
ABSTRACT
Modulation of autophagy has been increasingly regarded as a promising cancer therapeutic approach. In this study, we screened several ginsenosides extracted from Panax ginseng and identified ginsenoside Ro (Ro) as a novel autophagy inhibitor. Ro blocked the autophagosome-lysosome fusion process by raising lysosomal pH and attenuating lysosomal cathepsin activity, resulting in the accumulation of the autophagosome marker MAP1LC3B/LC3B and SQSTM1/p62 (sequestosome 1) in various esophageal cancer cell lines. More detailed studies demonstrated that Ro activated ESR2 (estrogen receptor 2), which led to the activation of NCF1/p47PHOX (neutrophil cytosolic factor 1), a subunit of NADPH oxidase, and subsequent reactive oxygen species (ROS) production. Treatment with siRNAs or inhibitors of the ESR2-NCF1-ROS axis, such as N-acetyl-L-cysteine (NAC), diphenyleneiodonium chloride (DPI), apocynin (ACN), Tiron, and Fulvestrant apparently decreased Ro-induced LC3B-II, GFP-LC3B puncta, and SQSTM1, indicating that ROS instigates autophagic flux inhibition triggered by Ro. More importantly, suppression of autophagy by Ro sensitized 5-fluorouracil (5-Fu)-induced cell death in chemoresistant esophageal cancer cells. 5-Fu induced prosurvival autophagy, and by inhibiting such autophagy, siRNAs against BECN1/beclin 1, ATG5, ATG7, and LC3B enhanced 5-Fu-induced autophagy-associated and apoptosis-independent cell death. We observed that Ro potentiates 5-Fu cytotoxicity via delaying CHEK1 (checkpoint kinase 1) degradation and downregulating DNA replication process, resulting in the delayed DNA repair and the accumulation of DNA damage. In summary, these data suggest that Ro is a novel autophagy inhibitor and could function as a potent anticancer agent in combination therapy to overcome chemoresistance.
Introduction
Esophageal cancer is relatively common and one of the leading causes of cancer-related deaths worldwide.Citation1 The high mortality rate is attributed to a low rate of diagnosis at the early stage, a tendency to relapse, and drug resistance. Although the use of surgical resection and definitive chemotherapy as a single-modality treatment is initially effective, chemotherapy with cisplatin and 5-fluorouracil (5-Fu) results in poor long-term survival associated with high rates of recurrence and drug resistance.Citation2,3 Considerable efforts have been made to identify novel and reliable biomarkers for early diagnosis, more effective combination chemotherapy strategies, and possible mechanisms of chemoresistance.Citation4-6 A recent study indicates that autophagy is a survival mechanism that promotes chemoresistance and that regulators selectively inhibiting autophagy have the potential to improve chemotherapeutic regimes in esophageal cancer cells.Citation7
Autophagy is an evolutionarily conserved survival response to growth-limiting conditions, in which cytosolic proteins or damaged organelles are sequestered, degraded, and released for recycling.Citation8-10 Autophagy is generally initiated by the coordination of several autophagy-related proteins to form double-membrane vacuoles, the autophagosomes. Following fusion of the autophagosomes with lysosomes to form autolysosomes, the defective organelles, misfolded or aggregated proteins and certain long-lived proteins are degraded by the activation of lysosomal hydrolases. There is an increasing understanding that autophagy plays a controversial role in tumor cell death or cell survival.Citation11,12 On the one hand, autophagy functions as a suppression mechanism for tumor development, and defective autophagy in tumor cells induces aggregation of toxic proteins and oxidative stress, resulting in genomic instability and ultimately malignant transformation.Citation13,14 On the other hand, once the tumor is formed, autophagy enhances the survival of cancer cells in metabolic stress such as DNA damage and hypoxia, protects them from anoikis, and promotes tumor metastasis.Citation15-17 Autophagy is also involved in the resistance of cancer cells to chemotherapy.Citation18 Although there are reports demonstrating that overactivating autophagy can enhance therapeutic efficacy by triggering autophagy-dependent cell death or autophagic cell death of cancer cells, current accumulating evidence suggests that suppression of autophagy would be a logical approach to overcome resistance and sensitize cancer cells to chemotherapeutic agents and radiotherapy.Citation19,20
Natural products are a major source of drugs against a wide variety of diseases, including cancer and infectious diseases.Citation21 For instance, ginseng, a traditional medicine, is commonly used either by itself or in combination with other medicinal ingredients to modulate immune functions and metabolism, and is widely accepted as complementary and alternative medicine in cancer therapy.Citation22 Ginsenosides are the pharmacologically active component in ginseng and are composed of a dammarane skeleton with various sugar moieties attached at the C-3 and C-20 positions. Several ginsenosides such as ginsenoside Rg3, Rh2, Re, and F2, have been tested in vitro and in vivo as novel anticancer agents to induce apoptosis and inhibit cell proliferation and metastasis.Citation22,23 Many molecular mechanisms have been proposed for such anticancer activity, including autophagy regulation.Citation24-27 Moreover, autophagy contributes to the neuronal protective effects of ginsenoside Rb1 and compound K.Citation28,29 However, there is currently no evidence showing a direct connection between autophagy and ginsenoside-induced cell death. Additionally, the mechanism of autophagy modulation by ginsenosides is unclear.
In this study, we screened novel autophagic regulators from ginsenosides extracted from Panax ginseng (Renshen, Chinese ginseng). We identified ginsenoside Ro (Ro) as a new autophagy suppressor that inhibited autophagic flux by blocking the fusion process between autophagosome and lysosome via the ESR2 (estrogen receptor 2)-NCF1-ROS axis. Thus, Ro enhanced 5-Fu induced DNA damage and sensitized chemoresistant esophageal cancer cells to 5-Fu-mediated cell death. These results suggest that targeting the autophagic pathway represents a promising new strategy for cancer therapy and screening for novel autophagy modulators from natural products may be an efficient approach for anticancer drug discovery.
Results
Ro regulates autophagy in human esophageal cancer cells
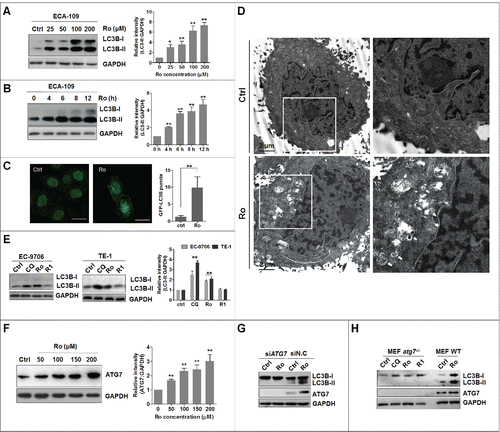
To identify novel autophagy modulators from ginsenosides, we examined LC3B (microtubule-associated protein 1 light chain 3 β) levels in ginsenoside-treated ECA-109 human esophageal cells. Interestingly, several new compounds (ginsenoside Ro, Ra1, Rh1, Rd, 20(R)-Rg2, and notoginsenoside Fc) induced LC3B-II accumulation (Fig. S1A). Further, fluorescence microscopy analyses confirmed their autophagy-modulating effects on ECA-109 cells transiently expressing green fluorescent protein (GFP)-tagged LC3B (Fig. S1B and S1C). Among all the tested ginsenosides, Ro exhibited preferential activity for inducing an increase in GFP-LC3B puncta and therefore was selected for further study to elucidate its mechanisms of action in modulating autophagy (Fig. S1D). Ro increased not only endogenous LC3B-II levels in a time- and dose-dependent manner in ECA-109 cells (), but also GFP-LC3B punctation (). Transmission electron microscopy was used to observe the accumulation of autophagic vacuoles in Ro-treated cells, which revealed an increased number of autophagic vacuoles compared with control cells (). We also tested the effect of Ro on other human esophageal cell lines including EC-9706 and TE-1 cells (). Consistently, Ro treatment resulted in a dramatic accumulation of LC3B-II in these cell lines, whereas R1, a negative control, showed no effect on the level of endogenous LC3B-II. Moreover, exposure to Ro increased the level of ATG7 (autophagy-related 7) in a concentration-dependent manner (). Knockdown of ATG7 by siRNA suppressed the Ro-mediated increase in LC3B-II level (). In addition, Ro did not induce accumulation of LC3B-II in atg7-knockout, mouse embryonic fibroblast (atg7 -/- MEF) cells, whereas it functioned as a potent autophagy regulator in the wild-type (WT) counterparts (WT MEF) (). This further suggested an essential role of ATG7 in Ro-mediated autophagy modulation.
Figure 1. Identification of Ro as a novel autophagy modulator. (A and B) Ro induces autophagosome formation in ECA-109 cells in a dose-dependent and time-dependent manner, as revealed by western blotting. Cells were treated with Ro (50 μM) for the indicated time periods, or treated with Ro at the indicated doses for 12 h. ImageJ densitometric analysis of the LC3B-II:GAPDH ratio from LC3B immunoblots (mean ± SD of 3 independent experiments). * indicates a significant difference from the controls. **, P< 0.005; *, P < 0.05; the Student t test. (C) Effect of Ro on GFP-LC3B punctation. ECA-109 cells transfected with GFP-LC3B were treated with Ro for 12 h, and the distribution of GFP-LC3B was examined by confocal microscopy. Percentage of cells with GFP-LC3B puncta was quantified by analyzing the number of GFP-LC3B dots in the cells. At least 50 cells were counted in each of the conditions. Data are shown as the mean ± SD of 3 independent experiments. Scale bar: 10 μm. (D) ECA-109 cells were treated with Ro (100 μM) for 12 h, fixed and examined using transmission electron microscopy. Higher power magnification of the image of Ro-treated cells revealed autophagosomes (arrows). Scale bar: 2 μm. (E) Ro increases the conversion of LC3B-II in other esophageal cancer cells. TE-1 and EC-9706 cells were treated with CQ (20 μM), Ro (50 μM), and R1 (50 μM) for 12 h, and cell lysates were analyzed by western blotting for endogenous LC3B. CQ was used as a positive control. ImageJ densitometric analysis of the LC3B-II:GAPDH ratio from LC3B immunoblots (mean ± SD of 3 independent experiments). (F) Ro increases the protein level of ATG7. ImageJ densitometric analysis of the ATG7:GAPDH ratio from ATG7 immunoblots (mean ± SD of 3 independent experiments). (G) ECA-109 cells transfected with siRNA against ATG7 were treated with Ro (50 μM) for 12 h, and cell lysates were analyzed by western blot. siN.C, nonsense control siRNA. (H) atg7 knockout and wild-type MEF cells were treated with Ro, R1 or CQ for 12 h, and cell lysates were collected and subjected to western blot.
Ro inhibits autophagic flux
To distinguish whether the increase in GFP-LC3B puncta or LC3B-II level was due to increased autophagosome generation or rather a blockage in the autophagosome-lysosome fusion process, we performed an autophagic flux assay by measuring the total cellular amount of SQSTM1/p62 (sequestosome 1). SQSTM1 is delivered to the lysosome for degradation and a steady-state level of this protein reflects the autophagic status.Citation30,31 Immunoblot analysis showed that Ro induced a remarkable increase in SQSTM1 levels in a time- and dose-dependent manner (). Quantitative real time-polymerase chain reaction (PCR) assay was performed to evaluate the mRNA level of SQSTM1 after Ro treatment and exclude the possibility that SQSTM1 accumulation was due to transcriptional activation (). We also detected the protein level of SQSTM1 after treatment with other autophagy-inducing ginsenosides and found that all autophagy-inducing ginsenosides showed effects similar to those of Ro (Fig. S2A), without affecting SQSTM1 mRNA expression (Fig. S2B).
Figure 2. Ro inhibits autophagic flux. (A) Ro increases SQSTM1 accumulation. ECA-109 cells were treated with different concentrations of Ro for 12 h, or treated with Ro (50 μM) over a specific duration, and the protein level of SQSTM1 was analyzed. Relative SQSTM1 mRNA expression levels (compared with GAPDH) were analyzed by quantitative real-time PCR. ImageJ densitometric analysis of the SQSTM1:GAPDH ratio from SQSTM1 immunoblots (mean ± SD of 3 independent experiments) (**, P < 0.005). (B) CQ fails to enhance Ro-mediated SQSTM1 increment. ECA-109 cells were treated with Ro (50 μM) for 12 h in the presence or absence of CQ (50 μM) as indicated. N.S., No significance. (C) Ro inhibits autophagic flux in EC-9706 and TE-1 cells, whereas the effects of R1 on SQSTM1 were not observed. (D) ECA-109 cells were transfected with mRFP-GFP-LC3B plasmid (2 μg). Twenty-four h after the transfection, the cells were treated with 50 μM Ro or 20 μM CQ, or starved in EBSS for 12 h. Cells were then fixed and subjected to confocal microscopy. Scale bar: 10 μm. The numbers of yellow LC3B and red LC3B dots per cell in each condition were quantified. More than 50 cells were counted in each of the conditions and the data (mean ± SD) are representative of 2 independent experiments (**, P < 0.005). (E) Confocal microscopy showing the colocalization between SQSTM1 and LC3B. The cells were transfected with mRFP-GFP-LC3B plasmid (2 μg), treated with Ro or CQ for 12 h, fixed, and then stained with anti-SQSTM1 (blue). More than 50 cells were counted in each of the conditions (mean ± SD) (**, P < 0.005). Scale bar: 10 μm. The smaller panels in E are 1.5 × zoom.
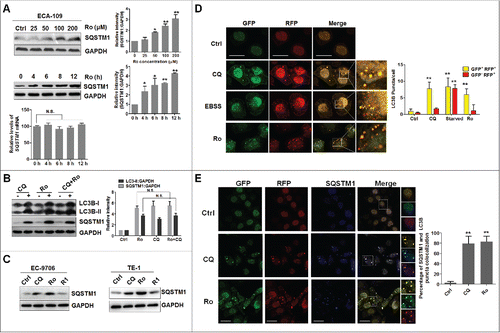
Chloroquine (CQ),Citation32 an autophagic inhibitor that significantly increases the amount of LC3B-II and SQSTM1, failed to further enhance the accumulation of SQSTM1 and LC3B-II induced by Ro, indicating that Ro inhibits autophagic flux (). The autophagic inhibitory effect of Ro was confirmed in TE-1 and EC-9706 cells (). In addition to using autophagic degradation of SQSTM1, we also used ECA-109 cells transfected with the tandem fluorescent-tagged LC3B (monomeric red fluorescent protein [mRFP]-GFP-LC3B) plasmid. GFP and RFP exhibit differing pH sensitivity, wherein GFP is quantitatively quenched in acidic compartments while RFP is stable. Therefore this probe capitalizes on the pH difference between the acidic autolysosome and the neutral autophagosome to monitor flux of LC3B from autophagosomes (GFP-positive RFP-positive; yellow dots) to autolysosomes (GFP-negative RFP-positive; red dots).Citation33 As shown in , the number of yellow and red puncta increased in the Earle's balanced salt solution (EBSS)-treated cells (positive control), indicating that autophagy flux had increased. In the presence of Ro, we observed an enhanced formation of yellow puncta without any significant increase in the number of red puncta, as well as CQ-treated cells. Moreover, the sizes of autophagosomes in the starved group were significantly smaller than those in the Ro-treated group. In addition, SQSTM1 was colocalized with GFP and RFP in the presence of Ro and CQ, indicating that autophagy was arrested at the autophagosome level (). Taken together, these results clearly suggest that Ro, similar to CQ, blocks the fusion between autophagosomes and autolysosomes.
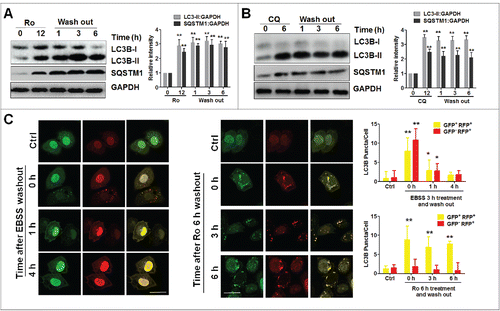
Next, we tested whether the autophagic inhibitory effect was reversible. Apparently, the block between autophagosomes and autolysosomes was not relieved even 6 h after removal of Ro from the medium; instead, both LC3B-II and SQSTM1 continued to accumulate (). Similarly, the flux inhibition by CQ through the autophagic pathway was not reversed (). Notably, starvation-induced autophagy was completely relieved at 4 h, as numbers of both, yellow (GFP+ RFP+) and red (GFP− RFP+) dots were diminished in ECA-109 cells expressing mRFP-GFP-LC3B after replacing EBSS with normal medium (). However, the yellow puncta still persisted irrespective of the presence of Ro, whereas red puncta were not observed (). These data indicate that the effect of Ro on autophagy inhibition is irreversible.
Figure 3. Ro irreversibly inhibits autophagic flux. (A and B) After ECA-109 cells treated with Ro (50 μM) for 12 h or CQ (20 μM) for 6 h, the compounds were washed out and proteins were extracted at the indicated time points. The SQSTM1:GAPDH and LC3B-II:GAPDH ratios were calculated from 3 independent experiments. (C) ECA-109 cells transfected with mRFP-GFP-LC3B plasmid were either treated with 50 μM Ro or starved in EBSS for 3 h. Cells were then fixed at the indicated times and subjected to confocal microscopy. The number of yellow or red LC3B puncta was also quantified. At least 50 cells from 5 representative fields were counted in each independent experiment. * indicates a significant difference from the controls. **, P < 0.005; *, P < 0.05. Scale bar: 10 μm.
Ro treatment inhibits lysosomal activity
We then explored the mechanism underlying Ro-mediated autophagy arrest. To assess whether Ro affected autophagosome-lysosome fusion, we examined the colocalization between LC3B-II and LysoTracker Red, a dye specific for live cell lysosomes. As a positive control, starvation remarkably increased the accumulation of LC3B-II puncta, which were well colocalized with LysoTracker Red, suggesting that autolysosome formation progressed normally during starvation upon autophagy activation (). However, such colocalization was abolished in the presence of Ro or CQ.
Figure 4. Ro interferes with autophagosome-lysosome fusion by raising lysosomal pH and downregulating lysosomal cathepsins. (A) Effect of Ro on autophagosome maturation. ECA-109 cells were either treated with 50 μM Ro, 20 μM CQ, or starved in EBSS for 12 h, before being stained with LysoTracker Red. The cells were then fixed, stained with LC3B, followed by confocal microscopy. Scale bar: 10 μm. (B) Ro affects AO staining of acidic compartments. ECA-109 cells were incubated with or without Ro, CQ, or starved in EBSS for 12 h before adding AO. In both, the control and EBSS-treated cells, the cytoplasm and nucleus essentially displayed green fluorescence, whereas the acidic compartments, including the lysosomes, displayed red fluorescence. Acidification of the latter compartments was inhibited by Ro and CQ. The ratio of red and green in the cytosolic region was also calculated. At least 50 cells were counted and the ratio of red and green in the cytosolic region was calculated (**, P < 0.005). Scale bar: 10 μm. (C) Ro and CQ inhibit EGF-induced EGFR degradation in ECA-109 cells. The cells pretreated with Ro or CQ for 12 h were incubated with EGF (200 ng/ml) for 2 h, the medium were washed out, and samples were collected at the indicated times. (D) Ro increases the number of free GFP fragments in ECA-109 cells transfected with GFP-LC3B. Immunoblot analysis was performed, for GFP and GAPDH levels in cells treated with 100 μM Ro for the indicated times. As a control, cells were either incubated in complete medium (ctrl), treated with 20 μM CQ, or starved for 12 h (EBSS). Three independent experiments were performed and a representative blot was shown. (E) Western blotting analysis for the processing of endogenous CTSB and CTSD. p/i, precursor/intermediate; m, mature form of CTSB and CTSD. (F) Relative CTSB and CTSD mRNA levels (compared with GAPDH) were analyzed by quantitative real-time PCR. Cells were treated with DMSO or Ro (100 μM) at 8, 12, and 24 h, as indicated. (G) Enzymatic activity of CTSB and CTSD in Ro-treated ECA-109 cells. Enzymatic activity was analyzed by fluorogenic kits. *indicates a significant difference from the controls; **, P < 0.005; *, P < 0.05.
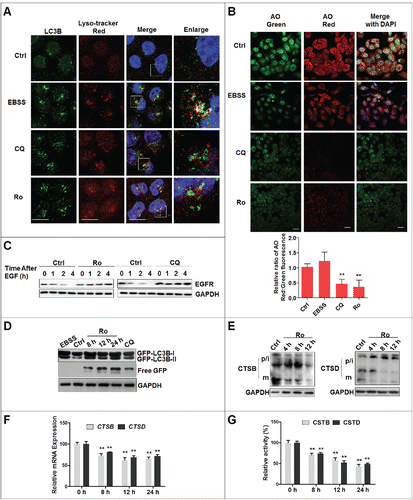
Since the autophagic flux depends on low pH,Citation34 we analyzed the lysosomal pH using an acridine orange (AO) assay. AO is a fluorescent nucleic acid dye that accumulates in acidic spaces such as lysosomes. Under the low pH conditions in lysosomes, the dye emits red light when excited by blue light. Unlike the control and starvation cells, the red fluorescent signal was greatly reduced in both, CQ- and Ro-treated cells (). The ratio of fluorescent intensity from red and green channels in the cytosolic region was also calculated and the data demonstrated that the reduction of the red fluorescence signal was due to an increase in lysosomal pH and not a decrease in AO loading.
Next, to examine whether Ro affected lysosomal function, an epidermal growth factor receptor (EGFR) assay was performed (). Upon stimulation with epidermal growth factor (EGF), the EGF-EGFR complex undergoes internalization through clathrin-meditated endocytosis and is targeted by lysosomes for degradation or recycling.Citation35 As shown in , Ro and CQ inhibited EGF-triggered EGFR degradation.
Interestingly, analysis of the chimeric protein in ECA-109 cells expressing GFP-LC3B by western blot showed that Ro treatment resulted in the accumulation of free GFP fragments, which was also observed in CQ-treated cells ().Citation36 However, free GFP fragments were barely detectable in starved cells where the lysosomal activity was high. These results suggest that the lysosomal proteolytic activity is impaired in Ro-treated cells.
The processing of cathepsins from the precursor form to its mature form has been used as a marker for lysosomal activity.Citation37 Next, we investigated the effect of Ro treatment on the expression and activation of lysosomal CTSB (cathepsin B) and CTSD (cathepsin D). Cathepsins are synthesized as inactive membrane-associated precursors, the latter of which are further cleaved to generate an active form within endosomes or lysosomes.Citation38 Western blot assay demonstrated that Ro dramatically downregulated the mature form and the total protein level of CSTB and CSTD (). Additionally, the mRNA expression of CSTB and CSTD were reduced (). Finally the enzymatic activity of CTSB and CTSD were measured by fluorogenic substrate assays. Both, CTSB and CTSD activities were reduced in a time-dependent manner upon Ro treatment (). In summary, these observations suggest that Ro inhibits lysosomal proteolytic activity by altering lysosomal pH and downregulating lysosomal cathepsins.
5-fluorouracil induces protective autophagy in esophageal cancer cells
Recent studies have shown that modulation of autophagic flux can sensitize cancer cells to agents that cause DNA damage.Citation39-42 Besides, esophageal cancer patients exhibit a higher level of autophagy.Citation43 Therefore, we tested the role of autophagy in 5-Fu-mediated cell death. First, we examined the cytotoxicity of 5-Fu on ECA-109 and TE-1 cells. 5-Fu treatment caused dose-dependent cell death with half maximal inhibitory concentration (IC50) of 60 μg/ml for ECA-109 cells and 78 μg/ml for TE-1 cells (Fig. S3A), which was consistent with previous reports.Citation44,45 Unexpectedly, these cells were more 5-Fu-resistant than other chemosensitive cells such as OE19 (IC50 = 3.03 μg/ml) and OE33 (IC50 = 1.24 μg/ml),Citation46 implying that ECA-109 and TE-1 cells were 5-Fu-resistant. A recent study showed that 5-Fu-induced cell death in chemoresistant esophageal cancer cells is apoptosis-independent, and that autophagy exerts a protective effect.Citation7 Therefore, we examined the effect of 5-Fu on autophagy induction. 5-Fu induced LC3B-II accumulation and decreased SQSTM1 protein level in a time- and dose-dependent manner in ECA-109 cells () as well as in TE-1 and EC-9706 cells (), indicating that 5-Fu-induced autophagy and increased autophagic flux. Next, siRNAs targeting autophagy-related genes were used to investigate the role of autophagy in 5-Fu-induced cell death. Suppression of autophagy by knockdown of ATG5, ATG7, LC3B, and BECN1 in ECA-109 cells significantly augmented 5-Fu-induced cell death (), further suggesting that autophagy may help cancer cells to avoid 5-Fu-induced, exacerbated DNA toxicity. In addition, inhibition of autophagy by 3-MA (10 mM), a class I phosphoinositide 3-kinase and class III phosphatidylinositol 3-kinase (PtdIns3K) inhibitor used widely as a pharmacological inhibitor in autophagy studies, also reduced 5-Fu-mediated LC3B-II accumulation and slightly augmented 5-Fu cytotoxicity (Fig. S3B).
Figure 5. 5-Fu induces prosurvival autophagy. (A) 5-Fu promotes autophagic flux in a time- and dose-dependent manner in ECA-109 cells. The cells were treated with the indicated doses of 5-Fu for 24 h or 100 μg 5-Fu for indicated durations. Cell lysates were collected and subjected to western blotting to detect LC3B conversion and SQSTM1 levels. ImageJ densitometric analysis of the LC3B-II:GAPDH or SQSTM1:GAPDH ratio (mean ± SD of 3 independent experiments). (B and C) 5-Fu induces LC3B-II accumulation and SQSTM1 degradation in EC-9706 and TE-1 cells. Cells were treated with 100 μg 5-Fu for 24 h and cell lysates were collected. (D) Suppression of autophagy augments 5-Fu-induced cell death. ECA-109 cells were transfected with 2 μg siRNAs against BECN1, ATG5, ATG7, and LC3B for 24 h, followed by 100 μg 5-Fu for 48 h. Cell viability was measured with the MTT assay. siN.C, nonsense control siRNA. * indicates a significant difference from the controls. **, P < 0.005; *, P < 0.05.
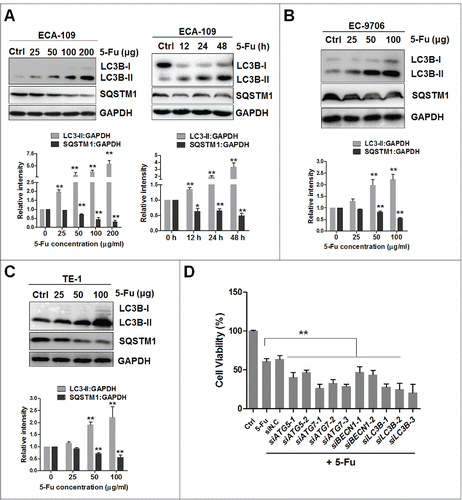
Finally, we also tested the effect of other chemotherapeutic agents including cisplatin and carboplatin, on autophagy. Unlike 5-Fu, neither cisplatin nor carboplatin induced protective autophagy in esophageal cancer cells (Fig. S3C), which was consistent with a previous report.Citation47 These results demonstrated a complicated regulatory network including autophagy, which is utilized by cancer cells to overcome survival stress and achieve drug resistance.
Ro sensitizes 5-fluorouracil-induced cell death in a caspase-independent manner
Our data suggest that autophagy serves as a prosurvival function in 5-Fu-induced cell death, whereas Ro interferes in the process of fusion between the autophagosome and lysosome. Therefore, we tested whether the suppression of autophagic flux by Ro sensitized chemoresistant cells to 5-Fu-induced cell death. Apparently, Ro raised LC3B-II levels induced by 5-Fu () and sensitized both, ECA-109 and TE-1 cells to 5-Fu-induced cell death ( and Fig. S4A). Similar results were obtained with CQ-treated cells (Fig. S4B and S4C), indicating that cells with compromised autophagic flux show higher sensitivity to DNA-damaging agent, 5-Fu. In addition, we examined the autophagy inhibition effect of Ro on cisplatin- and carboplatin-induced cell death (Fig. S4D). We found that Ro treatment did not enhance the cytotoxicity of cisplatin or carboplatin, which was consistent with the result that cisplatin and carboplatin did not induce prosurvival autophagy (Fig. S3C). Indeed, cisplatin induced protective autophagy in resistance cells and inhibition of autophagy by small molecules such as CQ could significantly sensitize cisplatin-induced apoptosis.Citation47 Therefore, targeting autophagy may be a potential therapeutic strategy in cancer treatment, especially in drug-resistant cancer, where anticancer agent-induced autophagy exerted a protective function.
Figure 6. Ro blocks 5-Fu-induced autophagy and sensitizes cell death. (A) Ro increases 5-Fu-induced LC3B-II and SQSTM1 protein levels. ECA-109 cells were treated with 100 μM Ro and 50 μg 5-Fu for 24 h. ImageJ densitometric analysis of the LC3B-II:GAPDH or SQSTM1:GAPDH ratio (mean ± SD of 3 independent experiments). (B) Ro renders chemoresistant esophageal cancer cells ECA-109 and TE-1, more sensitive to 5-Fu-induced cell death. Data (mean ± SD) are representative of 3 independent experiments (**, P < 0.005). (C) 5-Fu-induced cell death is apoptosis-independent. ECA-109 cells were treated with 5-Fu and Ro or CQ for 48 h. Western blot analysis was performed using the specified antibodies. The cells were treated with 10 μM SNX-2112, a HSP90 inhibitor, for 48 h, and lysates used as a positive control (P.C) of apoptosis induction. (D) ECA-109 cells were treated with 5-Fu, Ro, and CQ in the presence or absence of caspase inhibitor (20 μM Z-VAD-FMK) for 48 h. Cell viability was measured with the MTT assay. * indicates a significant difference from the controls. N.S., No significance. **, P < 0.005; *, P < 0.05. (E) ECA-109 cells treated with 5-Fu in the presence or absence of Ro for 48 h and the accumulation of autophagic vacuoles was examined using a transmission electron microscopy and highlighted in areas E1 to E4.
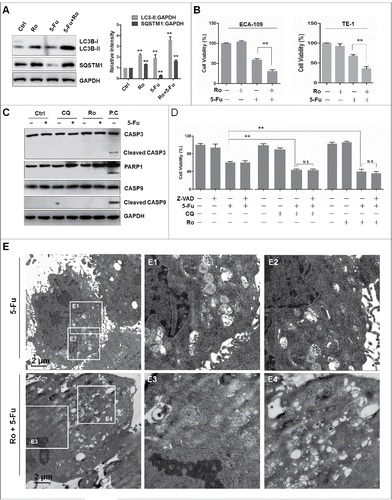
We next examined whether apoptosis was induced by the combination of Ro and 5-Fu in ECA-109 cells by western blot assay. As shown in , cleavage of CASP3 (caspase 3) was not seen in some of the dying cells treated with Ro and 5-Fu when compared to the positive control SNX-2112, a HSP90 inhibitor, which induced CASP3 cleavage. Additionally, cleaved CASP9 (caspase 9) and PARP1 (poly[ADP-ribose] polymerase 1) were not observed after treatment with 5-Fu and Ro or CQ. Consistently, SNX-2112-induced cleavage of CASP3 was detected by fluorescence microscopy analysis while signals were not observed in 5-Fu- and Ro- or CQ-treated cells (Fig. S4E). Interestingly, the conformational change of the F-actin cytoskeleton induced by SNX-2112 was different from those triggered by 5-Fu and Ro, suggesting a different mechanism of cell death induced by 5-Fu. Additionally, when the cells were treated with a general caspase inhibitor (Z-VAD-FMK), no effect was observed on cell death induced by the combination of Ro and 5-Fu (). Thus, cell death induced by Ro and 5-Fu seems to be largely a caspase-independent process. Moreover, transmission electron microscopy revealed that 5-Fu induced the accumulation of autophagosomes, whereas Ro treatment enhanced the formation of several autophagic vacuoles ( and Fig. S4F), implying the involvement of autophagy in 5-Fu-induced cell death. Furthermore, we tested whether autophagic flux inhibition by Ro further enhanced the sensitivity of normal esophageal cells (HEEC cells) to 5-Fu. We found that treatment with Ro dose-dependently increased the protein levels of LC3B-II and SQSTM1 (Fig. S5A). However, the cytotoxicity of 5-Fu was not enhanced by Ro in normal cells (Fig. S5B), further suggesting a specific role for Ro in cancer therapy. Taken together, these results suggest that Ro promotes caspase-independent cell death in chemoresistant esophageal cancer cells.
Ro renders chemoresistant cells more sensitive to 5-Fu-induced genotoxicity via DNA damage accumulation
Next, we investigated the correlation between Ro-mediated autophagic flux inhibition and the enhancement of cell death. Recent studies have demonstrated a protective effect of autophagy during DNA damage.Citation48-50 Autophagy participates in regulating the degradation or cytosol quality control of the main proteins maintaining DNA stability, such as CHEK1 (checkpoint kinase 1). Timely degradation of CHEK1 allows disengagement of DNA repair proteins and cell cycle progression after DNA repair, whereas persistence of CHEK1 leads to the accumulation of DNA damage.Citation51,52 Therefore, we examined the connection between autophagic flux deficiency and DNA damage accumulation. Exposure to 5-Fu significantly induced DNA damage, measured as higher levels of Ser139 phosphorylated H2AFX (H2A histone family member X; γH2AFX), a DNA double-strand break (DSB) marker, than in control cells (). Besides, the enhanced sensitivity to 5-Fu in Ro-treated cells was associated with increased levels of DNA damage ().
Figure 7. Blockage of autophagy by Ro enhances DNA damage and reduces DNA repair. (A) Ro enhances γH2AFX triggered by 5-Fu. ECA-109 cells were treated with 5-Fu in the presence or absence of Ro (100 μM) for 48 h. (B) Ro delays 5-Fu-induced DNA repair. The cells were treated with 100 μg 5-Fu in the presence or absence of Ro for the indicated time courses. Decay of γH2AFX in both groups of cells are shown. ImageJ densitometric analysis was performed, for the CHEK1:GAPDH ratio from CHEK1 immunoblots (mean ± SD of 3 independent experiments). (C) Cell cycle arrest induced by Ro. Cell cycle distribution of the cells subjected to FACS. Values are average and range of 2 different experiments. (D) Levels of CHEK1 in 5-Fu-treated cells in the presence or absence of Ro or CQ. (E) Ro delays CHEK1 degradation. The levels of p-CHEK1 and CHEK1 were quantified. ImageJ densitometric analysis of the p-CHEK1:GAPDH or CHEK1:GAPDH ratio from p-CHEK1 or CHEK1 immunoblots, respectively (mean ± SD of 3 independent experiments). (F) Immunoblot analyzing the activation of DNA damage response. (G) Relative mRNA levels (compared with GAPDH) of several DNA replication-related genes. * indicates a significant difference from the controls. **, P < 0.005; *, P < 0.05.
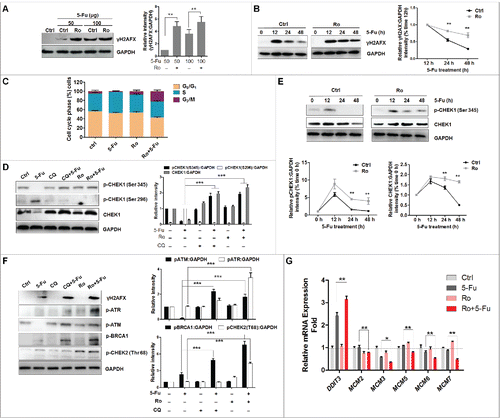
To determine whether higher levels of DNA DSBs in Ro-treated cells were due to increased DNA damage or delayed repair, we performed a time-course analysis 48 h post 5-Fu treatment (). While γH2AFX levels gradually decreased in control cells as a result of DNA repair, the decrease in Ro-treated cells was markedly slow. These results suggest that the higher content of DNA DSBs in cells treated with Ro was due for the most part, to inefficient DNA repair. Next, we analyzed cell cycle progression to determine the effect of Ro treatment on the cell cycle arrest that normally allows time for DNA repair. Unexpectedly, we found that a higher percentage of Ro-treated cells was arrested at G2 stage as compared to control or 5-Fu cells ().Citation49 In agreement with this finding, 5-Fu treatment largely decreased the levels of DNA damage-mediated phosphorylation of CHEK1 (Ser345) and total CHEK1, one of the best-characterized gatekeepers of the G2/M phase.Citation53 Although both Ro and CQ alone have little effect, the levels of phosphorylated (p)-CHEK1 and CHEK1 were significantly higher in the presence of 5-Fu and Ro or CQ when compared to control or 5-Fu-treated cells ().
In contrast, CHEK1 autophosphorylation (Ser296) was decreased by treatment with Ro. In addition, the gradual decrease with time, in levels of p-CHEK1 and CHEK1 observed in control cells after 5-Fu treatment was markedly delayed in Ro-treated cells (), which was consistent with the previous reports that CHEK1 underwent lysosomal degradation. These results suggested that persistence of CHEK1 induced by Ro-mediated autophagic flux inhibition causes a delay in DNA repair, the interruption of cell cycle progression and accumulation of DNA damage. To further elucidate the mechanisms leading to Ro-mediated increase in DNA damage, we examined phosphorylation of several upstream kinases of CHEK1, such as ATM, ATR, and BRCA1. Western blot results showed that Ro treatment increased the activation of ATM, ATR, and BRCA1 (). Activation of DNA damage-mediated phosphorylation of CHEK2 was also observed. Therefore, Ro induced the whole spectrum of the DNA damage response pathway. Finally, expression of DDIT3 (DNA damage inducible transcript 3), a transcription factor upregulated in response to a variety of cellular stress,Citation54 was upregulated in the presence of Ro and 5-Fu when compared with 5-Fu alone, whereas several genes regulating replication progression, including MCM2, MCM3, MCM5, MCM6, and MCM7, were downregulated in culture with combinations of 5-Fu and Ro (). These results indicate that the function of DNA replication machinery was interrupted. Collectively, these results support that inhibiting the lysosomal degradation of CHEK1 by Ro renders cells more sensitive to 5-Fu-induced cell death via delayed DNA repair and cell cycle progression, and accumulated DNA damage.
Ro induces intracellular ROS-mediated autophagy inhibition and cell death
To understand the mechanism by which Ro blocks the process of autophagosome and lysosome fusion and leads to higher DNA damage () and cell death (), we analyzed the possible role of reactive oxygen species (ROS) in Ro-promoted cell death. Accumulating evidences suggest that ROS are important triggers of autophagy under various circumstances.Citation55,56 We first measured the level of ROS in ECA-109 cells using the oxidation-sensitive probe dichloro-dihydro-fluorescein diacetate (DCFH-DA) by flow cytometry (). Both, Ro and CQ treatment increased intracellular ROS production and raised the lysosomal pH, which were significantly reduced by N-acetyl-L-cysteine (NAC), a ROS scavenger (). Besides, NAC substantially attenuated Ro-mediated autophagy inhibition in ECA-109 and TE-1 cells and blocked cell death enhanced by the combination of Ro and 5-Fu (). This indicated that accumulation of ROS by Ro triggered cell death. Addition of the generalized superoxide scavenger Tiron also lowered LC3B-II levels and SQSTM1 accumulation and partially restored autophagic flux inhibited by Ro and CQ (). Tiron treatment also normalized Ro- or CQ-induced GFP-LC3B-positive autophagosome accumulation (). This suggested that Ro impairs autophagosome turnover in a superoxide-dependent manner.
Figure 8. Ro induces intracellular ROS-mediated autophagy inhibition and cell death. (A) ECA-109 cells were treated with 50 μM Ro or 20 μM CQ for 12 h in the presence or absence of 3 mM NAC and stained with DCFH-DA. Intracellular ROS levels were analyzed using flow cytometry. Bar graph represents fold-increase of ROS from 3 independent experiments expressed as means ± SEM. (B) Effect of NAC on AO staining. The cells were treated with NAC and Ro or CQ for 12 h, fixed, and stained with AO. The ratio of red and green in the cytosolic region was calculated to evaluate lysosomal pH. (C) NAC reduces LC3B-II and SQSTM1 in TE-1 and ECA-109 cells. (D) NAC reduces 5-Fu-induced cell death enhanced by Ro in ECA-109 cells. The cells were treated with the indicated concentrations of NAC in combination with Ro or 5-Fu for 48 h. Cell viability was measured by MTT assay. (E) Immunoblot analysis of LC3B-II and SQSTM1 in ECA-109 cells incubated with vehicle or Ro in the presence or absence of 10 mM Tiron and with or without 20 µM CQ for 12 h. (F) Confocal microcopy analyses the effect of Tiron on Ro-trigged GFP-LC3B punctation. ECA-109 cells were transfected with GFP-LC3B for 24 h, treated with Tiron or Ro for 12 h, fixed, and analyzed by confocal microcopy. At least 40 cells were counted from 5 representative fields to determine the average number of GFP-LC3B per cell. (G) Relative activity of NADPH oxidase. ECA-109 cells were treated with Ro for 12 h and the NADPH oxidase activity was measured by lucigenin assay (mean ± SD of 3 independent experiments versus control). (H and I) Effect of 10 μM DPI or NAC on the accumulation of LC3B-II, SQSTM1, and GFP-LC3B. (J) Ro upregulates NCF1 expression in a dose-dependent manner. The cells were treated with Ro at the indicated concentration for 12 h. ImageJ densitometric analysis of the NCF1:GAPDH ratio from NCF1 immunoblots (mean ± SD of 3 independent experiments). (K) After treatment with 50 μM Ro for 12 h, immunofluorescence signals of NCF1 (green) were observed using a confocal microscope. Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (L to O) Effects of 10 μM ACN on LC3B-II and SQSTM1 accumulation (L), NADPH oxidase activity (M), ROS production (N), and lysosomal pH increase in ECA-109 cells (O). Data presented are representative of 3 independent experiments. * indicates a significant difference from the controls. **, P< 0.005; *, P < 0.05.
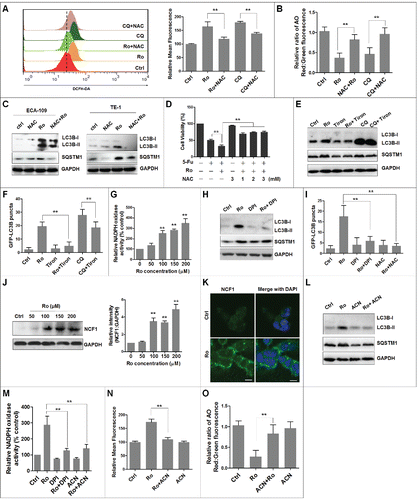
As an important source of superoxides, we explored if NADPH oxidase, particularly CYBB/Nox2 (cytochrome b-245, β polypeptide), modulated Ro-induced autophagic flux inhibition.Citation57 First, NADPH oxidase activity was measured by lucigenin assay and we found that Ro treatment significantly enhanced NADPH oxidases activity in a dose-dependent manner (). Exposure of cells to diphenyleneiodonium chloride (DPI), a NADPH oxidase inhibitor, decreased the formation of punctate GFP-LC3B, and increased LC3B-II and SQSTM1 levels (). We then examined activation of the superoxide-generating enzyme NCF1, a component of the CYBB complex. Western blot assay and confocal microscopy images showed that Ro increased the expression of NCF1 in a dose-dependent manner and mobilized its translocation from cytosol to the plasma membrane (). Moreover, pretreatment with apocynin (ACN), a specific CYBB inhibitor, considerably reduced autophagic flux inhibition and the NAPDH oxidase activity (). ACN also decreased ROS production () and restored lysosomal acidification downregulated by Ro-mediated ROS production (). This implied that NCF1-ROS is upstream of autophagy. Based on these results, we suggest that Ro-triggered accumulation of ROS inhibits autophagy and sensitizes cells to 5-Fu mediated cell death.
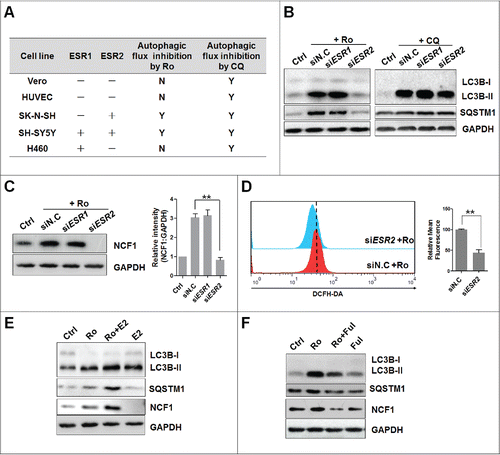
ESR2 (estrogen receptor 2) regulates NCF1 activity in Ro-treated cells
Next, we explored molecular signals linking Ro treatment to NCF1 activation, by focusing on ESR1 (estrogen receptor 1) and ESR2. To determine any possible role of ESR in autophagic flux inhibition by Ro, we used a panel of cell lines with varying combinations of ESR expression (ESR1 and ESR2).Citation58-61 Interestingly, Ro inhibited autophagic flux only in ESR2-expressing cell lines, SH-SY5Y and SK-N-SH, whereas no pattern was observed relating ESR expression to CQ-induced autophagy inhibition of cell lines ( and Fig. S6A). As additional evidence, siRNA-mediated deletion of ESR2 completely suppressed Ro-increased enhancement of LC3B-II and SQSTM1 protein levels in ECA-109 cells ( and Fig. S6B). Either no or only low effects of siRNA targeting ESR1 on CQ or Ro treatment were observed. In addition, knockdown of ESR2 blunted NCF1 expression, as well as ROS production in Ro-treated ECA-109 cells (). Furthermore, in silico molecular modeling method was used to determine whether Ro bound directly to ESR2 and activated its downstream signals. However, we failed to dock Ro into the ligand-binding pocket of ESR2 using software, including MOE and AutoDock, which suggested that Ro exerted estrogen-like activities via ligand-independent activation of ESR2. In agreement with this inference, preinhibiting ESR2 activity with specific inhibitor fulvestrant, reversed Ro-induced LC3B-II, SQSTM1, and NCF1 accumulation while preactivation of ESR2 with its ligand estradiol led to an opposite effect (). Together, these data suggest that ESR2 mediates the activation of CYBB and impairment of autophagic flux in response to Ro.
Figure 9. Activation of NCF1 and ROS requires ESR2. (A) Estrogen receptor expression and autophagy status in cell lines. −, no expression; +, positive expression based on analysis by western blot; Y, accumulation of LC3B-II and SQSTM1 induced by Ro (50 μM) or CQ (20 μM); N, cells resistant to Ro- or CQ-mediated autophagic flux inhibition. (B) Knockdown of ESR2 reduces Ro-induced LC3B-II and SQSTM1 accumulation. ECA-109 cells were transfected with siRNAs against ESR1 or ESR2 for 24 h and treated with Ro or CQ for another 12 h. siN.C, nonsense control siRNA. (C and D) Knockdown of ESR2 reduces NCF1 expression and ROS production. Three independent experiments were performed and a representative bolt was shown. ImageJ densitometric analysis of the NCF1:GAPDH ratio from NCF1 immunoblots (mean ± SD of 3 independent experiments) (**, P< 0.005). (E) Estradiol promotes Ro-induced LC3B, SQSTM1, and NCF1 accumulation. ECA-109 cells were pretreated with estradiol (E2) (100 ng/ml) for 2 h and then incubated with Ro for 12 h. (F) Fulvestrant compromises autophagic flux inhibition by Ro. Treatment cells with 10 μM Fulvestrant (Ful) for 2 h before adding Ro. Three independent experiments were performed and a representative result was shown.
Discussion
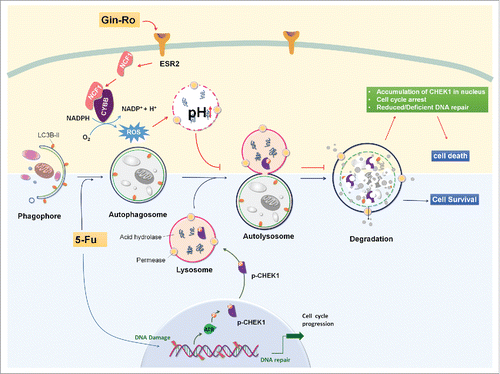
Autophagy, especially induced by chemotherapeutic agents, generally serves as a prosurvival mechanism. Consequently, suppression of autophagy has been increasingly recognized as a novel cancer therapeutic approach to induce autophagy-associated cell death or sensitize tumor cells to clinical drug-mediated cell death.Citation40,62-64 However, very limited information is currently available regarding the detailed mechanisms linking autophagic flux inhibition to cell death. In this study, we investigated the inhibitory effect of Ro on autophagy and its consequence on the crosstalk between CHEK1-mediated DNA damage checkpoint pathway and 5-Fu-induced cell death (). Autophagy served as a prosurvival function in 5-Fu-induced DNA damage via regulation of the degradation or cytosol quality control of CHEK1, one of the main proteins controlling cell cycle and DNA stability, to construct a beneficial niche for cancer cell proliferation. On the contrary, Ro activated the ESR2-NCF1-ROS signal pathway to impair autophagosome-lysosome fusion and lysosomal proteolytic activity, to suppress CHEK1 degradation and enhance the CHEK1-mediated DNA damage checkpoint signaling, which thereby sensitized chemoresistant cancer cells to 5-Fu-induced cell death.
Figure 10. Schematic illustration of Ro inhibiting autophagic flux and sensitizing chemoresistant esophageal cancer cells to 5-Fu-induced cell death. Upon 5-Fu treatment, autophagy is activated to serve as a prosurvival function partly through the selective degradation of CHEK1 to assure timely release of cell cycle arrest after DNA repair. When the fusion process of autophagosome and lysosome is blocked by Ro via the ESR2-NCF1-ROS pathway, high level of CHEK1 persists, leading to accumulation of DNA damage and aberrant cell cycle arrest at G2/M phase. Genes important for DNA replication are also downregulated by Ro. As a consequence, 5-Fu-mediated cell death is enhanced by Ro in a apoptosis-independent and autophagy-associated manner.
We first identified Ro, as well as ginsenosides Ra1, Rh1, Rd, 20(R)-Rg2, and notoginsenoside Fc, as novel autophagy inhibitors. Next, we presented confirmatory evidence that Ro interferes with autophagic flux by blocking autophagosome-lysosome fusion and inhibiting lysosomal proteolytic activity. This was demonstrated in several different approaches including measuring LC3B-II and SQSTM1 protein levels, detecting GFP-LC3B punctation and GFP-RFP-LC3B fluorescence and colocalization, determining the lysosomal pH or cathepsin enzyme activity, and analyzing EGFR degradation. Notably, results with Ro were similar to those with CQ, which inhibits autophagosome-lysosome fusion. We then demonstrated that interference with autophagic flux by Ro leads to the accumulation of DNA damage and reduced DNA repair, which renders cells more sensitive to 5-Fu-induced cell death. Moreover, detailed studies to explore the underlying mechanism revealed that the effect of Ro on the fusion process and autophagy-associated cell death requires the ligand-independent activation of ESR2, sequentially increased NCF1 activity, and ROS production. Data from this study thus expand our current knowledge about the bioactivity of Ro considering that only anti-inflammatory and anti-hepatic activities of Ro have been documented to date.Citation65,66 These observations also collectively provide a novel therapeutic strategy for human esophageal cancer treatment. Besides, the underlying mechanisms of autophagy inhibition mediated by other ginsenosides (Ra1, Rh1, Rd, 20(R)-Rg2, and notoginsenoside Fc) deserves further investigation to completely understand whether ginsenosides inhibit autophagy via a universal mechanism.
The results presented here demonstrate that the process of fusion between autophagosomes and lysosomes is preferably impaired by the action of Ro. Currently, it is generally considered that autophagosome-lysosome fusion depends on the pH of the acidic compartments.Citation34 Our data from AO staining indicated that Ro-mediated blockage of autophagosome-lysosome fusion might be due to a rise in lysosomal pH (). We also tested the effect of Ro on other lysosomal positioning proteins that may affect autophagosome-lysosome fusion, such as transcription factor EB and RAB5A,Citation67,68 and found that expression of these proteins were unchanged (data not shown). Thus, other factors may also contribute to the inhibitory effect of Ro on autophagosome-lysosome fusion. Besides, like CQ, Ro treatment blocked the degradation of GFP -LC3B, consistent with previously reported data that generation of free GFP fragments is the first consequence in the step-wise degradation of GFP-LC3B and the GFP moiety is probably degraded rather than quenched under lysosomal conditions.Citation36 In addition, activity of the cathepsin proteases CSTB and CSTD, the least-studied hydrolases within the lysosomal compartment, determines the efficiency of lysosomal degradation and autophagic flux.Citation37,38 Because CSTB and CSTD are involved in various pathologies and oncogenic processes in cancer,Citation69,70 inhibiting CSTB and CSTD activities represents a promising strategy for cancer therapy. The autophagic-lysosomal pathway is the main proteolytic system modified in esophageal cancer patients and the lysosomal protease activities are increased significantly in such patients, suggesting the involvement of the autophagic-lysosomal proteolytic system during development of cachexia in humans with esophageal cancer.Citation43 Therefore, modulation of the enzymatic activity of these systems, especially cathepsins, can be an efficient therapeutic approach for esophageal cancer. Indeed, using cathepsin substrate assay, CTSB and CSTD maturation immunoblot and real-time PCR, we confirmed that Ro dramatically downregulated the expression and activities of the most abundant cathepsins, CSTB and CSTD, leading to the inhibition of lysosomal activity, which in turn sensitized esophageal cancer cells to anticancer drug-induced cell cytotoxicity.
An important finding of this study is the NCF1-ROS-mediated impairment of lysosomal acidification and pH-dependent enzyme activity by Ro. An association between ROS accumulation and autophagy modulation has been previously described, implying that autophagy serves as a means for clearance of ROS and is both, a cell-survival and cell-death response.Citation55,56 Elevated oxidative stress is associated with impaired lysosomal maturation and disrupted autophagosome-lysosome fusion, possibly because it increases lysosomal pH.Citation71 Our data also indicate that ROS production trigged by Ro treatment raises the lysosomal pH and disrupts the fusion process, leading to an increase in LC3B-II and SQSTM1 as well as 5-Fu-mediated cell death (). Therefore, caution is required when considering a connection between ROS and autophagy induction. As an important source of ROS, the present study suggests a role of CYBB activation in ROS-derived autophagic flux impairment. Although we cannot exclude the involvement of other ROS sources (that is, the mitochondria), Ro treatment increased the expression and translocation of NCF1 in a dose-dependent manner (). NADPH oxidase is associated with lysosomal membranes that locally generate superoxide, which might modulate lysosomal acidification.Citation72-74 CYBB-derived ROS regulate autophagosome pH and inhibit autophagosome coalescence and fusion with the lysosome.Citation75-77 In agreement with previous reports, we found that inhibition of the CYBB completely reduces ROS production in parallel with normalizing lysosomal pH and lowering autophagosome abundance by Ro (). Recent studies also identify NADPH oxidase4 in the regulation of cardiac autophagy, particularly during energy stress, implying that the activation of specific NADPH oxidase isoforms depends on different stress stimuli.Citation78
In addition, for the first time, we uncovered that CYBB activation and ROS production is mediated by the activation of ESR2 upon Ro treatment. The siRNA knockdown assay performed with a panel of cell lines expressing various ESR isoforms demonstrated that ESR2, but not ESR1, plays a vital role in autophagy inhibition, independent of ligand binding (). This is consistent with previous reports that ginsenosides have an estrogenic effect on ESR activation.Citation22 Though the effect of ESR activation on ROS production has been widely reported,Citation79,80 the interaction between ESR and NADPH oxidase is barely discussed. On the contrary, scant evidence in the literature supports an inhibitory effect for ESR in NADPH oxidase activation and superoxide production.Citation81,82 Further efforts are required to elucidate the mechanism by which ESR2 activation stimulates NCF1 activity. Besides, the foundation of the indirect activation of ESR2 and possible Ro targets remain to be clarified.
Another interesting finding in our study is that, unlike cisplatin and carboplatin, 5Fu induces prosurvival autophagy, and inhibiting such autophagy enhances 5-Fu-induced cell death. In fact, our results are generally consistent with some previous reports.Citation7,83,84 The results from recent studies indicate that chemoresistant cells exhibit a prosurvival autophagy, and cell death induced by 5-Fu resembles the morphology of autophagic cell death.Citation7,85 Besides, esophageal cancer patients exhibit a higher level of autophagy.Citation43 Consistently, we found that ECA-109 and TE-1 cells are more resistant to 5-Fu than other chemosensitive esophageal cancer cells such as OE19 and OE33 cells (Fig. S3A) Citation44-46 and that 5-Fu induces prosurvival autophagy, and caspase-independent and autophagy-associated cell death (). Direct suppression of autophagy by knockdown of ATG5, ATG7, LC3B, and BECN1 apparently enhanced 5-Fu-mediated cell death (). Inhibition of autophagy by 3-MA (at a high concentration of 10 mM), a class III PtdIns3K inhibitor that is widely used as a pharmacological inhibitor in the autophagy studies, also slightly augmented the cytotoxicity of 5-Fu (Fig. S3B). Although there is increasing evidence showing that blockage of autophagy by 3-MA can reduce the protective effect of autophagy and potentiate the chemosensitivity and radiosensitivity of esophageal squamous cell carcinoma,Citation86,87 it is not clear if the potentiating effects of 3-MA on chemotherapy efficacy come from autophagy inhibition. Other studies have also reported that 3-MA can inhibit both class III PtdIns3K and class I phosphoinositide 3-kinase and exhibit a dual role in modulation of autophagy.Citation88,89 Additionally, the synergism between 3-MA and chemotherapeutic drugs seems to be independent of autophagy.Citation90 Therefore caution should be exercised in the application of 3-MA in cancer combinatory study. Remarkably, disruption of the late stages of autophagy leads to excessive accumulation of autophagic vacuoles containing harmful, undegraded material and has the potential to turn autophagy into a destructive process.Citation91-93 It is therefore conceivable that Ro, though inhibiting autophagic flux, is able to markedly sensitize chemoresistant cancer cells to 5-Fu-induced cell death. These findings suggest that Ro can be further developed as a potent cancer therapeutic agent by targeting autophagy, especially in those cancers where chemotherapeutic drugs induce protective autophagy that contributes to drug resistance.
Another important finding from our studies is the relevance of CHEK1-mediated DNA damage checkpoints and autophagy-associated cell death. Upon DNA damage induced by genotoxic agents such as 5-Fu, CHEK1 is activated to delay cell cycle progression to allow enough time for DNA repair. During completion of repair, CHEK1 is selectively degraded in lysosomes.Citation51,52 During this process, autophagy exerts a protective effect and is necessary for the degradation of specific DNA repair proteins.Citation94,95 We found that following Ro treatment, the degradation of phosphorylated (Ser 345) and total CHEK1 induced by 5-Fu is largely impaired or delayed due to compromised lysosomal proteolytic activity (). Consequently, the persistence of elevated levels of p-CHEK1 resulted in a higher proportion of cells being arrested in the G2/M phase, along with accumulation of DNA damage, associated with prolonged clearance of γH2AFX ().Citation49 The immunoblotting assay confirmed that the activation of ATM or ATR-initiated DNA damage response, the upstream of CHEK1, was another source of the Ro-mediated increase in DNA damage (). Moreover, expression of several DNA replication-related genes was downregulated in the presence of Ro, which could contribute to increased DNA damage and the chemo-sensitizing effect of autophagic flux inhibition in combination with 5-Fu (). Although we attributed the increased DNA damage and cell death to a consequence of Ro-mediated failure of autophagic degradation, further experiments should be performed to explore the specific molecular mechanism by which accumulated CHEK1 triggers the increased DNA damage-associated cell death. Other functional alterations in DNA repair, such as the deficiency of homologous recombination or nonhomologous end-joining for repair of DNA DSBs and the downregulation of DNA replication process (), should be clarified. For instance, DNA mismatch repair (MMR) plays an important role in the cytotoxic effect of 5-Fu. In response to 5-Fu treatment, MMR has been implicated in initially activating a G2/M cell cycle arrest followed by the activation of autophagy.Citation96,97 The duration of CHEK1-mediated G2/M arrest determines the level of autophagy following MMR Citation98 and a synergistic cytotoxic effect of 5-Fu combined with CHEK1 activity inhibition may result.Citation99 In addition, the relationship between ROS and DNA damage should also be examined since ROS has been implicated in oxidative injury of DNA and phosphorylation and stabilization of CHEK1, while autophagy inhibition by stimulating ROS formation has been reported to promote 5-Fu-induced cell death.Citation100
In summary, data from our study shed light on the intricate relationship between CYBB, ROS, autophagy, DNA damage, and cell death in cancer cells. We showed for the first time that ginsenoside Ro suppresses autophagy by interfering with autophagosome-lysosome fusion and lysosomal proteolytic activity via the ESR2-NCF1-ROS axis. Subsequently, such autophagic inhibition reduces CHEK1 degradation, enhances CHEK1-mediated DNA damage checkpoint arrest, and thereby sensitizes tumor cells to 5-Fu-induced cell death. Our findings support the theory that autophagy-modulating small molecules such as Ro may be effective therapeutic options for cancer treatment. Targeting DNA damage response, as well as cell-cycle checkpoint kinases, may also be a promising approach to overcome chemoresistance.
Materials and methods
Cell lines and cell culture
ECA-109, TE-1, and H460 cells were cultured in RPMI 1640 (Gibco, C11875500BT) supplemented with 10% fetal bovine serum (Gibco, 1027-106) and 1% penicillin/streptomycin (Invitrogen, 15140163) at 37°C in a humid atmosphere with 5% CO2. EC-9706, SK-N-SH, MEF, and atg7-knockout MEF (atg7−/−) cells were maintained in minimum essential medium (Gibco, 41500018). Vero, SH-SY5Y, HEEC, and HUVEC cells were cultured in Dulbecco's modified Eagle's medium (Gibco, 41965120). For nutrient starvation, tumor cells were washed 3 times with phosphate-buffered saline (PBS; Gibco, 10010049) buffer and then cultured in EBSS (Invitrogen, 14155063) for the indicated time periods.
Chemicals and reagents
Tiron (sc-253699), diphenyleneiodonium chloride (sc-202584), and apocynin (sc-203321) were purchased from Santa Cruz Biotechnology. CQ (C6628) and NAC (A9165) were purchased from Sigma-Aldrich. Z-VAD-FMK (C1202) was purchased from Beyotime. Estradiol (S4046) and Fulvestrant (S1191) were purchased from Selleck. EGF (epidermal growth factor) (PHG0314) and plasmid mRFP-GFP-LC3B (36239) was purchased from Invitrogen.
Anti-GFP (11814460001) was obtained from Roche. Anti-LC3B rabbit polyclonal (L7543) and anti-SQSTM/p62 antibody (P0067) were supplied by Sigma-Aldrich. Anti-ATG7 (ab53255) and anti-ATG5 antibody (ab78073) were from Abcam. Anti-NCF1 (4301s), anti-LC3B (3868), anti-p-ATM (5883), anti-p-ATR (2853), anti-p-BRCA1 (9009), anti-p-CHEK1 (Ser345) (2348), anti-p-H2AFX (Ser139; 9718), anti-p-CHEK2 (Thr68; 2197), anti-CHEK1 (2360), anti-p-CHEK1 (Ser296; 2349), anti-GAPDH (5174), anti-EGFR (4267), anti-CASP3 (9662), anti-cleaved CASP3 (9664), anti-CASP9 (9504), anti-PARP1 (9542), anti-ESR1/estrogen receptor α (8644), anti-ESR2/estrogen receptor β (5513), anti-mouse IgG (7076), and anti-rabbit IgG (7074) horseradish peroxidase (HRP)-linked antibodies were purchased from Cell Signaling Technology. Anti-CTSD/cathepsin D (PB0019) and anti-CTSB/cathepsin B (BA0428) antibodies were from Boster. Alexa Fluor 488 goat anti-rabbit IgG (H + L) was from Life Technologies (A-11034). Alexa Fluor 488 anti-mouse IgG (H + L) (4408), Alexa Fluor 647 anti-mouse IgG (H + L) (4410) and HRP-linked anti-mouse (7076) or anti-rabbit IgG (7074) were from Cell Signaling Technology. DAPI (D9542) and TRITC-phalloidin (P1951) were purchased from Sigma-Aldrich. All inhibitors were used in a noncytotoxic concentration.
RNA extraction and real-time PCR
For the mRNA expression level assay, total RNA was extracted with TRIzol reagent (Invitrogen, 15596-026) according to the manufacturer's protocol and 1 μg RNA was then reverse-transcribed with a PrimeScript RT reagent kit (TaKaRa, RR047A). A real-time PCR assay was performed using a Bio-Rad CFX96 real-time PCR system (singapore). The results were analyzed with CFX manager software (Bio-Rad). The sequences of primer pairs are shown in Table S1. mRNA transcription levels were standardized against housekeeping gene GAPDH.
Cell viability assay
The cell viability of chemical inhibitors and siRNA was determined with a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (Sigma-Aldrich, M2128) assay. In brief, cells were cultured in each well of 96-well plates with varying concentrations of drugs for 48 h. Following incubation with MTT (0.5 mg/ml) for 4 h, the supernatant fraction from each well was discarded and the insoluble formazan product was dissolved in 100 μl dimethyl sulfoxide (DMSO). The optical density was measured at 570 nm, with a reference wavelength of 630 nm on a multiscanner autoreader (Epoch, Bio-tek, Winooski, VT, USA). The OD570 for control cells was taken as 100%, viability.
Western blotting
Cells were lysed in RIPA buffer (Beyotime, P0013B). The lysates were normalized to equal amounts of protein, and the proteins were separated by 6% to 15% gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane, and probed with the indicated primary antibodies. Proteins were detected by incubation with species-specific HRP-conjugated secondary antibodies. Immunoreactive bands were visualized using enhanced chemiluminescence blotting detection reagents (Thermo, 34080). ImageJ software was used to quantify the band intensity. For each point, the signal intensity for each protein was normalized to that measured for GAPDH and was expressed as the fold-increase relative to the control.
AO staining
Cell staining with AO (Sigma-Aldrich, A6014) was performed as previously described,Citation61 adding a final concentration of 5 μg/ml for a period of 30 min (37°C, 5% CO2). Tumor cells were incubated with the indicated compounds for 12 h or starved with EBSS before AO was added. After washing with PBS 3 times, images were captured using a confocal microscope (Zeiss 510 META, Carl Zeiss Microimaging, Thornwood, NY, USA) equipped with an argon laser (excitation wavelength 488 nm), and a 40× objective lens. AO produces red fluorescence (emission peak at approximately 650 nm) in lysosomal compartments, and green fluorescence (emission between 530 and 550 nm) in the cytosolic and nuclear compartments. The red and green intensity ratio in the exclusive nuclear region was analyzed by ImageJ software.
Cell cycle analysis
To determine the effects of Ro or 5-Fu on the cell cycle distribution, tumor cells were treated with Ro (100 μM) or 5-Fu (100 μg/ml) for 48 h, fixed with 70% (v/v) ethanol, and stained with 50 μg/ml propidium iodide (Sigma, P4170) containing 0.1 mg/ml RNase (Sigma, R6513) and 5% Triton X-100 (Sigma,T8787) for 30 min at 37°C. The percentage of cells in different phases of the cycle was analyzed using a flow cytometer (BD FACSCalibur, Piscataway, NJ, USA) and inbuilt software.
Measurement of intracellular ROS
Tumor cells treated with the indicated compounds for 12 h or transfected with siRNAs were stained with 10 μM DCFH-DA (Sigma-Aldrich, D6883) for 30 min. ROS generation was determined at 525 nm with a flow cytometer (BD FACSCalibur, USA, Piscataway, New Jersey, USA).
NADPH oxidase activity assay
NADPH oxidase activity assay was performed using a modified procedure.Citation101 Equal amounts of proteins in Krebs-HEPES buffer (NOXYGEN, NOX-07.6.1; pH 7.4) containing protease and phosphatase inhibitor cocktail (Sigma-Aldrich, P5726) were incubated with 10 mM lucigenin (Santa Cruz Biotechnology, 2315-97-1) at 37°C for 10 min. Then, 100 μM NADPH (Sigma, 000000010107824001) was added with or without inhibitors and incubated for another 5 min at 37°C. Chemiluminescence was measured with a Fluostar Optima microplate reader (Molecular Devices SpectraMax I3, Sunnyvale, CA, USA). Lucigenin activity (RLU/mg protein) of control cells was arbitrarily set at 100%.
RNA interference and transfection
siRNAs targeting autophagy and estrogen receptors, and nonsense control siRNAs were purchased from GenePharma. Tumor cells were transfected with 2 μg siRNA against BECN1, ATG5, ATG7, LC3B, ESR1, ESR2, or nonsense control siRNA using Lipofectamine 2000 (Invitrogen, 11668-019) according to the manufacturer's instructions. Further experiments were performed after transfection for 24 h. The sequences of siRNAs are provided in Table S2.
Immunofluorescence staining and analysis
Cells were washed in PBS, fixed in 4% paraformaldehyde-PBS for 15 min and permeabilized with 0.1% Triton X-100 for 5 min. The samples were blocked in 5% bovine serum albumin (Gibco, 1027-106) and incubated with anti-LC3B antibody (1:1000; Sigma, L7543), anti-SQSTM1 antibody (1:5000), anti-NCF1 (1:1000), or anti-cleaved-CASP3 (1:1000). The stained cells were washed and incubated with Alexa Fluor-conjugated secondary antibodies (1:1000). Unless otherwise specified, staining was performed at room temperature for 1 h. Additionally, 1 mg/ml DAPI-PBS and 5 μM TRITC-phalloidin were added to label nuclei (15 min) and F-actin (40 min), respectively. Images were captured with a Zeiss LSM510 Meta confocal system under a 40× oil immersion objective (Carl Zeiss Microimaging, Thornwood, New York, USA).
For LysoTracker Red staining, cells were treated with chemicals (as indicated) for 12 h at 37°C and then incubated with 50 nM LysoTracker Red (Beyotime, C1043) for an additional 1 h. Cells were fixed with 4% paraformaldehyde, stained with DAPI, and analyzed by fluorescence microscopy.
Cathepsin activity assay
The catalytic activities of CSTB (cathepsin B) and CSTD (cathepsin D) were measured using fluorometric methods (Abnova, KA0766, KA0767). Briefly, 2 × 10Citation6 cells were collected by centrifugation and lysed in 50 μl chilled cell lysis buffer (Cell Signaling Technology, 9806). Then the cell lysate was transferred into 96-well plates, mixed with 50 μl reaction buffer and 2 μl substrate, and incubated at 37°C for 2 h. The samples were read in a fluorometer with 400 nm excitation and 505 nm emission filters. Fold-increase in cathepsin activity was determined by comparing the relative fluorescence units (RFU) with the level of the untreated sample.
GFP-LC3B or mRFP-GFP-LC3B assay
Cultured cells were transfected with GFP-LC3B or mRFP-GFP-LC3B (2 μg) for 24 h and incubated with different chemicals or starved for the indicated times. After treatment, the cells were fixed with 4% paraformaldehyde in PBS. Images were captured using a fluorescence microscope (Olympus co. Ltd, IX71, Tokyo, Japan) or LSM 510 META confocal microscope (Zeiss). For screening of autophagy modulators, cells were treated with different chemicals for 12 h and the average number of GFP-LC3B puncta per cell was determined. Autophagic flux analysis was measured by confocal counting of the cells with GFP+ mRFP+ (yellow) and GFP− RFP+ (red) LC3B-puncta. At least 50 cells from 5 representative fields were counted in each independent experiment.
Transmission electron microscopy assay
ECA-109 cells were treated as indicated and were fixed in 4% glutaraldehyde at 4°C overnight. After dehydration, ultrathin sections were embedded and stained with uranyl acetate/lead citrate. Images were captured under a transmission electron microscope (FEI Tcenai G2 20, Hillsboro, Oregon, USA).
Statistical analysis
Data shown in this study are represented as mean value ± standard deviation values, which use the results from at least 3 independent experiments. The Student 2-tailed t test was used for all statistical analysis, with the level of significance set at **, P < 0.005; *, P < 0.05.
Abbreviations
| ACN | = | apocynin |
| AO | = | acridine orange |
| ATG5 | = | autophagy-related 5 |
| ATG7 | = | autophagy-related 7 |
| BECN1 | = | Beclin 1 |
| CASP3 | = | caspase 3 |
| CASP9 | = | caspase 9 |
| CHEK | = | checkpoint kinase |
| CQ | = | chloroquine |
| CTSB | = | cathepsin B |
| CTSD | = | cathepsin D |
| CYBB | = | cytochrome b-245 β polypeptide |
| DDIT3 | = | DNA damage inducible transcript 3 |
| DMEM | = | Dulbecco's modified Eagle's medium |
| DPI | = | diphenyleneiodonium chloride |
| DSB | = | DNA double-strand break |
| EBSS | = | Earle's balanced salt solution |
| EGF | = | epidermal growth factor |
| EGFR | = | epidermal growth factor receptor |
| ESR1 | = | estrogen receptor 1 |
| ESR2 | = | estrogen receptor 2 |
| E2 | = | estradiol |
| GFP | = | green fluorescent protein |
| H2AFX | = | H2A histone family member X |
| MAP1LC3B/LC3B | = | microtubule associated protein 1 light chain 3 β |
| MEF | = | mouse embryonic fibroblast |
| MMR | = | DNA mismatch repair |
| NAC | = | N-acetyl-L-cysteine |
| NCF1 | = | neutrophil cytosolic factor 1 |
| PARP1 | = | poly(ADP-ribose) polymerase 1 |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| RFP | = | red fluorescence protein |
| Ro | = | ginsenoside Ro |
| ROS | = | reactive oxygen species |
| siRNA | = | small interfering RNA |
| SQSTM1 | = | sequestosome 1 |
| WT | = | wild type |
| γH2AFX | = | phosphorylated H2AFX |
| 5-Fu | = | 5-fluorouracil |
Disclosure of potential conflicts of interest
The authors declare that they have no potential conflicts of interest.
1192751_Supplemental_Material.docx
Download MS Word (6.8 MB)Funding
This work was supported by grants from the Shenzhen strategic emerging industry development project funding (JCYJ20130329105207152, SFG2013-180, KQCX20140522111508785), Shenzhen and Hong Kong collaborative innovation of science and technology plan (SGLH20120926161415784), Natural micro-molecule drug innovation engineering laboratory funding (Shenfagai [2013] 180), and the China Postdoctoral Science Foundation (Grant 2015M570726).
Related Research Data
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90; PMID:21296855; http://dx.doi.org/10.3322/caac.20107
- Tew WP, Kelsen DP, Ilson DH. Targeted therapies for esophageal cancer. Oncologist 2005; 10:590-601; PMID:16177283; http://dx.doi.org/10.1634/theoncologist.10-8-590
- Juergens RA, Forastiere A. Combined modality therapy of esophageal cancer. J Natl Compr Cancer Netw 2008; 6:851-861.
- Burmeister BH, Walpole ET, D'Arcy N, Burmeister EA, Cox S, Thomson DB, Harvey JA, Smithers BM. A phase II trial of chemoradiation therapy with weekly oxaliplatin and protracted infusion of 5-fluorouracil for esophageal cancer. Invest New Drugs 2009; 27: 275-279; PMID:18841327; http://dx.doi.org/10.1007/s10637-008-9178-4
- Miyazaki T, Ojima H, Fukuchi M, Sakai M, Sohda M, Tanaka N, Suzuki S, Ieta K, Saito K, Sano A, et al. Phase II Study of Docetaxel, Nedaplatin, and 5-Fluorouracil combined chemotherapy for advanced esophageal cancer. Ann Surg Oncol 2015; 22:3653-8. Feb 18. [Epub ahead of print]; PMID:25691281; http://dx.doi.org/10.1245/s10434-015-4440-4
- Song S, Honjo S, Jin J, Chang SS, Scott AW, Chen Q, Kalhor N, Correa AM, Hofstetter WL, Albarracin CT, et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and confers chemoresistance in esophageal cancer. Clin Cancer Res 2015; 21:2580-90. Mar 4. [Epub ahead of print]; PMID:25739674; http://dx.doi.org/10.1158/1078-0432.CCR-14-2191
- O'Donovan TR, O'Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011; 7(5):509-524; PMID:21325880; http://dx.doi.org/10.4161/auto.7.5.15066
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102-1109; PMID:17909521; http://dx.doi.org/10.1038/ncb1007-1102
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-1075; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biol Ther 2011; 11:157-168; PMID:21228626; http://dx.doi.org/10.4161/cbt.11.2.14622
- Livesey KM, Tang D, Zeh HJ, Lotze MT. Autophagy inhibition in combination cancer treatment. Curr Opin Investig Drugs 2009; 10:1269-1279; PMID:19943199
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, White E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21(13):1621-1635; PMID:17606641; http://dx.doi.org/10.1101/gad.1565707
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21(11):1367-1381; PMID:17510285; http://dx.doi.org/10.1101/gad.1545107
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51-64; PMID:16843265; http://dx.doi.org/10.1016/j.ccr.2006.06.001
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283(16):10892-10903; PMID:18281291; http://dx.doi.org/10.1074/jbc.M800102200
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19(3):797-806; PMID:18094039; http://dx.doi.org/10.1091/mbc.E07-10-1092
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726-734; PMID:16148885; http://dx.doi.org/10.1038/nrc1692
- Qian W, Liu J, Jin J, Ni W, Xu W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk Res 2007; 31:29-39.
- Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clongenic survival. Mol Cell 2011; 42:23-35; PMID:21353614; http://dx.doi.org/10.1016/j.molcel.2011.02.009
- Mishra BB, Tiwari VK. Natural products: an evolving role in future drug discovery. Eur J Med Chem 2011; 46(10):4769-4807; PMID:21889825; http://dx.doi.org/10.1016/j.ejmech.2011.07.057
- Wong AS, Che CM, Leung KW. Recent advances in ginseng as cancer therapeutics: a functional and mechanistic overview. Nat Prod Rep 2015; 32(2):256-72; PMID:25347695; http://dx.doi.org/10.1039/C4NP00080C
- Shin JY, Lee JM, Shin HS, Park SY, Yang JE, Cho SK, Yi TH. Anti-cancer effect of gnsenoside F2 against glioblastoma multiforme in xenograft model in SD rats. J Ginseng Res 2012; 36(1):86-92; PMID:23717108; http://dx.doi.org/10.5142/jgr.2012.36.1.86
- Mai TT, Moon J, Song Y, Viet PQ, Phuc PV, Lee JM, Yi TH, Cho M, Cho SK. Ginsenoside F2 induces apoptosis accompanied by protective autophagy in breast cancer stem cells. Cancer Lett 2012; 321(2):144-153; PMID:22326284; http://dx.doi.org/10.1016/j.canlet.2012.01.045
- Kim DG, Jung KH, Lee DG, Yoon JH, Choi KS, Kwon SW, Shen HM, Morgan MJ, Hong SS, Kim YS. 20(S)-Ginsenoside Rg3 is a novel inhibitor of autophagy and sensitizes hepatocellular carcinoma to doxorubicin. Oncotarget 2014; 5(12):4438-4451; PMID:24970805; http://dx.doi.org/10.18632/oncotarget.2034
- Ko H, Kim YJ, Park JS, Park JH, Yang HO. Autophagy inhibition enhances apoptosis induced by ginsenoside Rk1 in hepatocellular carcinoma cells. Biosci Biotechnol Biochem 2009; 73(10):2183-2189; PMID:19809182; http://dx.doi.org/10.1271/bbb.90250
- Kim AD, Kang KA, Kim HS, Kim DH, Choi YH, Lee SJ, Kim HS, Hyun JW. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis 2013; 4:e750.
- Chen Z, Lu T, Yue X, Wei N, Jiang Y, Chen M, Ni G, Liu X, Xu G. Neuroprotective effect of ginsenoside Rb1 on glutamate-induced neurotoxicity: with emphasis on autophagy. Neurosci Lett 2010; 482(3):264-268; PMID:20667501; http://dx.doi.org/10.1016/j.neulet.2010.07.052
- Guo J, Chang L, Zhang X, Pei S, Yu M, Gao J. Ginsenoside compound K promotes β-amyloid peptide clearance in primary astrocytes via autophagy enhancement. Exp Ther Med 2014; 8(4):1271-1274; PMID:25187838
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-614; PMID:16286508; http://dx.doi.org/10.1083/jcb.200507002
- Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, Cumming RC, Harris GL, Nezis IP, Schubert DR, Simonsen A, et al. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 2011; 7:572-583; PMID:21325881; http://dx.doi.org/10.4161/auto.7.6.14943
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445544; PMID:NOT_FOUND
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-460; PMID:17534139; http://dx.doi.org/10.4161/auto.4451
- Kawai A, Uchiyama H, Takano S, Nakamura N, Ohkuma S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007; 3:154-157; PMID:17204842; http://dx.doi.org/10.4161/auto.3634
- Zheng K, Kitazato K, Wang Y. Viruses exploit the function of epidermal growth factor receptor. Rev Med Virol 2014; 24(4):274-286; PMID:24888553; http://dx.doi.org/10.1002/rmv.1796
- Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 2011; 7:188-204; PMID:21107021; http://dx.doi.org/10.4161/auto.7.2.14181
- Katunuma N. Posttranslational processing and modification of cathepsins and cystatins. J Signal Transduct 2010; 2010:375345; PMID:21637353; http://dx.doi.org/10.1155/2010/375345
- Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta 2012; 1824(1):22-33; PMID:21914490; http://dx.doi.org/10.1016/j.bbapap.2011.08.016
- Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10(9):1533-11541; PMID:21878654; http://dx.doi.org/10.1158/1535-7163.MCT-11-0047
- Zhou J, Hu SE, Tan SH, Cao R, Chen Y, Xia D, Zhu X, Yang XF, Ong CN, Shen HM. Andrographolide sensitizes cisplatin-induced apoptosis via suppression of autophagosome-lysosome fusion in human cancer cells. Autophagy 2012; 8(3):338-349; PMID:22302005; http://dx.doi.org/10.4161/auto.18721
- Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, Morgan MJ, Mirsky DM, Handler MH, Foreman NK, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov 2014; 4(7):773-780; PMID:24823863; http://dx.doi.org/10.1158/2159-8290.CD-14-0049
- Eum KH, Lee M. Targeting the autophagy pathway using ectopic expression of Beclin 1 in combination with rapamycin in drug-resistant v-Ha-ras-transformed NIH 3T3 cells. Mol Cells 2011; 31(3):231-238; PMID:21350938; http://dx.doi.org/10.1007/s10059-011-0034-6
- Tardif N, Klaude M, Lundell L, Thorell A, Rooyackers O. Autophagic-lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am J Clin Nutr 2013; 98(6):1485-1492; PMID:24108784; http://dx.doi.org/10.3945/ajcn.113.063859
- Liu Y, Wang X, Wang Y, Zhang Y, Zheng K, Yan H, Zhang L, Chen W, Wang X, Liu Q, et al. Combination of SNX-2112 with 5-FU exhibits antagonistic effect in esophageal cancer cells. Int J Oncol 2015; 46(1):299-307; PMID:25333998
- Dun J, Chen X, Gao H, Zhang Y, Zhang H, Zhang Y. Resveratrol synergistically augments anti-tumor effect of 5-FU in vitro and in vivo by increasing S-phase arrest and tumor apoptosis. Exp Biol Med (Maywood) 2015 Mar 2. pii: 1535370215573396. [Epub ahead of print].
- Han S, Lee CW, Trevino JG, Hughes SJ, Sarosi GA Jr. Autocrine extra-pancreatic trypsin 3 secretion promotes cell proliferation and survival in esophageal adenocarcinoma. PLoS One 2013; 8(10):e76667; PMID:24146905; http://dx.doi.org/10.1371/journal.pone.0076667
- Bao L, Jaramillo MC, Zhang Z, Zheng Y, Yao M, Zhang DD, Yi X. Induction of autophagy contributes to cisplatin resistance in human ovarian cancer cells. Mol Med Rep 2015; 11(1):91-98; PMID:25322694
- Wang Y, Hu Z, Liu Z, Chen R, Peng H, Guo J, Chen X, Zhang H. MTOR inhibition attenuates DNA damage and apoptosis through autophagy-mediated suppression of CREB1. Autophagy 2013; 9(12):2069-2086; PMID:24189100; http://dx.doi.org/10.4161/auto.26447
- Eapen VV, Haber JE. DNA damage signaling triggers the cytoplasm-to-vacuole pathway of autophagy to regulate cell cycle progression. Autophagy 2013; 9(3):440-441; PMID:23322149; http://dx.doi.org/10.4161/auto.23280
- Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011; 471(7336):74-79; PMID:21368826; http://dx.doi.org/10.1038/nature09803
- Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun 2015; 6:6823; PMID:25880015; http://dx.doi.org/10.1038/ncomms7823
- Liu EY, Xu N, O'Prey J, Lao LY, Joshi S, Long JS, O'Prey M, Croft DR, Beaumatin F, Baudot AD, et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci U S A 2015; 112(3):773-778; PMID:25568088; http://dx.doi.org/10.1073/pnas.1409563112
- Liu, Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev 2000; 14:1448-1459; PMID:10859164; http://dx.doi.org/10.1101/gad.840500
- Friedman AD. GADD153/CHOP, a DNA damage-inducible protein, reduced CAAT/enhancer binding protein activities and increased apoptosis in 32D c13 myeloid cells. Cancer Res 1996; 56(14):3250-3256; PMID:8764117
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 2011; 36(1):30-38; PMID:20728362; http://dx.doi.org/10.1016/j.tibs.2010.07.007
- Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 2010; 6(7):838-854; PMID:20505317; http://dx.doi.org/10.4161/auto.6.7.12113
- Landry WD, Cotter TG. ROS signalling, NADPH oxidases and cancer. Biochem Soc Trans 2014; 42(4):934-938; PMID:25109982; http://dx.doi.org/10.1042/BST20140060
- Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, Hoffstrom BG, Dewald LE, Schornberg KL, et al. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med 2013; 5(190):190ra79; PMID:23785035; http://dx.doi.org/10.1126/scitranslmed.3005471
- Evans MJ, Harris HA, Miller CP, Karathanasis SK, Adelman SJ. Estrogen receptors a and b have similar activities in multiple endothelial cell pathways. Endocrinology 2002; 143:3785-3795; PMID:12239089; http://dx.doi.org/10.1210/en.2002-220356
- Ba F, Pang PK, Davidge ST, Benishin CG. The neuroprotective effects of estrogen in SK-N-SH neuroblastoma cell cultures. Neurochem Int 2004; 44(6):401-411; PMID:14687605; http://dx.doi.org/10.1016/j.neuint.2003.08.004
- Grassi D, Bellini MJ, Acaz-Fonseca E, Panzica G, Garcia-Segura LM. Estradiol and testosterone regulate arginine-vasopressin expression in SH-SY5Y human female neuroblastoma cells through estrogen receptors-α and -β. Endocrinology 2013; 154(6):2092-2100; PMID:23584859; http://dx.doi.org/10.1210/en.2012-2137
- Tang B, Li Q, Zhao XH, Wang HG, Li N, Fang Y, Wang K, Jia YP, Zhu P, Gu J, et al. Shiga toxins induce autophagic cell death in intestinal epithelial cells via the endoplasmic reticulum stress pathway. Autophagy 2015; 11(2):344-354; PMID:25831014; http://dx.doi.org/10.1080/15548627.2015.1023682
- Lao Y, Wan G, Liu Z, Wang X, Ruan P, Xu W, Xu D, Xie W, Zhang Y, Xu H, et al. The natural compound oblongifolin C inhibits autophagic flux and enhances antitumor efficacy of nutrient deprivation. Autophagy 2014; 10(5):736-749; PMID:24642486; http://dx.doi.org/10.4161/auto.28034
- Yue W, Hamaï A, Tonelli G, Bauvy C, Nicolas V, Tharinger H, Codogno P, Mehrpour M. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy 2013; 5:714-729; PMID:23519090; http://dx.doi.org/10.4161/auto.23997
- Matsuda H, Samukawa K, Kubo M. Anti-inflammatory activity of ginsenoside Ro. Planta Med 1990; 56(1):19-23; http://dx.doi.org/10.1055/s-2006-960875
- Matsuda H, Samukawa K, Kubo M. Anti-hepatitic activity of ginsenoside Ro. Planta Med 1991; 57(6):523-526; http://dx.doi.org/10.1055/s-2006-960198
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:142933; http://dx.doi.org/10.1126/science.1204592
- Lu Y, Dong S, Hao B, Li C, Zhu K, Guo W, Wang Q, Cheung KH, Wong CW, Wu WT, et al. Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating RAB5A. Autophagy 2014; 10(11):1895-1905; PMID:25483964; http://dx.doi.org/10.4161/auto.32200
- Gondi CS, Rao JS. Cathepsin B as a cancer target. Expert Opin Ther Targets 2013; 17:281-291; PMID:23293836; http://dx.doi.org/10.1517/14728222.2013.740461
- Turk B, Stoka V, Rozman-Pungercar J, Cirman T, Droga-Mazovec G, Oresić K, Turk V. Apoptotic pathways: involvement of lysosomal proteases. Biol Chem 2002; 383:1035-1044; PMID:12437086
- Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci 2010; 30:12535-12544; PMID:20844148; http://dx.doi.org/10.1523/JNEUROSCI.1920-10.2010
- Lam GY, Huang J, Brumell JH. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin. Immunopathol 2010; 32:415-430; http://dx.doi.org/10.1007/s00281-010-0221-0
- Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Duménil AM, Seabra MC, Raposo G, Amigorena S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006; 126:205-218; PMID:16839887; http://dx.doi.org/10.1016/j.cell.2006.05.035
- Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A 2009; 106:6226-6231; PMID:19339495; http://dx.doi.org/10.1073/pnas.0811045106
- Jaishy B, Zhang Q, Chung HS, Riehle C, Soto J, Jenkins S, Abel P, Cowart LA, Van Eyk JE, Abel ED. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. J Lipid Res 2015; 56(3):546-561; PMID:25529920; http://dx.doi.org/10.1194/jlr.M055152
- Pal R, Palmieri M, Loehr JA, Li S, Abo-Zahrah R, Monroe TO, Thakur PB, Sardiello M, Rodney GG. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat Commun 2014; 5:4425; PMID:25028121
- Kim Y, Kim YS, Kim DE, Lee JS, Song JH, Kim HG, Cho DH, Jeong SY, Jin DH, Jang SJ, et al. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy 2013; 9(12):2126-2139; PMID:24322755; http://dx.doi.org/10.4161/auto.26308
- Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M, Sadoshima J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2α/activating transcription factor 4 pathway. Circ Res 2013; 113(11):1253-1264; PMID:24081881; http://dx.doi.org/10.1161/CIRCRESAHA.113.301787
- Razandi M, Pedram A, Jordan VC, Fuqua S, Levin ER. Tamoxifen regulates cell fate through mitochondrial estrogen receptor beta in breast cancer. Oncogene 2013; 2(27):3274-3285; http://dx.doi.org/10.1038/onc.2012.335
- Kumari Kanchan R, Tripathi C, Singh Baghel K, Kumar Dwivedi S, Kumar B, Sanyal S, Sharma S, Mitra K, Garg V, Singh K, et al. Estrogen receptor potentiates mTORC2 signaling in breast cancer cells by upregulating superoxide anions. Free Radic Biol Med 2012; 53(10):1929-1941; PMID:23000059; http://dx.doi.org/10.1016/j.freeradbiomed.2012.08.595
- Meng X, Wang M, Wang X, Sun G, Ye J, Xu H, Sun X. Suppression of NADPH oxidase- and mitochondrion-derived superoxide by Notoginsenoside R1 protects against cerebral ischemia-reperfusion injury through estrogen receptor-dependent activation of Akt/Nrf2 pathways. Free Radic Res 2014; 48(7):823-838; PMID:24720662; http://dx.doi.org/10.3109/10715762.2014.911853
- Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, Vadlamudi RK, Brann DW. Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J Neurosci 2009; 29(44):13823-13836; PMID:19889994; http://dx.doi.org/10.1523/JNEUROSCI.3574-09.2009
- Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010; 10:370; PMID:20630104; http://dx.doi.org/10.1186/1471-2407-10-370
- Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol 2009; 16(3):761-771; PMID:19116755; http://dx.doi.org/10.1245/s10434-008-0260-0
- Nyhan MJ, O'Donovan TR, Elzinga B, Crowley LC, O'Sullivan GC, McKenna SL. The BH3 mimetic HA14-1 enhances 5-fluorouracil-induced autophagy and type II cell death in oesophageal cancer cells. Br J Cancer 2012; 106(4):711-718; PMID:22240779; http://dx.doi.org/10.1038/bjc.2011.604
- Liu KS, Zhang Y, Ding WC, Wang SX, Xiang YF, Yang P, Chen ZP, Zheng K, Liu Z, Xia M, et al. The selective Hsp90 inhibitor BJ-B11 exhibits potent antitumor activity via induction of cell cycle arrest, apoptosis and autophagy in Eca-109 human esophageal squamous carcinoma cells. Int J Oncol 2012; 41(6):2276-2284; PMID:23076967
- Chen YS, Song HX, Lu Y, Li X, Chen T, Zhang Y, Xue JX, Liu H, Kan B, Yang G, et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis Esophagus 2011; 24(6):437-443; PMID:21166739; http://dx.doi.org/10.1111/j.1442-2050.2010.01156.x
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285(14):10850-10861; PMID:20123989; http://dx.doi.org/10.1074/jbc.M109.080796
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275(2):992-998; PMID:10625637; http://dx.doi.org/10.1074/jbc.275.2.992
- Sheng Y, Sun B, Guo WT, Zhang YH, Liu X, Xing Y, Dong DL. 3-Methyladenine induces cell death and its interaction with chemotherapeutic drugs is independent of autophagy. Biochem Biophys Res Commun 2013; 432(1):5-9; PMID:23395679; http://dx.doi.org/10.1016/j.bbrc.2013.01.106
- Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol 2008; 183:101-116; PMID:18838554; http://dx.doi.org/10.1083/jcb.200801099
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004; 11:448-457; PMID:14713959; http://dx.doi.org/10.1038/sj.cdd.4401359
- Morgan MJ, Gamez G, Menke C, Hernandez A, Thorburn J, Gidan F, Staskiewicz L, Morgan S, Cummings C, Maycotte P, et al. Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy 2014; 10:1814-1826; PMID:25136801; http://dx.doi.org/10.4161/auto.32135
- Bae H, Guan JL. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res 2011; 9:1232-1241; PMID:21807966; http://dx.doi.org/10.1158/1541-7786.MCR-11-0098
- Dotiwala F, Eapen VV, Harrison JC, Arbel-Eden A, Ranade V, Yoshida S, Haber JE. DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase. Proc. Natl Acad. Sci. USA 2013; 110, E41-E49; http://dx.doi.org/10.1073/pnas.1218065109
- Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res 1998; 4:1-6; PMID:9516945
- Zeng X, Kinsella TJ. A novel role for DNA mismatch repair and the autophagic processing of chemotherapy drugs in human tumor cells. Autophagy 2007; 3:368-370; PMID:17426439; http://dx.doi.org/10.4161/auto.4205
- Zeng X, Kinsella TJ. BNIP3 is essential for mediating 6-thioguanine- and 5-fluorouracil-induced autophagy following DNA mismatch repair processing. Cell Res 2010; 20(6):665-675; PMID:20368736; http://dx.doi.org/10.1038/cr.2010.40
- Ashwell S, Janetka JW, Zabludoff S. Keeping checkpoint kinases in line: new selective inhibitors in clinical trials. Expert Opin Investing Drugs 2008; 17:1331-1340; http://dx.doi.org/10.1517/13543784.17.9.1331
- Pan X, Zhang X, Sun H, Zhang J, Yan M, Zhang H. Autophagy inhibition promotes 5-fluorouraci-induced apoptosis by stimulating ROS formation in human non-small cell lung cancer A549 cells. PLoS One 2013; 8(2):e56679; PMID:23441212; http://dx.doi.org/10.1371/journal.pone.0056679
- Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, Iwai-Kanai E, Ikeda K, Ueyama T, Ogata T, et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc Natl Acad Sci U S A 2014; 111:3116-3121; PMID:24516131; http://dx.doi.org/10.1073/pnas.1318951111