections of 2 μl each, separated by 120 s. The raw data were processed using the one binding site model in the MicroCal ORIGIN software. Prior to analysis, data were corrected by subtracting the dilution heat of the ligands.
Gene knockdown assay
N2a cells were transfected with mouse Atg5, Becn1, or Tfeb siRNA and the nontarget siRNA using Lipofectamine RNAiMAX (Invitrogen, 13778030) and incubated at 37°C for 72 h. For stable TFEB knockdown, HeLa cells were infected with lentivirus expressing nontarget shRNA (Sigma-Aldrich, SHC002V) or TFEB shRNACitation52 (Sigma-Aldrich, TRCN000013111) for 48 h and then incubated with medium containing puromycin (1.5 μg/ml) until resistant colonies were identified.
Animals and treatments
All animal care and experimental procedures were approved by the Hong Kong Baptist University Committee on the Use of Human and Animal Subjects in Teaching and Research. Adult male Sprague-Dawley (SD) rats weighing 350 to 400 g were maintained on ad libitum food and water with a 12-h light/dark cycle in a controlled environment. Rats (n = 6 per group) were orally administered by gavage with C1 (10 mg/kg and 25 mg/kg per day) or vehicle (1% sodium carbonyl methylcellulose [CMC-Na; Sigma-Aldrich, C5678]) for 24 h. At the end of the treatment, an additional dosage of C1 was given for 6 h before the rats were killed. Livers and major brain regions dissected according to the previous protocolCitation53 were snap-frozen in liquid nitrogen.
Quantitative real-time PCR
Total RNA was extracted from cells and tissues using the RNeasy Plus Mini Kit (Qiagen, 74134). Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies, 4368814). Autophagy and lysosome gene primers were retrieved from Origene and from a previous studyCitation14 and synthesized by Life Technologies. The oligonucleotide sequences are listed in Table S2. Real-time PCR was carried out with the Fast SYBRR Green Master Mix (Life Technologies, 4385612) using the ViiA™ 7 Real-Time PCR System (Life Technologies, Carlsbad, CA, USA). Fold changes were calculated using the ΔΔCT method, and the results were normalized against an internal control (GAPDH or ACTB).
Western blotting and immunoprecipitation
Cells were lysed on ice in 1x Lysis Buffer (Life Technologies, 9803) with complete protease inhibitor mixture (Roche Applied Science, 04693124001) and phosphatase inhibitor (Biotool, B15001). Animal tissues were homogenized in 9 volumes of ice-cold PBS supplemented with protease inhibitors.Citation46 For determining the level of SQSTM1, 1% SDS (Sigma-Aldrich, L3771) was added to the lysis buffer, which allowed identification of the entire cellular pool of SQSTM1.Citation33 Cytosolic and nuclear fractions were isolated using protocols similar to those described previously.Citation10 Anti-Flag or anti-TFEB antibody was added to the whole cell lysates and Dynabeads® Protein G (Life Technologies, 10003D) was used for immunoprecipitation. Proteins were separated by 10 to 15% SDS-PAGE, transferred, and blotted with the antibodies described. The blots were then incubated with secondary antibodies or the Clean-Blot IP Detection Reagent (Thermo Scientific, 21230) at room temperature for 1 h. The protein signals were detected by the ECL kit (Pierce, 32106) and quantified using ImageJ software.
Immunocytochemistry
Cells were seeded on coverslips placed in 24-well plates. For autophagy flux assay, cells were transfected with a tfLC3 plasmidCitation34 for 24 h and then treated with the indicated compounds. For lysosomal degradation assay, cells were preincubated with 10 μg/ml of DQ-BSA-Red for 1 h, washed and then treated with the indicated compounds. After drug treatment, slices were fixed with 3.7% paraformaldehyde, permeabilized in 0.2% Triton X-100 (Sigma-Aldrich, T8787) and blocked with 5% BSA. After blocking, the slices were stained with anti-TFEB (1:200), anti-Flag (1:500) or anti-LC3B (1:500) antibodies overnight at 4°C. Alexa Fluor® 488 (green) or Alexa Fluor® 594 (red) secondary antibodies (1:500) were added for 1 h at room temperature. After nuclear staining with DAPI, the slices were mounted with FluorSave reagent (Calbiochem, 345789). Cells were visualized using the Eclipse 80i fluorescence microscope (Nikon Instruments Inc., Melville, NY, USA) and the DeltaVision Deconvolution Microscope (GE Healthcare, Pittsburgh, PA, USA). LC3B puncta was quantified according to our previous protocol.Citation54 Colocalization of LC3B and DQ-BSA puncta was quantified as pearson correlation coefficient by using the Pearson-Spearman Correlation colocalization ImageJ plug-in.Citation55
Statistical analysis
Each experiment was performed at least 3 times, and the results were presented as mean ± SD. One-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls test using the SigmaPlot 11.0 software packages. A probability value of P < 0.05 was considered to be statistically significant.
Abbreviations
| AD | = | Alzheimer disease |
| ALP | = | autophagy-lysosome pathway |
| AMPK | = | AMP-activated protein kinase |
| ATG5 | = | autophagy-related 5 |
| BBB | = | blood-brain barrier |
| BECN1 | = | Beclin 1, autophagy related |
| CHX | = | cycloheximide |
| CLEAR | = | coordinated lysosomal expression and regulation |
| CMC-Na | = | sodium carbonyl methylcellulose |
| CQ | = | chloroquine |
| CTSD | = | cathepsin D |
| DQ-BSA | = | dequenched-bovine serum albumin |
| HD | = | Huntington disease |
| ITC | = | isothermal titration calorimetry |
| KD | = | knockdown |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| LDH | = | lactate dehydrogenase |
| LSDs | = | lysosomal storage disorders |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3B |
| MAPK | = | mitogen-activated protein kinase |
| MTORC1 | = | mechanistic target of rapamycin (serine/threonine kinase) complex 1 |
| N2a | = | neuro-2a |
| PD | = | Parkinson disease |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| RPS6KB1/p70S6K | = | ribosomal protein S6 kinase, polypeptide 1 |
| SQSTM1/p62 | = | sequestosome 1 |
| TFEB | = | transcription factor EB |
| tfLC3 | = | tandem fluorescent mRFP-GFP-LC3 |
| ULK1 | = | unc-51 like kinase 1 |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1179404_Supplemental_Material.docx
Download MS Word (2.3 MB)Acknowledgments
We thank Tamotsu Yoshimori (Osaka University) for providing the tfLC3 plasmid; Xianxiu QIU and Yanxiang ZHAO (The Hong Kong Polytechnic University) for assisting ITC assay; Jasmine Ka Man YU (Hong Kong University of Science and Technology) for assisting cloning and protein purification; Martha Dahlen for English editing.
Funding
This work was supported by the Hong Kong General Research Fund (RGC/HKBU-121009/14 to M.L.), the Research Grants Council & Natural Science Foundation Council of China (RGC-N_HKBU 213/11 to M.L. and N_HKUST621/13 to Z.G.), the Innovation and Technology Fund (ITF) of Hong Kong Government (ITS/274/12 to M.L.), the Interdisciplinary Research Matching Scheme (IRMS) (IRMS/12–13/1A to M.L.), the Matching Proof-of-Concept Fund (MPCF) (MPCF 04–12/13 and MPCF-008-2014/2015 to M.L.) and RC Start up grant for new academics (38-40-183 to J.X.S.) of Hong Kong Baptist University and Natural Science Foundation of China (21172101 to B.Z.).
Related Research Data
References
- Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta 2009; 1793:664-73; PMID:18706940; http://dx.doi.org/10.1016/j.bbamcr.2008.07.014
- Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy 2012; 8:719-30; PMID:22647656; http://dx.doi.org/10.4161/auto.19469
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson disease. Brain 2008; 131:1969-78; PMID:18187492; http://dx.doi.org/10.1093/brain/awm318
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; http://dx.doi.org/10.1038/nm.3232
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/10.1126/science.1204592
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473-7; PMID:19556463; http://dx.doi.org/10.1126/science.1174447
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 2013; 15:647-58; PMID:23604321; http://dx.doi.org/10.1038/ncb2718
- Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012; 8:903-14; PMID:22576015; http://dx.doi.org/10.4161/auto.19653
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31:1095-108; PMID:22343943; http://dx.doi.org/10.1038/emboj.2012.32
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5:ra42; PMID:22692423; http://dx.doi.org/10.1126/scisignal.2002790
- Moskot M, Montefusco S, Jakobkiewicz-Banecka J, Mozolewski P, Wegrzyn A, Di Bernardo D, Węgrzyn G, Medina DL, Ballabio A, Gabig-Cimińska M. The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J Biol Chem 2014; 289:17054-69; PMID:24770416; http://dx.doi.org/10.1074/jbc.M114.555300
- Spampanato C, Feeney E, Li L, Cardone M, Lim JA, Annunziata F, Zare H, Polishchuk R, Puertollano R, Parenti G, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 2013; 5:691-706; PMID:23606558; http://dx.doi.org/10.1002/emmm.201202176
- Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell 2011; 21:421-30; PMID:21889421; http://dx.doi.org/10.1016/j.devcel.2011.07.016
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A 2013; 110:E1817-26; PMID:23610405; http://dx.doi.org/10.1073/pnas.1305623110
- Parr C, Carzaniga R, Gentleman SM, Van Leuven F, Walter J, Sastre M. Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-β precursor protein. Mol Cell Biol 2012; 32:4410-8; PMID:22927642; http://dx.doi.org/10.1128/MCB.00930-12
- Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, Swartzlander DB, Palmieri M, di Ronza A, Lee VM, et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med 2014; 6(9):1142-60; PMID:25069841; http://dx.doi.org/10.15252/emmm.201303671
- Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, et al. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci 2014; 34:9607-20; PMID:25031402; http://dx.doi.org/10.1523/JNEUROSCI.3788-13.2014
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med 2012; 4:142ra97; PMID:22786682; http://dx.doi.org/10.1126/scitranslmed.3003799
- Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm 2007; 4:807-18; PMID:17999464; http://dx.doi.org/10.1021/mp700113r
- Han J, Pan XY, Xu Y, Xiao Y, An Y, Tie L, Pan Y, Li XJ. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy 2012; 8:812-25; PMID:22622204; http://dx.doi.org/10.4161/auto.19471
- Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol 2007; 72:29-39; PMID:17395690; http://dx.doi.org/10.1124/mol.106.033167
- Shehzad A, Khan S, Shehzad O, Lee YS. Curcumin therapeutic promises and bioavailability in colorectal cancer. Drugs Today (Barc) 2010; 46:523-32; PMID:20683505; http://dx.doi.org/10.1358/dot.2010.46.7.1509560
- Subramaniam D, May R, Sureban SM, Lee KB, George R, Kuppusamy P, Ramanujam RP, Hideg K, Dieckgraefe BK, Houchen CW, et al. Diphenyl difluoroketone: a curcumin derivative with potent in vivo anticancer activity. Cancer Res 2008; 68:1962-9; PMID:18339878; http://dx.doi.org/10.1158/0008-5472.CAN-07-6011
- Ohori H, Yamakoshi H, Tomizawa M, Shibuya M, Kakudo Y, Takahashi A, Takahashi S, Kato S, Suzuki T, Ishioka C, et al. Synthesis and biological analysis of new curcumin analogues bearing an enhanced potential for the medicinal treatment of cancer. Mol Cancer Ther 2006; 5:2563-71; PMID:17041101; http://dx.doi.org/10.1158/1535-7163.MCT-06-0174
- Liang G, Zhou H, Wang Y, Gurley EC, Feng B, Chen L, Xiao J, Yang S, Li X. Inhibition of LPS-induced production of inflammatory factors in the macrophages by mono-carbonyl analogues of curcumin. J Cell Mol Med 2009; 13:3370-9; PMID:19243473; http://dx.doi.org/10.1111/j.1582-4934.2009.00711.x
- Mosley CA, Liotta DC, Snyder JP. Highly active anticancer curcumin analogues. Adv Exp Med Biol 2007; 595:77-103; PMID:17569206; http://dx.doi.org/10.1007/978-0-387-46401-5_2
- Zheng A, Li H, Wang X, Feng Z, Xu J, Cao K, Zhou B, Wu J, Liu J. Anticancer Effect of a Curcumin Derivative B63: ROS Production and Mitochondrial Dysfunction. Curr Cancer Drug Targets 2014; 14:156-66; PMID:24274397.
- Zhang X, Zhang HQ, Zhu GH, Wang YH, Yu XC, Zhu XB, Liang G, Xiao J, Li XK. A novel mono-carbonyl analogue of curcumin induces apoptosis in ovarian carcinoma cells via endoplasmic reticulum stress and reactive oxygen species production. Mol Med Rep 2012; 5:739-44; PMID:22159410; http://dx.doi.org/10.3892/mmr.2011.700
- Beevers CS, Chen L, Liu L, Luo Y, Webster NJ, Huang S. Curcumin disrupts the Mammalian target of rapamycin-raptor complex. Cancer Res 2009; 69:1000-8; PMID:19176385; http://dx.doi.org/10.1158/0008-5472.CAN-08-2367
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 2009; 284:8023-32; PMID:19150980; http://dx.doi.org/10.1074/jbc.M900301200
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011; 30:4297-306; PMID:21516121; http://dx.doi.org/10.1038/onc.2011.137
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139;
- Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010; 13:805-11; PMID:20581817; http://dx.doi.org/10.1038/nn.2575
- Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013; 14:283-96; PMID:23609508; http://dx.doi.org/10.1038/nrm3565
- Cortes CJ, Miranda HC, Frankowski H, Batlevi Y, Young JE, Le A, Ivanov N, Sopher BL, Carromeu C, Muotri AR, et al. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat Neurosci 2014; 17:1180-9; PMID:25108912; http://dx.doi.org/10.1038/nn.3787
- Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Tripoli DL, Czerniewski L, Ballabio A, et al. Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Abeta Generation and Amyloid Plaque Pathogenesis. J Neurosci 2015; 35:12137-51; PMID:26338325; http://dx.doi.org/10.1523/JNEUROSCI.0705-15.2015
- Zhou J, Tan SH, Nicolas V, Bauvy C, Yang ND, Zhang J, Xue Y, Codogno P, Shen HM. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res 2013; 23:508-23; PMID:23337583; http://dx.doi.org/10.1038/cr.2013.11
- Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf 2013; 12:177-86; PMID:23252795; http://dx.doi.org/10.1517/14740338.2013.752814
- Jia H, Liggins JR, Chow WS. Entropy and biological systems: experimentally-investigated entropy-driven stacking of plant photosynthetic membranes. Sci Rep 2014; 4:4142; PMID:24561561; http://dx.doi.org/10.1038/srep04142
- Perozzo R, Folkers G, Scapozza L. Thermodynamics of protein-ligand interactions: history, presence, and future aspects. J Recept Signal Transduct Res 2004; 24:1-52; PMID:15344878
- Martina JA, Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol 2013; 200:475-91; PMID:23401004; http://dx.doi.org/10.1083/jcb.201209135
- Ferron M, Settembre C, Shimazu J, Lacombe J, Kato S, Rawlings DJ, Ballabio A, Karsenty G. A RANKL-PKCbeta-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev 2013; 27:955-69; PMID:23599343; http://dx.doi.org/10.1101/gad.213827.113
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011; 20:3852-66; PMID:21752829; http://dx.doi.org/10.1093/hmg/ddr306
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/10.1091/mbc.E03-09-0704
- He C, Sumpter R, Jr., Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 2012; 8:1548-51; PMID:22892563; http://dx.doi.org/10.4161/auto.21327
- Xiao J, Wang Y, Peng J, Guo L, Hu J, Cao M, Zhang X, Zhang H, Wang Z, Li X, et al. A synthetic compound, 1,5-bis(2-methoxyphenyl)penta-1,4-dien- 3-one (B63), induces apoptosis and activates endoplasmic reticulum stress in non-small cell lung cancer cells. Int J Cancer 2012; 131:1455-65; PMID:22189907; http://dx.doi.org/10.1002/ijc.27406
- Dai F, Liu GY, Li Y, Yan WJ, Wang Q, Yang J, Lu DL, Ding DJ, Lin D, Zhou B. Insights into the importance for designing curcumin-inspired anticancer agents by a prooxidant strategy: The case of diarylpentanoids. Free Radic Biol Med 2015; 85:127-37; PMID:25912482; http://dx.doi.org/10.1016/j.freeradbiomed.2015.04.017
- Liu H, Naismith J. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnology 2008; 8:91; http://dx.doi.org/10.1186/1472-6750-8-91;
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 2005; 41:207-34; PMID:15915565
- Martina JA, Diab HI, Lishu L, Jeong AL, Patange S, Raben N, Puertollano R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal 2014; 7:ra9; PMID:24448649; http://dx.doi.org/10.1126/scisignal.2004754
- Chiu K, Lau WM, Lau HT, So KF, Chang RC. Micro-dissection of rat brain for RNA or protein extraction from specific brain region. J Vis Exp 2007; 7:e269; PMID:18989440; http://dx.doi.org/10.3791/269.
- Lu J, He L, Behrends C, Araki M, Araki K, Jun Wang Q, Catanzaro JM, Friedman SL, Zong WX, Fiel MI, et al. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat Commun 2014; 5:3920; PMID:24849286; http://dx.doi.org/10.1038/ncomms4920
- French AP, Mills S, Swarup R, Bennett MJ, Pridmore TP. Colocalization of fluorescent markers in confocal microscope images of plant cells. Nat Protoc 2008; 3:619-28; PMID:18388944; http://dx.doi.org/10.1038/nprot.2008.31