
ABSTRACT
Reactive oxygen species (ROS) have been commonly accepted as inducers of autophagy, and autophagy in turn is activated to relieve oxidative stress. Yet, whether and how oxidative stress, generated in various human pathologies, regulates autophagy remains unknown. Here, we mechanistically studied the role of TRPM2 (transient receptor potential cation channel subfamily M member 2)-mediated Ca2+ influx in oxidative stress-mediated autophagy regulation. On the one hand, we demonstrated that oxidative stress triggered TRPM2-dependent Ca2+ influx to inhibit the induction of early autophagy, which renders cells more susceptible to death. On the other hand, oxidative stress induced autophagy (and not cell death) in the absence of the TRPM2-mediated Ca2+ influx. Moreover, in response to oxidative stress, TRPM2-mediated Ca2+ influx activated CAMK2 (calcium/calmodulin dependent protein kinase II) at levels of both phosphorylation and oxidation, and the activated CAMK2 subsequently phosphorylated BECN1/Beclin 1 on Ser295. Ser295 phosphorylation of BECN1 in turn decreased the association between BECN1 and PIK3C3/VPS34, but induced binding between BECN1 and BCL2. Clinically, acetaminophen (APAP) overdose is the most common cause of acute liver failure worldwide. We demonstrated that APAP overdose also activated ROS-TRPM2-CAMK2-BECN1 signaling to suppress autophagy, thereby causing primary hepatocytes to be more vulnerable to death. Inhibiting the TRPM2-Ca2+-CAMK2 cascade significantly mitigated APAP-induced liver injury. In summary, our data clearly demonstrate that oxidative stress activates the TRPM2-Ca2+-CAMK2 cascade to phosphorylate BECN1 resulting in autophagy inhibition.
Introduction
Physiological levels of reactive oxygen species (ROS) participate in cell signaling, whereas high levels of ROS may irreversibly damage lipids, proteins, and DNA, and thus interfere with critical cellular functions. Complex antioxidant defense mechanisms, including both enzymatic and nonenzymatic systems, can protect cells from oxidative damage by strictly regulating ROS levels in vivo. Whereas maintaining a balance between ROS formation and antioxidant defense is essential to cell survival and growth, an imbalance generates oxidative stress, which has been implicated in numerous human diseases.Citation1 For example, an overdose of APAP, a popular over-the-counter analgesic and antipyretic drug, results in severe liver and renal damage.Citation2 APAP toxicity is due to its reactive metabolite, N-acetyl-p-benzoquinone imine (NAPQI) being catalyzed by hepatic CYP/cytochrome P450. Overproduction of NAPQI from APAP overdose depletes glutathione levels and NPAQI binds to many cellular proteins, resulting in an accumulation of both ROS and reactive nitrogen species. The resultant oxidative stress is one of main causes of liver cell death.Citation3 Obviously, being able to differentiate between the molecular effectors of ROS that are required for normal cellular processes, and those that lead to pathologies is essential for the development of selective therapies for treating disease and at the same time, sparing healthy tissue.Citation1
Autophagy, an essential catabolic degradation process for cell survival,Citation4-7 acts as an important cellular antioxidant pathway to relieve oxidative stress. When autophagy is defective, there is an accumulation of ROS and mitochondrial damage; the latter is itself a source of ROS and thus a feedback loop of increasing levels of ROS is formed. Along this line, ROS are known to induce early autophagy.Citation8 For example, ROS activate ATM (ataxia telangiectasia mutated) serine/threonine kinase and STK11/LKB1 (serine/threonine kinase 11), which in turn activate TSC2 (tuberous sclerosis 2) and inhibit MTOR (mechanistic target of rapamycin [serine/threonine kinase]), thereby activating AMP-activated protein kinase (AMPK) to induce autophagy.Citation9 Alternatively, oxidative inactivation of ATG4 (autophagy-related 4) increases the formation of autophagosomes by promoting MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) lipidation.Citation10 ROS can also induce autophagy via the MAPK (mitogen-activated protein kinase) signaling pathways for cell survival.Citation8 Paradoxically, oxidative stress generated in various pathologies normally results in cell or tissue damage. Whether and how oxidative stress regulates autophagy in these pathologies remains unresolved.Citation11
TRPM2, a nonselective Ca2+ permeable cation channel, has a unique C-terminal diphosphoribose hydrolase (Nudix-like) domain, which is where intracellular adenosine diphosphate ribose (ADPR), the main activator of TRPM2, binds. TRPM2 can also be positively regulated by Ca2+, cyclic adenosine diphosphoribose, H2O2, and nicotinic acid adenine dinucleotide phosphate, and is negatively regulated by AMP and acidic pH.Citation12,13 TRPM2-mediated Ca2+ influx has been implicated in several physiological and pathophysiological processes, including insulin secretion and endothelium permeability, as well as dendritic cell maturation and chemotaxis.Citation14 Since TRPM2 can be activated by oxidative stress, it has recently emerged as a potential therapeutic target in fighting oxidative stress-related diseases, including diabetes, myocardial infarction, and neurodegenerative diseases.Citation15
While functional TRPM2 is ubiquitously expressed in animal tissues, the expression and/or function of TRPM2 in many commonly used cell lines, especially in various cancer lines, is compromised, rendering these cell lines refractory to oxidative stress-induced cell death.Citation16-18 Therefore, we speculate that the ability of oxidative stress to activate TRPM2-Ca2+ signaling might allow cells to switch from autophagic cell survival to apoptotic or necrotic cell death. Here, we mechanistically studied the role of ROS-TRPM2-mediated Ca2+ influx in autophagy regulation and demonstrated that oxidative stress triggers Ca2+ influx via TRPM2 to activate CAMK2 (calcium/calmodulin dependent protein kinase II), and activated CAMK2 subsequently phosphorylates BECN1/Beclin 1 to inhibit autophagy, which ultimately renders cells or tissues more vulnerable to death or damage.
Results
Oxidative stress triggers Ca2+ influx via TRPM2 to inhibit autophagy
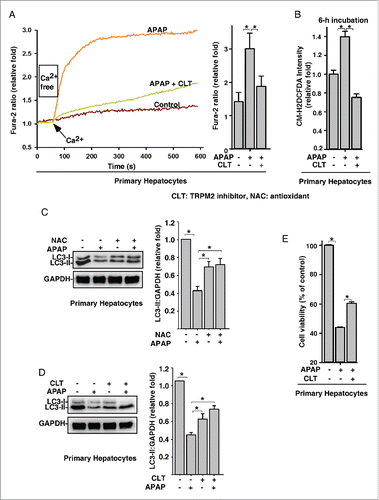
Oxidative stress is a major cause of APAP overdose-induced hepatotoxicity, which is the leading cause of acute liver damage worldwide. Since autophagy inhibition exacerbates APAP-induced hepatotoxicity,Citation19 we first assessed whether APAP overdose regulates autophagy in primary mouse hepatocytes. While LC3-II levels were increased by 2.5 mM or 5 mM APAP, higher concentrations of APAP (≥15 mM) markedly inhibited LC3-II levels in hepatocytes (Fig. S1), suggesting that APAP overdose actually inhibits autophagy. Since TRPM2 is the first known oxidative stress-activated Ca2+ channel and is expressed in liver,Citation20 we next examined the role of TRPM2-mediated Ca2+ influx in APAP overdose-induced autophagy inhibition in hepatocytes. Similar to the effects of H2O2 in primary mouse hepatocytes (Fig. S1B and S1C), APAP treatment significantly increased intracellular Ca2+ levels in hepatocytes, and this increase was abolished by pretreatment with the TRPM2 antagonist, clotrimazole (CLT) (). Not surprisingly, APAP treatment also significantly increased intracellular ROS in mouse hepatocytes (Fig. S1D). This APAP-induced ROS increase was compatible to that induced by exogenous H2O2 treatment (Fig. S1E), and was abolished by pretreatment with CLT (). Likewise, APAP overdose-induced LC3-II decrease was abolished by pretreatment with the antioxidant N-acetylcysteine (NAC) (), and CLT (). Moreover, APAP overdose markedly decreased the viability of hepatocytes (Fig. S1F), whereas pretreatment with CLT significantly alleviated APAP overdose-induced hepatotoxicity (). Collectively, these data suggest that APAP overdose induces oxidative stress to trigger Ca2+ influx via TRPM2, which subsequently inhibits autophagy and induces hepatotoxicity.
Figure 1. Acetaminophen overdose-induced oxidative stress triggers Ca2+ influx via TRPM2 to regulate autophagy in primary hepatocytes. (A) APAP (15 mM) treatment markedly increased the intracellular Ca2+ in mouse hepatocytes, but this effect was abolished by pretreatment with CLT (10 μM). Quantification of intracellular Ca2+ peak values are expressed as mean ± SD, n = 3 (15–20 cells in each independent experiment). *, P < 0.05. (B) APAP (15 mM) treatment significantly increased the levels of intracellular ROS in mouse hepatocytes, and this increase in ROS was abolished by pretreatment with CLT. Quantification of intracellular ROS/control group are expressed as mean ± SD, n = 3. *, P < 0.05. (C) and (D) APAP (15 mM) decreased LC3-II levels, and this effect of APAP was abolished by pretreatment with NAC (15 mM) (C) or CLT (10 μM) (D). Data quantification was expressed as mean ± SD, n = 3. *, P < 0.05. (E) CLT (10 μM) markedly reversed the toxicity of APAP (15 mM) on hepatocytes.
To confirm the role of TRPM2-mediated Ca2+ entry in oxidative stress-mediated autophagy inhibition, we infected HeLa cells, which do not express functional TRPM2, with lentiviruses expressing Flag-tagged full-length human TRPM2 (). As expected, H2O2 induced a significant increase in intracellular Ca2+ in TRPM2-expressing HeLa cells (Fig. S2B) when compared with control HeLa cells (Fig. S2A). This intracellular Ca2+ increase was abolished by pretreatment with the TRPM2 antagonists CLT or 8-Br-ADPR (), or with EGTA (Fig. S2C). Conversely, overexpressing TRPM2P1018L (an inactive TRPM2 variant identified during the high incidence of amyotrophic lateral sclerosis and parkinsonism-dementia complex in Guam),Citation21 failed to augment H2O2-triggered Ca2+ influx (Fig. S2D). These data indicate that oxidative stress can activate TRPM2 to trigger Ca2+ influx in HeLa cells. In addition, H2O2 markedly decreased cell viability in TRPM2-expressing HeLa cells but not in the nonexpressing controls, and this H2O2-induced cell death was abolished by pretreatment with CLT (). We also infected control and TRPM2-expressing HeLa cells with lentiviruses carrying expression cassettes that encode tandem fluorescence-tagged MAP1LC3-II.Citation22 LC3-II-positive autophagosomes are labeled with both GFP and RFP and thus show up as yellow puncta; then following fusion with lysosomes, the autolysosomes show up as red puncta because GFP loses its fluorescence in the acidic pH. Consistently, H2O2 (75 μM) induced the yellow and red LC3-II puncta in control HeLa cells but not in the TRPM2-overexpressing cells (). Western blotting also confirmed that H2O2 (75 μM) increased LC3-II levels in control HeLa cells, whereas it markedly decreased LC3-II levels in TRPM2-expressing cells (); this decrease was abolished by pretreatment with the TRPM2 antagonist CLT (Fig. S2E), or with CAT (catalase) (Fig. S2F). Again, these data suggest that oxidative stress activates TRPM2-Ca2+ signaling to inhibit autophagy.
Figure 2. Oxidative stress triggers Ca2+ influx via TRPM2 to inhibit autophagy in HeLa cells. (A) Western blot and immunofluorescence image showing the expression of Flag-TRPM2 in HeLa cells that stably express Flag-TRPM2. Scale bar: 5 μm. (B) In TRPM2-expressing HeLa cells, the intracellular Ca2+ increases induced by H2O2 (75 μM) were blocked by CLT (10 μM) or 8-Br-ADPR (200 μM). Quantification of intracellular Ca2+ peak values are expressed as mean ± SD, n = 4 (60–80 cells for each independent experiment). *, P < 0.05. (C) H2O2 (200 μM) markedly decreased cell viability in TRPM2-expressing but not control HeLa cells, and this effect was reversed by pretreatment with CLT (10 μM), as determined by MTT assay. (D) H2O2 (75 μM) induced both yellow and red-only LC3 puncta in control, but not TRPM2-expressing, tandem fluorescence-tagged LC3 HeLa cells. (E) H2O2 (75 μM) induced LC3-II levels in control HeLa cells, but decreased LC3-II in TRPM2-expressing HeLa cells. (F) and (G) In the presence and absence of H2O2 (75 μM), bafilomycin A1 (BAF) (10 nM) markedly increased LC3-II levels (F) and yellow RFP-GFP-LC3-II puncta (G) in control HeLa cells, whereas H2O2 (75 μM) markedly decreased BAF-induced LC3-II levels (F) and yellow RFP-GFP-LC3-II puncta (G) in TRPM2-expressing HeLa cells. Quantification of yellow or red-only puncta/cell in (D) and (G) is expressed as mean ± SE, n = 3 (total of 40–50 cells), *, P < 0.05. Scale bar: 5 μm. (H) Pepstatin (10 μM) markedly increased LC3-II levels in control HeLa cells, whereas H2O2 (75 μM) markedly decreased pepstatin-induced LC3-II levels in TRPM2-expressing HeLa cells.
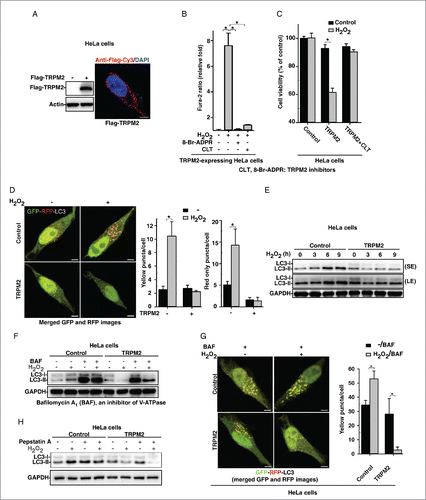
To further clarify the role of TRPM2-Ca2+ signaling in autophagy regulation, we treated cells with H2O2 in the presence or absence of bafilomycin A1 (BAF), an inhibitor of the vacuolar proton pump that blocks the fusion of autophagosomes with lysosomes.Citation23 As expected, BAF arrested autophagic flux as shown by the increase in LC3-II levels and the accumulation of yellow LC3-II puncta in both control and TRPM2-expressing HeLa cells (). Strikingly, in TRPM2-expressing HeLa cells, H2O2 treatment markedly decreased LC3-II levels and LC3-II yellow puncta induced by BAF (). Likewise, treatment of cells with pepstatin A, a potent lysosomal protease inhibitor, prevented the degradation of LC3-II, and H2O2 treatment in TRPM2-expressing HeLa cells markedly decreased pepstatin-induced LC3-II increases (). These data indicate that oxidative stress indeed triggers Ca2+ influx via TRPM2 to inhibit the induction of autophagy. Similarly, H2O2 markedly induced the conjugation of ATG12–ATG5 (Fig. S2G) in control but not in TRPM2-expressing HeLa cells. Imaging HeLa cells via transmission electron microscopy showed that large numbers of autophagic vacuoles were formed in H2O2-treated control cells but not in TRPM2-expressing cells (Fig. S2H). Likewise, hypoxia, another oxidative stress inducer, which activates autophagy in various other cell types,Citation24 activated autophagy in control HeLa cells but markedly inhibited autophagy in TRPM2-expressing HeLa cells (Fig. S2I and S2J). Taken together, it is clear that oxidative stress triggers Ca2+ influx via TRPM2 to inhibit early autophagy induction.
CALM/calmodulin and CAMK2 are required for ROS-TRPM2-Ca2+-mediated autophagy inhibition
Constitutive ITPR/InsP3R (inositol 1,4,5-trisphosphate receptor)-mediated Ca2+ release into mitochondria suppresses autophagy.Citation25 Interestingly, H2O2 treatment also markedly increased the Ca2+ content in the mitochondria in TRPM2-expressing but not control HeLa cells (Fig. S3A). However, treatment of cells with Ru360, a specific mitochondrial Ca2+ uptake inhibitor,Citation26 or xestospongin C, an inositol trisphosphate pathway-specific blocker, to prevent the mitochondrial Ca2+ overload, failed to affect the H2O2-mediated inhibition of autophagy in TRPM2-expressing HeLa cells, although Ru360 or xestospongin C alone did increase basal LC3-II levels and LC3-II puncta in both control and TRPM2-expressing HeLa cells (Fig. S3B–S3D, and data not shown), which was similar to the previous report.Citation26 Accordingly, these data demonstrated that mitochondrial Ca2+ overload is not responsible for ROS-TRPM2-Ca2+-mediated inhibition of autophagy.
It has also been previously shown that CAPN/calpain, a Ca2+-dependent cysteine protease, can cleave ATG5 and BECN1 to inhibit autophagy, and thus promote apoptosis.Citation27-29 Indeed, treatment of cells with ALLM or MDL, 2 potent CAPN inhibitors, increased basal autophagy in both control and TRPM2-expressing HeLa cells. However, neither of these inhibitors affected the H2O2-mediated decrease of LC3-II levels or LC3 puncta in TRPM2-expressing cells (Fig. S4A–S4D). These data indicate that CAPN is also not involved in the ROS-TRPM2-Ca2+-mediated inhibition of autophagy.
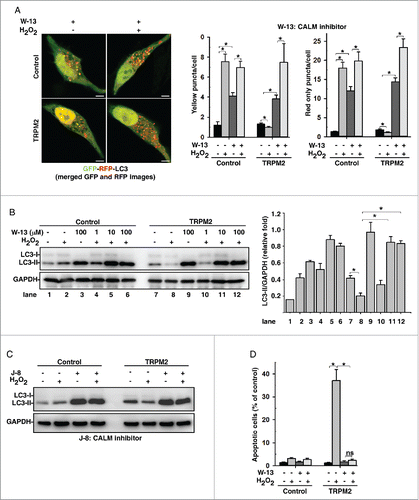
Other than CAPN, CALM/calmodulin, which contains 4 “EF” hand motifs for Ca2+ binding, is one of the most ubiquitous Ca2+-sensing proteins. The Ca2+-CALM complex interacts with a large number of proteins to regulate their function.Citation30 Not surprisingly, treatment of cells with a CALM inhibitor, W-13, completely abolished the inhibitory effects of H2O2 on autophagy in TRPM2-expressing HeLa cells, but did not affect H2O2-induced autophagy in control HeLa cells (). Similar results have been observed in cells treated with 2 other CALM inhibitors, J-8 hydrochloride () and W-7 (Fig. S5A). Likewise, W-13 treatment also completely abolished H2O2-induced cell death in TRPM2-expressing HeLa cells (). Obviously, CALM acts downstream of oxidative stress and TRPM2-Ca2+ cascade to inhibit autophagy and promote cell death.
Figure 3. CALM/calmodulin is required for ROS-TRPM2-Ca2+-mediated autophagy inhibition. (A) and (B) Treatment with W-13 (100 μM), a CALM inhibitor, rescued the H2O2 (75 μM)-mediated decrease in LC3-II (B) and induced the formation of RFP-GFP-LC3-II puncta (A) in TRPM2-expressing HeLa cells. Quantification of both yellow and red-only puncta/cell are expressed as mean ± SD, n = 3 (total of 40–50 cells), *, P < 0.05. Scale bar: 5 μm. (C) Treatment with J-8 hydrochloride (100 μM), a CALM inhibitor, rescued the H2O2 (75 μM)-mediated decrease in LC3-II in TRPM2-expressing HeLa cells. (D) Treatment of cells with W-13 (100 μM), a CALM inhibitor, abolished H2O2-induced apoptosis in TRPM2-expressing HeLa cells. Quantification of TUNEL assay is presented as TUNEL-positive cells: DAPI-stained cells ± SD, n = 3 (100–150 cells per experiment), *, P < 0.05.
One of the important targets of the Ca2+-CALM complex is PPP3/calcineurin, a Ca2+-activated serine/threonine protein phosphatase. PPP3 plays a key role in relaying both local and global Ca2+ signals to effectors that regulate both the immediate cellular responses as well as the somewhat slower gene transcription, e.g. TFEB (transcription factor EB), an autophagy regulator.Citation31 However, application of the PPP3 inhibitors FK506 or cyclosporine A failed to change the inhibitory effects of H2O2 on autophagy in TRPM2-expressing HeLa cells (Fig. S5B and S5C), although these 2 drugs efficiently blocked starvation-induced TFEB nuclear translocation (Fig. S5D). These data suggest that PPP3 is not involved in the ROS-TRPM2-Ca2+-mediated inhibition of autophagy.
In addition to PPP3, CAMK2 is another key Ca2+-CALM effector, which is a ubiquitously expressed serine/threonine protein kinase that transduces both local and global Ca2+ signals to a number of target proteins, e.g., ion channels and transcription factors.Citation32 Interestingly, treatment of cells with KN-93, a CAMK2 inhibitor, blocked the effects of H2O2-mediated decreases of LC3-II level () and LC3-II puncta (Fig. S6A) in TRPM2-expressing cells. Likewise, KN-93 treatment markedly inhibited H2O2-induced cell death in TRPM2-expressing cells ( and S6B), suggesting that CAMK2 is the mediator of ROS-TRPM2-Ca2+-induced inhibition of autophagy. Indeed, overexpression of a dominant-negative mutant of CAMK2, CAMK2AK42R, blocked H2O2-induced autophagy inhibition (). Moreover, knockdown of CAMK2G (Fig. S6C) abolished H2O2-induced autophagy inhibition in TRPM2-expressing HeLa cells () as well as cell death (). Therefore, these data clearly establish CAMK2 as the true effector of the TRPM2-Ca2+-CALM cascade to inhibit autophagy in response to oxidative stress.
Figure 4. CAMK2 is required for ROS-TRPM2-Ca2+-mediated autophagy inhibition. (A) Treatment with KN-93, a CAMK2 inhibitor, rescued the H2O2-mediated decrease in LC3-II in TRPM2-expressing HeLa cells. (B) Treatment of cells with KN-93 (80 μM) significantly inhibited H2O2-induced cytotoxicity in TRPM2-expressing HeLa cells. (C) and (D) Expression of a dominant-negative form of CAMK2A, CAMK2K42R, rescued the H2O2 (75 μM)-mediated decrease in LC3-II (C) and induced the formation of RFP-LC3-II puncta (D) in TRPM2 expressing HeLa cells. Quantification of red puncta/cell is expressed as mean ± SD, n = 3 (total 30–40 cells), *P < 0.05. Scale bar: 5 μm. (E) and (F) CAMK2G knockdown rescued the H2O2 (75 μM)-mediated decrease in LC3-II (F) and significantly alleviated H2O2-induced cytotoxicity in TRPM2-expressing HeLa cells (G).]
![Figure 4. CAMK2 is required for ROS-TRPM2-Ca2+-mediated autophagy inhibition. (A) Treatment with KN-93, a CAMK2 inhibitor, rescued the H2O2-mediated decrease in LC3-II in TRPM2-expressing HeLa cells. (B) Treatment of cells with KN-93 (80 μM) significantly inhibited H2O2-induced cytotoxicity in TRPM2-expressing HeLa cells. (C) and (D) Expression of a dominant-negative form of CAMK2A, CAMK2K42R, rescued the H2O2 (75 μM)-mediated decrease in LC3-II (C) and induced the formation of RFP-LC3-II puncta (D) in TRPM2 expressing HeLa cells. Quantification of red puncta/cell is expressed as mean ± SD, n = 3 (total 30–40 cells), *P < 0.05. Scale bar: 5 μm. (E) and (F) CAMK2G knockdown rescued the H2O2 (75 μM)-mediated decrease in LC3-II (F) and significantly alleviated H2O2-induced cytotoxicity in TRPM2-expressing HeLa cells (G).]](/cms/asset/1fcd9723-3b90-4382-ad88-f9b0cb284780/kaup_a_1187365_f0004_c.gif)
Oxidative stress activates CAMK2 to phosphorylate BECN1, thereby inhibiting autophagy and promoting cell death
Upon activation by Ca2+-CALM binding, CAMK2 undergoes an immediate autophosphorylation on Thr286 or Thr287 (depending on the isoform). In TRPM2-expressing cells, H2O2 treatment did transiently activate CAMK2 as shown by the immunoblot analysis with an anti-phospho-CAMK2-Thr286/287 antibody (). Unexpectedly, overexpression of autonomous CAMK2AT286D mutant alone failed to inhibit autophagy (Fig. S7A), suggesting that as well as T286 phosphorylation, an additional mechanism is required for its ability to inhibit autophagy. Along this line, CAMK2 is oxidized on 2 adjacent methionine residues (or on a cysteine-methionine pair in CAMK2A) in the regulatory domain.Citation33 Indeed, H2O2 only induced the oxidation of CAMK2 in TRPM2-expressing HeLa cells but not in control HeLa cells as shown by the immunostaining analysis with an anti-oxidized CAMK2-(C) M281/282 antibody (, S7B, and S7C). Notably, strong staining signals were found in the nucleus of all cells (, S7B, and S7C), which are likely nonspecific since CAMK2 is a cytosolic protein. Moreover, overexpression of a CAMK2AC281V,M282V oxidation-defective mutant blocked the H2O2-induced LC3-II level decrease in TRPM2-expressing HeLa cells (). Collectively, these data indicate that oxidative stress triggers Ca2+ influx via TRPM2 to activate CAMK2 both at the level of phosphorylation and oxidation, which subsequently inhibits autophagy and renders cells more susceptible to death.
Figure 5. Oxidative stress activates CAMK2 to inhibit autophagy in TRPM2-expressing HeLa cells. (A) H2O2 (75 μM) increased the T286 autophosphorylation of CAMK2 in TRPM2-expressing, but not control, HeLa cells. Data quantification was expressed as mean ± SD, n = 3. *, P < 0.05. (B) H2O2 (75 μM) treatment markedly increased the oxidation of CAMK2 in TRPM2-expressing HeLa cells as shown by the immunofluorescent staining analysis with an oxidative-specific antibody against CAMK2AM281V,M282V. (C) Expression of an oxidation-defective mutant of CAMK2, CAMK2AC281V,M282V, rescued the H2O2 (75 μM)-mediated decrease in LC3-II. (D) H2O2 (75 μM) treatment markedly induced the colocalization of GFP-CAMK2 with HSPA5/GRP78, an ER marker in saponin-permeabilized TRPM2-expressing HeLa cells. Scale bar: 5 μm.
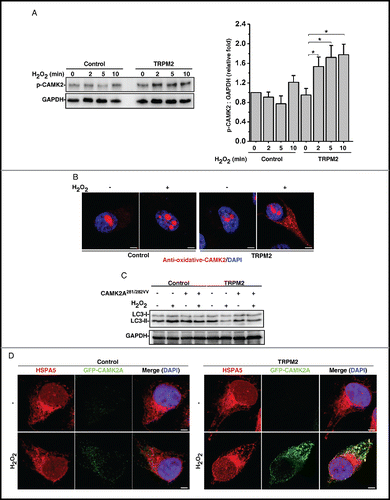
We next examined whether oxidative stress might induce the localization change of CAMK2. GFP-CAMK2A-expressing HeLa cells were treated with H2O2 followed by saponin to remove the majority of soluble cytosolic GFP-CAMK2A. Strikingly, H2O2 markedly induced the GFP-CAMK2A puncta, some of which were colocalized with HSPA5/GRP78 (heat shock protein family A [Hsp70] member 5), an ER marker, only in TRPM2-expressing HeLa cells (). Notably, the induced GFP-CAMK2A puncta were not colocalized with Golgi or lysosomal markers (Fig. S7D and S7E). This suggests that CAMK2 might regulate some ER-associated proteins to modulate autophagy.
CAMK2 can phosphorylate many proteins in vitro, and its conserved phosphorylation motif has been identified as either HydXRXXS/T or HydXRNBXS/T, where Hyd, X and NB represent a hydrophobic, any, and a non-basic amino acid residue, respectively.Citation34 We, therefore, performed a bioinformatics search to determine if any proteins involved in the regulation of autophagy contain this motif. Interestingly, we found that BECN1 contains this motif and Ser295 is a potential phosphorylation site, which is also evolutionarily conserved (). We speculate that ER-localized active CAMK2 induced by the ROS-TRPM2 cascade can directly phosphorylate BECN1 in the ER. An in vitro CAMK2 “cold” kinase assay was thus performed by incubating GFP-CAMK2 (immuno-purified from TRPM2-expressing HeLa cells treated with or without H2O2) with BECN1 or BECN1S295A (immuno-purified from HeLa cells), followed by immunoblot analysis with an anti-phospho-BECN1 (S295) antibody. As expected, activated CAMK2 directly phosphorylated wild-type BECN1, but not BECN1S295A, in vitro (). Moreover, immunostaining and immunoblot analysis using this anti-phospho-BECN1 (S295) antibody confirmed that Ser295 in BECN1 is phosphorylated upon H2O2 treatment in TRPM2-expressing cells (). Notably, Ser295-phosphorylated BECN1 was localized in the ER in TRPM2-expressing cells, as shown by its colocalization with HSPA5 (). Not surprisingly, H2O2 markedly induced the binding between CAMK2 and BECN1 in TRPM2-expressing cells (), but not in control HeLa cells (Fig. S8A). Consistently, the expression of BECN1S295A, which eliminates the potential phosphorylation, blocked the H2O2-induced LC3-II decrease in TRPM2-expressing HeLa cells (). Conversely, expression of BECN1S295D, which mimics the phosphorylation, rendered control HeLa cells insensitive to H2O2-induced LC3-II increases, but had no effect on the H2O2-induced LC3-II decrease in TRPM2-expressing HeLa cells (). Likewise, the expression of BECN1S295A markedly inhibited H2O2-induced cell death in TRPM2-expressing cells (Fig. S8B). Collectively, these data indicate that CAMK2 phosphorylates Ser295 in BECN1 to inhibit autophagy under oxidative stress.
Figure 6. Oxidative stress activates CAMK2 to phosphorylate BECN1 resulting in autophagy inhibition. (A) BECN1 contains a highly conserved CAMK2 consensus phosphorylation site, Ser295. (B) GFP-CAMK2A, immuno-purified from TRPM2-expressing HeLa cells treated with H2O2, directly phosphorylated BECN1, not BECN1S295A, in vitro. (C) H2O2 (75 μM) induced the phosphorylation of BECN1 on Ser295 as shown by the immunoblot analysis using a phospho-specific antibody against Ser295 of BECN1. (D) Ser295 in BECN1 was phosphorylated upon H2O2 (75 μM) treatment in TRPM2-expressing cells as shown by the immunofluorescent staining analysis with a phospho-specific antibody against BECN1 (Ser295), which was also colocalized with HSPA5. Scale bar: 5 μm. (E) H2O2 (75 μM) induced the binding between BECN1 and CAMK2 in TRPM2-expressing HeLa cells. (F) and (G) Expression of the BECN1S295A mutant rescued the H2O2 (75 μM)-mediated decrease in LC3-II in TRPM2-expressing HeLa cells (F), while expression of the BECN1S295D mutant inhibited the H2O2-induced increase in LC3-II in control HeLa cells (G). (H) and (I) H2O2 (75 μM) inhibited binding between BECN1 and PIK3C3 (G) but induced binding between BECN1 and BCL2 (H) in TRPM2-expressing HeLa cells.
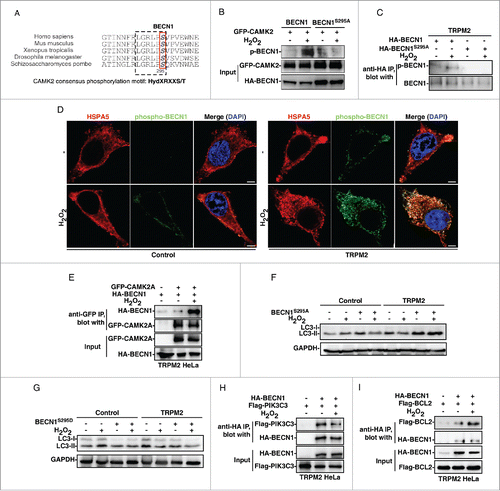
The binding of BECN1 with the lipid kinase PIK3C3 (the mammalian ortholog of yeast VPS34), is essential for the formation of BECN1-PIK3C3-PIK3R4/VPS15 core complexes, thereby inducing autophagy.Citation35 Indeed, H2O2 treatment only decreased the interaction of BECN1 with PIK3C3 in TRPM2-expressing cells (), but not in control cells (Fig. S8C). On the one hand, these data suggest that phosphorylated BECN1, induced by the ROS-TRPM2-CAMK2 cascade, dissociates from and inactivates PIK3C3, thereby inhibiting autophagy. On the other hand, BECN1 can bind to BCL2 or BCL2L1/BCL-XL (BCL2 like 1), which leads to the disassociation of BAX (BCL2-associated X protein) from BCL2, and thus induces apoptosis. Citation35 Indeed, H2O2 treatment markedly increased the binding between BECN1 and BCL2 in TRPM2-expressing HeLa cells (), but not in control HeLa cells (Fig. S8D). Obviously, these data suggest that the increase of association between phosphorylated BECN1 and BCL2 is, at least partially, responsible for the observed cell death induced by H2O2 in TRPM2-expressing cells. Taken together, it is clear that the ROS-TRPM2-CAMK2 cascade phosphorylates BECN1 to inhibit autophagy, which subsequently renders cells more susceptible to death.
The pleckstrin homology domain containing protein AKT also recognizes the RXXS/T motif and thus might potentially phosphorylate Ser295 of BECN1 to inhibit autophagy.Citation36 Interestingly, AKT was markedly activated by H2O2 in TRPM2-expressing HeLa cells but not in control HeLa cells (Fig. S8E). Surprisingly, treatment of cells with a potent AKT inhibitor, MK-2206, completely inactivated AKT but failed to affect the inhibitory effects of H2O2 on autophagy in TRPM2-expressing HeLa cells (Fig. S8F). Notably, MK-2206 treatment alone markedly increased the basal LC3-II level, which is likely due to the inactivation of the AKT-MTOR cascade, but this MK-2206-induced LC3-II increase was actually abolished by H2O2 treatment in TRPM2-expressing HeLa cells but not in control HeLa cells (Fig. S8F). Thus, it is clear that AKT is not involved in ROS-TRPM2-Ca2+-induced inhibition of autophagy.
Oxidative stress can activate MAPK8/JNK1 (mitogen-activated protein kinase 8), and MAPK8 activation induces autophagyCitation37 We also found that H2O2 activated MAPK8 in both control and TRPM2-expressing HeLa cells, but treatment of cells with SP600125, a potent MAPK8 inhibitor, only blocked H2O2-induced LC3-II increase in control HeLa cells, but had no effects on H2O2-mediated LC3-II decrease in TRPM2-expressing HeLa cells (Fig. S8G). These data suggest that MAPK8 is not involved in TRPM2-Ca2+-mediated autophagy inhibition in response to oxidative stress.
ROS-TRPM2-Ca2+-CAMK2-mediated autophagy inhibition is involved in acetaminophen-induced hepatotoxicity
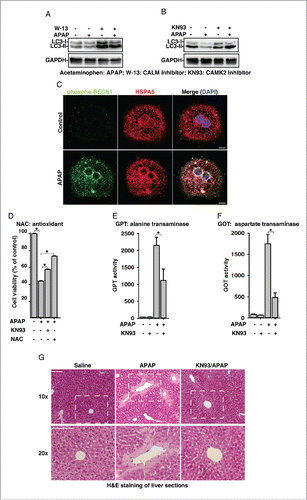
We next assessed whether APAP overdose also activates the TRPM2-CAMK2-BECN1 signaling to inhibit autophagy and thus cause hepatotoxicity. As expected, treatment of mouse primary hepatocytes with W-13 (a CALM inhibitor) or KN-93 (a CAMK2 inhibitor) abolished the APAP overdose-induced decreases of LC3-II levels ( and ). Moreover, APAP treatment markedly induced the S295 phosphorylation of BECN1 in hepatocytes, and this phosphorylated BECN1 was also associated with the ER as indicated by its colocalization with HSPA5/GRP78 (). Likewise, pretreatment with NAC or KN-93 significantly reversed the toxicity of APAP on hepatocytes (), which is similar to the effects of CLT, a TRPM2 inhibitor (). Finally, we examined whether inhibiting TRPM2-CAMK2 signaling alleviates APAP overdose-induced liver damage in vivo. C57BL/6 mice were administered with a single dose of APAP (500 mg/kg body weight) in the presence or absence of KN-93. Twenty-four h after drug treatment, APAP treatment alone markedly induced GPT/ALT (glutamic-pyruvate transaminase [alanine aminotransferase]) and GOT/AST (glutamic-oxaloacetic transaminase) levels in mice, and these increases were significantly reduced by the treatment with KN-93 (). Consistently, APAP treatment induced extensive cell death in mice liver as shown by H&E staining of liver sections, and KN-93 treatment markedly alleviated this liver injury ( and S9). Collectively, these data demonstrate that APAP overdose activates the ROS-TRPM2-Ca2+-CAMK2-BECN1 cascade to inhibit autophagy, which is at least partially responsible for the induced liver damage.
Figure 7. Acetaminophen (APAP) activates the ROS-TRPM2-CAMK2 signaling cascade and thus inhibits autophagy and induces cell death in primary mouse hepatocytes. (A) and (B) APAP (15 mM) decreased LC3-II levels, and this effect of APAP was abolished by pretreatment with W-13 (10 μM) (A) or KN-93 (10 μM) (B) in mouse hepatocytes. (C) APAP (15 mM) treatment markedly induced the phosphorylation of Ser295 on BECN1 in hepatocytes, showing the colocalization with PIK3C3. Scale bar: 10 μm. (D) KN-93 (10 μM) or NAC (15 mM) markedly reversed the toxicity of APAP (15 mM) on hepatocytes. (E) and (F) KN-93 (4 mg/kg) treatment significantly inhibited APAP (500 mg/kg)-induced serum GPT (E) and GOT (F) levels in mouse. (G) KN-93 (4 mg/kg) treatment markedly alleviated APAP (500 mg/kg)-induced liver damage as shown by H&E staining of liver sections 24 h after drug treatment. Scale bar: 100 μm.
Discussion
Here, we mechanistically studied how oxidative stress regulates autophagy. We demonstrated that oxidative stress activates a TRPM2-induced Ca2+ influx to inhibit autophagy and thus cause cells to become more susceptible to damage. Conversely, in the absence of TRPM2-mediated Ca2+ influx, oxidative stress actually induced autophagy to render cells refractory to death (). These data clearly establish TRPM2 as one of the molecular entities determining cell autophagic response to oxidative stress. We further demonstrated that oxidative stress triggers TRPM2-mediated Ca2+ influx to activate CAMK2 by a combination of phosphorylation and oxidation (). The activated form of CAMK2 then phosphorylates BECN1 on Ser295. Phosphorylated BECN1 subsequently dissociates from PIK3C3 to bind with BCL2, and thereby inhibits autophagy (). Collectively, our data uncover a novel mechanism underlying oxidative stress-mediated autophagy inhibition; that is, oxidative stress activates the TRPM2-Ca2+-CAMK2 cascade to phosphorylate BECN1, thereby inhibiting autophagy and promoting cell death ().
Figure 8. Model of the role of the TRPM2-Ca2+-CAMK2-BECN1 cascade in oxidative stress-mediated autophagy inhibition.
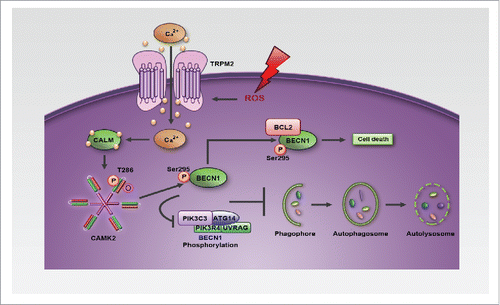
Oxidative stress has been implicated in numerous human diseases yet physiological levels of ROS are essential for various normal cellular processes. Not surprisingly, simple application of antioxidants for treatment or prevention of these pathologies normally fails or generates mixed results.Citation38 Therefore, dissecting the molecular mechanisms underlying oxidative stress-induced pathologies is essential to develop effective therapies for treating these diseases. Here we demonstrated that oxidative stress generated by APAP overdose activates the TRPM2-Ca2+-CAMK2-BECN1 cascade to inhibit autophagy and thus promote hepatotoxicity (). Moreover, we found that inhibiting this cascade markedly alleviated APAP overdose-induced liver damage (). Notably, APAP overdose is the leading cause of acute liver failure worldwide. In addition to the liver, other organs, such as the heart, brain, and kidney, are also susceptible to oxidative stress-induced tissue damage,Citation39 and TRPM2 is widely expressed in those organs.Citation14 It is, therefore, of interest to assess whether oxidative stress generated in these pathologies, e.g. myocardial infarction or stroke, also activates the TRPM2-Ca2+-CAMK2-BECN1 cascade to inhibit autophagy, thus leading to tissue damage in these organs. If so, then developing specific inhibitors, which target the TRPM2-Ca2+-CAMK2-BECN1 cascade, should offer better treatment regimens for these human pathologies.
BECN1, originally identified as a BCL2 binding protein, is a multifunctional protein, which is especially important in the regulation of autophagy and apoptosis.Citation40 Here, we found that CAMK2 phosphorylated BECN1 on Ser295 () to inhibit autophagy. When BECN1 is phosphorylated on Ser295, it dissociates from PIK3C3/VPS34 (), and thus presumably inhibits PIK3C3 activity to arrest autophagy. In contrast, Ser295-phosphorylated BECN1 associates with BCL2 (), likely resulting in more free BAX proteins, which might account for the enhanced cell death. Notably, Ser295 is located in the evolutionarily conserved domain (ECD), which is responsible for interaction with PIK3C3. We speculate that the negative charge in the ECD might disrupt the interaction of BECN1 with PIK3C3, allowing more BECN1 binding with BCL2. Interestingly, STK4/MST1 (serine/threonine kinase 4) induces the phosphorylation of Thr108, which is located in the BCL2 homology domain (BH3) of BECN1, and this phosphorylated Thr108 interacts with a positively charged His113 on the α3 helix of BCL2L1 at the entrance of the hydrophobic groove. This interaction increases the binding affinity between BECN1 and BCL2, thus leaving less BECN1 for PIK3C3 and resulting in autophagy inhibition.Citation41 Intriguingly, DAPK (death-associated protein kinase) phosphorylates BECN1 on Thr119, which is also located at the BH3 domain, and this promotes the dissociation of BECN1 from BCL2L1 and thus induces autophagy.Citation42 Considering the central role of BECN1 in autophagy and cell death, it is expected that other novel phosphorylation sites or posttranslational modifications might be identified in the near future.
CAMK2, encoded by 4 genes (CAMK2A, CAMK2B, CAMK2D, and CAMK2G), is an essential signaling molecule in a variety of tissues. An increase in cytosolic Ca2+ induces the binding Ca2+-CALM to the regulatory domain of CAMK2, which keeps this domain away from the kinase domain. As a result the enzyme is activated and this results in the intersubunit autophosphorylation on T286 of the regulatory domain. Phosphorylation of T286 further prevents the re-association of the kinase domain with the regulatory region to create a Ca2+-autonomous activity.Citation43 Interestingly, oxidation of a pair of regulatory domain cysteine-methionine residues sustains the CAMK2 activity even in the absence of Ca2+-CALM.Citation33 Here, we demonstrated that oxidative stress actually induces both the phosphorylation () and oxidation of CAMK2, and the oxidation of CAMK2 is dependent on TRPM2-induced Ca2+ influx (, S7B and S7C). Moreover, oxidation of CAMK2 is required for oxidative stress-induced autophagy inhibition (). It remains to be determined whether CAMK2 in the both oxidized and phosphorylated form exhibits higher kinase activity than phosphorylated- or oxidized-only CAMK2. Alternatively, the oxidized methionine-cysteine pair in CAMK2 might create a binding motif for BECN1, thus allowing its phosphorylation by the catalytic domain of CAMK2. It is also possible that the oxidation of CAMK2 changes its subcellular localization to allow its colocalization with BECN1 in the ER (). Also notably, expression of BECN1S295A only partially rescues oxidative stress-induced cell death (Fig. S8B), suggesting that CAMK2 phosphorylates additional proteins besides BECN1 to regulate cell death. Thus, the search for these CAMK2 effectors in cell death regulation by oxidative stress must continue.
Materials and methods
Cell culture
HeLa (ATCC, CCL-2) and HEK 293T (ATCC, CRL-11268) cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen Life Technologies, 12800017) with 10% fetal bovine serum (Invitrogen Life Technologies, 16000-044) and 1% penicillin-streptomycin (Invitrogen Life Technologies, 15140122). Under normal conditions, all cells were maintained at 37°C with 5% CO2 and 95% humidity, and they were passaged every 2 d.
Animal studies
C57BL/6 male mice were used for this study. Mice were divided into 4 groups and treated with the indicated drugs through intraperitoneal injection: saline control; APAP (500 mg/kg in saline; Sigma-Aldrich, A7085); KN-93 (4 mg/kg; Santa Cruz Biotechnology, sc-202199); and APAP (500 mg/kg in saline) 1 h after injection of KN-93 (4 mg/kg) (n = 4–6 mice per treatment group). Mice were killed after 24 h. The animal experimental procedures were approved by the Animal Committee of City University of Hong Kong and were carried out according to the NIH Guide for the Care and Use of Laboratory Animals.
Isolation and culture of primary mouse hepatocytes
Primary hepatocytes were isolated from 6-wk-old C57BL/6 male mice (17–20 g), as previously described.Citation44 Briefly, the portal vine and inferior vena cava were cannulated and the liver was perfused in situ with MMP2/collagenase IV (Invitrogen Life Technologies, 17104–019) at 37°C for 10 min. The liver was then minced and suspended in HBSS (Invitrogen Life Technologies, 14025092), after which the suspension was filtered using Falcon cell strainers (100 μm; Becton Dickinson Biosciences, 352360) and centrifuged at 50 × g for 5 min. Hepatocytes were subsequently plated in culture dishes pre-coated with COL1A/collagen I (Invitrogen Life Technologies, A1048301). Cells were cultured in William's medium E (Invitrogen Life Technologies, 32551–020) with 10% fetal bovine serum and 1% penicillin-streptomycin, and were maintained at 37°C with 5% CO2 and 95% humidity.
Antibodies and reagents
Flag-BCL2 (Addgene, 18003; Clark Distelhorst lab), Flag-PIK3C3 (Addgene, 24398; Qing Zhong lab), GFP-CAMK2A (Addgene, 21226; Tobias Meyer lab) and CAMK2AK42R (Addgene, 21221; Tobias Meyer lab) constructs were purchased from Addgene. The antibodies (and dilutions) used in the western blot analyses were as follows: anti-LC3 (1:1000; NB100-2220) was from Novus Biologicals; anti-GAPDH (1:3000; G8795) and anti-Flag (1:2000; F1804) were from Sigma-Aldrich; anti-ACTB/β-Actin (1:5000; MAB1501) was from Chemicon International; anti-BECN1 (1:1000; 3738), anti-PIK3C3 (1:1000; 4263) and anti-phospho-AKT (1:1000; 9271) were from Cell Signaling Technology; anti-HA (1:1000; sc805), anti-TRPM2 (1:500; sc-19198) and anti-phospho-CAMK2A (1:1000; sc-12886) were from Santa Cruz Biotechnology; anti-GFP (1:1000; 75–131) was from NIH NeuroMab; and anti-phospho-BECN1 (S295) (1:500; MBS502207) was from MyBioSource, Inc. The antibodies (and dilution or concentration) used for immunocytochemistry were as follows: anti-Flag-Cy3 (10 µg/ml; A9594) was from Sigma-Aldrich, anti-oxidized-CAMK2 (1:1000; 07–1387) was from Merck Millipore, anti-HSPA5 (1:500; sc-1050) was from Santa Cruz Biotechnology, anti-LAMP1 (1:200; L1418) and anti-GOGLA2/GM130 (1:500; 2296) were from Sigma-Aldrich. DAPI (1:3000; 62248), CM-H2DCFDA (C6827) and tetramethylrhodamine (TMRM; T-668) were purchased from Invitrogen Life Technologies. H2O2 (freshly prepared just prior to each experiment; 1072980250) was purchased from Merck Millipore. CAT (catalase; C1345), tacrolimus (FK506; tlrl-fk5), bafilomycin A1 (B1793), rapamycin (R0395), BAPTA-AM (16609), acetaminophen (A7085), and clotrimazole (CLT; C6019) were purchased from Sigma-Aldrich. W-13 (sc-222413), KN-93 (sc-202199), Ru360 (557440), ALLM (sc-201268), cyclosporin A (CsAsc-3503), and MDL28170 (sc-201301) were purchased from Santa Cruz Biotechnology.
Molecular cloning
A His6-Flag, RFP, or RFP-GFP was engineered into pENTR-GFP-C1 vector (Addgene, 17396; Eric Campeau lab) to generate pENTR-His-Flag-C1, pENTR-RFP-C1, or pENTR-RFP-GFP-C1 construct, respectively. To generate the pLenti-His6-Flag-TRPM2 construct, the full-length human TRPM2 gene was subcloned into a pENTR-His6-Flag-C1 vector between the XbaI and XhoI sites. The His-Flag-TRPM2 sequence was then recombined into the pLenti-CMV-puro-DEST vector using the LR reaction, according to the manufacturer's instructions. To generate the pLenti-GFP-tagged, RFP-tagged, or mRFP-GFP tandem tagged MAP1LC3 (tf-LC3) construct, MAP1LC3 was amplified and incorporated into pENTR-GFP-C1, pENTR-RFP-C1, or pENTR-RFP-GFP-C1 empty vectors, respectively. Then the GFP-LC3, RFP-LC3 or tf-LC3 was recombined into the pLenti-CMV-puro-DEST vector (Addgene, 17452; Eric Campeau lab).
Mutagenesis
TRPM2P1018L, BECN1S295A, BECN1S295D, and CAMK2AC281V,M282V were made by site-directed mutagenesis (Stratagene) as described previously. Citation45 The primers used are listed in Table S1.
shRNA and lentivirus production and infection
Two or 3 optimal 21-mers were selected in human CAMK2A, CAMK2G or TRPM2 (Table S1). One 21-mer was selected in the GFP nucleotide sequence as a control. These sequences were then cloned into the pLKO.1 vector (Addgene, 8453; Bob Weinberg lab) for expressing shRNA. The lentivirus production and infection were performed as described previously.Citation46 The shRNA knockdown efficiencies or specific gene expression were investigated by protein gel blot analysis or quantitative real-time RT-PCR (qRT-PCR) analysis.
Western blot analysis and immunoprecipitation
Western blot analysis and immunoprecipitation were performed as described previously.Citation45
Reverse transcription and quantitative real-time PCR
qRT-PCR was performed as described previously.Citation47 The primers for detecting CAMK2A, CAMK2G, GAPDH, and TRPM2 are listed in Table S1. The relative gene expression was normalized to the level of GAPDH expression.
Ca2+ measurements
The cytosolic Ca2+ measurements in HeLa cells, or mouse hepatocytes were performed as described previously.Citation48
For the mitochondrial Ca2+ measurements, HeLa cells were plated on 18-mm coverslips in 12-well plates and then transfected with a Mito-GEM-GECO1 plasmid, which expresses a mitochondria targeting Ca2+ indicator,Citation49 using lipofectamine 2000 (Invitrogen Life Technologies, 11668019) according to the manufacturer's instructions. After 4-h incubation, the medium was changed to regular culture medium and the cells were cultured for an additional 24 h. The coverslips were transferred to Ca2+ measurement chambers and images were obtained every 20 sec using a 40× oil objective lens by the LSM500 software.
Immunocytochemistry
Immunocytochemistry was performed as described previously.Citation50
Transmission electron microscopy
Cell preparation for transmission electron microscopy was performed as described previously.Citation51
Cell viability analysis
Cell viability was assessed using either the TUNEL assay or the MTT assay as described previously.Citation52
Intracellular ROS detection
The intracellular level of ROS in HeLa cells and hepatocytes was assessed with CM-H2DCFDA (Invitrogen Life Technologies, C6827), after which the fluorescence was recorded by flow cytometry or via a microplate reader, respectively. In brief, HeLa cells were collected, washed twice with DMEM, and incubated with 5 μM CM-H2DCFDA at 37°C for 30 min. The cells were then washed twice with DMEM and analyzed with the BD FACSAria I Cell Sorter to determine the fluorescence intensity at excitation and emission wavelengths of 488 nm and 530 nm, respectively.
In addition, hepatocytes were plated in a 96-well plate, and treated with different concentrations of acetaminophen in the presence or absence of different inhibitors for the indicated time. The cells were then washed with HBSS, and incubated with 10 μM CM-H2DCFDA at 37°C for 30 min. After incubation, the fluorescence intensity of the stained cells was detected with a Tecan Infinite M200 microplate reader.
Statistical analysis
Data were presented as mean ± SE. The statistical significance of differences was calculated by unpaired Student t test where P < 0.05 was considered to be significant.
Abbreviations
| ADPR | = | adenosine diphosphate ribose |
| APAP | = | acetaminophen |
| ATG | = | autophagy-related |
| BAF | = | bafilomycin A1 |
| BECN1 | = | Beclin 1, autophagy related |
| CALM | = | calmodulin (phosphorylase kinase, delta) |
| PPP | = | calcineurin |
| CAMK2 | = | calcium/calmodulin dependent protein kinase II |
| CAPN | = | calpain |
| CLT | = | clotrimazole |
| GOT/AST | = | glutamic-oxaloacetic transaminase |
| GPT/ALT | = | glutamic-pyruvate transaminase (alanine aminotransferase) |
| H2O2 | = | hydrogen peroxide |
| HSPA5/GRP78 | = | heat shock protein family A member 5 |
| MAPK8/JNK1 | = | mitogen-activated protein kinase 8 |
| MAP1LC3/LC3 | = | microtubule associated protein 1 light chain 3 |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| NAC | = | N-acetylcysteine |
| NAPQI | = | N-acetyl-p-benzoquinone imine |
| ROS | = | reactive oxygen species |
| TRPM2 | = | transient receptor potential cation channel subfamily M member 2 |
| RFP | = | red fluorescence protein |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1187365_Supplemental_Material.pdf
Download PDF (3.4 MB)Acknowledgments
We thank members of Yue lab for their advice on the preparation of this manuscript.
Funding
This work was supported by Hong Kong Research Grant Council (RGC) grants (782709M, 785911M, 769912M, 785213M, and 17126614M), ITS/261/14, and Guangdong-Hong Kong Joint Grant (#20160503) to JY, and 766911M to QH.
Related Research Data
References
- Li L, Ishdorj G, Gibson SB. Reactive oxygen species regulation of autophagy in cancer: implications for cancer treatment. Free Radic Biol Med 2012; 53:1399-410; PMID:22820461; http://dx.doi.org/10.1016/j.freeradbiomed.2012.07.011
- Thomas SH. Paracetamol (acetaminophen) poisoning. Pharmacology & therapeutics 1993; 60:91-120; PMID:8127925; http://dx.doi.org/10.1016/0163-7258(93)90023-7
- McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res 2013; 30:2174-87; PMID:23462933; http://dx.doi.org/10.1007/s11095-013-1007-6
- Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014; 15:81-94; PMID:24401948; http://dx.doi.org/10.1038/nrm3735
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/10.1038/ncb0910-814
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823-30; PMID:20811354; http://dx.doi.org/10.1038/ncb0910-823
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 2015; 22:377-88; PMID:25257172; http://dx.doi.org/10.1038/cdd.2014.150
- Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A 2010; 107:4153-8; PMID:20160076; http://dx.doi.org/10.1073/pnas.0913860107
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749-60; PMID:17347651; http://dx.doi.org/10.1038/sj.emboj.7601623
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 2011; 36:30-8; PMID:20728362; http://dx.doi.org/10.1016/j.tibs.2010.07.007
- Yu P, Wang Q, Zhang LH, Lee HC, Zhang L, Yue J. A cell permeable NPE caged ADP-ribose for studying TRPM2. PloS one 2012; 7:e51028; PMID:23236422; http://dx.doi.org/10.1371/journal.pone.0051028
- Magnone M, Bauer I, Poggi A, Mannino E, Sturla L, Brini M, Zocchi E, De Flora A, Nencioni A, Bruzzone S. NAD+ levels control Ca2+ store replenishment and mitogen-induced increase of cytosolic Ca2+ by Cyclic ADP-ribose-dependent TRPM2 channel gating in human T lymphocytes. J Biol Chem 2012; 287:21067-81; PMID:22547068; http://dx.doi.org/10.1074/jbc.M111.324269
- Sumoza-Toledo A, Penner R. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol 2011; 589:1515-25; PMID:21135052; http://dx.doi.org/10.1113/jphysiol.2010.201855
- Takahashi N, Kozai D, Kobayashi R, Ebert M, Mori Y. Roles of TRPM2 in oxidative stress. Cell Calcium 2011; 50:279-87; PMID:21616534; http://dx.doi.org/10.1016/j.ceca.2011.04.006
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 2002; 9:163-73; PMID:11804595; http://dx.doi.org/10.1016/S1097-2765(01)00438-5
- Liu X, Cotrim A, Teos L, Zheng C, Swaim W, Mitchell J, Mori Y, Ambudkar I. Loss of TRPM2 function protects against irradiation-induced salivary gland dysfunction. Nat Commun 2013; 4:1515; PMID:23443543; http://dx.doi.org/10.1038/ncomms2526
- Homma Y, Nomiya A, Tagaya M, Oyama T, Takagaki K, Nishimatsu H, Igawa Y. Increased mRNA expression of genes involved in pronociceptive inflammatory reactions in bladder tissue of interstitial cystitis. J Urol 2013; 190:1925-31; PMID:23727186; http://dx.doi.org/10.1016/j.juro.2013.05.049
- Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012; 55:222-32; PMID:21932416; http://dx.doi.org/10.1002/hep.24690
- Kheradpezhouh E, Ma L, Morphett A, Barritt GJ, Rychkov GY. TRPM2 channels mediate acetaminophen-induced liver damage. Proc Natl Acad Sci U S A 2014; 111:3176-81; PMID:24569808; http://dx.doi.org/10.1073/pnas.1322657111
- Hermosura MC, Cui AM, Go RC, Davenport B, Shetler CM, Heizer JW, Schmitz C, Mocz G, Garruto RM, Perraud AL. Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc Natl Acad Sci U S A 2008; 105:18029-34; PMID:19004782; http://dx.doi.org/10.1073/pnas.0808218105
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/10.4161/auto.4451
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Structure and Function 1998; 23:33-42; PMID:9639028; http://dx.doi.org/10.1247/csf.23.33
- Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxidants & Redox Signaling 2009; 11:777-90; PMID:18828708; http://dx.doi.org/10.1089/ars.2008.2270
- Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Schmitz C, Mocz G, Garruto RM, Perraud AL. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270-83; PMID:20655468; http://dx.doi.org/10.1016/j.cell.2010.06.007
- Garcia-Rivas Gde J, Carvajal K, Correa F, Zazueta C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. British J Pharmacol 2006; 149:829-37; PMID:17031386; http://dx.doi.org/10.1038/sj.bjp.0706932
- Shi M, Zhang T, Sun L, Luo Y, Liu DH, Xie ST, Song XY, Wang GF, Chen XL, Zhou BC, et al. Calpain, Atg5 and Bak play important roles in the crosstalk between apoptosis and autophagy induced by influx of extracellular calcium. Apoptosis 2013; 18:435-51; PMID:23242420; http://dx.doi.org/10.1007/s10495-012-0786-2
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8:1124-32; PMID:16998475; http://dx.doi.org/10.1038/ncb1482
- Xia HG, Zhang L, Chen G, Zhang T, Liu J, Jin M, Ma X, Ma D, Yuan J. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy 2010; 6:61-6; PMID:19901552; http://dx.doi.org/10.4161/auto.6.1.10326
- Berridge MJ. Calcium signalling remodelling and disease. Biochemical Society transactions 2012; 40:297-309; PMID:22435804; http://dx.doi.org/10.1042/BST20110766
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 2015; 17:288-99; PMID:25720963; http://dx.doi.org/10.1038/ncb3114
- Soderling TR, Chang B, Brickey D. Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 2001; 276:3719-22; PMID:11096120; http://dx.doi.org/10.1074/jbc.R000013200
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008; 133:462-74; PMID:18455987; http://dx.doi.org/10.1016/j.cell.2008.02.048
- White RR, Kwon YG, Taing M, Lawrence DS, Edelman AM. Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J Biol Chem 1998; 273:3166-72; PMID:9452427; http://dx.doi.org/10.1074/jbc.273.6.3166
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell death and differentiation 2011; 18:571-80; PMID:21311563; http://dx.doi.org/10.1038/cdd.2010.191
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012; 338:956-9; PMID:23112296; http://dx.doi.org/10.1126/science.1225967
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/10.1016/j.molcel.2008.06.001
- Giustarini D, Dalle-Donne I, Tsikas D, Rossi R. Oxidative stress and human diseases: Origin, link, measurement, mechanisms, and biomarkers. Crit Rev Clin Lab Sci 2009; 46:241-81; PMID:19958214; http://dx.doi.org/10.3109/10408360903142326
- Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000; 50:279-89; PMID:11327322; http://dx.doi.org/10.1080/15216540051081010
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008; 4:600-6; PMID:18497563; http://dx.doi.org/10.4161/auto.6260
- Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med 2013; 19:1478-88; PMID:24141421; http://dx.doi.org/10.1038/nm.3322
- Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep 2009; 10:285-92; PMID:19180116; http://dx.doi.org/10.1038/embor.2008.246
- Griffith LC. Regulation of calcium/calmodulin-dependent protein kinase II activation by intramolecular and intermolecular interactions. J Neurosci 2004; 24:8394-8; PMID:15456810; http://dx.doi.org/10.1523/JNEUROSCI.3604-04.2004
- Shen L, Hillebrand A, Wang DQ, Liu M. Isolation and primary culture of rat hepatic cells. Journal of visualized experiments: JoVE 2012; 64:3917
- Yue J, Ferrell JE, Jr. Mechanistic studies of the mitotic activation of Mos. Mol Cell Biol 2006; 26:5300-9; PMID:16809767; http://dx.doi.org/10.1128/MCB.00273-06
- Lu Y, Hao BX, Graeff R, Wong CW, Wu WT, Yue J. Two pore channel 2 (TPC2) inhibits autophagosomal-lysosomal fusion by alkalinizing lysosomal pH. J Biol Chem 2013; 288:24247-63; PMID:23836916; http://dx.doi.org/10.1074/jbc.M113.484253
- Wei WJ, Sun HY, Ting KY, Zhang LH, Lee HC, Li GR, Yue J. Inhibition of cardiomyocytes differentiation of mouse embryonic stem cells by CD38/cADPR/Ca2+ signaling pathway. J Biol Chem 2012; 287:35599-611; PMID:22908234; http://dx.doi.org/10.1074/jbc.M112.392530
- Li S, Hao B, Lu Y, Yu P, Lee HC, Yue J. Intracellular alkalinization induces cytosolic Ca2+ increases by inhibiting sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). PloS one 2012; 7:e31905; PMID:22384096; http://dx.doi.org/10.1371/journal.pone.0031905
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, et al. An expanded palette of genetically encoded Ca(2)(+) indicators. Science 2011; 333:1888-91; PMID:21903779; http://dx.doi.org/10.1126/science.1208592
- Lu Y, Dong S, Hao B, Li C, Zhu K, Guo W, Wang Q, Cheung KH, Wong CW, Wu WT, et al. Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating RAB5A. Autophagy 2014; 10:1895-905; PMID:25483964; http://dx.doi.org/10.4161/auto.32200
- Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, Wong WM, Wang L, Su H, Chu TH, et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med 2007; 13:1228-33; PMID:17906634; http://dx.doi.org/10.1038/nm1664
- Hao B, Lu Y, Wang Q, Guo W, Cheung KH, Yue J. Role of STIM1 in survival and neural differentiation of mouse embryonic stem cells independent of Orai1-mediated Ca2+ entry. Stem Cell Res 2014; 12:452-66; PMID:24424349; http://dx.doi.org/10.1016/j.scr.2013.12.005