
ABSTRACT
Macroautophagy/autophagy protects against cellular stress. Renal sublethal injury-triggered tubular epithelial cell cycle arrest at G2/M is associated with interstitial fibrosis. However, the role of autophagy in renal fibrosis is elusive. Here, we hypothesized that autophagy activity in tubular epithelial cells is pivotal for inhibition of cell cycle G2/M arrest and subsequent fibrogenic response. In both renal epithelial cells stimulated by angiotensin II (AGT II) and the murine kidney after unilateral ureteral obstruction (UUO), we observed that occurrence of autophagy preceded increased production of COL1 (collagen, type I). Pharmacological enhancement of autophagy by rapamycin suppressed COL1 accumulation and renal fibrosis. In contrast, genetic ablation of autophagy by proximal tubular epithelial cell-specific deletion of Atg5, with reduction of the LC3-II protein level and degradation of SQSTM1/p62, showed marked cell cycle arrest at the G2/M phase, robust COL1 deposition, and severe interstitial fibrosis in a UUO model, as compared with wild-type mice. In vitro, AGT II exposure triggered autophagy preferentially in the G1/S phase, and increased COL1 expression in the G2/M phase in renal epithelial cells. Stimulation of Atg5-deficient primary proximal tubular cells with AGT II also resulted in elevated G2/M arrest and COL1 production. Pharmacological or genetic inhibition of autophagy increased AGT II-mediated G2/M arrest. Enhanced expression of ATG5, but not the autophagy-deficient ATG5 mutant K130R, rescued the G2/M arrest, suggesting the regulation of cell cycle progression by ATG5 is autophagy dependent. In conclusion, Atg5-mediated autophagy in proximal epithelial cells is a critical host-defense mechanism that prevents renal fibrosis by blocking G2/M arrest.
Introduction
Renal tubulointerstitial fibrosis, characterized by excessive deposition of extracellular matrix components, is the final outcome of chronic kidney disease, irrespective of the diverse etiologies, which accounts for a growing burden of morbidity and mortality around the world.Citation1 The pathogenesis of renal fibrosis is complex and involves multiple cell types and cytokines. Under heavy and prolonged insults, renal epithelial cells can release a variety of cytokines that affect interstitial fibroblasts in a paracrine fashion to promote their proliferation and extracellular matrix (ECM) production.Citation2 Although there has been enormous progress in understanding the molecular mechanisms of renal fibrosis over the past few decades, the event that initiates this process remains elusive.
The cell cycle is a predominant physiological process that controls tissue homeostasis. Upon repetitive sublethal injury, renal tubular epithelial cells undergo repair and activate an intricate network of pathways that includes cell cycle checkpoints, proliferation, and cell death.Citation3 Using a variety of approaches to produce different severities of acute kidney damage, Yang et al. showed that a significant increase in the proportion of proximal tubular epithelial cells arrested in the G2/M phase correlated with the progression of fibrosis in critical or sustained kidney injury.Citation4 Conversely, reversal of the G2/M arrest attenuated production of cytokines and fibrosis, suggesting a direct molecular link between cell cycle dysregulation in tubular epithelial cells and fibrosis.
Autophagy, an evolutionarily conserved and genetically controlled pathway, plays a critical role in preserving cellular function.Citation5 Recent studies have indicated protective effects of autophagy in renal fibrosis. Specifically, kidneys from mice deficient in the autophagic protein BECN1/Beclin 1 exhibit a profibrotic phenotype. Moreover, genetic disruption of Becn1 or downregulation of it by specific siRNA in primary mouse mesangial cells of glomeruli, leads to enhanced expression of COL1.Citation6 Similarly, blockage of autophagy through pharmacological inhibitors or genetic ablation of autophagy-specific genes augments the progression of renal fibrosis, demonstrating ECM accumulation, increased mature TGFB protein levels, and mitochondrial dysfunction.Citation7 Nonetheless, the molecular mechanisms of autophagy in renal fibrosis have not been completely decoded.
Recent progress in different model systems has shown a connection between autophagy and the cell cycle.Citation8 It is evident that starved mouse embryonic fibroblasts lacking Atg7 fail to undergo trp53/p53-mediated cell cycle arrest.Citation9 Furthermore, it was found that autophagy is most active in the G1/S cell cycle, but is inhibited in mitosis.Citation10 Although both autophagy and epithelial cell cycle arrest have been independently associated with renal fibrosis, it is unknown whether and how these 2 processes interface during fibrotic processes.
In this study, we explored the interplay between autophagy and the cell cycle in the evolution of renal fibrosis. Our results indicate that Atg5-deficient proximal tubular epithelial cells enhance cell cycle arrest at G2/M, elevate COL1 production and ultimately accelerate renal fibrosis in unilateral ureteral obstruction (UUO) mice.
Results
Autophagy in tubular cells precedes COL1 deposition in obstructed kidneys
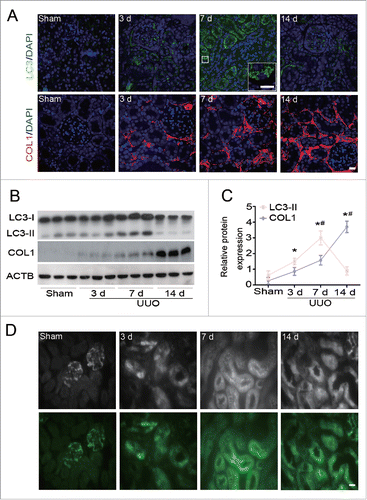
To examine whether autophagy and collagen accumulation are linked in the development of kidney fibrosis, we first analyzed the induction of autophagy and COL1 deposition after UUO in wild-type C57BL/6 mice. Immunofluorescence staining revealed that LC3-II, a marker of autophagy,Citation11,12 was barely detected in the kidney of sham-operated mice. In contrast, intense dot-like LC3 staining puncta were initially increased in renal tubular cells of obstructed kidneys at d 3, peaked at d 7, and nearly declined to basal levels at d 14 after UUO (). Meanwhile, the deposition of COL1, a major component of the ECM, increased progressively over time with the obstruction and was most prominent at d 14 after UUO (). Immunoblot analysis further confirmed obstruction-associated changes in the expression of LC3-II and COL1 during UUO (). Moreover, the induction of autophagy was verified by transgenic mice expressing green fluorescent protein (GFP)-LC3, demonstrating an obvious accumulation of GFP-LC3 puncta with a similar curve after UUO (). These observations reveal that autophagy likely occurs before the accumulation of extracellular matrix during kidney fibrogenesis.
Figure 1. Dynamics of autophagy and COL1 accumulation after UUO. (A) Representative images of immunofluorescence staining for LC3 (green) and COL1 (red) on kidney sections after 3,7, and 14 d of UUO compared with sham-operated C57BL/6 mice. Nuclei are highlighted with DAPI. Insert shows higher magnification images of LC3 puncta. Scale bar: 20 μm and 10 μm (insert). (B) Immunoblot analyses of LC3 and COL1 in the sham-operated and UUO kidneys of C57BL/6 mice. ACTB was used as loading control. (C) Densitometry of LC3-II and COL1 in immunoblots. Data are means ± SEM (n = 6); *, P < 0.01 vs. sham; #, P < 0.05 vs. other groups in UUO. (D) Representative images of LC3-positive dots in the kidney of GFP-Lc3 transgenic mice in different groups as indicated. Fluorescent signals were depicted as black and white in the upper panels. Scale bar: 20 μm.
Pharmacological activation of autophagy by rapamycin attenuates UUO-induced tubulointerstitial fibrosis (TIF)
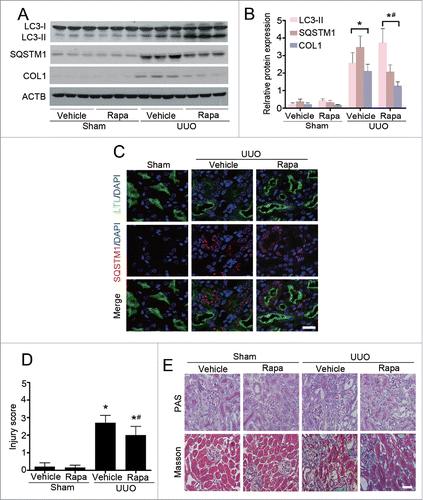
Rapamycin, the best characterized pharmacological inducer of autophagy, acts by inhibiting the mammalian target of rapamycin (serine/threonine kinase) complex 1 (MTORC1).Citation13 To determine whether autophagy is associated with the evolution of kidney fibrosis, C57BL/6 mice were treated with daily rapamycin (1 mg/kg) or vehicle, starting 1 d prior to UUO operation. Compared with vehicle-treated control mice, rapamycin administration significantly increased the level of LC3-II and concomitant degradation of SQSTM1/p62, a receptor and selective substrate of autophagy,Citation14 in the obstructed kidneys at d 7 after UUO (). Notably, activation of autophagy by rapamycin markedly attenuated UUO-induced expression of COL1 (). Consistent with the immunoblotting data, co-immunofluorescence with SQSTM1/p62 (red) and lotus tetragonolobus lectin (LTL, green), a marker of proximal tubules, revealed that abundant SQSTM1/p62-positive aggregates were located predominantly in the tubular cells of vehicle-treated mice following UUO, whereas the accumulation of SQSTM1/p62 was alleviated in mice treated with rapamycin (). Accordingly, histological examination showed the degree of tubular injury () and the severity of interstitial fibrosis () were markedly reduced by rapamycin administration. In sham-operated kidneys, there was no statistical difference in morphological characterization between vehicle- and rapamycin-treated mice. Taken together, pharmacological induction of autophagy using rapamycin significantly ameliorated UUO-induced TIF, indicating renoprotective roles of autophagy in the pathogenesis of renal fibrosis.
Figure 2. Enhancement of autophagy by rapamycin (Rapa) ameliorates renal interstitial fibrosis in C57BL/6 mice. (A) Immunoblot analyses of LC3, SQSTM1/p62, and COL1 in the obstructed kidneys in different groups as indicated. (B) Quantitative determination of the relative abundance of the indicated proteins among different groups. Data are means ± SEM (n = 6); *, P < 0.001 vs. sham; #, P < 0.05 vs. UUO mice treated with vehicle. (C) Coimmunostaining of LTL (green) and SQSTM1/p62 (red) in kidney sections. Scale bar: 20 μm. (D) Semiquantitative analysis of tubular damage in the obstructed kidneys. Data are means ±SEM (n = 6); *, P < 0.05 vs. sham; #, P < 0.05 vs. UUO mice treated with vehicle. (E) Representative micrographs from indicated groups with either periodic acid-Schiff (PAS; upper panels) or Masson's trichrome staining (lower panels). Scale bar: 20 μm.
Proximal tubular epithelial cell-specific Atg5 deletion aggravates TIF
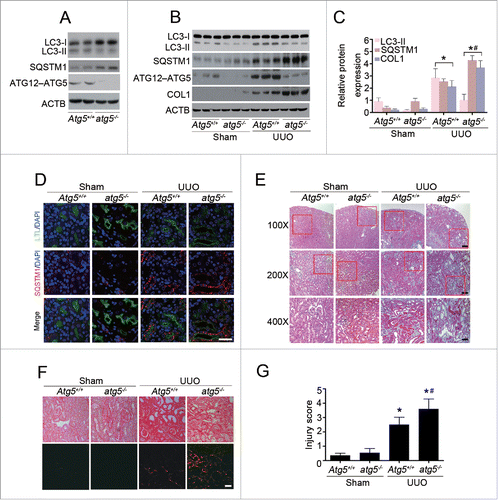
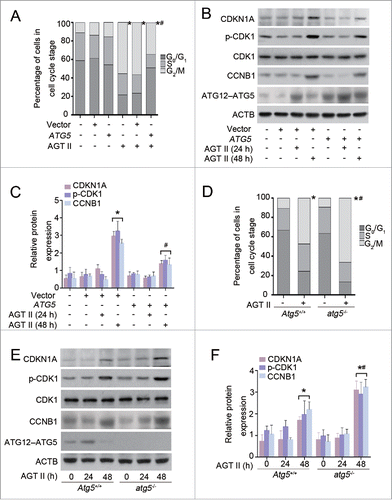
A series of autophagy-related genes are involved in the process of autophagy, and the ATG5 protein is required for the initiation of autophagosome formation. To further investigate the effects of autophagy on TIF, we generated mice with selective Atg5 deletion (atg5−/−) in proximal tubular epithelial cells (TECs) by crossing Atg5flox/flox mice with transgenic mice expressing Cre recombinase under the control of the Kap (kidney androgen regulated protein) promoter (Kap-cre), as previously described.Citation15 We first performed immunoblot analysis to confirm a significantly decreased ATG12–ATG5 conjugation expression with reduced LC3-II accumulation and SQSTM1/p62 degradation in the kidney cortex of atg5−/− mice, as compared with their wild-type (Atg5+/+) littermates (). Kidney fibrosis was induced by UUO in 8-wk-old male mice. As shown in , kidney tissue of wild-type mice showed a significant increase in protein level of LC3-II at d 7 after UUO, whereas this response was markedly abrogated in the atg5−/− mice. Consistently, a substantial reduction of SQSTM1/p62 degradation was observed in fibrotic kidneys of atg5−/− animals. The impaired autophagy in proximal tubular epithelial cells was concurrently accompanied by increased COL1 expression. Likewise, the amount and size of cytoplasmic SQSTM1/p62-positive aggregates were significantly greater in proximal tubular cells of atg5−/− mice than those of Atg5+/+ mice, as evaluated by immunofluorescence staining of renal tissue after UUO (). Histological examination further confirmed dramatically elevated interstitial fibrosis () and collagen deposition (), with more severe tubular injury () in the obstructed kidneys of atg5−/− mice in contrast to Atg5+/+ mice. These pathological changes were observed throughout the whole cortex, although the degree of TIF severity was not distributed evenly. Collectively, these findings indicate that aberrant activation of autophagy in proximal tubular cells is critical for promoting kidney fibrosis.
Figure 3. Proximal tubular epithelial cell-specific Atg5 deletion exacerbates UUO-induced renal fibrosis. (A) Immunoblot analyses of ATG12–ATG5 conjugation, LC3 and SQSTM1/p62 in the kidneys of Atg5+/+ and atg5−/− mice. (B) Immunoblot analyses of proteins in the kidneys from sham-operated and at d 7 after UUO in Atg5+/+ and atg5−/− mice. (C) Relative expression levels of the indicated proteins normalized to ACTB in Atg5+/+ and atg5−/− mice. Data are means ± SEM (n = 6); *, P < 0.001 vs. sham; #, P < 0.05 vs. Atg5+/+ mice with UUO. (D) Coimmunostaining of LTL (green) and SQSTM1/p62 (red) on kidney sections. Scale bar: 20 μm. (E) Representative micrographs of kidney histology with Masson's trichrome staining from Atg5+/+ and atg5−/− mice. Scale bar: 80 (upper panels), 40 (middle panels), and 20 (lower panels) μm. (F) Representative images of Sirius red-stained sections of sham-operated and 7 d-obstructed Atg5+/+ and atg5−/− mice. Scale bar: 20 μm. (G) Semiquantitative analysis of tubular damage in the obstructed kidneys. Data are means ± SEM (n = 6); *, P < 0.05 vs. sham; #, P < 0.05 vs. Atg5+/+ mice with UUO.
Atg5 deficiency abolishes the protective effect of rapamycin against renal fibrosis
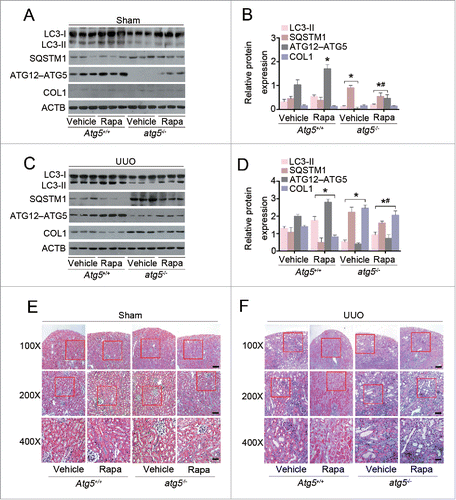
Next, we sought to determine whether UUO-induced renal fibrosis in Atg5-deficient mice was rescued by rapamycin. As shown in , in the sham-operated kidneys, treatment of Atg5+/+ mice with rapamycin caused a significant increase in the expression levels of ATG12–ATG5 conjugation compared with those in vehicle-treated Atg5+/+ mice. Rapamycin-treated atg5−/− mice demonstrated a very slight increase of endogenous ATG12–ATG5 conjugation and a decrease in SQSTM1/p62 as compared to those in vehicle-treated atg5−/− mice, but the difference was not statistically significant. Administration of rapamycin resulted in dramatic differences in protein levels of LC3-II, SQSTM1/p62, and ATG12–ATG5 conjugation between Atg5+/+ and atg5−/− mice. As expected, in obstructed kidneys at d 7, administration of rapamycin significantly upregulated autophagy and reduced COL1 expression when compared with those in vehicle-treated Atg5+/+ mice. Whereas, the protective effects of rapamycin were not significant in atg5−/− mice (). Similarly, obstructed kidneys from Atg5+/+ mice demonstrated less TIF in rapamycin-treated versus vehicle-treated mice, as indicated by a positive blue color in Masson's trichrome-stained sections. In contrast, these changes were not apparent in the obstructed kidneys from atg5−/− mice treated with or without rapamycin (). Our results suggest that the protective effect of rapamycin against renal fibrosis appear to be at least in part mediated through enhanced autophagy via ATG5.
Figure 4. Atg5 deficiency abolishes the protective effect of rapamycin against renal fibrosis in mice. (A) Immunoblot analyses of proteins in the sham-operated kidneys of Atg5+/+ and atg5−/− mice treated with or without rapamycin. (B) Relative expression levels of the indicated proteins normalized to ACTB in Atg5+/+ and atg5−/− mice treated with or without rapamycin. Data are means ± SEM (n = 6); *, P < 0.001 vs. vehicle-treated Atg5+/+ mice; #, P < 0.05 vs. rapamycin-treated Atg5+/+ mice. (C) Immunoblot analyses of proteins in the kidneys at day 7 after UUO in Atg5+/+ and atg5−/− mice treated with or without rapamycin. (D) Relative expression levels of the indicated proteins normalized to ACTB in Atg5+/+ and atg5−/- mice treated with or without rapamycin. Data are means ± SEM (n = 6); *, P < 0.001 vs. vehicle-treated Atg5+/+ mice; #, P < 0.05 vs. rapamycin-treated Atg5+/+ mice. (E) Representative micrographs of kidney histology with Masson's trichrome staining from Atg5+/+ and atg5−/− mice. Scale bar: 80 (upper panels), 40 (middle panels), and 20 (lower panels) μm.
Autophagy deficiency associates with enhanced tubular cell G2/M arrest in the fibrotic kidney
Because autophagy is involved in cell cycle progression in several cancer types,Citation16 the relationship among autophagy, the cell cycle and ECM deposition was examined in a UUO-induced fibrosis model. To this end, we first analyzed cells in proliferation and arrested at the G2/M phase of the cell cycle by costaining MKI67 with phosphorylation of histone H3 at Ser10 (p-H3) to evaluate the cell cycle distribution. Compared with sham-operated C57BL/6 mice, the percentage of TECs in the G2/M phase (p-H3 positive) among all proliferative TECs (MKI67 positive) elevated to 68.7 ± 10.1 on d 7 and was even increased to 78.7 ± 4.0 on d 14 after UUO (). The UUO-induced time-dependent G2/M phase arrest in TECs was virtually identical to those observed for ECM accumulation, indicating that G2/M arrest correlates with kidney fibrogenesis.
Figure 5. Tubular cell G2/M arrest is increased in UUO-induced fibrotic kidney. (A) Dual immunofluorescence of MKI67 (red) and p-H3 (green) in sham-operated and UUO kidneys on d 7 and 14 in C57BL/6 mice. DAPI (blue) was used for nuclear staining. Scale bar: 20 μm. (B) The percentage of MKI67+ p-H3+ cells among total MKI67+ tubular cells in different groups. Data represented as mean ± SEM (n = 6); *, P < 0.05 vs. sham. #, P < 0.05 vs. UUO 7 d. (C) Representative immunofluorescence staining of p-H3 (red), COL1 (green), and DAPI (blue) on kidney sections of Atg5-deficient mice from different groups. Scale bar: 20 μm. (D) Quantification of tubular cells with p-H3 positive. Data represented as mean ± SEM (n = 6); *, P < 0.05 vs. sham; #, P < 0.01 vs. obstructed kidney of Atg5+/+ mice.
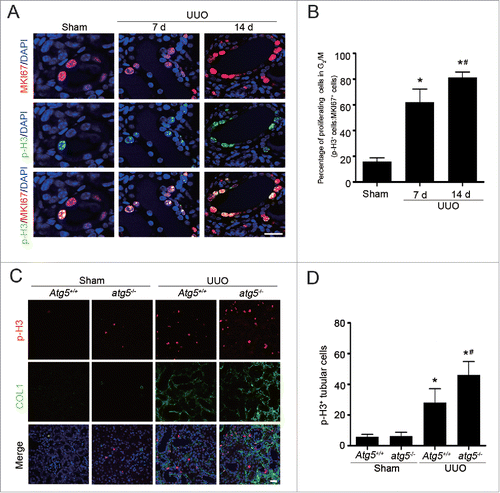
Next, we attempted to verify our findings utilizing renal tubular epithelial cell-specific atg5 knockout mice following UUO. As shown in , very few TECs labeled positively with p-H3 antibody in both sham-operated atg5−/− and Atg5+/+ mice. At 7 d following UUO, increased p-H3-positive cells with more COL1 accumulation were evident in Atg5+/+ animals, whereas the responses were substantially aggravated in the atg5−/− mice (). Therefore, ATG5 has a negative influence on cell cycle progression, and inhibition of autophagy may facilitate kidney fibrosis through a G2/M arrest-dependent mechanism in TECs.
Autophagy blocks AGT II-induced COL1 expression in vitro
To ascertain the role of autophagy in ECM accumulation, the renal epithelial cell line HK-2 was treated with AGT II, an important mediator of tubulointerstitial fibrosis. As shown in , a time-course experiment in which HK-2 cells were treated with AGT II (10−6 mol/L) for various durations (0–48 h) resulted in increased levels of LC3-II starting from 6 h, reaching the highest levels at 24 h and reducing to baseline at 48 h. Whereas, the expression of COL1 was significantly upregulated after 48 h of treatment. These data are in agreement with in vivo findings that renal epithelial cells make attempts to trigger autophagy, followed by COL1 expression after 24 h. Additionally, pre-incubation with the lysosomal protease inhibitors E64d and pepstatin A (E/P), which block the lysosome-dependent autophagic degradation, appeared to increase LC3-II level in cells untreated with AGT II, and further to a greater content in cells exposed to AGT II at 24 and 48 h (). Consistently, pretreatment with E/P increased the accumulation of LC3 puncta by immunofluorescence staining in AGT II-treated cells (). Our data suggest AGT II stimulation causes a significant autophagy flux.
Figure 6. Overexpression of ATG5 augments AGT II-mediated autophagy in vitro. (A) Serum-deprived HK-2 cells were treated with 10−6 mol/L of AGT II for the indicated time period. Cell lysates were probed with antibodies against LC3 and COL1. ACTB was used as a loading control. (B) Densitometry of LC3-II and COL1 proteins in immunoblots (relative to ACTB). Data are means ± SEM (n = 3); *, P <0.001 vs. control group. #, P <0.001 vs. AGT II-treated cells for 12 or 24 h. (C) Cells were treated with AGT II at 10−6 mol/L for 24 and 48 h in the presence or absence of E64d and pepstatin A (E/P). Induction of autophagy was assessed by analyzing the expression of LC3. ACTB was used as a loading control. (D) Densitometry of LC3-II in immunoblots (relative to ACTB). Data are means ± SEM (n = 3); *, P < 0.001 vs. control group; #, P < 0.05 vs. AGT II-treated cells at the same time point in the absence of E/P. (E) Cells were treated with AGT II (10−6 mol/L) for 24 and 48 h in the absence or presence of E/P. Formation of LC3 puncta was analyzed by immunofluorescence under confocal microscopy (×400). (F) Cells were transfected with either pcDNA3.1-ATG5 or pcDNA3.1-HA vector followed by incubation with 10−6 mol/L of AGT II for 24 h. LC3 and ATG12–ATG5 conjugation protein levels were examined by Western blotting. ACTB was used as a loading control. (G) LC3 and ATG5 contents were quantitatively analyzed using a densitometer. Values are mean ± SEM (n = 3); *, P < 0.001 vs. control group in the same condition; #, P < 0.05 vs. AGT II-treated cells in the corresponding condition without E/P; §, P < 0.05 vs. cells transfected with empty vector in the corresponding condition.
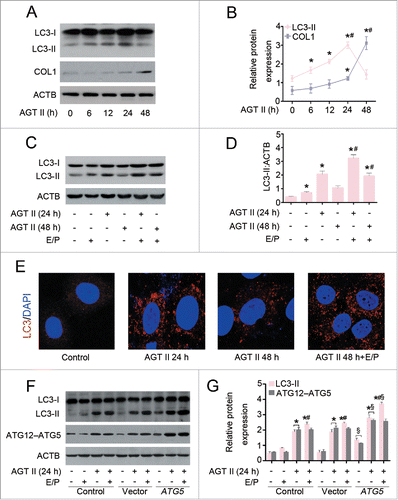
Given that atg5−/− mice showed an increase in COL1 accumulation and interstitial fibrosis () compared with wild-type mice, we sought to determine the role of ATG5 in AGT II-induced COL1 production in vitro. To this end, we transfected HK-2 cells with either ATG5 expression or control vectors. As shown in , enhanced expression of ATG5 alone induced a small increase in basal LC3-II levels, and this response became pronounced in the presence of E/P. Therefore, elevated LC3-II in ATG5-overexpressing cells does not appear to be a result of inhibition of autophagic flux. ATG5 overexpression markedly increased LC3-II content upon AGT II stimulation for 24 h both in the presence and absence of protease inhibitors, compared with the control vector. In contrast, the protein level of COL1 was dramatically reduced after 48 h of AGT II exposure in ATG5-overexpressing cells ().
Figure 7. ATG5 inhibits AGT II-induced COL1 expression in vitro. (A) HK-2 cells were transfected with either pcDNA3.1-ATG5 or pcDNA3.1-HA vector before stimulation with 10−6 mol/L of AGT II for 24 or 48 h. Cell lysates were analyzed by immunoblotting with the indicated antibodies. (B) Densitometry of LC3-II and COL1 proteins in immunoblots. Data are means ± SEM (n = 3); *, P < 0.05 vs. negative and empty control; #, P < 0.05 vs. control and AGT II-treated cells for 24 h with or without ATG5 overexpression; §, P < 0.05 vs. control and AGT II-treated cells for 48 h without ATG5 overexpression. (C) Expression of LC3, ATG12–ATG5 conjugation, SQSTM1/p62, and LRP2/MEGALIN in primary TECs from Atg5+/+ and atg5−/− mice were assessed by Western blot analysis. (D) Primary TECs from Atg5+/+ and atg5−/− mice were stimulated with 10−6 mol/L of AGT II for 24 or 48 h. Cell lysates were probed with antibodies against the indicated proteins.
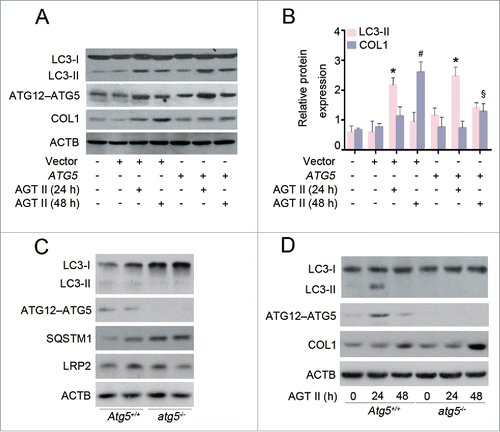
Further, we characterized the effect of ATG5 specifically on COL1 expression by using primary renal proximal TECs isolated from 3-wk-old Atg5+/+ or atg5−/− mice. As shown in , the expression of ATG12–ATG5 conjugation and LC3-II were almost completely suppressed, and SQSTM1/p62 accumulated in primary atg5−/− TECs. As expected, the protein levels of LC3-II and COL1 in primary TECs from Atg5+/+ mice were similar in pattern to those in HK-2 cells after AGT II treatment for 24 and 48 h. Remarkably, AGT II-stimulated COL1 production was exacerbated in primary atg5−/− TECs (). Taken together, these data suggest that Atg5-deficient TECs are more sensitive to induction of COL1 expression in response to AGT II.
ATG5 prevents ECM deposition through blocking G2/M arrest
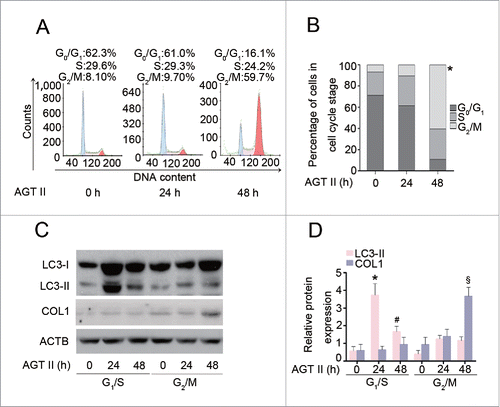
As our observations indicated that AGT II induces autophagy and COL1 production at 2 distinct time points, we sought to determine whether these appearances of 2 biological events were due to the heterogeneity of cellular populations. As shown in , stimulation of HK-2 cells with AGT II for 24 h did not alter cell cycle progression. However, AGT II treatment at 48 h caused a striking increase in the percentage of cells in G2/M phase by 50% as compared to the cells untreated or treated for 24 h. Moreover, we synchronized HK-2 cells at G1/S or G2/M phase by incubation with hydroxyurea or nocodazole, respectively.Citation17 Immunoblot analysis showed that the expression of LC3-II in G1/S-phase-synchronized cells was higher than that in G2/M-phase-synchronized cells after exposure to AGT II for 24 and 48 h. Interestingly, synchronization of cells at the G1/S phase almost completely blocked AGT II-induced COL1 production. In contrast, a dramatic increase of COL1 expression was observed in cells synchronized at the G2/M phase after 48 h of treatment with AGT II (). These data suggest that the cell cycle state determines cell fate propensity, and that AGT II induces autophagy preferentially in the G1/S phase.
Figure 8. AGT II-induced autophagy is dependent on cell cycle status in HK-2 cells. Cells were treated with10−6 mol/L of AGT II for the indicated time period. (A) Cells were exposed to AGT II, followed by PI staining and flow cytometry. (B) Cell cycle distribution among different groups. Data are means ± SEM (n = 3); *, P < 0.05 vs. control and AGT II-treated cells for 24 h. Symbol indicates the fraction of G2/M phase cells. (C) Cells synchronized at G1/S or G2/M phase were treated with AGT II for the indicated time period. Cell lysates were probed with antibodies against LC3 and COL1. (D) Densitometry of LC3-II and COL1 proteins in immunoblots. Data are means ± SEM (n = 3); *, P < 0.05 vs. respective control group; #, P < 0.05 vs. control and cells synchronized at G1/S phase treated with AGT II for 24 h; §, P < 0.05 vs. cells synchronized at G2/M phase treated with AGT II for 0–24 h, or cells synchronized at G1/S phase treated with or without AGT II.
Next, we investigated the functional role of ATG5 in cell cycle progression. Compared with empty vector, ATG5 overexpression significantly reduced the proportion of cells in G2/M arrest (), and was accompanied by a decrease in the levels of CDKN1A/p21, phosphorylated (p)-CDK1 and CCNB1 (cyclin B1) after treatment with AGT II for 48 h (). In the absence of AGT II, neither empty vector nor ATG5 plasmid alone had effects on the distribution of the cell cycle and expression of cell cycle regulatory proteins in HK-2 cells (). Further, treatment of primary atg5−/− TECs with AGT II for 48 h greatly led to an accumulation of cells in the G2/M phase with a concomitant reduction of cells in the G0/G1 phase in contrast to primary Atg5+/+ TECs. Quantitative analysis revealed a 20% increase of the cell population in the G2/M phase in atg5−/− TECs compared to that in wild-type TECs (). The changes in expression of regulatory proteins were consistent with the cell cycle distribution (). Taken together, our results indicate that ATG5 may attenuate COL1 production primarily by blocking G2/M cell cycle arrest.
Figure 9. Overexpression of ATG5 attenuates AGT II-induced G2/M cell cycle arrest in HK-2 cells and primary TECs. (A) HK-2 cells were transiently transfected with either pcDNA3.1-ATG5 or pcDNA3.1-HA vector followed by treatment with 10−6 mol/L of AGT II for 48 h, and then cell cycle distribution was analyzed by flow cytometry. Data are means ± SEM (n = 3); *, P < 0.01 vs. AGT II-untreated cells; #, P < 0.05 vs. AGT II-treated cells without ATG5 overexpression. Symbols indicate the fraction of G2/M phase cells. (B) HK-2 cells were treated as described above. Expression of CDKN1A/p21, p-CDK1, and CCNB1 (cyclin B1) was examined by immunoblot. (C) Protein levels were quantified with scanning densitometry and standardized to ACTB. Data are expressed as mean ± SEM (n = 3); *, P < 0.01 vs. control or cells treated with AGT II for 24 h; #, P <0.05 vs. cells treated with AGT II for 48 h without ATG5 overexpression. (D) Primary renal proximal TECs from Atg5+/+ and atg5−/− mice were stimulated with 10−6 mol/L of AGT II for 48 h. Cell cycle distribution was assessed by flow cytometry. Data are expressed as mean ± SEM (n = 3); *, P < 0.01 vs. respective control; #, P < 0.05 vs. AGT II-treated cells from Atg5+/+ mice. Symbols indicate the fraction of G2/M phase cells. (E) Primary renal proximal TECs were treated as described in . Protein expression was detected by immunoblotting. (F) The expression of CDKN1A/p21, p-CDK1, and CCNB1 was quantitatively analyzed using a densitometer. Values are mean ± SEM (n = 3); *, P < 0.05 vs. respective control or cells treated with AGT II for 24 h; #, P < 0.05 vs. cells from Atg5+/+ mice treated with AGT II for 48 h.
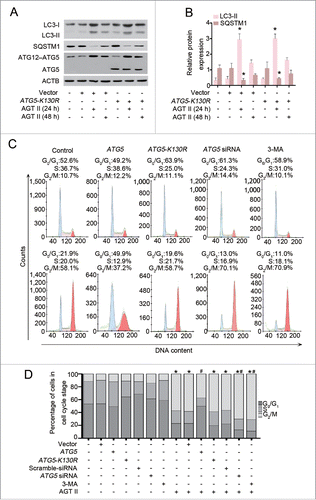
ATG5-mediated inhibition of G2/M arrest is autophagy-dependent
Aside from its function as a critical regulator of autophagy induction, ATG5 has been associated with proliferation and apoptosis under stress conditions.Citation18-20 These previous findings encouraged us to investigate whether autophagy induction is required for ATG5-mediated cell cycle progression. To this end, HK-2 cells were transfected with a plasmid expressing a mutant form of ATG5 (ATG5K130R), which is unable to conjugate with ATG12 and loses autophagic activity.Citation21 Indeed, we found that overexpression of ATG5K130R did not enhance formation of the ATG12–ATG5 conjugate with or without AGT II exposure, and had no influence on AGT II-induced autophagy, as assessed by LC3-II immunoblotting (). Interestingly, when compared with empty vector, overexpression of ATG5 reversed the percentage of G2/M-arrested cells following AGT II treatment for 48 h, whereas ATG5K130R expression failed to show such inhibitory effects. On the contrary, inhibition of autophagy by pharmacological inhibitor, 3-methyladenine (3-MA), or specific knockdown of ATG5 with its siRNA, significantly increased G2/M arrest under the same conditions (). In the absence of AGT II, empty vector, ATG5, ATG5-K130R, ATG5 siRNA, or 3-MA did not affect cell cycle progression. Collectively, these data indicate that blockage of G2/M arrest by ATG5 is dependent on its function in autophagy.
Figure 10. Autophagy induction is required for ATG5-mediated cell cycle progression. (A) HK-2 cells were transiently transfected with plasmids encoding pcDNA3.1-ATG5-K130R or pcDNA3.1-HA followed by incubation with 10−6 mol/L of AGT II for 24 or 48 h. Expression of LC3, SQSTM1/p62, and ATG5 (ATG12-conjugated and monomeric) were examined by immunoblot. (B) Relative expression levels of the indicated proteins normalized to ACTB. Data are mean ± SEM (n = 3); *, P < 0.05 vs. negative control, empty vector, or ATG5-K130R control vector. (C) HK-2 cells were transiently transfected with plasmids encoding pcDNA3.1-ATG5, pcDNA3.1-ATG5-K130R or pcDNA3.1-HA, or scramble siRNA, ATG5 siRNA, or cells pretreated with 3-MA, and then cell cycle distribution was analyzed in the absence (upper panels) or presence (lower panels) of AGT II for 48 h. (D) HK-2 cells were treated as indicated and the histograms represent the distribution of cells cycle among different groups. Data are means ± SEM (n = 3); *, P < 0.05 vs. cells in the absence of AGT II; #, P < 0.05 vs. cells treated with AGT II without ATG5 overexpression. Symbols indicate the fraction of G2/M phase cells.
Discussion
The present study demonstrates activation of autophagy in renal proximal tubular cells and its protective effect in renal fibrogenesis. Pharmacological enhancement of autophagy by rapamycin reduced COL1 accumulation and TIF, whereas deficient autophagy by ablation of the Atg5 gene in proximal tubular epithelial cells significantly aggravated cell cycle arrest in the G2/M phase, COL1 production, and fibrosis in a UUO model, as compared with wild-type mice. Rapamycin failed to protect against renal fibrosis in Atg5-deficient mice. The elevation of G2/M arrest in Atg5-deficient cells was dependent on the role of ATG5 in autophagy, suggesting that ATG5-mediated autophagy may be an important determinant of G2/M cell cycle regulation and subsequent renal fibrosis.
Emerging evidence from studies in renal cells in culture and experimental animals indicates that autophagic dysfunction is involved in the pathogenesis of renal injury caused by ischemia/reperfusion,Citation22,23 diabetes,Citation24,25 agingCitation26,27 and anticancer agents.Citation28-30 The functional role of autophagy in renal fibrosis is currently under investigation, and it has been proposed that autophagy serves as a protective mechanism against fibrosis. Sung et al. reported that autophagy may function to promote intracellular degradation of COL1 induced by TGFB1 in cultured glomerular mesangial cells.Citation6 By using autophagy-deficient lc3 null (lc3−/−) and Becn1 heterozygous (Becn1+/−) mice and GFP-Lc3 transgenic mice, the same group has recently shown that autophagy is induced primarily in renal tubular epithelial cells. Both lc3−/− and Becn1+/− mice develop more severe renal fibrosis in UUO than those of wild-type littermates. Furthermore, autophagy may inhibit the development of kidney fibrosis via degradation of TGFB.Citation7 Our present studies also demonstrated that both autophagy in tubular epithelial cells, as evidenced by punctate localization of GFP-LC3 and increased expression of LC3-II formation, and interstitial fibrosis occurred in obstructed kidneys. Pretreatment with rapamycin significantly protected animals against TIF. Importantly, we noted that autophagy preceded COL1 production within both in vitro and in vivo experimental models. These time-dependent events led us to further explore the mechanism of autophagy action in renal fibrosis. Despite the fact that the role of epithelial-to-mesenchymal transition in the development of renal fibrosis has been questioned,Citation31 injured epithelial cells have been shown to contribute to fibrosis by synthesis and secretion of pro-fibrotic factors.Citation4 Therefore, autophagy activity in tubular epithelial cells after injury may play a role in renal fibrosis.
Unlike previous studies, we used proximal tubular epithelial cell-specific Atg5-knockout mice, but not global genetic knockout approaches, to directly determine the role of renal epithelial cell autophagy in fibrogenesis. The results revealed that Atg5 deficiency facilitated accumulation of SQSTM1/p62 in tubular epithelial cells and enhanced susceptibility to COL1 accumulation and renal fibrosis in obstructive nephropathy. ATG5 acts downstream of MTOR within autophagy. In our study, rapamycin treatment increased endogenous ATG5 protein levels in atg5−/− mice compared to vehicle-treated atg5−/− mice. However, the protective effect of rapamycin against fibrosis in atg5−/− mice was reduced when compared with Atg5+/+ mice. This response may be due, at least in part, to inadequate enhancement of ATG5 resulting in insufficient autophagy. In the in vitro study, we showed that overexpression of ATG5 largely reduced AGT II-induced COL1 expression in HK-2 cells, and the ATG5-mediated inhibition could be eliminated in primary atg5−/− TECs. To our knowledge, this is the first study to emphasize that autophagy and renal fibrosis appear to be sequentially linked, and modulation of ATG5-mediated proximal tubular epithelial cell-specific autophagy may be a valuable therapeutic approach to prevent, delay or halt progression of renal fibrosis.
The cell cycle is tightly controlled by precise mechanisms, where some autophagy genes contribute to cell cycle progression.Citation9,18,32 However, autophagy as an upstream regulator of cell cycle progression in renal fibrosis has not been confirmed. In our present study, we showed that synchronization of cells at G2/M phase dramatically enhanced AGT II-induced COL1 expression in HK-2 cells. In contrast, cells synchronized at the G1/S phase did not show this response. Notably, proximal tubular epithelial cell-specific Atg5 deficiency sensitized cells to greater G2/M arrest both in vivo and in vitro, which coincided with marked increase in the levels of CDKN1A/p21, p-CDK1 and CCNB1 (cyclin B1), COL1 content, and renal fibrosis. Therefore, our results suggest that there exists a crosstalk between autophagy and cell cycle regulation and that ATG5 in the tubular epithelial cells may retard the development of renal fibrosis through reducing cell cycle arrest in G2/M phase.
ATG proteins may also modulate physiological and pathophysiological processes via autophagy-independent mechanisms.Citation18 However, whether ATG5-mediated autophagy is required for renal protection against fibrosis remains to be investigated. The present study showed that enhanced expression of wild-type ATG5 increased AGT II-mediated autophagy and reduced G2/M arrest. Inhibition of autophagy by a pharmacological inhibitor (3-MA) or specific downregulation of ATG5 by its specific siRNA significantly increased G2/M arrest under the same conditions. In contrast, overexpression of ATG5K130R failed to interfere with cell cycle progression, due to an inability to conjugate with ATG12 and an inability to launch autophagy. Our findings suggest that the effects of ATG5 on cell cycle regulation appear to rely on its autophagic capacity. Although it is not yet clear which mechanism(s) ATG5 uses to facilitate progression through the G2/M phase of the cell cycle, our data indicate that ATG5 exerts this effect dependent on its function in autophagy.
In summary, our studies demonstrate an unknown relationship among autophagy, the cell cycle and renal fibrosis. ATG5 is a critical host-defense mechanism that prevents renal fibrosis by modulating G2/M arrest in proximal epithelial cells and its subsequent effect on COL1 production, and this renal protection is dependent on its autophagic activity. In view of the role of ATG5 in renal fibrosis, our findings provide novel insights into the mechanism underlying induction of autophagy and development of TIF. Manipulation of ATG5-mediated autophagy in proximal epithelial cells may be a promising avenue for preventing the progression of renal fibrosis.
Materials and methods
Reagents
Reagents were obtained from the following sources: AGT II/angiotensin II (A9525), 3-MA (M9281), hydroxyurea (H8627), nocodazole (M1404), E64d (E8640) and pepstatin A (P5318) were from Sigma. Antibodies against LC3B (3868), ATG5 (2630) and CDKN1A/p21 (2947) were from Cell Signaling Technology. Anti-CDK1 (ab18), anti-p-CDK1 (ab133463), anti-CCNB1/cyclin B1 (ab2949), anti-LRP2/MEGALIN (ab129198) anti-p-H3 antibody (ab7031) and anti-MKI67 (ab16667) antibodies were purchased from Abcam. Anti-COL1/collagen I (1310–01) was obtained from SouthernBiotect. Anti-SQSTM1/p62 (GTX100685) was from GeneTex. Fluorescein-labeled Lotus Tetragonolobus Lectin (FL-1321) was from Vector laboratories. Alexa Fluor® 488 goat anti-rabbit IgG antibody (659082), Alexa Fluor® 488 goat anti-mouse IgG antibody (A11001), Alexa Fluor® 546 goat anti-mouse IgG antibody (A11003) and Alexa Fluor®546 goat anti-rabbit IgG antibodies (A11010) were from Invitrogen.
Animals
Wild-type C57BL/6 mice were obtained from the Sun Yat-Sen University Animal Center (Guangzhou, China). GFP-Lc3 transgenic mice and Atg5flox/flox mice were purchased from the RIKEN BioResource Center (Tsukuba, Ibaraki, Japan). The Kap-cre mice expressing Cre recombinase under the control of the Kap promoter were from the Jackson Laboratory (Bar Harbor, Maine, USA). All animal studies were approved by the Animal Care and Use Committee of the Sun Yat-Sen University.
Proximal tubule-specific atg5−/− mice were generated by crossing an Atg5flox/flox mouse with a Kap-cre mouse.Citation15 All mice were maintained by backcross breeding to C57BL/6 mice. Genotyping was identified by polymerase chain reaction (PCR) analysis using transgene-specific primers, as described previously.Citation33-34 Male mice between 8 and 10 wk of age were used in the present study.
For the UUO model, mice underwent ligation of the left ureter with silk sutures, and sham-operated mice were used as controls.Citation35 To activate autophagy, rapamycin (Sigma, R0395) was dissolved in vehicle (0.01% [v/v] dimethyl sulfoxide (Sigma, D2650]) followed by daily intraperitoneal injection in C57BL/6, atg5−/−, or Atg5+/+ mice (1 mg/kg/d) 1 d before and until UUO or sham surgery. Control mice received the same dose of dimethyl sulfoxide. Mice (n = 6) were sacrificed at 3, 7, and 14 d after UUO. Both contralateral and obstructed kidneys were harvested for further studies.
Kidney histology analyses
The kidney tissues from C57BL/6, Atg5+/+ and atg5−/− mice were fixed with 4% paraformaldehyde (Sigma, P6148). Paraffin-embedded 3-μm kidney sections were stained with periodic acid-Schiff (Sigma, 395B-1KT), Masson's trichrome (Baso, BA4079A) or picrosirius red (Polysciences, 24901). The degree of tubular injury was assessed semiquantitatively by the point-counting method in a blinded fashion.Citation35 Samples from GFP-Lc3 transgenic mice were analyzed as described.Citation33
Briefly, mice were transcardially perfused with 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4). Kidney tissues were further fixed in the same buffer for an additional 4 h, immersed in 15% sucrose (Sigma, S0389) for 4 h, followed by 30% immersion in sucrose at 4°C overnight for cryoprotection. The sections (5–7 μm) were embedded and frozen with optimal cutting temperature (OCT) compound (Tissue-Tek, 4583) before being sectioned with a cryostat, air-dried, and stored at −20°C. Frozen tissue sections were mounted on a glass slide and viewed under a fluorescence microscope.
Immunofluorescence staining of the kidney was performed using an established procedure.Citation35 Briefly, the 4-μm paraffin-embedded tissue sections were deparaffinized, rehydrated and incubated with the indicated primary antibodies at 4°C overnight, followed by their corresponding secondary antibodies for 1 h at room temperature.
For immunofluorescent analysis, cells were seeded onto glass coverslips, exposed to different experimental conditions, and fixed in 4% phosphate-buffered saline (PBS; Boster, PYG0021)-buffered paraformaldehyde. Formation of autophagic vesicles was monitored by LC3 puncta formation in cells. DAPI (Sigma-Aldrich, D9542) was used to stain the nuclei. All images were collected by laser scanning confocal microscopy (Zeiss LSM 510 META; Carl Zeiss, Oberkochen, Germany).
Western blotting analysis
Kidney cortex or cultured cells were homogenized in lysis buffer (Sigma, R0278) containing Complete Mini Protease Inhibitor Cocktail (Roche Applied Science, 05892791001). Protein concentration was measured with the Bradford assay. Samples were loaded and separated by 10–15% SDS polyacrylamide gels, electrotransferred onto a nitrocellulose membrane, blotted with the indicated antibodies, and then detected by enhanced chemiluminescence (Amersham Pharmacia Biotech, RPN2232).Citation35 Densitometric analysis was performed with an image analysis program (FluorchemTM 8900, Alpha Innotech).
Cell culture and treatments
A human proximal tubule epithelial cell line (HK-2) was purchased from American Type Culture Collection (ATCC®CRL-2190TM) and cultured at 37°C, 5% CO2 in DMEM/F-12 medium (GIBCO, 11330–057) supplemented with 10% fetal bovine serum (GIBCO, 10099) and antibiotics.
Mouse TECs were isolated and cultured as described previously.Citation15 Briefly, kidneys were harvested from 3-wk-old male Atg5+/+ or atg5−/− mice under sterile conditions. The kidney cortex was diced into small pieces (<1 mm3), incubated in DMEM medium containing collagenase (Sigma, C1639) at 37°C for 20 min, and sieved through 2 nylon sieves (pore size 250 μm and 70 μm). The filtrated cells were resuspended cultured at 37°C, 5% CO2 in DMEM/F-12 medium supplemented with 10% fetal bovine serum and antibiotics.
HK-2 and primary tubular epithelial cells between passages 3 to 5 were grown to approximately 80% confluence, followed by serum deprivation for 24 h prior to experimental manipulation. To induce autophagy, cells were treated with human AGT II (10−6 mol/L) for different time points. In some experiments, cells were pre-treated with 0.5 mmol/L hydroxyurea for 20 h, 0.25 ug/ml nocodazole for 20 h or 10 µg/ml E64d-pepstatin A for 60 min, and then stimulated with AGT II.
Plasmid constructs and transfection
The full-length ATG5 gene was amplified from genomic DNA of the HK-2 cell line by PCR and subcloned into the pcDNA3.1 vector (Invitrogen,V79020). The ATG5-K130R mutant replacing Lys130 with Arg was generated by PCR and cloned into the Xhol and Xbal sites of pcDNA3.1-HA.Citation36 The primers used for PCR were ATG5: sense 5′-GCGTCGACTCCTGGAAGAATGACAGAT-3′, antisense 5′-CGCCTCGAGTCCTTCAATCTGTTGGCT-3′ and ATG5-K130R mutant: sense 5′-CATGTATGAGAGAAGCTGATGCT-3′, antisense 3′-AGCATCAGCTTCTCTCATACATGA-5′. pcDNA3.1-HA mammalian expression vector was from Invitrogen Life Technologies (V79020). Scrambled and ATG5-specific siRNA products were from Shanghai GenePharma Co., Ltd (Shanghai, China). ATG5 siRNA knockdown was performed by transient transfection with validated ATG5 siRNA (sense, GCAACUCUGGAUGGGAUUGTT; antisense, CAAUCCCAUCCAGAGUUGCTT). Scrambled siRNA was used as control. Cells were transfected with ATG5, ATG5-K130R mutant, or ATG5 siRNA using Lipofectamine™ LTX and Plus reagent in Opti-mem (Invitrogen, 15338–100) according to the manufacturer's instructions. Overexpression efficacy was assessed by western blotting.
Cell cycle distribution analysis
Propidium iodide (PI; Sigma, P4170) staining was used to analyze DNA content and cell cycle profile. After treatment, cells were washed twice with ice-cold PBS, harvested, and fixed with 70% ethanol at 4°C overnight. Cells were resuspended in PBS, permeabilized with 0.125% Triton X-100 (Sigma-Aldrich, T9284) and stained with 40 μg/ml PtdIns for 30 min on ice. Cell cycle distributions were analyzed by flow cytometry (FACS Calibur, BD Biosciences).
Statistical analysis
The data are expressed as mean ± SEM. Comparison among groups was assessed by the t test or ANOVA using the statistical software program SPSS 18.0 for Windows (Armonk, NY) and P values < 0.05 were considered statistically significant.
Abbreviations
| ATG5 | = | autophagy-related 5 |
| CDKN1A/p21 | = | cyclin-dependent kinase inhibitor 1A |
| COL1 | = | collagen, type I |
| ECM | = | extracellular matrix |
| E/P | = | E64d and pepstatin A |
| KAP | = | kidney androgen regulated protein |
| LC3-II | = | phosphatidylethanolamine-conjugated isoform of microtubule-associated protein 1 light chain 3 |
| LTL | = | lotus tetragonolobus lectin |
| 3-MA | = | 3-methyladenine |
| MKI67 | = | antigen identified by monoclonal antibody Ki 67 |
| p-CDK1 | = | phosphorylated form of cyclin-dependent kinase 1 at Y15 |
| p-H3 | = | phosphorylated form of histone H3 at Ser10 |
| SQSTM1/p62 | = | sequestosome 1 |
| TECs | = | tubular epithelial cells |
| TIF | = | tubulointerstitial fibrosis |
| UUO | = | unilateral ureteral obstruction |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81070553, 81270783, 81100491), the Natural Science Foundation of Guangdong province (10251008901000003), the National Basic Research Program of China (2011CB504005, 2012CB517700-2012CB517706), and Guangdong Translational Medicine Public Platform (4202037).
References
- Radhakrishnan J, Remuzzi G, Saran R, Williams DE, Rios-Burrows N, Powe N, Bruck K, Wanner C, Stel VS, Venuthurupalli SK, et al. Taming the chronic kidney disease epidemic: a global view of surveillance efforts. Kidney Int 2014; 86:246–50; PMID:24897034; http://dx.doi.org/10.1038/ki.2014.190
- Canaud G, Bonventre JV. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol Dial Transplant 2015; 30(4):575–83; PMID:25016609; http://dx.doi.org/10.1093/ndt/gfu230
- Thomasova D, Anders HJ. Cell cycle control in the kidney. Nephrol Dial Transplant 2015; 30(10):1622–30; PMID:25538161; http://dx.doi.org/10.1093/ndt/gfu395
- Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 2010; 16:535–43, 1p-143p; http://dx.doi.org/10.1038/nm.2144
- Hale AN, Ledbetter DJ, Gawriluk TR, Rucker ER. Autophagy: regulation and role in development. Autophagy 2013; 9:951–72; PMID:24121596; http://dx.doi.org/10.4161/auto.24273
- Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J Biol Chem 2012; 287:11677–88; PMID:22351764; http://dx.doi.org/10.1074/jbc.M111.308460
- Ding Y, Kim SL, Lee SY, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 2014; 25:2835–46; PMID:24854279; http://dx.doi.org/10.1681/ASN.2013101068
- An Z, Tassa A, Thomas C, Zhong R, Xiao G, Fotedar R, Tu BP, Klionsky DJ, Levine B. Autophagy is required for G(1)/G(0) quiescence in response to nitrogen starvation in Saccharomyces cerevisiae. Autophagy 2014; 10:1702–11; PMID:25126732; http://dx.doi.org/10.4161/auto.32122
- Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, Finkel T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012; 336:225–8; PMID:22499945; http://dx.doi.org/10.1126/science.1218395
- Tasdemir E, Maiuri MC, Tajeddine N, Vitale I, Criollo A, Vicencio JM, Hickman JA, Geneste O, Kroemer G. Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle 2007; 6:2263–7; PMID:17890908; http://dx.doi.org/10.4161/cc.6.18.4681
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1–222
- Mizushima N, Yoshimori T, Levine, B. Methods in mammalian autophagy research. Cell 2010; 140(3):313–26; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004; 18:1926–45; PMID:15314020; http://dx.doi.org/10.1101/gad.1212704
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062–75; PMID:19524509; http://dx.doi.org/10.1016/j.cell.2009.03.048
- Kimura T, Takabatake Y, Takahashi A, Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 2011; 22:902–13; PMID:21493778; http://dx.doi.org/10.1681/ASN.2010070705
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21:1367–81; PMID:17510285; http://dx.doi.org/10.1101/gad.1545107
- Wang H, Yu S, Davis AT, Ahmed K. Cell cycle dependent regulation of protein kinase CK2 signaling to the nuclear matrix. J Cell Biochem 2003; 88:812–22; PMID:12577315; http://dx.doi.org/10.1002/jcb.10438
- Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, Simon HU. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun 2013; 4:2130; PMID:23945651; http://dx.doi.org/10.1038/ncomms3130
- Shi M, Zhang T, Sun L, Luo Y, Liu DH, Xie ST, Song XY, Wang GF, Chen XL, Zhou BC, et al. Calpain, Atg5 and Bak play important roles in the crosstalk between apoptosis and autophagy induced by influx of extracellular calcium. Apoptosis 2013; 18:435–51; PMID:23242420; http://dx.doi.org/10.1007/s10495-012-0786-2
- Codogno P, Meijer AJ. Atg5: more than an autophagy factor. Nat Cell Biol 2006; 8:1045–7; PMID:17013414; http://dx.doi.org/10.1038/ncb1006-1045
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature 1998; 395:395–8; PMID:9759731; http://dx.doi.org/10.1038/26506
- Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol 2010; 176:1181–92; PMID:20075199; http://dx.doi.org/10.2353/ajpath.2010.090594
- Liu S, Hartleben B, Kretz O, Wiech T, Igarashi P, Mizushima N, Walz G, Huber TB. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012; 8:826–37; PMID:22617445; http://dx.doi.org/10.4161/auto.19419
- Ding Y, Choi ME. Autophagy in diabetic nephropathy. J Endocrinol 2015; 224:R15–30; PMID:25349246; http://dx.doi.org/10.1530/JOE-14-0437
- Yamahara K, Yasuda M, Kume S, Koya D, Maegawa H, Uzu T. The role of autophagy in the pathogenesis of diabetic nephropathy. J Diabetes Res 2013; 2013:193757; PMID:24455746; http://dx.doi.org/10.1155/2013/193757
- Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 2010; 120:1084–96; PMID:20200449; http://dx.doi.org/10.1172/JCI39492
- Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S, Sugimoto T, Haneda M, Kashiwagi A, Koya D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest 2010; 120:1043–55; PMID:20335657; http://dx.doi.org/10.1172/JCI41376
- Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol 2012; 180:517–25; PMID:22265049; http://dx.doi.org/10.1016/j.ajpath.2011.11.001
- Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 2012; 82:1271–83; PMID:22854643; http://dx.doi.org/10.1038/ki.2012.261
- Kaushal GP, Kaushal V, Herzog C, Yang C. Autophagy delays apoptosis in renal tubular epithelial cells in cisplatin cytotoxicity. Autophagy 2008; 4:710–2; PMID:18497570; http://dx.doi.org/10.4161/auto.6309
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010; 176:85–97; PMID:20008127; http://dx.doi.org/10.2353/ajpath.2010.090517
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809–20; PMID:14638851; http://dx.doi.org/10.1172/JCI20039
- Mizushima N. Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol 2009; 452:13–23; PMID:19200873; http://dx.doi.org/10.1016/S0076-6879(08)03602-1
- Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, Nishiyama A, Ichikawa I. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol 2012; 23:1181–9; PMID:22518004; http://dx.doi.org/10.1681/ASN.2011121159
- Mao H, Li Z, Zhou Y, Li Z, Zhuang S, An X, Zhang B, Chen W, Nie J, Wang Z, et al. HSP72 attenuates renal tubular cell apoptosis and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 2008; 295:F202–14; PMID:18417540; http://dx.doi.org/10.1152/ajprenal.00468.2007
- Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 2005; 280:20722–9; PMID:15778222; http://dx.doi.org/10.1074/jbc.M413934200