
ABSTRACT
Adenosine monophosphate-activated protein kinase (AMPK) is a crucial energy sensor that maintains cellular energy homeostasis. AMPK plays a critical role in macroautophagy/autophagy, and autophagy facilitates hepatitis B virus (HBV) replication. To date, the intrinsic link among AMPK, autophagy and HBV production remains to be elucidated. Here, we demonstrate that PRKAA (a catalytic subunit of AMPK) is activated in response to HBV-induced oxidative stress, which in turn decreases the production of HBV. Mechanistic studies reveal that the autophagy machinery is associated with the inhibitory effect of PRKAA/AMPK on HBV production. Activation of PRKAA/AMPK promotes autolysosome-dependent degradation through stimulation of cellular ATP levels, which then leads to the depletion of autophagic vacuoles. Taken together, our data suggest that the activation of AMPK might be a stress response of host cells to restrict virus production through promotion of autophagic degradation. These findings therefore indicate that AMPK could provide a potential therapeutic target for HBV infection.
Introduction
Hepatitis B virus (HBV) is a strictly hepatotropic DNA virus of the Hepadnaviridae family.Citation1 Chronic hepatitis B virus infection represents a severe public health burden in many parts of the world.Citation2 HBV has recently been characterized as a “metabolovirus” due to the modulation of host metabolic patterns to provide energy and building blocks for HBV replication.Citation3 Adenosine monophosphate-activated protein kinase (AMPK) is a highly conserved intracellular energy sensor that acts as a guard to maintain cellular energy stores by either switching on catabolic pathways that produce ATP or switching off anabolic pathways that consume ATP.Citation4,5 However, whether AMPK plays a role in HBV replication remains poorly understood.
AMPK is a metabolic checkpoint that plays a crucial role in the modulation of the autophagy process.Citation6 Autophagy is initiated in response to poor-nutrient or low-energy conditions, acting as a survival mechanism to ensure availability of critical metabolic intermediates.Citation7 AMPK triggers the initiation of autophagy through regulation of early autophagic events, such as phosphorylation and activation of ULK1, inhibition of MTOR and regulation of PIK3C3/VPS34 complexes.Citation7-8 Interestingly, AMPK is also implicated in a late stage of autophagy, such as autophagic proteolysis.Citation9 However, the underlying mechanism remains to be elucidated.
Autophagy plays a critical role in HBV replication.Citation10 HBV can activate the early autophagic pathway to promote viral DNA replication.Citation10,11 Additionally, studies performed in HBV-transgenic mice with liver-specific knockout of Atg5 further suggest that the autophagy machinery is required for efficient HBV DNA replication in vivo.Citation12 Intriguingly, recent studies reveal that HBV can induce the early stages of autophagy but block autophagic degradation to benefit its propagation, suggesting that autophagic degradation may negatively regulate HBV replication.Citation10,13
In this study, we show that PRKAA/AMPK is activated in host cells in response to HBV-induced reactive oxygen species (ROS) accumulation, which in turn represses HBV production. Further studies reveal that active PRKAA/AMPK increases the cellular ATP levels that promote autophagic degradation, leading to the decreased production of HBV particles. Collectively, our data have delineated a novel molecular mechanism whereby oxidative stress-induced AMPK activation negatively regulates the production of HBV particles by promotion of autophagic degradation, suggesting that AMPK activators could be utilized at least as adjuvant anti-HBV agents.
Results
PRKAA/AMPK is activated by HBV-induced ROS accumulation
HBV switches the metabolic pattern of host cells to provide energy and building blocks for its replication.Citation3 To determine whether AMPK, an important cellular metabolic checkpoint, is involved in HBV replication, we assessed PRKAA/AMPK activity in HepG2.2.15 cells that stably express HBV (hereafter termed as HBV-producing cells) by immunoblot analysis of the phosphorylated form of PRKAA. The results revealed that HBV-producing cells, as indicated by the expression of hepatitis B virus core antigen (HBcAg), displayed significantly increased levels of phosphorylated PRKAA (Thr172) ().
Figure 1. PRKAA is activated in response to HBV-induced ROS accumulation. (A) HepAD38 cells were grown with tetracycline (Tet+) or without tetracycline for 10 d. Control cells (HepG2 or HepAD38 [Tet+] cells), and HBV-producing cells (HepG2.2.15 or HepAD38 cells) were lysed and analyzed by immunoblot with the indicated antibodies. Relative intensity of the band was quantified by normalization to PRKAA using ImageJ software. p-, phosphorylated. (B) The phosphorylation levels of PRKAA (Thr172) in HBV-infected (HBV+) and HBV noninfected (HBV−) liver samples were determined by immunoblot. Densitometry quantification of the band intensities in Fig. S2 was carried out using ImageJ software and was shown as a percentage of relative densitometry normalized to ACTB. The mean ± SD densities were displayed in relation to HBV noninfected (HBV−) tissues. (C) The ROS level was monitored with an oxidant-sensitive fluorescent probe, DCFH-DA. Data were shown as mean ± SD of 3 independent experiments. (D) Cells were mock-treated or treated with NAC (10 mM) for 2 h followed by immunoblot analysis. (E) Cells were treated with DMSO or STO-609 (10 μg/mL) for 2 h followed by immunoblot analysis. Relative intensity of the indicated protein bands was quantified by normalization to PRKAA using ImageJ software. *, p < 0.05; **, p < 0.01.
![Figure 1. PRKAA is activated in response to HBV-induced ROS accumulation. (A) HepAD38 cells were grown with tetracycline (Tet+) or without tetracycline for 10 d. Control cells (HepG2 or HepAD38 [Tet+] cells), and HBV-producing cells (HepG2.2.15 or HepAD38 cells) were lysed and analyzed by immunoblot with the indicated antibodies. Relative intensity of the band was quantified by normalization to PRKAA using ImageJ software. p-, phosphorylated. (B) The phosphorylation levels of PRKAA (Thr172) in HBV-infected (HBV+) and HBV noninfected (HBV−) liver samples were determined by immunoblot. Densitometry quantification of the band intensities in Fig. S2 was carried out using ImageJ software and was shown as a percentage of relative densitometry normalized to ACTB. The mean ± SD densities were displayed in relation to HBV noninfected (HBV−) tissues. (C) The ROS level was monitored with an oxidant-sensitive fluorescent probe, DCFH-DA. Data were shown as mean ± SD of 3 independent experiments. (D) Cells were mock-treated or treated with NAC (10 mM) for 2 h followed by immunoblot analysis. (E) Cells were treated with DMSO or STO-609 (10 μg/mL) for 2 h followed by immunoblot analysis. Relative intensity of the indicated protein bands was quantified by normalization to PRKAA using ImageJ software. *, p < 0.05; **, p < 0.01.](/cms/asset/361b5077-9bf9-48fd-a3e7-4873e495ecc9/kaup_a_1191857_f0001_b.gif)
To elucidate whether this difference was specific to HBV replication or due to physiological differences between HepG2.2.15 cells and the parental HepG2 cells, a second cell line (HepAD38, a HepG2-derived cell line expressing HBV under tetracycline-off controlCitation14) was studied. As shown in and S1, the phosphorylation level of PRKAA (Thr172) was increased in the HBV-producing cells (HepAD38) compared to the levels in cells maintained in tetracycline to suppress virus production (HepAD38 [Tet+]). These data demonstrated that PRKAA/AMPK was activated in response to HBV replication.
To further investigate the correlation of the activation of PRKAA/AMPK with HBV replication, we measured the activity of PRKAA (Thr172) in liver tissues from HBV-infected and HBV noninfected patients. Consistent with the data obtained in the cell lines, the phosphorylation levels of PRKAA were significantly increased in HBV-infected tissues compared with HBV noninfected tissues ( and S2), indicating an activation of PRKAA upon HBV infection. Taken together, these results suggest that PRKAA/AMPK was activated in response to HBV replication.
Chronic HBV infection causes sustained oxidative stress in host cells.Citation15 In addition, PRKAA/AMPK could be activated in response to cellular oxidative stress.Citation16 The results revealed that HBV replication induced mitochondrial ROS, as NAC (N-acetyl-L-cysteine, a general ROS scavenger) and rotenone (an inhibitor of mitochondrial respiratory chain complex I, which blocks mitochondrial superoxide production) abolished the increased ROS in HBV-producing cells, while apocynin (an NADPH oxidase inhibitor) and NDGA (a LOX-specific inhibitor) failed to prevent ROS productionCitation17 ( and S3A–S3D). To determine whether the activation of PRKAA/AMPK in HBV-producing cells was mediated by virus-induced ROS, phosphorylation of PRKAA was examined in HBV-producing cells treated with the reducing agent NAC (). These results showed that NAC treatment diminished PRKAA activation in HBV-producing cells, indicating that ROS was required for HBV-induced PRKAA activation. ROS could activate AMPK through CAMKK2/CaMKKβ or STK11/LKB1, 2 key upstream kinases activating AMPK.Citation18,19 Our results showed that knockdown of STK11 by siRNA did not alter the phosphorylation level of PRKAA (Fig. S4), but inhibition of CAMKK2 by STO-609 attenuated PRKAA activation (), indicating that CAMKK2 was involved in ROS-induced activation of PRKAA/AMPK in HBV-producing cells.
A recent study has demonstrated that TXN (thioredoxin) plays a crucial role in PRKAA/AMPK activation by preventing ROS-induced PRKAA aggregation.Citation20 As shown in Fig. S5A, H2O2 or diamide (a thiol oxidizing compound) induced a mobility shift of PRKAA in HepG2 cells, which could be reversed by dithiothreitol, a reducing agent that breaks disulfide bonds. However, there was no significant mobility shift of PRKAA in response to HBV-induced oxidative stress (Fig. S5B). To determine whether TXN functioned as the reducing force protecting PRKAA from oxidative aggregation, we first evaluated the redox status of cysteines in TXN using maleimide-polyethylene glycol (Mal-PEG), which covalently binds to the reduced form of thiols. The levels of the Mal-PEG-labeled reduced form of TXN decreased significantly in HBV-producing cells compared with parental cells (Fig. S5C). However, reduced TXN remained detectable in HBV-producing cells, suggesting that TXN successfully protected PRKAA from oxidative aggregation. Moreover, overexpression of TXNIP (thioredoxin interacting protein), which binds to the redox active site of TXN and inhibits its thioredoxin activity,Citation21 markedly suppressed the activity of PRKAA, as indicated by the decreased phosphorylation levels of PRKAA (Fig. S5D). Taken together, these results confirmed that PRKAA/AMPK was activated in response to HBV-induced ROS accumulation, depending on the oxidoreductase activity of TXN (Fig. S5E).
Active PRKAA/AMPK negatively regulates the production of HBV
To determine the role of PRKAA/AMPK in HBV production, we treated the HBV-producing cells with AICAR (an AMPK agonist,Citation22 Figs. S6A–S6B and S7A) and analyzed the levels of the extracellular HBV DNA using real-time PCR. As depicted in , the levels of extracellular virus were significantly decreased in AICAR-treated cells. Additionally, compound C (an AMPK inhibitorCitation23) resulted in a 2-fold increase in extracellular HBV (Figs. S6C–S6D and S7B), suggesting that PRKAA/AMPK activity negatively regulated HBV production. Consistent with compound C treatment, genetic depletion of PRKAA caused an increased production of the viral particles ( and S8), confirming that PRKAA/AMPK played an important role in viral production. To further investigate the correlation between HBV replication and PRKAA activation, we examined the expression of HBcAg, which is a hepatitis B viral protein and an indicator of active viral replication. As shown in Fig. S9, HBcAg expression was significantly higher in cells treated with compound C or transfected with PRKAA-specific siRNA compared with that in corresponding control groups, while AICAR treatment decreased the HBcAg levels.
Figure 2. PRKAA activation restricts HBV production. (A, B) HepG2.2.15 and HepAD38 cells were incubated with DMSO, compound C (CC, 10 µM), or AICAR (1 mM) for 24 h (A), or transfected with either siScramble or siPRKAA1/2 for 48 h (B), respectively. HBV progeny DNA in the supernatant was quantified by real-time PCR. The values obtained from the control group were set at 1.0. *, p < 0.05; **, p < 0.01. (C, D) BALB/c mice were hydrodynamically injected with vector, HBV1.3 and vector, HBV1.3 and/or DN-PRKAA1. HBV serum titer was determined by quantitative real-time PCR at d 3 post injection (C). The expressions of HBcAg in liver tissues were determined by immunochemical analysis (D). Data are presented as mean ± SD (n = 6); **, p < 0.01; ***, p < 0.001.
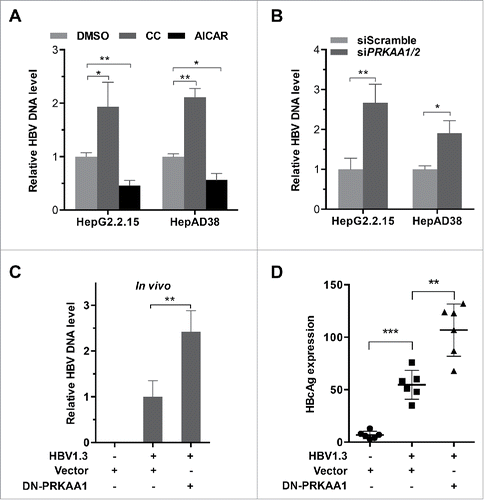
In addition, we validated the antiviral effect of PRKAA/AMPK in vivo. BALB/c mice were hydrodynamically co-injected with HBV1.3 and a plasmid encoding dominant-negative PRKAA1 (DN-PRKAA1) or vector control plasmid, respectively. Serum samples were harvested at d 3 post-injection and secreted HBV levels were analyzed. Consistent with the in vitro results, inhibition of the activity of PRKAA through overexpression of DN-PRKAA1 significantly enhanced the concentration of HBV particles in serum compared to the control mice (). Moreover, we also observed that DN-PRKAA1 increased the expression of HBcAg in HBV-infected mice ( and S10), suggesting that HBV replication was correlated with the activation of PRKAA/AMPK. Collectively, these data demonstrated that PRKAA/AMPK acted as a limitation factor against HBV replication in host cells.
Autophagy is associated with PRKAA/AMPK-mediated restriction of HBV production
PRKAA/AMPK plays a critical role in the initiation of autophagy.Citation24 Additionally, HBV induces autophagy, which in turn facilitates HBV genome replication and persistent infection.Citation11 Indeed, we found an increase of LC3B-II conversion (the phagophore and autophagosome-associated lipidated form of LC3B) and an elevated number of LC3B or GFP-LC3B (a green fluorescent autophagy reporter protein) puncta in HBV-producing cells ( and S11). Additionally, the HBV-producing mice liver tissues displayed higher expression levels of LC3B, suggesting an increased autophagic activity (Fig. S10). To investigate whether autophagy was involved in PRKAA/AMPK-regulated HBV production, we treated HBV-producing cells with 3-methyladenine (3-MA; an inhibitor of autophagyCitation25) in combination with compound C. We found that inhibition of autophagy by 3-MA abrogated the enhanced production of viral particles in response to PRKAA inactivation (). Additionally, we employed an ATG5-specific siRNA to decrease ATG5 expression (Fig. S12), thus blocking autophagic events. As shown in , siATG5 transfection reduced the level of HBV production induced by compound C treatment. Moreover, we found that siATG5 transfection could also reverse the increased production of viral particles induced by genetic depletion of PRKAA (). Taken together, these results demonstrated that inhibition of PRKAA/AMPK enhanced the production of HBV via autophagy.
Figure 3. Autophagy is involved in PRKAA-mediated regulation of HBV production. (A) Control cells (HepAD38 [Tet+] or HepG2 cells), and HBV-producing cells (HepAD38 or HepG2.2.15 cells) were lysed and analyzed by immunoblot. Relative intensity of LC3B-II was quantified by normalization to ACTB using ImageJ software. The mean ± SD densities were displayed in relation to HepG2. (B) Immunofluorescence analysis of LC3B puncta in HepG2, HepAD38 [Tet+], HepG2.2.15 and HepAD38 cells. The fluorescent signal was visualized using a Leica DM2500 microscope. The number of LC3B puncta (mean ± SD) was quantified by ImageJ software. Scale bar: 10 μm. **, p < 0.01. (C) Cells were treated with either DMSO or CC (10 µM) for 24 h after pretreatment for 2 h with 3-MA (5 mM). (D) HepG2.2.15 and HepAD38 cells were transfected with siScramble or siATG5 for 48 h, and then treated with DMSO or CC (10 µM) for 24 h. (E) HepG2.2.15 and HepAD38 cells were transfected with siScramble, siPRKAA1/2, or cotransfected with siPRKAA1/2 and siATG5. HBV progeny DNA in the supernatant was quantified by real-time PCR. The values obtained from the control group were set at 1.0. Values were means ± SD, n = 3 per group. *, p < 0.05; **, p < 0.01.
![Figure 3. Autophagy is involved in PRKAA-mediated regulation of HBV production. (A) Control cells (HepAD38 [Tet+] or HepG2 cells), and HBV-producing cells (HepAD38 or HepG2.2.15 cells) were lysed and analyzed by immunoblot. Relative intensity of LC3B-II was quantified by normalization to ACTB using ImageJ software. The mean ± SD densities were displayed in relation to HepG2. (B) Immunofluorescence analysis of LC3B puncta in HepG2, HepAD38 [Tet+], HepG2.2.15 and HepAD38 cells. The fluorescent signal was visualized using a Leica DM2500 microscope. The number of LC3B puncta (mean ± SD) was quantified by ImageJ software. Scale bar: 10 μm. **, p < 0.01. (C) Cells were treated with either DMSO or CC (10 µM) for 24 h after pretreatment for 2 h with 3-MA (5 mM). (D) HepG2.2.15 and HepAD38 cells were transfected with siScramble or siATG5 for 48 h, and then treated with DMSO or CC (10 µM) for 24 h. (E) HepG2.2.15 and HepAD38 cells were transfected with siScramble, siPRKAA1/2, or cotransfected with siPRKAA1/2 and siATG5. HBV progeny DNA in the supernatant was quantified by real-time PCR. The values obtained from the control group were set at 1.0. Values were means ± SD, n = 3 per group. *, p < 0.05; **, p < 0.01.](/cms/asset/dad9ccb2-18b7-4eb5-96c4-4fcc7a787162/kaup_a_1191857_f0003_oc.gif)
PRKAA/AMPK reduces autophagosome accumulation by promoting autophagic flux
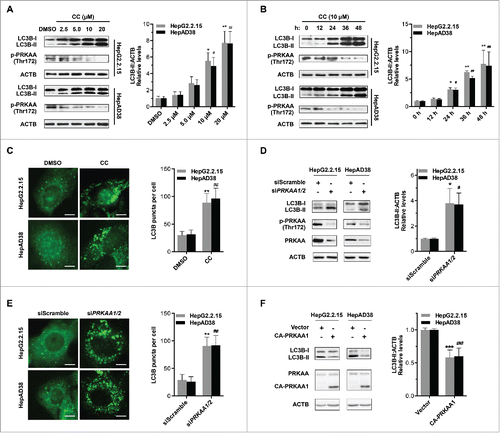
To further determine whether PRKAA/AMPK regulated HBV production by modulation of the autophagic pathway, we analyzed the role of PRKAA/AMPK in autophagy. It was shown that compound C treatment resulted in enhanced LC3B-II conversion in a dose- and time-dependent manner (), which was consistent with the increased numbers of LC3B puncta (). Additionally, we observed elevated levels of LC3B-II conversion () and increased numbers of LC3B puncta () in siPRKAA1/2-treated cells. Consistently, similar results were observed in vivo where DN-PRKAA1 induced a significant increase in the expression level of LC3B (Fig. S10). To further corroborate these findings, we investigated whether constitutively active PRKAA1 (CA-PRKAA1) could alter the levels of LC3B-II. As shown in , CA-PRKAA1-transfected cells displayed decreased levels of LC3B-II. These data indicated that activation of PRKAA/AMPK decreased the accumulation of autophagosomes in HBV-producing cells.
Figure 4. Inhibition of PRKAA contributes to autophagosome accumulation. (A) HepG2.2.15 or HepAD38 cells were treated with different concentrations of CC (0, 2.5, 5.0, 10, 20 μM) for 24 h, and cell lysates were subjected to immunoblot assay. (B) HepG2.2.15 or HepAD38 cells were treated with DMSO or 10 μM CC as indicated (0, 12, 24, 36 and 48 h) and subjected to immunoblot assay. (C) Immunofluorescence analysis of LC3B puncta in cells that were treated with DMSO or CC (10 μM) for 24 h. (D) Immunoblot analysis of total protein extracts from HepG2.2.15 and HepAD38 cells transfected with siScramble or siPRKAA1/2 for 48 h, respectively. Relative intensity of LC3B-II was quantified by normalization to ACTB using ImageJ software. (E) Immunofluorescence analysis of LC3B puncta in cells that were transfected with siScramble, or siPRKAA1/2 for 48 h. The number of LC3B puncta (mean ± SD) was quantified by ImageJ software. Values are means ± SD (n = 30). (F) Immunoblot analysis of total protein extracts from HepG2.2.15 and HepAD38 cells transfected with vector or plasmid encoding CA-PRKAA1 for 48 h. Relative intensity of LC3B-II was quantified by normalization to ACTB using ImageJ software. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (in HepG2.2.15); #, p < 0.05; ##, p < 0.01; ###, p < 0.001 (in HepAD38). Scale bar: 10 μm.
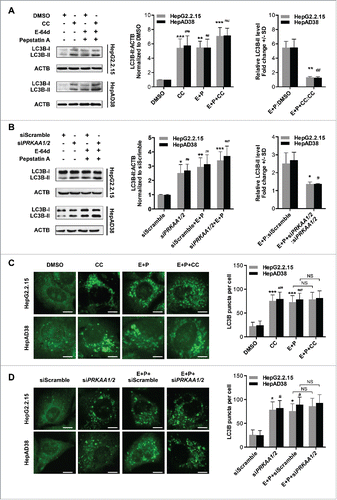
To evaluate whether the autophagic flux is affected by PRKAA/AMPK, we inhibited the activity of PRKAA using compound C or depleted the expression of PRKAA in the presence or absence of E-64d and pepstatin A (inhibitors of cysteine and aspartic proteases in lysosomesCitation26). Treatment with lysosomal protease inhibitors induced a significantly increased level of LC3B-II under normal conditions, but only a slight increase in the presence of compound C (5.47-fold vs. 1.32-fold in HepG2.2.15 cells, and 5.49-fold vs. 1.25-fold in HepAD38 cells; ) or siPRKAA1/2 (2.51-fold vs. 1.36-fold in HepG2.2.15 cells, and 2.69-fold vs. 1.37-fold in HepAD38 cells; ). Likewise, when the cells were treated with compound C or transfected with siPRKAA1/2, addition of E-64d and pepstatin A resulted in no obvious increase in the number of LC3B puncta (), indicating that inhibition of PRKAA blocked autophagic flux. Similar results were obtained by substituting lysosome protease inhibitors with chloroquine, a lysosomotropic agent that also blocks autophagic fluxCitation27 (Fig. S13). Taken together, these results demonstrated that the inhibition of PRKAA/AMPK-induced autophagosome accumulation was due to the blockage of autophagic flux in HBV-producing cells.
Figure 5. PRKAA activity is required for autophagic flux. (A) Immunoblot analysis of total protein extracts from cells treated with DMSO (0.1%), or CC (10 μM) in the absence or presence of E-64d (E, 10 µg/mL) and pepstatin A (P, 10 µg/mL) for 24 h. (B) HepG2.2.15 or HepAD38 cells were transfected with siScramble or siPRKAA1/2 for 48 h, and then treated with E-64d (E, 10 µg/mL) and pepstatin A (P, 10 µg/mL) for 24 h. The total protein extracts were subjected to immunoblot assay. Relative intensity of LC3B-II was quantified by normalization to ACTB by ImageJ software. Values were means ± SD (n = 3). (C) Immunofluorescence analysis of LC3B puncta in cells that were incubated with DMSO (0.1%), CC (10 µM), or CC in combination with E-64d and pepstatin A (E+P, 10 µg/mL each) for another 24 h. (D) Immunofluorescence analysis of LC3B puncta in cells that were transfected with siScramble or siPRKAA1/2, followed by incubation with E-64d and pepstatin A (E+P, 10 µg/mL each) for another 24 h. The fluorescent signal was visualized using a Leica DM2500 microscope. The number of LC3B puncta (mean ± SD) was quantified by ImageJ software. Values were means ± SD (n = 30). *, p < 0.05; **, p < 0.01; ***, p < 0.001 (in HepG2.2.15); #, p < 0.01; ##, p < 0.01; ###, p < 0.001 (in HepAD38); NS, non-significant. Scale bar: 10 μm.
PRKAA/AMPK activation supports autophagic degradation
We further investigated the mechanism by which PRKAA/AMPK regulated the autophagic flux. During autophagosome formation, SQSTM1/p62-mediated cargo incorporation occurs,Citation28 which can be monitored by imaging the colocalization of SQSTM1 and LC3B. As shown in , colocalization between LC3B and SQSTM1 was more prominent in compound C-treated or siPRKAA1/2-transfected cells compared with the control cells, suggesting that PRKAA-deficient cells exhibited a functional incorporation of SQSTM1-labeled cargo into autophagosomes. Additionally, the protease protection assay was performed to measure the completion of the autophagosomes induced by PRKAA inhibition. After completion of autophagosome formation, SQSTM1 is sequestered within autophagosomes and becomes resistant to proteinase K digestion, whereas membrane permeabilization by Triton X-100 enables SQSTM1 digestion.Citation29 Upon proteinase K treatment, we found that in the absence of Triton X-100, SQSTM1 was protected in compound C-treated cells, whereas addition of Triton X-100 promoted the digestion of SQSTM1 (Fig. S14), indicating that the incorporation of SQSTM1-labeled cargo within completed autophagosomes was actually functional in PRKAA-deficient cells.
Figure 6. Active PRKAA enables the degradation of autophagosomes. (A) Colocalization analysis of LC3B (green) and SQSTM1 (red) in HepG2.2.15 or HepAD38 cells treated with DMSO (0.1%), compound C (CC, 10 μM), or transfected with siScramble or siPRKAA1/2 for 48 h, respectively. Scale bar: 10 μm. (B) Colocalization analysis of LC3B (green) and LAMP1 (red) in HepG2.2.15 or HepAD38 cells treated with DMSO (0.1%), compound C (CC, 10 μM), or transfected with siScramble or siPRKAA1/2 for 48 h, respectively. Scale bar: 10 μm. (C) Colocalization analysis of SQSTM1 (green) and LAMP1 (red) in HepG2.2.15 or HepAD38 cells treated with DMSO (0.1%), compound C (CC, 10 μM), or transfected with siScramble or siPRKAA1/2 for 48 h, respectively. The quantitative colocalization analysis of 2 immunoreactivities was performed with ImageJ Colocalization Finder plug-in software. Scale bar: 10 μm. (D) Autolysosomes stained with DQ-BSA in cells treated with DMSO (0.1%), compound C (CC, 10 μM), bafilomycin A1 (100 nM), cultured in medium without fetal bovine serum (Starvation), or transfected with siPRKAA1/2, respectively. Accumulation of fluorescent signal, indicating the autolysosomal proteolysis of DQ-BSA, was recorded by microscopy. Scale bar: 20 μm. **, p < 0.01; ***, p < 0.001 (in HepG2.2.15); ##, p < 0.01; ###, p < 0.001 (in HepAD38).
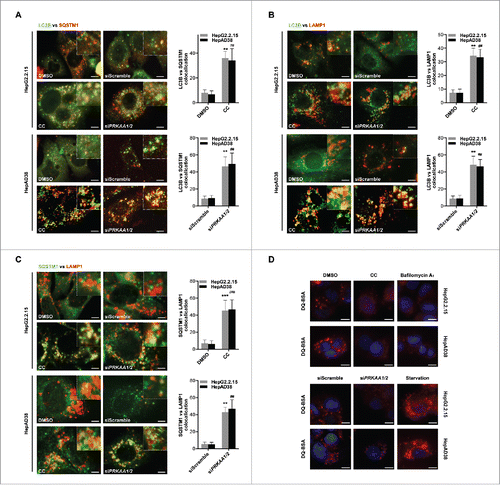
To examine whether the accumulation of autophagic vacuoles resulted from insufficient fusion of the autophagosomes with endosomes and/or lysosomes, we examined the colocalization of LC3B and LAMP1. As shown in , the density of LC3B-LAMP1 double-positive dots, which represent amphisome or autolysosomes, was increased in compound C-treated or siPRKAA1/2-transfected cells compared with the control cells, suggesting that the fusion of autophagosomes with endosomes or lysosomes was promoted in PRKAA-deficient cells. Next, we determined whether autolysosome formation was completed in PRKAA-deficient cells. We found that depletion of PRKAA enhanced the colocalization between SQSTM1 and LAMP1, indicating an increased sequestration of SQSTM1 within amphisomes or autolysosomes (). Additionally, we performed the self-quenched fluorophore DQ-BSA degradation assay to determine whether PRKAA activation influences the autophagic degradation rate. As shown in , the fluorescence intensity generated from lysosomal proteolysis of DQ-BSA was weaker in compound C-treated or siPRKAA1/2-transfected cells than that in the control cells, suggesting that the proteolysis activity of autolysosomes was greatly impaired in PRKAA-deficient cells. These results indicated that activation of PRKAA/AMPK was required for autolysosomal degradation.
ATP is required for PRKAA/AMPK-mediated promotion of autophagic degradation
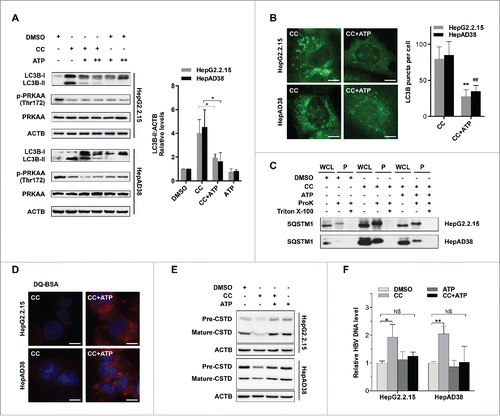
To investigate the mechanism underlying the blockage of autophagic proteolysis mediated by PRKAA/AMPK inactivation, we first checked the biogenesis and acidification capacity of lysosomes. We found that inhibition of PRKAA had no effect on the biogenesis or the acidification capacity of lysosomes (Figs. S15 and S16). PRKAA/AMPK conserves cellular ATP levels by switching off anabolic pathways that consume ATP and switching on catabolic pathways that generate ATP.Citation30 As shown in Fig. S17, inactivation of PRKAA in hepatocytes by compound C decreased the cellular ATP levels, whereas activation of PRKAA by either AICAR treatment or CA-PRKAA1 transfection elevated the levels of ATP. ATP can activate proteolysis in lysosomes of liver cells.Citation31 Thus, we presumed that PRKAA deficiency-induced impairment of autophagic proteolysis might result from decreased ATP levels. To verify this hypothesis, we added disodium ATP (5′-ATP-Na2) to replenish the cellular ATP levels. As depicted in , treatment with 5′-ATP-Na2 significantly mitigated the protein levels of LC3B-II in compound C-treated cells. Consistent with the immunoblot results (), immunofluorescence analysis revealed that 5′-ATP-Na2 could alleviate the PRKAA inhibition-induced accumulation of LC3B puncta (), suggesting that PRKAA/AMPK activity was essential for autophagic degradation by maintaining cellular ATP levels. In addition, treatment of 5′-ATP-Na2 could reverse the accumulation of SQSTM1 and inhibition of proteolysis activity caused by compound C treatment (). To investigate the mechanism of ATP-induced promotion of lysosomal degradation, we examined the number and acidification ability of lysosomes upon 5′-ATP-Na2 treatment. As shown in Fig. S15 and S16, ATP did not alter either the number or acidification ability of lysosomes. ATP can activate lysosomal proteases, including CTSD (cathepsin D).Citation32 The inactive form of CTSD (44 kDa) can be cleaved into active form (31 kDa) in mature lysosomes.Citation13 We found that inhibition of PRKAA with compound C reduced the mature form of CTSD (31 kDa), while addition of 5′-ATP-Na2 could restore the maturation of CTSD (), suggesting that ATP might promote lysosomal degradation through induction of CTSD maturation. Since ATP was required for the degradation of autophagic vacuoles, we then assessed the effect of ATP on HBV production. As shown in , addition of 5′-ATP-Na2 could reverse compound C-mediated enhanced production of HBV particles. These results suggested that PRKAA/AMPK activation might repress HBV production through promotion of autophagosome degradation.
Figure 7. ATP is required for PRKAA-mediated autophagosome degradation. (A) Immunoblot analysis for LC3B in HepAD38 cells and HepG2.2.15 cells treated with DMSO (0.1%, lane 1), compound C (CC, 10 μM, lane 2), CC plus 5′-ATP-Na2 (0.25 mM, lane 3), CC plus 5′-ATP-Na2 (0.5 mM, lane 4), 5′-ATP-Na2 (0.25 mM, lane 5), and 5′-ATP-Na2 (0.5 mM, lane 6), respectively. Quantification of the LC3B-II was quantified by normalization to ACTB by ImageJ software. (B) HepG2.2.15 and HepAD38 cells were incubated with CC (10 µM) or CC together with 5′-ATP-Na2 (ATP, 0.25 mM) for 24 h, and subjected to the immunofluorescence detection of LC3B. **, p < 0.01 (in HepG2.2.15); ##, p < 0.01 (in HepAD38). Scale bar: 10 μm. (C) HepG2.2.15 and HepAD38 cells were incubated with DMSO, CC (10 µM) or CC together with 5′-ATP-Na2 (ATP, 0.25 mM) for 24 h, and followed by the proteinase K (ProK) protection assay for SQSTM1. WCL, whole cell lysate; P, pellet fraction. (D) Autolysosomes stained with DQ-BSA in cells treated with DMSO (0.1%), compound C (CC, 10 μM), or CC together with 5′-ATP-Na2 (ATP, 0.25 mM) for 24 h, respectively. Scale bar: 20 μm. (E) HepG2.2.15 and HepAD38 cells were incubated with DMSO, CC (10 µM) or CC together with 5′-ATP-Na2 (ATP, 0.25 mM) for 24 h, and subjected to immunoblot analysis for CTSD. (F) HepG2.2.15 and HepAD38 cells were incubated with DMSO, CC (10 µM), 5′-ATP-Na2 (ATP, 0.25 mM) or CC in combination with 5′-ATP-Na2 for 24 h. HBV titer in the supernatant was quantified by real-time PCR. The values obtained from DMSO-treated samples were set at 1.0. Values were means ± standard error. n = 3 per group. *, p < 0.05; **, p <0.01; NS, non-significant.
Discussion
Viruses hijack metabolism in host cells to acquire energy and building blocks for their replication.Citation33 Metabolic reprogramming can cause dysfunction of the mitochondrial respiratory chain, resulting in ROS overproduction.Citation34 Sustained oxidative stress is a hallmark of chronic HBV infection, which is associated with many liver diseases, including fibrosis, cirrhosis and hepatocellular carcinoma.Citation35 AMPK plays a crucial role in maintaining cellular energy homeostasis.Citation36 Recent studies have indicated that the AMPK activity can be regulated by redox modification under oxidative stress.Citation20 In this study, we found that AMPK was activated by HBV-induced oxidative stress, indicating that virus-induced ROS accumulation could exert a feedback regulation on metabolic stress. TXN, an important reducing enzyme that catalyzes disulfide reduction, serves as a vital cofactor for AMPK activation by preventing the oxidative aggregation of AMPK.Citation20 Our data revealed that HBV-producing cells maintained relatively high levels of reduced TXN to block the excessive oxidative modification, suggesting that the antioxidant system of host cells was delicately controlled to mediate the activation of metabolic sensors, such as AMPK, thus relieving the metabolic stress induced by robust virus replication.
Autophagy is emerging as an important component of host defense during the infection of certain viruses (such as herpes simplex virus).Citation37 In contrast, other viruses, including HBV, can manipulate the autophagic process to facilitate their replication.Citation38 HBV induces the early autophagic pathway in hepatocyte cells,Citation11 while increasing evidence indicates that the late stages of autophagy (e.g., autophagic degradation) are blocked in HBV-producing cells,Citation10,11,13 suggesting that autophagosome accumulation resulted from impaired autophagic flux may be beneficial for HBV production. HBV small surface proteins, a key component of virion maturation, can colocalize and interact with LC3B during HBV replication, suggesting that autophagosomes or other autophagic vacuoles might provide a physical scaffold for HBV envelopment.Citation10 Together with this observation, our study reveals that elimination of autophagosomes impairs the production of HBV, supporting a notion that the early and late stages of autophagy may have distinct effects on HBV replication.
AMPK plays an essential role in the induction of autophagy under energy-deprived conditions.Citation39 AMPK activates the proautophagic PIK3C3/VPS34 complex by phosphorylating BECN1/Beclin 1 to trigger autophagy.Citation8 In addition, AMPK can initiate autophagy via a double-pronged mechanism, by which AMPK inhibits MTORC1 or directly activates ULK1.Citation7 Besides the effect of AMPK on the early stage of autophagy, our results showed that AMPK could also enhance the degradation ability of autolysosomes. This observation was supported by a previous report that inhibition of AMPK activity could impair autophagic proteolysis.Citation9 Collectively, these findings revealed that AMPK activity was required for a late stage of autophagy. HBV X protein can inhibit autophagic degradation by impairing lysosomal maturation.Citation13 Consistently, we found that activation of AMPK elevated cellular ATP levels and induced the maturation of lysosomal proteases concomitantly, and therefore promoted autophagic degradation. Taken together, our study revealed a novel regulatory mechanism through which AMPK could restrict HBV replication through enhancement of autophagic proteolysis.
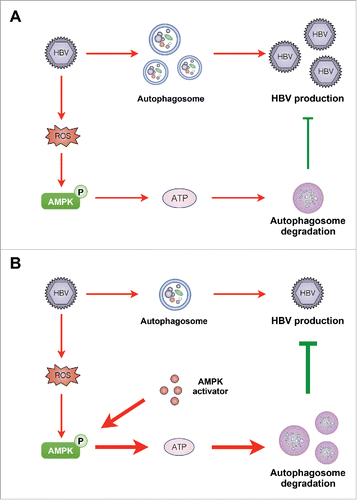
In this study, we explored the interplay among AMPK, autophagy and HBV. As depicted in , activation of AMPK inhibited HBV production through promotion of autophagic degradation. Our data indicate that AMPK activation may be an intrinsic response of host cells to restrict virus production, suggesting that AMPK activators hold promise for further improvement of anti-HBV therapy.
Figure 8. Diagram showing the proposed mechanism by which PRKAA/AMPK inhibits HBV production through promotion of autophagosome degradation. (A) Under normal conditions, AMPK is activated in response to HBV replication-induced production of cellular ROS. The activated AMPK promotes autophagic degradation and restricts the production of HBV. (B) Upon treatment with AMPK activator, increased activity of AMPK elevates the cellular ATP levels, leading to the promotion of autophagic degradation and elimination of HBV production.
Materials and methods
Antibodies and reagents
The following reagents were used: Compound C (EMD Millipore, 171260); AICAR (Beyotime, S1515); rapamycin (Sigma, R8781), bafilomycin A1 (Sigma, B1793), E-64d (Sigma, E8640), pepstatin A (Sigma, P4265), chloroquine (Sigma, C6628), 5′-ATP-Na2 (Sigma, A26209), 2-deoxyglucose (Sigma, D8375), and protease inhibitor cocktail (Sigma, P8340). NDGA (Sigma, N2036). MitoSOX (Thermo Fisher Scientific, M36008). Diamide (Sigma, D3648). Dithiothreitol (Sigma, 10197777001). LysoTracker Red (Beyotime, C1046).
Antibodies used in this study were: ACTB (Santa Cruz Biotechnology, sc-69879), ATG5 (Cell Signaling Technology, 2630), goat anti-mouse IgG (H+L) secondary antibody, HRP conjugate (Thermo Fisher Scientific, 31430), goat anti-rabbit IgG (H+L) secondary antibody, HRP conjugate (Thermo Fisher Scientific, 31460), HBcAg (EMD Millipore, MAB16990), LAMP1 (Santa Cruz Biotechnology, sc-20011), LC3B (MBL, PM036), phospho-PRKAA/AMPKα (Thr172; Cell Signaling Technology, 2535), PRKAA/AMPKα (Cell Signaling Technology, 5832), Phospho-ULK1 (Ser555; Cell Signaling Technology, 5869), SQSTM1 (MBL, M162-3), TXNIP (Santa Cruz Biotechnology, sc-33099), ULK1 (Cell Signaling Technology, 8054).
Cell culture and treatment
HepG2, HepG2.2.15 and HepAD38 cells were cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, 12800017) supplemented with 10% fetal bovine serum (HyClone, SH30088.03), penicillin (107 U/L) and streptomycin (10 mg/L; HyClone, SV30010) at 37°C in a humidified chamber containing 5% CO2. For HepG2.2.15 cells, the medium was further supplied with 200 μg/ml of G418 (Thermo Fisher Scientific, 11811023), and the HepAD38 cell line that supports HBV replication by removal of tetracycline (Sigma, 87128) was grown as described previously.Citation40 For glucose starvation experiments, cells were cultured in DMEM without fetal bovine serum (Thermo Fisher Scientific, 12100046).
Real-Time PCR
For extracellular HBV, culture medium was collected and briefly centrifuged to remove cell debris. To determine the extracellular HBV titer, the viral DNA was extracted from culture supernatant using the care HBV PCR assay II kit (Qiagen Biotechnology, 1061051), and then processed for real-time PCR analysis using the Bio-Rad CFX96 Real-Time PCR System, following the manufacturer's instructions.
Transfection and oligonucleotides
Plasmids were transfected using the X-tremeGENE HP DNA Transfection Reagent (Roche, 06366236001) according to the manufacturer's protocol. For siRNA transfections, cells were seeded the day before transfection and then transfected with 100 nM siRNA duplexes or control scrambled RNA using X-tremeGENE siRNA Transfection Reagent (Roche, 04476093001) according to the manufacturer's protocol. Transfection reagent-DNA complexes or transfection reagent-siRNA complexes were made by incubating transfection reagent with DNA or siRNA in Opti-MEM Reduced-Serum Medium (Thermo Fisher Scientific, 31985062) at room temperature. At 48 h after transfection, the cells were used for subsequent assays. For each transfection, the following sequences were used: siRNA for ATG5 (sense 5′-GCC TGC TGA GGA GCT CTC CAT-3′ and antisense 5′-AAG GAA GAG CTG TGA CTC C-3′) were synthesized by Genepharma; PRKAA/AMPKα siRNA (pools of 3 to 5 target-specific 19-25 nucleotide siRNAs; Santa Cruz Biotechnology, sc-45312) specific to PRKAA1/2 or a non-targeting siRNA were purchased from Santa Cruz Biotechnology; dominant-negative PRKAA1 (DN-PRKAA1/DN-AMPKα1) plasmid was a gift from Prof. Jae Bum Kim (Institute of Molecular Biology and Genetics, Department of Biological Sciences, Seoul National University, Korea),Citation41 and pLVX-puro-TXNIP plasmid was a gift from Prof. Yu Qiang (Laboratory of Cancer Biology and Pharmacology, Genome Institute of Singapore, Singapore).Citation42 Constitutively active PRKAA1 (CA-PRKAA1/CA-AMPKα1) was generated by introducing a T172D mutation into PRKAA1 truncated at residue 312.Citation4
Protein extraction and immunoblotting
Cells were solubilized in lysis buffer (50 mM Tris-HCl, pH 7.4, 1 mM EDTA, 150 mM NaCl, 0.25% Na-deoxycholate (Sigma, V900388), 1% NP-40 (Sigma, 74385), 0.10% SDS (Sigma, L5750), 1% TritonX-100 (Sigma, T8787-50ML) on ice and lysates were centrifuged at 5000×g for 20 min. Supernatant fractions were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (EMD Millipore, ISEQ00010). Following incubation with primary and secondary antibodies, immunoreactive bands were detected by ECL (EMD Millipore, WBKLS0500) according to the manufacturer's instructions.
Immunofluorescence
Cells grown on poly-L-lysine-coated coverslips were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS; Beyotime, C0221A) and incubated with 0.1% Triton X-100 for permeabilization. Immunolabeling with the anti-LC3B polyclonal antibody was performed by incubation at room temperature. Secondary labeling was performed with DyLight 594-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific, 35560), DyLight 488-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific, 35552), DyLight 594-conjugated goat anti-mouse IgG (Thermo Fisher Scientific, 35510) or DyLight 488-conjugated goat anti-mouse IgG (Thermo Fisher Scientific, 35502). Stained coverslips were mounted with Prolong Gold with DAPI (Thermo Fisher Scientific, P36931). Imaging was performed with a Leica DM2500 microscope with LAS-AF imaging software. Quantification of images was obtained with ImageJ and a set of defined intensity thresholds that were applied to all images. Colocalization is shown in merged as black pixels using the “colocalization” plugin in ImageJ.
GFP-LC3B fluorescence analysis
Cells were plated onto glass coverslips, and transfected the following day with a plasmid encoding GFP-LC3B. The cells were fixed with 2% paraformaldehyde for 20 min and rinsed with PBS twice. Cells were mounted and visualized using a Leica DM2500 microscope with LAS-AF imaging software. Five different microscopy images were randomly chosen for quantification analysis of GFP-LC3B-positive autophagosomes.
Protease protection assay
A protease protection assay was performed as described previously.Citation29 Briefly, cultured cells were incubated with DMSO or compound C for 24 h, harvested, and suspended in homogenization buffer (20 mM HEPES-KOH, pH 7.4, 0.22 M mannitol (Sigma, M4125-500G), 0.07 M sucrose (Sigma, S7903-1KG), and phosphatase inhibitor cocktail (Sigma, P8340-5ML). Cells were then passed 10 times through a 27-gauge needle using a syringe and centrifuged at 9,300x g to obtain the pellet fraction. The pellet fraction was resuspended in homogenization buffer and then treated with 100 µg/ml proteinase K (Sigma, P2308-5ML) with or without 0.5% Triton X-100. After a 20-min incubation on ice, 10% TCA was added, and the samples were centrifuged at 9,300x g for 5 min. The pellet fraction was washed with ice-cold acetone, resuspended in SDS-PAGE sample buffer, and boiled immediately.
DQ-BSA degradation assay
DQ Red BSA is a fluorogenic substrate that requires enzymatic cleavage in lysosomal/autolysosomal compartments to generate a highly fluorescent product.Citation43 Cultured cells were incubated with 10 µg/ml DQ Red BSA (Thermo Fisher Scientific, D-12051) for 30 min. Cells were then washed with PBS, fixed, and stained with DAPI. The fluorescent signal from lysosomal proteolysis of DQ Red BSA was recorded with a Leica DM2500 microscope using LAS-AF imaging software (Leica, Germany).
Mouse model with HBV replication
Male BALB/C mice at 6-9 wk of age were provided by the Beijing HFK Bioscience Co., Ltd. Ethics approval was obtained from the Institutional Ethics Committee of Sichuan University. Eighteen mice were randomly divided into 3 groups: vector-injected group, HBV1.3 and vector-injected group, HBV1.3 and dominant-negative PRKAA1 (DN-PRKAA1)-injected group. The plasmids were injected into the tail vein in 1.6 ml saline within 5-8 sec (hydrodynamic in vivo transfection). The animals were sacrificed at day 3 after injection, and sera and liver collected for real-time PCR or immunohistochemical staining.
Determination of the free sulfhydryl accessibility
To detect free sulfhydryls, direct labeling of cellular proteins with maleimide-polyethylene glycol (M.W. 2000; Aladdin Industrial Corporation, M110240) was performed as described.60 Cells were lysed on ice with modified RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 1% NP-40, pH 8.0) containing 20 mM Mal-PEG, but in the absence of any other thiol-modifying reagents or protease inhibitors. Cell lysates were incubated on ice for 30 min and then centrifuged for 30 min at 20,000×g at 4°C. The recovered supernatant fractions were mixed with sample buffer containing 50 mM dithiothreitol (to neutralize excess Mal-PEG). Finally, proteins modified by Mal-PEG were analyzed by immunoblotting.
Lysosomal pH measurement
Measurement of lysosomal pH was performed using the LysoSensor™ reagent (LysoSensor™ Yellow/Blue DND-160; Thermo Fisher Scientific, L7545), which exhibits a pH-dependent increase in fluorescence intensity upon acidification. Briefly, cells were loaded with the lysosomal pH indicator probes for 5 min. Quantitative comparison was performed on parallel cells in a 96-well plate, and the fluorescence was measured with VarioskanFlash (Thermo Fisher Scientific, Inc.). Light emitted (440, 540 nm) in response to excitation at 340 nm and 380 nm was measured, respectively.
Determination of ATP Levels
The concentration of cellular ATP was measured using a commercially available firefly luciferase assay kit (Beyotime Institute of Biotechnology, S0026) according to the manufacturer's instructions. The luminescent signal was measured on a VarioskanFlash, and the values were normalized to total cellular protein, and converted to percentage of control.
Immunohistochemical analysis
Tissues were formalin fixed and paraffin embedded, and sections were consecutively cut (4-μm thickness) for immunohistochemistry analysis as reported previously.Citation44,45 Briefly, the paraffin-sections were dewaxed, rehydrated, and incubated in 3% H2O2 for 10 min in the dark at room temperature to quench the endogenous peroxidase activity. Antigen retrieval was performed in citrate buffer (pH 6.0) using the autoclave sterilizer method. Subsequently, the sections were blocked with normal rabbit serum (Fuzhou Maixin Biotechnology, SP KIT-B4) diluted in PBS (pH 7.4) for 20 min at 37°C, followed by incubation at 4°C overnight with the primary antibodies. After rinsing in fresh PBS for 15 min, slides were incubated with horseradish peroxidase-linked secondary antibody (Fuzhou Maixin Biotechnology, KIT-5030) at 37°C for 40 min, followed by reaction with diaminobenzidine (Fuzhou Maixin Biotechnology, DAB-0031) and counterstaining with Mayer hematoxylin (Beyotime, C0107). Immunohistochemical staining was assessed and scored by calculating the fraction of positive cells (0-100%) and the immunostaining intensity (0 = negative, 1 = weak, 2 = moderate, 3 = strong). The score was calculated by multiplying the fraction score and the intensity score, producing a total range of 0 to 300.
Statistical analysis
All data shown are reported as mean ± SD. Data analysis was performed using Prism 5.0 (Graph-Pad Software, Inc., La Jolla, CA). The statistical significance of the difference between experimental groups in instances of single comparisons was determined using the 2-tailed unpaired Student t test of the means. Comparisons where P < 0.05 were deemed significant.
Abbreviations
| 3-MA | = | 3-methyladenine |
| ACTB/β-actin | = | actin, β |
| AICAR | = | 5-aminoimidazole-4-carboxamide 1-β-D-ribofuranoside |
| AMPK | = | adenosine monophosphate-activated protein kinase |
| ATG5 | = | autophagy-related 5 |
| HBV | = | hepatitis B virus |
| CA- | = | constitutively active |
| CAMKK2/CaMKKβ | = | calcium/calmodulin-dependent protein kinase kinase 2 |
| CTSD | = | cathepsin D |
| DN- | = | dominant-negative |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| Mal-PEG | = | maleimide-polyethylene glycol |
| MAP1LC3B/LC3B | = | microtubule associated protein 1 light chain 3 β |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| NAC | = | N-acetyl-L-cysteine |
| PRKAA | = | protein kinase AMP-activated catalytic subunit α |
| ROS | = | reactive oxygen species |
| SQSTM1/p62 | = | sequestosome 1 |
| STK11/LKB1 | = | serine/threonine kinase 11 |
| TXN/Trx1 | = | thioredoxin |
| TXNIP | = | thioredoxin interacting protein |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1191857_Supplemental_Material.zip
Download Zip (9.2 MB)Funding
This work was supported by grants from the National 973 Basic Research Program of China (2013CB911300 and 2012CB518900), the Chinese NSFC (81225015 and 81430071 for Canhua Huang, 81501743 for Na Xie, 81502441 for Kefei Yuan, 81401951 for Yunlong Lei), and Sichuan Science-Technology Innovative Research Team for Young Scientist (2013TD0001).
Related Research Data
References
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000; 64:51-68; PMID:10704474; http://dx.doi.org/10.1128/MMBR.64.1.51-68.2000
- Yuen MF, Lai CL, Hepatitis B. in 2014: HBV research moves forward–receptors and reactivation. Nat Rev Gastroenterol Hepatol 2015; 12:70-2; PMID:25511530
- Geier A. Hepatitis B virus: the “metabolovirus” highjacks cholesterol and bile acid metabolism. Hepatology 2014; 60:1458-60; PMID:24829054
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 2005; 18:283-93; PMID:15866171; http://dx.doi.org/10.1016/j.molcel.2005.03.027
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012; 13:251-62; PMID:22436748; http://dx.doi.org/10.1038/nrm3311
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 2011; 25:1895-908; PMID:21937710; http://dx.doi.org/10.1101/gad.17420111
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013; 152:290-303; PMID:23332761; http://dx.doi.org/10.1016/j.cell.2012.12.016
- Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 2006; 281:34870-9; PMID:16990266; http://dx.doi.org/10.1074/jbc.M605488200
- Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, Ding H, Yuan Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol 2011; 85:6319-33; PMID:21507968; http://dx.doi.org/10.1128/JVI.02627-10
- Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A 2010; 107:4383-8; PMID:20142477; http://dx.doi.org/10.1073/pnas.0911373107
- Tian Y, Sir D, Kuo CF, Ann DK, Ou JH. Autophagy required for hepatitis B virus replication in transgenic mice. J Virol 2011; 85:13453-6; PMID:21957292; http://dx.doi.org/10.1128/JVI.06064-11
- Liu B, Fang M, Hu Y, Huang B, Li N, Chang C, Huang R, Xu X, Yang Z, Chen Z, et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 2014; 10:416-30; PMID:24401568; http://dx.doi.org/10.4161/auto.27286
- Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 1997; 41:1715-20; PMID:9257747
- Tell G, Vascotto C, Tiribelli C. Alterations in the redox state and liver damage: hints from the EASL Basic School of Hepatology. J Hepatol 2013; 58:365-74; PMID:23023012; http://dx.doi.org/10.1016/j.jhep.2012.09.018
- Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem 2010; 285:33154-64; PMID:20729205; http://dx.doi.org/10.1074/jbc.M110.143685
- Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol 2003; 161:933-44; PMID:12796479; http://dx.doi.org/10.1083/jcb.200211118
- Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol 2011; 31:3531-45; PMID:21670147; http://dx.doi.org/10.1128/MCB.05124-11
- Sinha RA, Singh BK, Zhou J, Wu Y, Farah BL, Ohba K, Lesmana R, Gooding J, Bay BH, Yen PM. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015; 11:1341-57; PMID:26103054; http://dx.doi.org/10.1080/15548627.2015.1061849
- Shao D, Oka S, Liu T, Zhai P, Ago T, Sciarretta S, Li H, Sadoshima J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab 2014; 19:232-45; PMID:24506865; http://dx.doi.org/10.1016/j.cmet.2013.12.013
- Spindel ON, World C, Berk BC. Thioredoxin interacting protein: redox dependent and independent regulatory mechanisms. Antioxid Redox Signal 2012; 16:587-96; PMID:21929372; http://dx.doi.org/10.1089/ars.2011.4137
- Vincent EE, Coelho PP, Blagih J, Griss T, Viollet B, Jones RG. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene 2015; 34:3627-39; PMID:25241895; http://dx.doi.org/10.1038/onc.2014.301
- Terry LJ, Vastag L, Rabinowitz JD, Shenk T. Human kinome profiling identifies a requirement for AMP-activated protein kinase during human cytomegalovirus infection. Proc Natl Acad Sci U S A 2012; 109:3071-6; PMID:22315427; http://dx.doi.org/10.1073/pnas.1200494109
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, Simon HU. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun 2013; 4:2130; PMID:23945651; http://dx.doi.org/10.1038/ncomms3130
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/10.1080/15548627.2015.1100356
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell 2011; 42:731-43; PMID:21700220; http://dx.doi.org/10.1016/j.molcel.2011.04.024
- Zhao H, Zhao YG, Wang X, Xu L, Miao L, Feng D, Chen Q, Kovács AL, Fan D, Zhang H. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol 2013; 200:731-41; PMID:23479740; http://dx.doi.org/10.1083/jcb.201211014
- O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013; 493:346-55; PMID:23325217; http://dx.doi.org/10.1038/nature11862
- Pillai S, Zull JE. ATP activation of protein degradation by extracts of crude and purified lysosomal preparations. Biochim Biophys Acta 1985; 843:92-100; PMID:3851674; http://dx.doi.org/10.1016/0304-4165(85)90054-6
- Pillai S, Zull JE. Effects of ATP, vanadate, and molybdate on cathepsin D-catalyzed proteolysis. J Biol Chem 1985; 260:8384-9; PMID:3891755
- Goodwin CM, Xu S, Munger J. Stealing the keys to the kitchen: Viral manipulation of the host cell metabolic network. Trends Microbiol 2015; 23:789-98; PMID:26439298; http://dx.doi.org/10.1016/j.tim.2015.08.007
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 2010; 107:8788-93; PMID:20421486; http://dx.doi.org/10.1073/pnas.1003428107
- Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer 2013; 13:123-35; PMID:23344543; http://dx.doi.org/10.1038/nrc3449
- Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 2015; 17:351-9; PMID:25774832; http://dx.doi.org/10.1038/ncb3124
- Orvedahl A, MacPherson S, Sumpter R, Jr., Talloczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010; 7:115-27; PMID:20159618; http://dx.doi.org/10.1016/j.chom.2010.01.007
- Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle 2010; 9:1295-307; PMID:20305376; http://dx.doi.org/10.4161/cc.9.7.11109
- Meijer AJ, Codogno P. Autophagy: regulation by energy sensing. Curr Biol 2011; 21:R227-9; PMID:21419990; http://dx.doi.org/10.1016/j.cub.2011.02.007
- Ma Y, Yu J, Chan HL, Chen YC, Wang H, Chen Y, Chan CY, Go MY, Tsai SN, Ngai SM, et al. Glucose-regulated protein 78 is an intracellular antiviral factor against hepatitis B virus. Mol Cell Proteomics 2009; 8:2582-94; PMID:19671925; http://dx.doi.org/10.1074/mcp.M900180-MCP200
- Jeong HW, Hsu KC, Lee JW, Ham M, Huh JY, Shin HJ, Kim WS, Kim JB. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am J Physiol Endocrinol Metab 2009; 296:E955-64; PMID:19208854; http://dx.doi.org/10.1152/ajpendo.90599.2008
- Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, Koh TL, Yu Q, Chng WJ. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood 2011; 118:2830-9; PMID:21734239; http://dx.doi.org/10.1182/blood-2010-07-294827
- Ha SD, Ham B, Mogridge J, Saftig P, Lin S, Kim SO. Cathepsin B-mediated autophagy flux facilitates the anthrax toxin receptor 2-mediated delivery of anthrax lethal factor into the cytoplasm. J Biol Chem 2010; 285:2120-9; PMID:19858192; http://dx.doi.org/10.1074/jbc.M109.065813
- Yuan K, Lei Y, Chen HN, Chen Y, Zhang T, Li K, Xie N, Wang K, Feng X, Pu Q, et al. HBV-induced ROS accumulation promotes hepatocarcinogenesis through Snail-mediated epigenetic silencing of SOCS3. Cell Death Differ 2016; 23:616-27; PMID:26794444; http://dx.doi.org/10.1038/cdd.2015.129
- Liu R, Li J, Zhang T, Zou L, Chen Y, Wang K, Lei Y, Yuan K, Li Y, Lan J, et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: involvement of abnormal cholesterol trafficking. Autophagy 2014; 10:1241-55; PMID:24905460; http://dx.doi.org/10.4161/auto.28912