
ABSTRACT
Severe hepatic inflammation is a common cause of acute or chronic liver disease. Macrophages are one of the key mediators which regulate the progress of hepatic inflammation. Increasing evidence shows that the TAM (TYRO3, AXL and MERTK) family of RTKs (receptor tyrosine kinases), which is expressed in macrophages, alleviates inflammatory responses through a negative feedback loop. However, the functional contribution of each TAM family member to the progression of hepatic inflammation remains elusive. In this study, we explore the role of individual TAM family proteins during autophagy induction and evaluate their contribution to hepatic inflammation. Among the TAM family of RTKs, AXL (AXL receptor tyrosine kinase) only induces autophagy in macrophages after interaction with its ligand, GAS6 (growth arrest specific 6). Based on our results, autophosphorylation of 2 tyrosine residues (Tyr815 and Tyr860) in the cytoplasmic domain of AXL in mice is required for autophagy induction and AXL-mediated autophagy induction is dependent on MAPK (mitogen-activated protein kinase)14 activity. Furthermore, induction of AXL-mediated autophagy prevents CASP1 (caspase 1)-dependent IL1B (interleukin 1, β) and IL18 (interleukin 18) maturation by inhibiting NLRP3 (NLR family, pyrin domain containing 3) inflammasome activation. In agreement with these observations, axl−/− mice show more severe symptoms than do wild-type (Axl+/+) mice following acute hepatic injury induced by administration of lipopolysaccharide (LPS) or carbon tetrachloride (CCl4). Hence, GAS6-AXL signaling-mediated autophagy induction in murine macrophages ameliorates hepatic inflammatory responses by inhibiting NLRP3 inflammasome activation.
Introduction
Liver disease is often initiated by severe hepatic inflammation which mediates progressive liver damage and fibrosis.Citation1,2 During hepatic inflammation, macrophages act as sentinels which initiate immune responses and provide important mediators for wound healing and metabolic functions.Citation3,4 Therefore, regulating the activity of macrophages provides an excellent therapeutic strategy to alleviate severe hepatic inflammation.
The TAM family of RTKs expressed in macrophages negatively regulates inflammatory responses by not only facilitating the clearance of apoptotic bodies,Citation5,6 but also by providing signals which inhibit inflammation.Citation7,8 Indeed, TAM-deficient (tyro3−/−, axl−/−, mertk−/−) mice or Mertk mutant mice develop spontaneous lymphoproliferative diseases such as the autoimmune disease lupus.Citation9,10
Previous observations show that the interaction between the TAM family of RTKs and their common ligand, GAS6, plays a protective role during liver inflammation. In a murine ischemia and reperfusion injury model, Gas6-deficient (gas6−/−) mice show more severe hepatic injury than do wild-type (Gas6+/+) mice, and treatment of gas6−/− mice with exogenous GAS6 rescues the phenotype.Citation11 In a murine model of CCl4-induced liver injury, Gas6 deficiency delays wound healing.Citation12 Since GAS6 is a common ligand of all TAM RTKs, the specific role of each TAM family member in the progress of hepatic inflammation remains to be determined.
Autophagy is a homeostatic degradative process that removes damaged organelles or turns over cytoplasmic constituents via lysosomal compartments in eukaryotic cells.Citation13 Although autophagy was initially identified to enhance cell survival, increasing evidence shows that autophagy is involved in a variety of biological events.Citation14,15 In particular, recent observations have demonstrated an inverse relationship between autophagy induction and maturation of NLRP3 inflammasomes in macrophages.Citation16-18 Therefore, autophagy may regulate proinflammatory cytokine production via inhibiting activation of the NLRP3 inflammasome in macrophages, which in turn may contribute to the symptoms of specific inflammatory diseases.Citation19,20
Given the above information, we explored the role of individual TAM family members during autophagy induction and evaluated their roles in hepatic inflammation. We found that the interaction between AXL and GAS6 induced autophagy via autophosphorylation of 2 tyrosine residues within the cytoplasmic domain of AXL in a manner dependent on MAPK14. Furthermore, GAS6-AXL-mediated autophagy induction inhibited NLRP3 inflammasome activation, which led to reduced production of IL1B and IL18. In accordance with these observations, axl−/− mice showed more severe symptoms than did wild-type (Axl+/+) mice in acute hepatic injury models. Therefore, GAS6-AXL signaling inhibits inflammatory responses by inducing autophagy.
Results
Interaction between GAS6 and AXL induces autophagy in macrophages by increasing mRNA transcript levels of Becn1, Atg5, and Map1lc3b
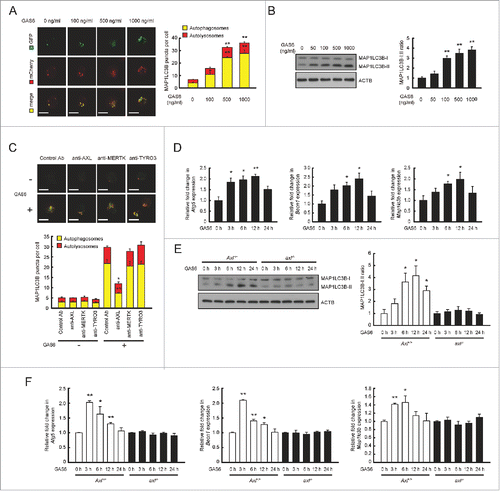
To determine the effect of TAM receptor signaling on autophagy induction, we first analyzed the surface expression of each TAM receptor on murine macrophages by flow cytometry. P388D1 cells expressed relatively higher levels of AXL and lower levels of MERTK but no TYRO3, whereas J774 cells expressed MERTK only (Fig. S1). Then, we treated P388D1 and J774 cells with GAS6, a high-affinity ligand for all TAM receptors, for 24 h.Citation9 To observe autophagy induction, we expressed a tandem fusion of MAP1LC3B (microtubule associated protein 1 light chain 3 β) protein to acid-insensitive mCherry together with GFP (mCherry-EGFP-MAP1LC3B) in these cells, and then performed morphometric analyses using confocal microscopy to monitor the formation of autophagosomes and autolysosomes.Citation21 In these analyses, autophagosomes were observable as yellow puncta (colocalization between mCherry and GFP) and autolysosomes appeared as red puncta (mCherry).Citation21 Also, we measured the ratio of MAP1LC3B-I to MAP1LC3B-II because the conversion of MAP1LC3B-I to MAP1LC3B-II is correlated with the formation of autophagosomes.Citation21,22 The number of yellow puncta and red puncta per cell increased only in GAS6-treated P388D1 cells (Fig. S2A). In agreement with this observation, GAS6-treated P388D1 cells showed increased conversion of MAP1LC3B-I to MAP1LC3B-II relative to untreated cells, whereas GAS6-treated J774 cells did not (Fig. S2B). This result was confirmed by treating P388D1 cells with various concentrations of GAS6 for 24 h. Autophagy was induced in GAS6-treated P388D1 cells in a dose-dependent manner ( and ).
Figure 1. Interaction between GAS6 and AXL induces autophagy in macrophages. (A) mCherry-EGFP-MAP1LC3B expressing P388D1 cells were treated with various concentrations of GAS6 for 24 h. After treatment, the formation of autophagosomes (yellow puncta) and autolysosomes (red puncta) was analyzed under a confocal microscope (left panel). Scale bar: 10 μm. Quantifications of autophagosome and autolysosome formation are shown in the right panel. (B) P388D1 cells were treated with various concentrations of GAS6 for 24 h. After treatment, cell lysates were subjected to immunoblot analyses using anti-MAP1LC3B antibody and anti-ACTB/actin, β antibody (left panel). The ratios of MAP1LC3B-I to MAP1LC3B-II are shown in the right panel. (C) mCherry-EGFP-MAP1LC3B-expressing P388D1 cells were treated with various TAM neutralizing antibodies in the presence or absence of GAS6 (100 ng/ml) for 24 h. Then, the formation of autophagosomes and autolysosomes was analyzed by confocal microscopy (upper panel). Scale bar: 10 μm. Quantifications of autophagosome and autolysosome formation are shown in the lower panel. Control antibody, isotype control; anti-AXL, AXL neutralizing antibody; anti-MERTK, MERTK neutralizing antibody; anti-TYRO3, TYRO3 neutralizing antibody; “−”, no treatment; “+”, treatment. (D) P388D1 cells were treated with GAS6 (100 ng/ml) for the indicated times. After treatment, qRT-PCR was performed using mRNA from each cell as a template. (E and F) Peritoneal macrophages isolated from Axl+/+ and axl−/− mice were treated with GAS6 (100 ng/ml) for the indicated times. After treatment, cell lysates were subjected to immunoblot analyses (E, left panel). The ratios of MAP1LC3B-I to MAP1LC3B-II are shown (E, right panel). Also, qRT-PCR was performed using mRNA from each cell as a template (F). Axl+/+, Axl+/+ peritoneal macrophages; axl−/−, axl−/− peritoneal macrophages. All data represent the mean ± SEM from 3 independent experiments. *P < 0.05; **P < 0.01.
We further tested whether the degree of autophagy induction with GAS6 treatment in macrophages is augmented by treatment with chloroquine (CQ, a lysosomal acidification inhibitor) or serum deprivation.Citation21 The results indicated that treatment with CQ and GAS6 increased the level of endogenous MAP1LC3B-II in macrophages, relative to treatment with GAS6 alone or CQ alone (Fig. S2C). However, serum deprivation along with GAS6 treatment had no additive effect on autophagy induction, compared with serum depleted cells (Fig. S2D).
Next, we treated P388D1 cells with neutralizing antibodies against AXL, MERTK, or TYRO3 during GAS6 incubation. Our results indicated that blocking GAS6-AXL signaling abolished autophagy induction in macrophages after GAS6 treatment ( and Fig. S3). ATG5 (Autophagy related 5), BECN1 (Beclin 1) and MAP1LC3B proteins are essential for induction of autophagy.Citation23 Therefore, we monitored the corresponding mRNA transcripts by qRT-PCR after treatment of P388D1 cells with GAS6. The expression levels of all of these transcripts increased within 24 h after GAS6 treatment ().
Autophagy induction via GAS6-AXL signaling was further confirmed in wild-type (Axl+/+) and axl−/− mice. Surface expression of AXL was confirmed on Axl+/+ macrophages, and was not present on axl−/− macrophages (Fig. S4). The surface expression levels of MERTK and TYRO3 were not different between Axl+/+ and axl−/− macrophages (Fig. S4). Upon GAS6 treatment, the conversion of MAP1LC3B-I to MAP1LC3B-II increased in Axl+/+ macrophages, relative to that in untreated cells (). However, no changes in the MAP1LC3B-I to MAP1LC3B-II ratio were observed in axl−/− macrophages after GAS6 treatment (). Also, we monitored mRNA transcripts of Atg5, Becn1 and Map1lc3b in GAS6-treated macrophages. Induction of these genes was observed in Axl+/+ macrophages, but not in axl−/− macrophages following GAS6 treatment (). Taken together, these results demonstrate that GAS6-AXL signaling mediates autophagy induction by increasing the expression of Atg5, Becn1 and Map1lc3b in macrophages.
Autophosphorylation of 2 tyrosine residues, Tyr815 and Tyr860, in the cytoplasmic domain of AXL is required for autophagy induction
We generated a mutant lacking the entire cytoplasmic domain of AXL (AXLCYΔ) (Table S2 and Fig. S5A). Wild-type AXL (WT AXL) or AXLCYΔ was expressed in J774 cells which lack endogenous AXL. The expression level of AXLCYΔ was comparable to that of WT AXL (Fig. S5B). Then, we treated these cells with GAS6 to test whether the cytoplasmic domain of AXL is required for autophagy induction. WT Axl transfectants showed enhanced conversion of MAP1LC3B-I to MAP1LC3B-II after GAS6 treatment, whereas AxlCYΔ transfectants did not (). To confirm this observation, we monitored mRNA transcripts of Atg5, Becn1 and Map1lc3b in WT Axl or AxlCYΔ transfectants after GAS6 treatment. Expression of these genes was induced only in GAS6-treated WT Axl transfectants, and not in GAS6-treated AxlCYΔ transfectants ().
Figure 2. Two autophosphorylation residues (Tyr815 and Tyr860) within the cytoplasmic domain of AXL are required for autophagy induction through GAS6-AXL signaling. (A and B) Wild-type AXL (WT AXL) and its cytoplasmic domain deletion mutant (AXLCYΔ) were expressed in J774 cells. Then, the transfected cells were treated with GAS6 (100 ng/ml) for 24 h. After treatment, cell lysates were subjected to immunoblot analyses using anti-MAP1LC3B antibody and anti-ACTB antibody (A, left panel). The ratios of MAP1LC3B-I to MAP1LC3B-II are shown (A, right panel). Also, qRT-PCR was performed using mRNA from each cell as a template (B). “−”, no treatment; “+”, treatment; Mock, empty vector transfectant. (C and D) Wild-type AXL (WT AXL) and several AXL mutants were expressed in J774 cells. Then, transfected cells were treated with GAS6 (100 ng/ml) for 24 h. After treatment, cell lysates were subjected to immunoblot analyses (C, left panel). The ratios of MAP1LC3B-I to MAP1LC3B-II are shown (C, right panel). Also, qRT-PCR was performed using mRNA from each cell as a template (D). AXLY773F, AXL mutant with Tyr773 to Phe773; AXLY815F, AXL mutant with Tyr815 to Phe815; AXLY860F, AXL mutant with Tyr860 to Phe860; and AXLY815F,Y860F, AXL double mutant with Tyr815 and Tyr860 to Phe815 and Phe860 respectively. “−”, no treatment; “+”, treatment. All data represent the mean ± SEM from 3 independent experiments. *P < 0.05; **P < 0.01.
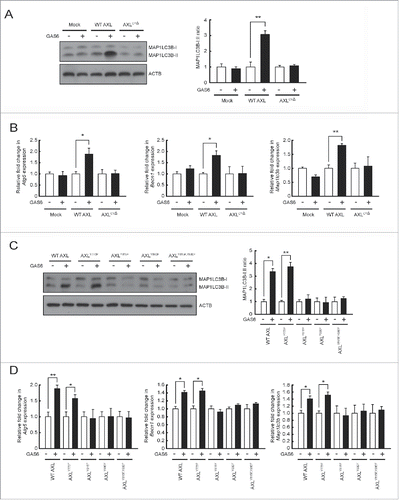
Three tyrosine residues (Tyr773, Tyr815, Tyr860) in the cytoplasmic domain of mouse AXL have been identified as potential autophosphorylation sites which mediate signal transduction.Citation24 To determine which tyrosine residue mediates the signal for autophagy induction, we made several mutants with amino acid substitutions (AXLY773F replaces Tyr773 with Phe773, AXLY815F replaces Tyr815 with Phe815 and AXLY860F replaces Tyr860 with Phe860) (Table S2 and Fig. S5A). After verifying the proper cell surface expression of each mutant protein on J774 cells (Fig. S5B), we compared the extent of autophagy induction of each mutant transfectant with that of the WT Axl transfectant after GAS6 treatment. The conversion of MAP1LC3B-I to MAP1LC3B-II in GAS6-treated WT Axl and AxlY773F (Tyr773 to Phe773) transfectants was increased relative to that in untreated cells, whereas the conversion of MAP1LC3B-I to MAP1LC3B-II in AxlY815F (Tyr815 to Phe815) and AxlY860F (Tyr860 to Phe860) transfectants was not (). To confirm this result, we monitored mRNA levels of Atg5, Becn1 and Map1lc3b in each transfectant after GAS6 treatment. These genes were induced in WT Axl and AxlY773F transfectants, but not in AxlY815F and AxlY860F transfectants ().
To further verify that Tyr815 and Tyr860 residues are important for autophagy induction via GAS6-AXL signaling, we also made a double amino acid substitution mutant (AXLY815F,Y860F replaces Tyr815 and Tyr860 with Phe815 and Phe860 respectively) (Table S2 and Fig. S5A and B) and tested whether this mutant is capable of autophagy induction after GAS6 treatment. The results clearly demonstrated that the conversion of MAP1LC3B-I to MAP1LC3B-II and the mRNA levels of Atg5, Becn1 and Map1lc3b were not different in the GAS6-treated AxlY815F,Y860F transfectant than in untreated cells ( and ).
Blocking the MAPK14 pathway inhibits AXL-mediated autophagy induction in macrophages
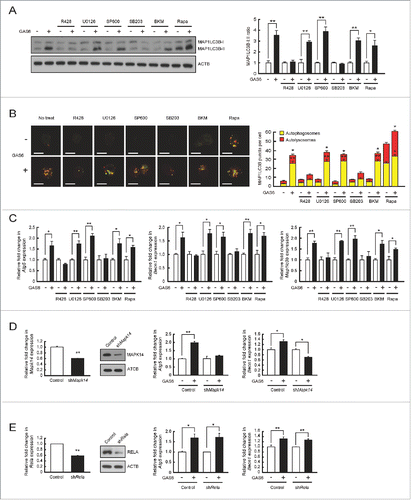
We tested several different kinase inhibitors to identify those that would block downstream effectors of GAS6-AXL signaling in order to determine which downstream pathway is involved in autophagy induction. After treatment of GAS6 with several kinase inhibitors in J774 cells expressing WT Axl, the extent of autophagy induction was measured by monitoring the conversion from MAP1LC3B-I to MAP1LC3B-II after GAS6 treatment. In these experiments, we also assessed the formation of autophagosomes and autolysosomes by detecting fluorescence puncta in WT Axl expressing mCherry-EGFP-MAP1LC3B. As expected, no autophagy induction was observed in cells after treatment with R428 (AXL inhibitor) and GAS6 ( and , and Fig. S6). However, treatment with U0126 (MAP2K1/2 [mitogen-activated protein kinase kinase 1/2] and MAPK1/3 inhibitor), SP600125 (MAPK8/9/10 inhibitor), BKM120 (PtdIns3K inhibitor) and rapamycin (MTOR [mechanistic target of rapamycin] inhibitor) had no effect on autophagy induction through GAS6-AXL signaling ( and and Fig. S6). Notably, treatment with SB203580 (MAPK11/14 inhibitor) abolished GAS6-AXL-mediated autophagy induction ( and and Fig. S6). In support of this result, Atg5, Becn1 and Map1lc3b mRNA levels were not increased in cells treated with R428 or SB203580 together with GAS6, whereas they were increased in cells treated with U0126, SP600125, BKM120, or rapamycin along with GAS6 ().
Figure 3. Inhibition of the MAPK14 pathway blocks GAS6-AXL signaling-mediated autophagy induction. (A to C) J774 cells expressing WT AXL or WT AXL and mCherry-EGFP-MAP1LC3B were treated with GAS6 (100 ng/ml) in the absence or presence of various kinase inhibitors for 24 h. After treatment, cell lysates were subjected to immunoblot analyses using anti-MAP1LC3B antibody and anti-ACTB antibody (A, left panel). The ratios of MAP1LC3B-I to MAP1LC3B-II are shown (A, right panel). Then, the formation of autophagosomes (yellow puncta) and autolysosomes (red puncta) was analyzed by confocal microscopy (B, left panel). Scale bar: 10 μm. Quantifications of autophagosome and autolysosome formation are shown in the right panel (B, right panel). Also, qRT-PCR was performed using mRNA from each cell as a template (C). R428, AXL inhibitor; U0126, MAP2K1/2 and MAPK1/3 inhibitor; SP600 (SP600125), MAPK8/9/10 inhibitor; SB203 (SB203580), MAPK11/14 inhibitor; BKM (BKM120), PtdIns3K inhibitor; Rapa (rapamycin), MTOR inhibitor; “−”, no treatment; “+”, treatment. (D) Control plasmid (Control) and shMap14 plasmid (shMap14) were transfected into WT AXL-expressing J774 cells. Then, qRT-PCR and immunoblot analyses were performed to measure the level of MAPK14 expression (left panel). After treatment with GAS6 (100 ng/ml) for 24 h, qRT-PCR was performed to detect Atg5 and Becn1 mRNA transcripts (right panel). “−”, no treatment; “+”, treatment. (E) Control plasmid (Control) and shRela plasmid (shRela) were transfected into WT AXL-expressing J774 cells. Then, qRT-PCR and immunoblot analyses were performed to measure the level of RELA expression (left panel). After treatment with GAS6 (100 ng/ml) for 24 h, qRT-PCR was performed to detect Atg5 and Becn1 mRNA transcripts (right panel). “−”, no treatment; “+”, treatment. All data represent the mean ± SEM from 3 independent experiments. *P < 0.05; **P < 0.01.
Since SB203580 inhibits both MAPK11 and MAPK14 activities,Citation25 we monitored mRNA transcripts of Mapk11 and Mapk14 in AXL-expressing J774 cells before or after treatment with GAS6. The mRNA transcript of Mapk14 was observed whereas mRNA transcript of Mapk11 was hardly detected in AXL-expressing J774 cells by RT-PCR analyses (Fig. S7A). Further, we analyzed the activation status of MAPK11 or MAPK14 using an activation-specific anti-phospho MAPK antibody in AXL-expressing J774 cells after treatment with GAS6 and SB203580. MAPK14 activation in AXL-expressing J774 cells was successfully blocked by treating SB203580 in a dose-dependent manner. However, no activation form of MAPK11 was detected in AXL-expressing J774 cells (Fig. S7B).
To confirm these results, we depleted MAPK14 in AXL-expressing J774 cells using shRNA and monitored mRNA transcripts of Atg5 and Becn1 after GAS6 treatment. The levels of MAPK14 expression were significantly decreased in the shMapk14 plasmid transfectant (shMapk14) when compared to levels in the control plasmid transfectant (control) (). Moreover, shRNA-mediated knockdown of MAPK14 clearly abolished the upregulation of Atg5 and Becn1 mRNA transcripts after GAS6 treatment (). Together, these results indicate that GAS6-AXL signaling mediated autophagy induction is dependent on the activity of MAPK14.
To rule out the possibility that NFKB (nuclear factor of kappa light polypeptide gene enhancer in B cells) is involved in GAS6-AXL-mediated autophagy induction, we also depleted Rela (v-rel avian reticuloendotheliosis viral oncogene homolog A), a key subunit of NFKB, in AXL-expressing J774 cells, using shRNA and measured the expression patterns of Atg5 and Becn1 after GAS6 treatment. The shRNA-mediated knockdown of Rela did not change the expression levels of Atg5 and Becn1 mRNA transcripts after GAS6 treatment ().
Autophagy induction by GAS6-AXL signaling mediates suppression of IL1Β and IL18 production by inhibiting NLRP3 inflammasome activation
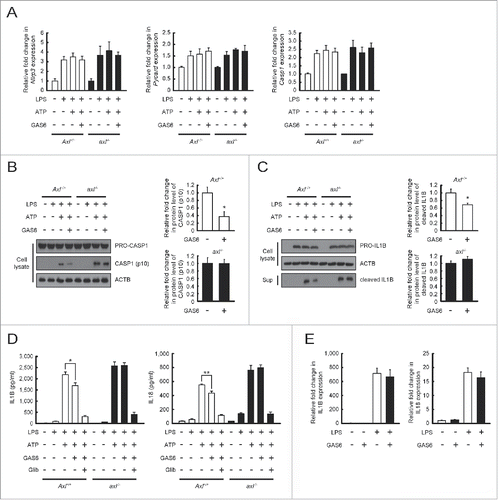
To evaluate whether autophagy induction by GAS6-AXL signaling inhibits NLRP3 inflammasome activation in murine macrophages, we pretreated Axl+/+ or axl−/− macrophages with LPS and GAS6, and then treated them with ATP, a well-characterized NLRP3 stimulator.Citation26,27
After NLRP3 inflammasome activation, the levels of Nlrp3, Pycard (PYD and CARD domain containing), and Casp1 mRNA transcripts were increased in both Axl+/+ and axl−/− macrophages. No significant change in the mRNA levels of these inflammasome components was observed between Axl+/+ and axl−/− macrophages regardless of the presence or absence of GAS6 (). Interestingly, GAS6-treated Axl+/+ macrophages were found to have reduced levels of cleaved CASP1 (p10, the active form of CASP1) compared to untreated Axl+/+ macrophages (). However, GAS6-treated axl−/− macrophages exhibited no change in the active form of CASP1 relative to untreated axl−/− macrophages (). Consistent with these results, GAS6-treated Axl+/+ macrophages had reduced levels of the cleaved form of IL1Β, which is produced from a precursor (PRO-IL1Β) by activated CASP1, in response to treatment with LPS and ATP than did untreated Axl+/+ macrophages (). However, GAS6-treated axl−/− macrophages and untreated axl−/− macrophages had comparable levels of the cleaved form of IL1B (). These findings were further confirmed by ELISA results showing decreases in IL1B and IL18 secretion in GAS6-treated Axl+/+ macrophages compared with untreated Axl+/+ macrophages (). As expected, no significant changes were observed in the secretion of these proinflammatory cytokines in GAS6-treated or untreated axl−/− macrophages (). In addition, glibenclamide (NLRP3 inflammasome inhibitor)Citation18 treatment effectively blocked secretion of IL1B and IL18 from macrophages treated with LPS and ATP (). After activation of the NLRP3 inflammasome, we measured mRNA transcripts of Il1b and Il18 by qRT-PCR to determine whether treatment of macrophages with GAS6 affects the expression of these genes. The levels of Il1b and Il18 mRNA transcripts in both Axl+/+ and axl−/− macrophages were not altered by treatment with GAS6 (). To confirm these results, we pretreated Atg7fl/fl Lyz2-Cre+(Atg7fl/fl CKO) or Atg7fl/fl Lyz2-Cre− (Atg7fl/fl CTRL) macrophagesCitation28,29 with LPS and GAS6, and then treated them with ATP. The results demonstrated that Atg7fl/fl CKO macrophages were defective in conversion of MAP1LC3B-I to MAP1LC3B-II after GAS6 treatment, whereas increased endogenous MAP1LC3B-II levels were found in Atg7fl/fl CTRL macrophages after GAS6 treatment (Fig. S8A). This result indicates that selective deletion of Atg7 in macrophages causes a defect in autophagy induction after GAS6 treatment. Furthermore, GAS6-treated Atg7fl/fl CKO macrophages had no changes in the active form of CASP1 and the cleaved form of IL1B relative to untreated Atg7fl/fl CKO macrophages (Fig. S8B and C). In contrast to the results from Atg7fl/fl CTRL macrophages, GAS6-treated Atg7fl/fl CTRL macrophages were found to have reduced levels of the active form of CASP1 and the cleaved form of IL1Β relative to untreated Atg7fl/fl CTRL macrophages (Fig. S8B and C). Supporting these results, levels of IL1B and IL18 secreted from GAS6-treated Atg7fl/fl CKO macrophages were the same as those from untreated Atg7fl/fl CKO macrophages, whereas the extent of IL1B and IL18 secretion from GAS6-treated Atg7fl/fl CTRL macrophages was significantly decreased (Fig. S8D). Collectively, these data strongly suggest that GAS6-AXL signaling-mediated autophagy induction inhibits the conversion of the active forms of IL1Β and IL18 through NLRP3 inflammasome-mediated activation of CASP1.
Figure 4. Autophagy induction through GAS6-AXL signaling reduces IL1B and IL18 secretion by inhibiting NLRP3 inflammasome-mediated CASP1 activation. (A to D) Peritoneal macrophages isolated from Axl+/+ and axl−/− mice were treated with LPS (100 ng/ml) for 6 h and then stimulated with ATP (5 mM) in the absence or presence of GAS6 (100 ng/ml) or NLRP3 inflammasome inhibitor (Glib, 100 μM). After treatment, qRT-PCR was performed using mRNA from each cell as a template (A). Also, cell lysates or culture supernatants were subjected to immunoblot analyses using anti-CASP1 antibody (B) and anti-IL1B antibody (C). Anti-ACTB antibody was used as an internal control for immunoblotting. Then, ELISAs were performed to detect IL1B and IL18 secretions in culture supernatants (D). “−”, no treatment; “+”, treatment; Axl+/+, Axl+/+ peritoneal macrophages; axl−/−, axl−/− peritoneal macrophages; Sup, culture supernatant; Glib, Glibenclamide. (E) Peritoneal macrophages isolated from Axl+/+ mice were treated with LPS (100 ng/ml) for 6 h in the absence or presence of GAS6 (100 ng/ml). After treatment, qRT-PCR was performed using mRNA from each cell as a template. All data represent the mean ± SEM from 3 independent experiments. *P < 0.05; **P < 0.01.
Previous observations demonstrated that STAT1 (signal transducer and activator of transcription 1)-dependent type I IFN (interferon) signaling suppresses CASP1 dependent IL1Β maturation by repressing NLRP3 inflammasome activation.Citation30 To determine whether NLRP3 inflammasome inhibition through GAS6-AXL signaling is dependent on the activity of type I IFN and STAT1, we blocked type I IFN signaling using an anti-IFNAR1 (interferon α and β receptor subunit 1) antibody (5A3 clone) and measured the degree of NLRP3 inflammasome activation in Axl+/+ and axl−/− macrophages. After treatment with anti-IFNAR1 antibody, phosphorylation of STAT1 (p-STAT1) was decreased in a dose-dependent manner in LPS-activated macrophages (Fig. S9A). Then, we activated NLRP3 inflammasomes in Axl+/+ and axl−/− macrophages by treatment with an anti-IFNAR1 antibody and GAS6. Our results indicated that treatment with the anti-IFNAR1 antibody during NLRP3 inflammasome activation did not change the conversion of PRO-IL1Β to mature IL1Β in GAS6-treated Axl+/+ or axl−/− macrophages (Fig. S9B). Consistent with this result, the decrease in IL1B secretion is dependent on GAS6-AXL signaling, but not on type I IFN signaling (Fig. S9C).
GAS6-AXL signaling-mediated autophagy induction in Kupffer cells ameliorates acute liver injury by inhibition of NLRP3 inflammasome activation
Since recent observations demonstrated that NLRP3 inflammasome activation in Kupffer cells contributes to severe liver inflammation,Citation31,32 we investigated whether axl−/− mice aggravate the symptom of acute liver inflammation induced by LPS or CCl4. Gas6 mRNA levels were increased in liver and spleen tissues within 12 h after LPS treatment (Fig. S10). Similarly, the levels of Nlrp3, Pycard and Casp1 mRNA transcripts were significantly increased in both Axl+/+ and axl−/− Kupffer cells within 12 h after LPS treatment (). These results indicate that LPS treatment leads to the induction of Gas6 and the activation of NLRP3 inflammasomes in Kupffer cells. After treatment with LPS, the conversion of MAP1LC3B-I to MAP1LC3B-II in Axl+/+ Kupffer cells was significantly increased relative to that in axl−/− Kupffer cells (). Consequently, LPS-treated Axl+/+ Kupffer cells contained the less active form of CASP1 (p10), whereas LPS-treated axl−/− kupffer cells did not (). Consistent with these results, IL1B and IL18 secretions in sera taken from LPS-treated Axl+/+ mice were significantly decreased compared with those in sera from LPS-treated axl−/− mice (). However, the levels of Il1b and Il18 mRNA transcripts in Axl+/+ and axl−/− Kupffer cells were comparable (Fig. S11).
Figure 5. Axl deficiency in mice aggravates hepatic inflammation after LPS challenge. Axl+/+ and axl−/− mice were injected intraperitoneally with vehicle (saline, Control) or LPS (2.5 μg/g). Mice were sacrificed and analyzed 12 h after treatment. (A) qRT-PCR was performed using mRNA from Axl+/+ or axl−/− Kupffer cells (Axl+/+ or axl−/−) as templates. (B) Cell lysates from Axl+/+ or axl−/− Kupffer cells were subjected to immunoblot analyses using anti-MAP1LC3B antibody and anti-ACTB antibody (left panel). The ratios of MAP1LC3B-I to MAP1LC3B-II are shown (right panel). (C) Cell lysates from Axl+/+ or axl−/− Kupffer cells were subjected to immunoblot analyses using anti-CASP1 antibody and anti-ACTB antibody. (D) ELISAs were performed to detect serum concentrations of IL1B and IL18 from Axl+/+ or axl−/− mice (Axl+/+ or axl−/−). (E) Hematoxylin and eosin (H&E) staining of liver sections (upper left) and liver injury scores (upper right) from Axl+/+ or axl−/− mice are shown in the upper panel. Immunohistochemisty of liver sections with anti-fibrin(ogen) antibody (lower left) and fibrin(ogen) deposition area (lower right) from Axl+/+ or axl−/− mice are shown in the lower panel. Fibrin(ogen) deposition area = stained area/total image area. Scale bar: 100 μm. (F) Immunohistochemistry of liver sections with anti-ACTA2 antibody, anti-ADGRE1/F4/80 antibody (ADGRE1), anti-MAP1LC3B antibody (MAP1LC3B) and anti-ATG5 antibody (ATG5) from Axl+/+ or axl−/− mice. Each staining area = stained area/total image area. DAPI staining was performed to visualize nuclei. Scale bar: 100 μm. “−”, vehicle treatment; “+”, LPS treatment. All data represent the mean ± SEM from 3 independent experiments. *P < 0.05; **P < 0.01.
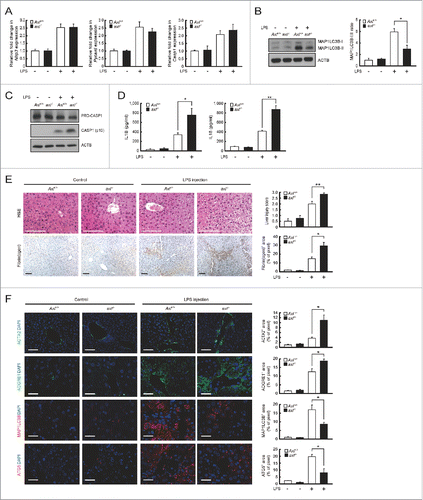
To confirm that NLRP3 inflammasome activation is involved in the increased inflammation seen in axl−/− mice, we used glibenclamide to inhibit NLRP3 inflammasome activation in this animal model.Citation33 The results showed that glibenclamide treatment diminished the active form of CASP1 (p10) in both LPS-treated Axl+/+ and axl−/− Kupffer cells (Fig. S12A). Also, glibenclamide treatment caused a significant decrease in IL1B and IL18 levels in sera taken from LPS-treated Axl+/+ or axl−/− mice (Fig. S12B).
Liver tissue specimens from Axl+/+ and axl−/− mice were obtained 12 h after LPS treatment. Normal histology was observed in the control groups without treatment (, upper panels). In contrast, marked histological changes (multiple erosions, inflammatory cell infiltration and bleeding) were observed in the LPS-treated groups (, upper panels). More severe tissue injury was observed in the LPS-treated axl−/− group than in the Axl+/+ group. Accordingly, liver injury score was higher in specimens from the LPS-treated axl−/− mouse group than in those from the LPS-treated Axl+/+ mouse group (, upper panels). Consistent with this, more severe kidney and lung tissue injury was also observed in LPS-treated axl−/− mice compared with Axl+/+ mice (Fig. S13).
The degree of fibrin deposition in the liver is a useful indicator of measuring the severity of acute liver injury.Citation34 The amount of fibrin deposition in LPS-treated axl−/− mice was significantly greater than in Axl+/+ mice (). In order to access the severity of liver injury, we also measured hepatic stellate cell (HSC) activation by staining with anti-ACTA2 (actin, α 2, smooth muscle, aorta) antibody (green fluorescence) and visualized the number of macrophages within the lesions by staining with anti-ADGRE1/F4/80 antibody (red fluorescence).Citation35 The results showed that activated HSCs and infiltrated macrophages at the sites of injury were more numerous in the livers of LPS-treated axl−/− mice than in those of Axl+/+ mice ().
The combined results indicate that axl−/− mice show more severe inflammation following LPS-induced liver injury because axl−/− mice cannot inhibit NLRP3 inflammasome activation due to deficits in GAS6-AXL signaling-mediated autophagy induction. Supporting this idea, immunohistochemical staining with anti-MAP1LC3B antibody and anti-ATG5 antibody showed that the number of cells expressing MAP1LC3B and ATG5 proteins was markedly elevated in liver tissue from LPS-treated Axl+/+ mice compared with that from LPS-treated axl−/− mice ().Citation36,37 The liver function of experimental animals was further determined by measuring serum levels of GPT (glutamic pyruvic transaminase, soluble) and GOT1 (glutamic-oxaloacetic transaminase 1, soluble). Serum levels of GPT and GOT1 were more elevated in the LPS-treated axl−/− group than in the LPS-treated Axl+/+ group (Fig. S14).
Using a more liver-specific injury model to evaluate the inhibitory effect of GAS6-AXL signaling on NLRP3 inflammasome activation, we performed the same sets of experiments in mice subjected to intraperitoneal injections of either CCl4 or vehicle. Gas6 mRNA transcript levels increased in liver tissue of CCl4-treated animals 24 h after treatment (Fig. S15). Also, Nlrp3, Pycard and Casp1 mRNA levels were elevated in Axl+/+ and axl−/− Kupffer cells 24 h after CCl4 treatment (). The conversion of MAP1LC3B-I to MAP1LC3B-II in Axl+/+ Kupffer cells was significantly increased relative to that in axl−/− Kupffer cells following treatment with CCl4 (). Consequently, Kupffer cells from CCl4-treated Axl+/+ mice expressed the less active form of CASP1 (p10), whereas axl−/− Kupffer cells from CCl4-treated mice did not (). Supporting this result, IL1Β and IL18 secretions 24 h after CCl4 treatment were significantly decreased in sera from Axl+/+ mice, compared with that from axl−/− mice (). However, the levels of Il1b and Il18 mRNA transcripts in Axl+/+ and axl−/− Kupffer cells were not significantly different (Fig. S16).
Figure 6. Axl-deficient mice display more severe hepatic inflammation after CCl4treatment. Axl+/+ and axl−/− mice were injected intraperitoneally with vehicle (corn oil, Control) or CCl4 (2 μl/g). Mice were sacrificed and analyzed 2 d after treatment. (A) qRT-PCR was performed using mRNA from Axl+/+ or axl−/− Kupffer cells (Axl+/+ or axl−/−) as templates. (B) Cell lysates from Axl+/+ or axl−/− Kupffer cells were subjected to immunoblot analyses using anti-MAP1LC3B antibody and anti-ACTB antibody (left panel). The ratios of MAP1LC3B-I to MAP1LC3B-II are shown (right panel). (C) Cell lysates from Axl+/+ or axl−/− Kupffer cells were subjected to immunoblot analyses using anti-CASP1 antibody and anti-ACTB antibody. (D) ELISAs were performed to detect serum concentrations of IL1B and IL18 from Axl+/+ or axl−/− mice (Axl+/+ or axl−/−). (E) Hematoxylin and eosin (H&E) staining of liver sections (upper left) and liver injury scores (upper right) from Axl+/+ or axl−/− mice are shown in the upper panel. Immunohistochemisty of liver sections with anti-fibrin(ogen) antibody (lower left) and fibrin(ogen) deposition area (lower right) from Axl+/+ or axl−/− mice are shown in the lower panel. Fibrin(ogen) deposition area = stained area/total image area. Scale bar: 100 μm. (F) Immunohistochemistry of liver sections with anti-ACTA2 antibody, anti- ADGRE1/F4/80 antibody (ADGRE1), anti-MAP1LC3B antibody (MAP1LC3B) and anti-ATG5 antibody (ATG5) from Axl+/+ or axl−/− mice. Each staining area = stained area/total image area. DAPI staining was performed to visualize nuclei. Scale bar: 100 μm. “−”, vehicle treatment; “+”, CCl4 treatment. All data represent the mean ± SEM from 3 independent experiments. *P < 0.05; **P < 0.01.
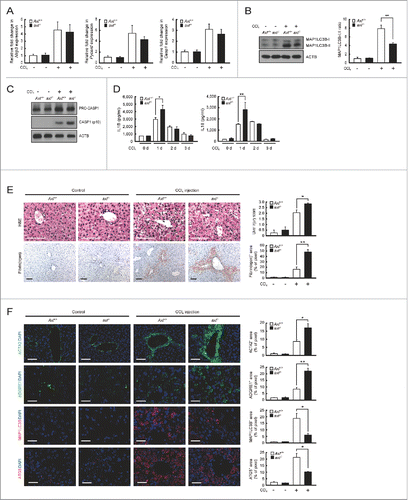
To investigate whether NLRP3 inflammasome activation participates in the increased inflammation observed in axl−/− mice, we used glibenclamide to inhibit the activation of NLRP3 inflammasome in this animal model.Citation32 Glibenclamide treatment in both CCl4-treated Axl+/+ and axl−/− Kupffer cells led to decreased conversion of active CASP1 (p10) (Fig. S17A). Also, the amounts of IL1B and IL18 in sera taken from CCl4-`treated Axl+/+ and axl−/− mice were significantly reduced by glibenclamide treatment (Fig. S17B).Citation33
Liver tissue specimens from axl−/− and Axl+/+ mice were collected 2 d after CCl4treatment. Specimens from the CCl4-treated axl−/− group had higher liver injury scores and a higher degree of fibrin deposition than did those from the CCl4-treated Axl+/+ group (). In agreement with these observations, infiltration of activated HSCs and ADGRE1-positive macrophages into injury sites was increased in livers from CCl4-treated axl−/− mice than in those of CCl4-treated Axl+/+ mice (). Also, the number of cells expressing MAP1LC3B and ATG5 proteins was elevated in livers from CCl4-treated Axl+/+ mice compared with those from CCl4-treated axl−/− mice (), and serum levels of GPT and GOT1 on d 2 post-CCl4 treatment were statistically higher in CCl4-treated axl−/− mice than in CCl4-treated Axl+/+ mice (Fig. S18). These combined results are consistent with the result from the LPS-induced liver injury model and demonstrate that GAS6-AXL signaling inhibits NLRP3 inflammasome activation through autophagy induction following CCl4-induced liver injury.
Discussion
This study provides novel evidence showing that GAS6-AXL signaling promotes autophagy induction in murine macrophages, which may depress acute liver inflammation by inhibiting NLRP3 inflammasome activation. Based on our study, AXL is the only molecule that induces autophagy among TAM receptors. This result was further confirmed by the observation that mRNA expression levels of Atg5, Becn1 and Map1lc3b, essential genes for autophagy induction,Citation23 increased when we triggered GAS6-AXL signaling. Consistent with this observation, autophagy induction via GAS6-AXL signaling was found to be defective in axl−/− macrophages.
We also provide molecular evidence showing that the cytoplasmic domain of AXL is critical for autophagy induction. Our experiments with several AXL mutants revealed that 2 autophosphorylated tyrosine residues (Tyr815 and Tyr860) in the cytoplasmic domain of AXL are required for autophagy induction. Our next question addressed which downstream signaling molecule mediates autophagy induction through GAS6-AXL signaling. GAS6-AXL signaling has been linked to multiple downstream pathways through GRB2 and PtdIns3K.Citation38 Previously identified downstream effector molecules of GRB2 and PtdIns3K that transmit GAS6-AXL signaling include MAPK14, MAP2K1/2, MAPK1/3, and MAPK8/9/10, which promote cell migration, proliferation, and survival of different cell types.Citation38 GAS6-AXL signaling also mediates activation of NFKB through PtdIns3K.Citation38,39 We found that treatment with the MAPK11/14 inhibitor alone was sufficient to block GAS6-AXL signaling-mediated autophagy induction. This observation was further confirmed by the result that inhibition of MAPK14 activity mediated throgh shRNA abolished GAS6-AXL signaling-mediated autophagy induction.
The NLRP3 inflammasome in macrophages is a protein complex which consists of NLRP3, PYCARD, and CASP1.Citation40,41 After recognition of several pathogen-associated molecular patterns, NLRP3 inflammasomes control the maturation and secretion of IL1Β and IL18 by activating CASP1.Citation40,41 Nlrp3, Pycard or Casp1 deficient mice show significant defects in secretion of IL1Β and IL18 after treatment with LPS and ATP.Citation26,27 Recently, an inverse relationship between autophagy induction and NLRP3 inflammasome activation has been proposed.Citation16-18 Indeed, mice lacking genes encoding several autophagy proteins such as ATG16L1, BECN1, and MAP1LC3B produce excessive amounts of IL1Β and IL18 in response to LPS or LPS and ATP.Citation16-18
To clarify the biological relationship between autophagy induction through GAS6-AXL signaling and NLRP3 inflammasome activation, we explored the possibility that autophagy induction through GAS6-AXL signaling may inhibit NLRP3 inflammasome activation in macrophages. Our results clearly indicated that autophagy induction through GAS6-AXL signaling inhibits NLRP3 inflammasome-dependent activation of CASP1. Previous observations show that autophagy inhibits NLRP3 inflammasome activation by reducing the concentration of reactive oxygen species (ROS).Citation17,18 Supporting this idea, several observations show that autophagy negatively regulates ROS generation,Citation16,42 and NLRP3 inflammasome activity is suppressed by ROS blockade.Citation41 In agreement with these reports, we detected less ROS in GAS6-treated Axl+/+ macrophages than in untreated Axl+/+ macrophages during NLRP3 inflammasome activation (Fig. S19A). Also, PYCARD multimer formation was decreased in Axl+/+ macrophages treated with GAS6, but not in axl−/− macrophages (Fig. S19B). Nevertheless, most previous observations have used specific gene-deficient mouse models to speculate on the relationship between autophagy and NLRP3 inflammasome activation.Citation16-18 However, our results provide direct evidence showing that autophagy formation by GAS6-AXL signaling indeed inhibits NLRP3 inflammasome activity.
Previously, 2 independent studies have shown that GAS6-AXL signaling inhibits the secretion of proinflammatory cytokines. The first study demonstrates that GAS6-AXL signaling induces the expression of Twist1 (twist family bHLH transcription factor 1), which inhibits the NFKB dependent transcription of Tnf.Citation7 The second study indicates that IFNA receptor-STAT1-dependent AXL signaling induces Socs1 and Socs3, which inhibit toll-like receptor and cytokine receptor signaling.Citation8 Our results showed that autophagy induced by GAS6-AXL signaling is NFKB-independent and that mRNA levels of Il1b and Il18 do not change after treatment with GAS6. Also, we showed that the inhibition of NLRP3 inflammasome activation via GAS6-AXL signaling is independent of IFNA-STAT1 signaling. Therefore, the proposed mechanism herein by which GAS6-AXL signaling inhibits proinflammatory cytokine secretion is different from those proposed in the other studies mentioned above.Citation7,8 Our results demonstrate a novel mechanism of GAS6-AXL signaling which blocks proinflammatory cytokine production by inhibiting NLRP3 inflammasome mediated CASP1 activation through autophagy induction. Therefore, 2 previous observations together with our results, strongly suggest that GAS6-AXL signaling is important in regulation of the inflammatory response using different effector molecules such as TWIST1, SOCS1, SOCS3 or MAPK14.Citation7,8
Increasing evidence demonstrates the importance of NLRP3 inflammasome regulation in Kupffer cells during liver inflammation. For example, NLRP3 inflammasome activation in Kupffer cells often causes severe liver inflammation.Citation31,32 Also, a recent study reports spontaneous generation of severe liver inflammation in myeloid cell-specific hyperactive Nlrp3 transgenic mice.Citation43 In the current study, we demonstrated the physiological relevance of GAS6-AXL signaling-mediated autophagy induction on NLRP3 inflammasome-medicated CASP1 activation in LPS- or CCl4-induced acute liver injury models. First, we demonstrated that GAS6 secretion and NLRP3 inflammasome activation in Kupffer cells are induced by LPS or CCl4 treatment in mice. Second, we showed that autophagy induction and NLRP3 inflammasome-mediated CASP1 activation are inversely correlated. Third, axl−/− mice were more susceptible to LPS- or CCl4-induced acute liver injury and showed enhanced expression of IL1B and IL18. The combined results suggest that deficits in GAS6-AXL signaling in axl−/− Kupffer cells result in insufficient inhibition of NLRP3 inflammasome activation and thus lead to more severe liver inflammation.
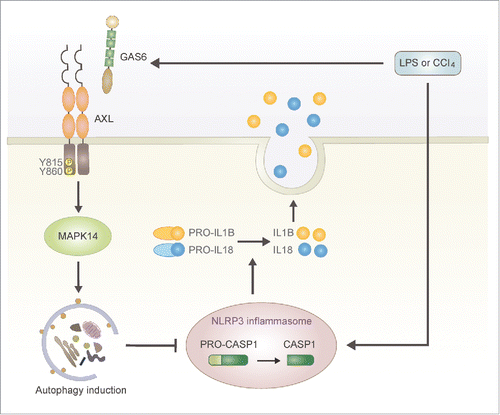
Based on our results, we propose a novel role for GAS6-AXL signaling during acute liver injury. During the early stages of liver injury induced by LPS or CCl4treatment, CASP1 is activated through the NLRP3 inflammasome in Kupffer cells and proinflammatory cytokines such as IL1B and IL18 are promptly secreted by kupffer cells (). At the same time GAS6 levels are substantially increased and the interaction between GAS6 and AXL leads to autophagy induction in Kupffer cells (). Then, the autophagy prevents further activation of CASP1 through inhibition of NLRP3 inflammasome activation in kupffer cells ().
Figure 7. Schematic summary for the role of GAS6-AXL signaling-mediated autophagy during LPS- or CCl4-mediated acute liver injury. Our results suggest that autophagy induced by GAS6-AXL signaling represses hepatic inflammatory response via a negative feedback loop. During the early stages of liver inflammation induced by LPS or CCl4, Kupffer cells promptly secrete mature IL1Β and IL18 through NLRP3 inflammasome-mediated activation of CASP1. Simultaneously, GAS6 induced by LPS or CCl4 binds to AXL, which subsequently leads to the induction of autophagy. Then, autophagy inhibits further secretion of mature IL1Β and IL18 by blocking NLRP3 inflammasome activation.
Although the effect of GAS6-AXL signaling in the liver seems to be antiinflammatory in conditions of acute inflammation, 2 previous observations have found that TAM signaling promotes liver fibrosis during chronic inflammation. The first study demonstrates that Gas6 deficiency attenuates hepatic steatosis by reducing macrophage infiltration and attenuating hepatic progenitor cell activation.Citation44 The second study indicates that GAS6-AXL signaling promotes liver fibrogenesis by hepatic stellate cell activation.Citation45 These results, along with ours, may reflect differential effects of TAM signaling that are dependent on particular downstream molecules in different target cells since multiple and divergent downstream effectors are involved in GAS6-AXL signaling.Citation38
In conclusion, this study shows that the GAS6-AXL signaling pathway mediates autophagy induction in macrophages through MAPK14, and that this effect depends on the Tyr815 and Tyr860 residues in the cytoplasmic domain of AXL. Furthermore, we show that GAS6-AXL signaling-mediated autophagy induction in Kupffer cells prevents more severe inflammation by inhibiting NLRP3 inflammasome activation in murine acute liver injury models induced by LPS or CCl4. These findings provide the first evidence that AXL signaling regulates autophagy in macrophages, and that autophagic activity in Kupffer cells is a key factor that determines the extent of secretion of proinflammatory cytokines such as IL1B and IL18 during acute liver injury.
Materials and methods
Antibodies and reagents
The antibodies used are the following: anti-MAP1LC3B antibody (Cell Signaling Technology, 2775), anti-MAPK11 antibody (Cell Signaling Technology, 2339), anti-MAPK14 antibody (Cell Signaling Technology, 9218), anti-phosphorylated MAPK antibody (Cell Signaling Technology, 9216), anti-ATG5 antibody (Cell Signaling Technology, 8540), anti-phosphorylated STAT1 antibody (Cell Signaling Technology, 9167), anti-ACTB/actin, β antibody (Sigma, A5441) and anti-ACTA2 antibody (mouse IgG2a; Sigma, A2547), anti-mouse AXL antibody (rat IgG2a; R&D systems, MAB8541), rat IgG2a isotype control (R&D systems, MAB006), anti-mouse MERTK antibody (rat IgG1; R&D systems, MAB591), rat IgG1 isotype control (R&D systems, MAB005), biotinylated anti-mouse TYRO3 antibody (rat IgG1; R&D systems, BAM759), rat IgG1 biotinylated isotype control (R&D systems, IC005B), anti-mouse AXL neutralizing antibody (goat polyclonal; R&D systems, AF854), anti-mouse MERTK neutralizing antibody (goat polyclonal; R&D systems, AF591), anti-mouse TYRO3 neutralizing antibody (goat polyclonal; R&D systems, AF759), goat IgG antibody isotype control (R&D systems, AB-108-C), FITC-conjugated anti-ITGAM/CD11b antibody (rat IgG2b; eBioscience, 11-0112), FITC-conjugated rat IgG2b isotype control (eBioscience, 11-4031), anti-mouse AXL antibody (Santa Cruz Biotechnology, sc-20741), anti-IL18 antibody (Santa Cruz Biotechnology, sc-7954), anti-RELA antibody (Santa Cruz Biotechnology, sc-372), anti-STAT1 antibody (Santa Cruz Biotechnology, sc-346), anti-CASP1 antibody (Santa Cruz Biotechnology, sc-514), anti-PYCARD antibody (Santa Cruz Biotechnology, sc-22514-R), goat anti-mouse IgG-HRP (Santa Cruz Biotechnology, sc-2005), goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, sc-2004), anti-ADGRE1/F4/80 antibody (rat IgG2a; Thermo Fisher Scientific, MF48000), Alexa Fluor 488-conjugated anti-mouse IgG (Thermo Fisher Scientific, A-11001), Alexa Fluor 488-conjugated anti-rat IgG (Thermo Fisher Scientific, A-11006), Alexa Fluor 594-conjugated anti-rabbit IgG (Thermo Fisher Scientific, A-11037), PE-conjugated anti-ADGRE1/F4/80 antibody (Thermo Fisher Scientific, MF48004), rabbit anti-mouse fibrin(ogen) antiserum (Molecular Innovations, ASMFBGN), anti-IL1Β antibody (BioVision, 5129-100), anti-mouse IFNA receptor antibody (mouse IgG1; BioXcell, BE0241), mouse IgG1 isotype control (BioXcell, BE0083), biotinylated goat anti-rat IgG (Vector Laboratories, BA-9400), Streptavidin-PE (BD Biosciences, 349023), and anti-PE microbeads (Miltenyi Biotec, 130-048-801). The reagents used are the following: Recombinant mouse GAS6 (R&D systems, 986-GS), R428 (AXL inhibitor; Selleck Chemicals, S2841), SP600125 (MAPK8/9/10 inhibitor; Sigma, S5567), glibenclamide (NLRP3 inflammasome inhibitor; Sigma, G0639), U0126 (MAP2K1/2 and MAPK1/3 inhibitor; InvivoGen, tlrl-u0126), SB203580 (MAPK11/14 inhibitor; InvivoGen, tlrl-sb20), BKM120 (PtdIns3K inhibitor; Selleck Chemicals, S2247), rapamycin (LC Laboratories, R5000), LPS (Sigma, L4391), CQ (Sigma, C6628), ATP (Sigma, A2383), CCl4 (Junsei Chemical, 33650-0330), MitoSOX Red mitochondrial superoxide indicator (Invitrogen, M36008).
Cells
Murine macrophage cell lines (P388D1 and J774) were maintained in DMEM (Invitrogen, 11965-084) supplemented with 5% fetal bovine serum (Hyclone, SH30071) and 1% penicillin/streptomycin (Invitrogen, 15140-122). The pBABE-puro mCherry-EGFP-LC3B plasmid (Addgene, 22418; deposited by Dr. Jay Debnath). mCherry-EGFP-MAP1LC3B-expressing P388D1 and J774 cell lines were established as previously described.Citation46
Peritoneal macrophages were isolated from axl−/−, Axl+/+, Atg7fl/fl Lyz2-Cre+ (Atg7fl/fl CKO) or Atg7fl/fl Lyz2-Cre− (Atg7fl/fl CTRL) mice (8-wk old) as previously described.Citation47 Briefly, mice were intraperitoneally injected with 1 ml of 3% Brewer thioglycollate (BD Biosciences, 211716). Three d after injection, cells were isolated by flushing out the peritoneal cavity using 10 ml of phosphate-buffered saline (PBS; 135 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) without calcium and magnesium. Cells were then plated on culture dishes and incubated at 37°C and 5% CO2 in DMEM/F-12 medium (Invitrogen, 11330-032) supplemented with 10% (v/v) fetal bovine serum and 1% penicillin/streptomycin for 4 h. Subsequently, nonadherent cells were removed by gently washing 3 times with warm PBS. Adherent cells were counted, stained with FITC-conjugated anti-ITGAM/CD11b antibody, and analyzed using flow cytometry. More than 90% of cells were ITGAM-positive.
Measurement of autophagy induction
Cells were treated with various concentrations of recombinant GAS6, CQ and/or serum deprivation for 24 h. Also, various combinations of neutralizing antibodies including anti-mouse AXL antibody (2 μg/ml), anti-mouse MERTK antibody (2 μg/ml), and anti-mouse TYRO3 antibody (2 μg/ml), or inhibitors including R428 (AXL inhibitor, 2.5 μM), SP600125 (MAPK8/9/10 inhibitor, 10 μM), U0126 (MAP2K1/2 and MAPK1/3 inhibitor, 5 μM), SB203580 (MAPK11/14 inhibitor, 2.5 μM), BKM120 (PtdIns3K inhibitor, 1 μM) and rapamycin (MTOR inhibitor, 100 nM) were added to block autophagy induction after GAS6 treatment. After treatment, autophagy induction in each cell was measured via immunoblotting and confocal microscopy. Also, Atg5, Becn1 and Map1lc3b mRNA levels were measured by quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR).
Western blot analyses
Cells were washed 3 times with PBS, centrifuged, and lysed with RIPA lysis solution (50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40 [Sigma, 74385], 0.5% sodium deoxycholate [Sigma, D6750], 0.1% SDS [Sigma, L4509]) containing 200 μg/ml of phenylmethylsulfonyl fluoride (Sigma, P7626), phosphatase inhibitor cocktail (Sigma, P0044) and protease inhibitor cocktail (Millipore, 535140). The cell lysates were subsequently resolved by 12% SDS-polyacrylamide gel electrophoresis, transferred onto Immobilon P membranes (Millipore, IPVH00010), and immunoblotted with appropriate antibodies. Immunoreactive bands were visualized using an ECL solution (GE Healthcare, RPN2232). To determine the MAP1LC3B-I to MAP1LC3B-II ratio, immunoreactive bands were quantified using ImageJ software version 1.43u (National Institutes of Health, Bethesda, MD, USA). Then, the values of MAP1LC3B-I to MAP1LC3B-II ratio were relative to the control value of the experiment. To quantify the relative expression of other proteins, immunoreactive bands were normalized to ACTB levels.
Confocal microscopy
To observe autophagy induction, mCherry-EGFP-MAP1LC3B-expressing P388D1 cells and J774 cells were treated with various combinations of recombinant GAS6, neutralizing antibody and/or inhibitors. The cells were then washed 3 times with PBS and fixed with 4% paraformaldehyde (Sigma, P6148) in PBS at room temperature for 10 min. After washing 3 times with PBS, cells were stained with a mounting solution containing DAPI (Vector Laboratories, H-1200) and imaged by confocal microscopy on an LSM 5 EXCITER (Carl-Zeiss, Oberkochen, Germany).
qRT-PCR
The mRNA transcripts of mouse Atg5, Becn1, Map1lc3b, Gas6, Il1b, Il18, Nlrp3, Pyard and Casp1 were quantitatively measured by real-time PCR analyses. The mRNA transcript of mouse Gapdh was used as an internal control for qRT-PCR. Briefly, total RNAs from cells or tissues were isolated using Trizol reagent according to the manufacturer's protocol (Thermo Fisher Scientific, 15596-026), and reverse transcribed into cDNA using the First-Strand cDNA Synthesis Kit with oligo-dT primers and Super-Script RT (Thermo Fisher Scientific, K1622). After cDNA synthesis, quantitative real-time PCR was conducted using a 7500 Real-time PCR system (Applied Biosystems, Foster City, CA, USA). The primer pairs used for qRT-PCR are listed in Table S1.
Cloning and expression of mutant Axl
Several amino acid substitution mutants of Axl were cloned with PCR from the mouse Axl gene (GenBank accession BC050914) and expressed in macrophages (J774 cell) via retroviral transduction. The cDNA of wild-type mouse Axl was isolated by RT-PCR using total RNAs of P388D1 as a template. The expression vectors encoding wild-type mouse Axl (WT Axl), the cytoplasmic domain deletion mutant of Axl (AxlCYΔ), Axl mutants with a single amino acid substitution (AxlY696F, AxlY697F, AxlY773F, AxlY815F, and AxlY860F), and an Axl mutant with a double amino acid substitution (AxlY815F,Y860F) were constructed by cloning PCR products using NotI and SalI restriction sites in the retroviral vector pLNCX2 (Clontech, 631503). PCR was performed using the cDNA of wild-type mouse Axl as a template. The primer pairs used for each expression vector are listed in Table S2. The schematic diagrams of individual proteins expressed from DNA constructs are shown in Supporting Fig. 5A. Each DNA construct was then transfected into 293GPG cells using Lipofectamine (Thermo Fisher Scientific, 11668-019). After 3 d, the supernatant from each transfectant was used to infect J774 cells using 1 μg/ml of polybrene (Sigma, H9268). To establish stable cell lines, each group of infected cells was selected with 1.5 mg/ml neomycin (Sigma, N1876). Proper cell surface expression of wild-type AXL and its mutants were observed by flow cytometry analyses.
Flow cytometry analyses
Flow cytometry analyses were performed to determine the cell surface expression of mouse AXL, MERTK, TYRO3 and ITGAM. For surface staining of AXL or MERTK, cells were stained with either purified anti-AXL antibody (rat IgG2a) or anti-MERTK antibody (rat IgG1), followed by biotinylated goat anti-rat IgG and streptavidin-PE. Rat IgG2a and rat IgG1 control were used as isotype controls. For surface staining of TYRO3, cells were stained with biotinylated anti-mouse TYRO3 antibody (rat IgG1) in combination with streptavidin-PE. Biotinylated rat IgG1 was used as an isotype control. To visualize ITGAM surface expression, cells were stained with FITC-conjugated anti-ITGAM antibody (rat IgG2b). FITC-conjugated rat IgG2b control was used as an isotype control. After washing several times with PBS, all stained cells were resuspended in PBS and analyzed by flow cytometry using a FACSCalibur with CellQuest software (BD Biosciences, San Jose, CA, USA).
RT-PCR
The mRNA transcripts of mouse Mapk11 and Mapk14 were measured by RT-PCR analyses. The cDNA synthesis was described as above. After cDNA synthesis, PCR was conducted using a SimpliAmp Thermal Cycler (Thermo Fisher Scientific). The resulting PCR products were loaded onto a 1.5% agarose gel containing ethidium bromide, and were visualized using ultraviolet light. The primer pairs used for qRT-PCR are listed in Table S3.
Co-immunoprecipitation
AXL-expressing J774 cells were washed in PBS and resuspended in cell lysis buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1% NP-40) containing 200 μg/ml of phenylmethylsulfonyl fluoride, phosphatase inhibitor cocktail and protease inhibitor cocktail. The lysates were incubated with anti-MAPK11 antibody or anti-MAPK14 antibody and precipitated with protein G sepharose™ Fast Flow (GE Healthcare, 17-0618-01). The precipitated proteins were resolved via SDS-PAGE, transferred to Immobilon-P membranes, and immunoblotted with anti-phosphorylated MAPK antibody using the ECL system. Anti-ACTB antibody was utilized as an internal control.
Inhibition of gene expression by shRNAs
The lentiviral plasmids expressing shRNAs directed against several distinct regions of Mapk14 (sc-29434) or Rela (sc-29411) were purchased from Santa Cruz Biotechnology together with control shRNA plasmids (sc-108060). Mouse Axl-expressing J774 cells were transfected with each shRNA-containing plasmid using Lipofectamine 2000 (Thermo Fisher Scientific, 11668-019) according to the manufacturer's instructions. After transfection, cells were selected with 4 μg/ml of puromycin (Sigma, P7255). Inhibition of MAPK14 expression and RELA expression were determined by immunoblotting with appropriate antibodies.
Measurement of NLRP3 inflammasome activation
Murine macrophages were treated with ATP, IFNA receptor blocking antibody, LPS, glibenclamide, and/or recombinant GAS6 as indicated and the cells were tested for inflammasome activation by immunoblotting and ELISA.
ELISAs
IL1Β and IL18 secretion levels were quantified by sandwich ELISAs using mouse IL1Β (eBioscience, BMS6002) and IL18 ELISA kits (eBioscience, BMS618/3) according to the manufacturer's instructions.
Animal studies
Axl deficient (axl−/−) mice were kindly provided by Dr. Lemke (Salk Institute, La Jolla, CA) and backcrossed into the C57BL/6J strain for more than 9 generations. Atg7-floxed mice (Atg7fl/fl, C57BL/6 background) were kindly provided by Dr. Komatsu (Tokyo Metropolitan Institute of Medica Science, Tokyo, Japan). Atg7fl/fl mice were crossed with B6.129P2-Lyz2tm1(cre)Ifo/J mice (Jackson Laboratory, 004781) where Cre recombinase expression is under the control of lysozyme M promoter to generate Atg7 conditional knockout mice with myeloid lineage cell-specific deletion of Atg7 (Atg7fl/fl CKO) and their wild-type counterparts (Atg7fl/fl CTRL). More information about the generation of Atg7fl/fl CKO and Atg7fl/fl CTRL mice was previously described.Citation28,29 Animal models of acute liver injury were created by injecting either LPS or CCl4 into mice.Citation48,49 To induce LPS-challenged acute liver injury, mice were intraperitoneally injected with 2.5 μg/g of PBS-dissolved LPS. Mice were sacrificed at 3, 6, and 12 h after LPS injection and mRNA transcripts of Gas6 from liver or spleen tissue of sacrificed mice were monitored by qRT-PCR. In addition, Kupffer cells, sera, and several tissues were harvested from mice sacrificed at 12 h after LPS injection to perform qRT-PCR analyses, western blot analyses, histological analyses, immunohistochemistry, and to measure serum GPT, GOT1, and cytokine secretions. For induction of CCl4-mediated acute liver injury, mice were injected intraperitoneally with 2 μl/g of CCl4 mixed with corn oil at a ratio of 1:9. Mice were sacrificed at 1, 2, and 3 d after CCl4 injection, mRNA transcripts of Gas6 from liver tissue of sacrificed mice were monitored by qRT-PCR. Also, Kupffer cells, sera, and liver were harvested from mice sacrificed at 1, 2, and 3 d after CCl4 injection to perform qRT-PCR analyses, western blot analyses, histological analyses, immunohistochemistry, and to measure serum GPT, GOT1, and cytokine secretions. To evaluate the effect of glibenclamide in acute liver injury model, mice were injected with LPS and 1 μg/g of DMSO-dissolved glibenclamide or CCl4 and glibenclamide. Kupffer cells and sera from mice were harvested to perform western blot analyses and to measure cytokine secretions. All animals received proper care in accordance with a protocol based on the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Korea University and Chungnam National University (protocol numbers: KUIACUC-2014-259 and CNUH-014-A0007).
Histological analyses and immunochemistry
Tissues were fixed with 10% neutral buffered formalin (Sigma, HT5011) and embedded in paraffin. Sections measuring 5 μm were cut using a Leica CM1800 cryostat (Leica Microsystems, Wetzlar, Germany), air dried at room temperature, and stained with hematoxylin and eosin (H&E). The severity of liver injury and necroinflammatory activity were scored using H&E stained sections according to standard methods as previously described.Citation50,51 The severity of liver injury observed in the tissue sections was scored as follows: 0, minimal or no evidence of injury; 1, mild injury consisting of cytoplasmic vacuolation and focal nuclear pyknosis; 2, moderate to severe injury with extensive nuclear pyknosis, cytoplasmic hypereosinophilia, and loss of intercellular borders; and 3, severe necrosis with disintegration of the hepatic cords, hemorrhage, and neutrophil infiltration. Ten different areas (× 100 fields) were examined for necroinflammatory activity. The necroinflammatory activity observed in the tissue sections was scored as follows: 0, no affected area; 1, less than 2 areas; 2, between 2 and 4 areas; 3, between 5 and 10 areas; 4, more than 10 areas with necroinflammatory activity.
For detection of fibrin deposition, livers were perfused through a portal vein with PBS and liver specimens were rapidly sampled, fixed in 10% zinc fixative (BD Biosciences, 550523), and embedded in paraffin. Tissue sections were incubated overnight with rabbit anti-mouse fibrinogen antiserum (1:5,000) followed by treatment with the rabbit Vectastain Elite ABC kit (Vector Laboratories, PK-6100). Antigen-antibody complexes were detected using a DAB Substrate Kit (Vector Laboratories, SK-4100). Immunohistochemical analysis was performed with an anti-ACTA2 antibody coupled with Alexa Fluor 488-conjugated anti-mouse IgG (green fluorescence), an anti-ADGRE1/F4/80 antibody coupled with Alexa Fluor 488-conjugated anti-rat IgG (green fluorescence), an anti-MAP1LC3B antibody coupled with Alexa Fluor 594-conjugated anti-rabbit IgG (red fluorescence) and an anti-ATG5 antibody coupled with Alexa Fluor 594-conjuated anti-rabbit IgG (red fluorescence) using a Bond Max autostainer according to the manufacturer's protocol (Leica Microsystems, Wetzlar, Germany).
Kupffer cell isolation
Kupffer cells were isolated from mice as described previously.Citation32 Briefly, the portal vein of anesthetized mice was cannulated, and the liver was perfused at 7 mL/min for 10 min with Gey balanced salt solution (Sigma, G9779) without Ca2+ at 37°C. Subsequently, perfusion with Gey balanced salt solution containing 1.5 mM CaCl2 and 0.5 mg/mL of collagenase type IV (Invitrogen, 17104019) was performed for 10 min. The digested liver was dissected out thoroughly on a petri dish and then the supernatant was filtered. The nonparenchymal cell fraction containing Kupffer cells was separated by 3 cycles of differential centrifugation (30 × g for 3 min). The supernatant was collected and centrifuged (400 × g for 3 min) to obtain nonparenchymal cell pellets. Isolated nonparenchymal cells were stained with PE-conjugated anti-ADGRE1/F4/80 antibody coupled with anti-PE microbeads and then Kupffer cells were isolated using a magnetic bead-associated cell sorting system (Miltenyi Biotec, Bergisch Gladbach, Germany). The purity of Kupffer cells was more than 90%, as assessed by flow cytometry analyses.
Measurement of plasma GPT and GOT1
Plasma GPT and GOT1 levels were measured by the standard colorimetric method using Asan GPT and GOT1 kits (Asan Pharmaceutical, AM101-K) according to the manufacturer's instructions.
Measurement of ROS
Measurement of mitochondrial superoxide was done using MitoSOX Red Mitochondrial Superoxide Indicator according to the manufacturer's instructions. Briefly, isolated mouse peritoneal macrophages were treated with LPS (100 ng/ml), ATP (5 mM) and/or recombinant GAS6 (100 ng/ml) for 6 h. After treatment, cells were incubated in Hank's balanced salt solution (Invitrogen, 14025-092) with 5 μM MitoSOX Red for 10 min at 37°C and washed with PBS. Then, cells were harvested and assessed by flow cytometry.
PYCARD pyroptosome detection
PYCARD pyroptosomes were detected as described previously.Citation52 Briefly, isolated mouse peritoneal macrophages were treated with LPS (100 ng/ml), ATP (5 mM) and/or recombinant GAS6 (100 ng/ml) for 6 h. After treatment, cells were harvested and resuspended in 0.5 ml of ice-cold buffer (20 mM HEPES-KOH, pH 7.5, 150 mM KCl, 1% NP-40, 0.1 mM PMSF and protease inhibitor cocktail), and lysed by shearing 10 times using a 21-gauge needle. Then the cell lysates were centrifuged at 6,000 × g for 10 min at 4°C, and the pellets were washed with PBS. The pellets were resuspended in 0.5 ml of 4 mM DSS in PBS and cross-linked in a shaking incubator for 30 min at 37°C, and then centrifuged at 6,000 × g for 10 min. The crosslinked pellets were resuspended in 30 μl SDS sample buffer, separated by 12% SDS-polyacrylamide gel electrophoresis and immunoblotted with anti-mouse PYCARD antibody.
Statistical analyses
Mean values were compared using a Student t test for independent variables. Significant differences as determined by P values of less than 0.05 and 0.01 are indicated with asterisks (*) and (**), respectively, on each graph.
Abbreviations
| ACTA2 | = | actin, α 2, smooth muscle, aorta |
| ACTB | = | actin, β |
| ADGRE1 | = | adhesion G protein-coupled receptor E1 |
| ATG5 | = | autophagy-related 5 |
| ATG7 | = | autophagy-related 7 |
| AXL | = | AXL receptor tyrosine kinase |
| AXLCYΔ | = | deletion mutant lacking the cytoplasmic domain of AXL |
| BECN1 | = | beclin 1, autophagy related |
| CASP1 | = | caspase 1 |
| CCl4 | = | carbon tetrachloride |
| CQ | = | chloroquine |
| GAS6 | = | growth arrest specific 6 |
| GOT1 | = | glutamic-oxaloacetic transaminase 1, soluble |
| GPT | = | glutamic pyruvic transaminase, soluble |
| HSC | = | hepatic stellate cell |
| IFN | = | interferon |
| IFNAR1 | = | interferon α and β receptor subunit 1 |
| IL1B | = | interleukin 1, β |
| IL18 | = | interleukin 18 |
| ITGAM | = | integrin α M |
| LPS | = | lipopolysaccharide |
| MAP1LC3B | = | microtubule-associated protein 1 light chain 3 β |
| MAPK | = | mitogen-activated protein kinase |
| MAP2K1/2 | = | mitogen-activated protein kinase kinase 1/2 |
| mCherry-EGFP-MAP1LC3B | = | tandem fusion of MAP1LC3B protein to acid-insensitive mCherry together with GFP |
| MERTK | = | c-mer proto-oncogene tyrosine kinase |
| MTOR | = | mechanistic target of rapamycin |
| NFKB | = | nuclear factor of kappa light polypeptide gene enhancer in B-cells |
| NLRP3 | = | NLR family, pyrin domain containing 3 |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| PYCARD | = | PYD and CARD domain containing |
| qRT-PCR | = | quantitative reverse-transcriptase polymerase chain reaction |
| RELA | = | v-rel avian reticuloendotheliosis viral oncogene homolog A |
| RTK | = | receptor tyrosine kinases |
| SMA | = | smooth muscle actin |
| ROS | = | reactive oxygen species |
| STAT1 | = | signal transducer and activator of transcription 1 |
| TAM | = | TYRO3, AXL and MERTK family of RTKs |
| TWIST1 | = | twist family bHLH transcription factor 1 |
| TYRO3 | = | TYRO3 protein tyrosine kinase 3 |
| WT AXL | = | wild-type AXL |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1235124_Supplemental_Material.zip
Download Zip (2.7 MB)Acknowledgments
We are very grateful to Dr. Lemke (Salk Institute, La Jolla, CA) for providing Axl deficient (axl−/−) mice, Dr. Komatsu (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) for providing Atg7-floxed mice (Atg7fl/fl) and Dr. Debnath (University of California San Francisco, San Francisco, CA) for providing a cDNA construct encoding mCherry-EGFP-MAP1LC3B (Addgene, 22418).
Funding
This work was supported by a grant from the Next-Generation Bio-Green 21 program (project no. PJ01113001), Rural Development Administration, Republic of Korea. We would also like to thank School of Life Sciences and Biotechnology for BK21 PLUS, Korea University for supporting the publication fee.
ORCID
Jihye Han http://orcid.org/0000-0002-9477-0270
Joonbeom Bae http://orcid.org/0000-0002-2502-9147
Chang-Yong Choi http://orcid.org/0000-0003-2139-3109
Sang-Pil Choi http://orcid.org/0000-0001-8797-8332
Hyung-Sik Kang http://orcid.org/0000-0002-2295-9293
Eun-Kyeong Jo http://orcid.org/0000-0002-9623-9037
Jongsun Park http://orcid.org/0000-0002-4690-1854
Young Sik Lee http://orcid.org/0000-0002-6709-3809
Hyun-Seuk Moon http://orcid.org/0000-0002-5216-2090
Chung-Gyu Park http://orcid.org/0000-0003-4083-8791
Myung-Shik Lee http://orcid.org/0000-0003-3292-1720
Taehoon Chun http://orcid.org/0000-0002-5940-8620
Related Research Data
References
- Rockey DC, Bell PD, Hill JA. Fibrosis- a common pathway to organ injury and failure. N Engl J Med 2015; 372:1138-49; PMID:25785971; http://dx.doi.org/10.1056/NEJMra1300575
- Seki E, Schwabe RF. Hepatic inflammation and Fibrosis: Functional links and key pathways. Hepatology 2015; 61:1066-79; PMID:25066777; http://dx.doi.org/10.1002/hep.27332
- Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol 2009; 27:147-63; PMID:19302037; http://dx.doi.org/10.1146/annurev.immunol.021908.132629
- Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012; 22:787-95; http://dx.doi.org/10.1172/JCI59643
- Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001; 411:207-11; PMID:11346799; http://dx.doi.org/10.1038/35075603
- Zagórska A, Través PG, Lew ED, Dransfield I, Lemke G. Diversification of TAM receptor tyrosine kinase function. Nat Immunol 2014; 15:920-8; http://dx.doi.org/10.1038/ni.2986
- Sharif MN, Sosic D, Rothlin CV, Kelly E, Lemke G, Olson EN, Ivashkiv LB. Twist mediates suppression of inflammation by type I IFNs and Axl. J Exp Med 2006; 203:1891-901; PMID:16831897; http://dx.doi.org/10.1084/jem.20051725
- Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007; 131:1124-36; PMID:18083102; http://dx.doi.org/10.1016/j.cell.2007.10.034
- Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001; 293:306-11; PMID:11452127; http://dx.doi.org/10.1126/science.1061663
- Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med 2002; 196:135-40; PMID:12093878; http://dx.doi.org/10.1084/jem.20012094
- Llacuna L, Bárcena C, Bellido-Martín L, Fernández L, Stefanovic M, Marí M, García-Ruiz C, Fernández-Checa JC, García de Frutos P, Morales A. Growth arrest-specific protin 6 is hepatoprotective against murine ischemia/reperfusion injury. Hepatology 2010; 52:1371-9; PMID:20730776; http://dx.doi.org/10.1002/hep.23833
- Lafdil F, Chobert MN, Deveaux V, Zafrani ES, Mavier P, Nakano T, Laperche T, Brouillet A. Growth arrest-specific protein 6 deficiency impairs liver tissue repair after acute toxic hepatitis in mice. J Hepatol 2009; 51:55-66; PMID:19443073; http://dx.doi.org/10.1016/j.jhep.2009.02.030
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717-21; PMID:10634780
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 2007; 7:767-77; PMID:17767194; http://dx.doi.org/10.1038/nri2161
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID:18849965; http://dx.doi.org/10.1038/nature07383
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221-5; PMID:21124315; http://dx.doi.org/10.1038/nature09663
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12:222-30; PMID:21151103; http://dx.doi.org/10.1038/ni.1980
- Beth L, Noboru M, Herbert WV. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722-37; PMID:24064518; http://dx.doi.org/10.1038/nri3532
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/10.1080/15548627.2015.1100356
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/10.4161/auto.4600
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID:21801009; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005
- Paccez JD, Vogelsang M, Parker MI, Zerbini LF. The receptor tyrosine kinase Axl in cancer: biological functions and therapeutic implications. Int J Cancer 2014; 134:1024-33; PMID:23649974; http://dx.doi.org/10.1002/ijc.28246
- Eyers PA, Craxton M, Morrice N, Cohen P, Goedert M. Conversion of SB203580-insensitive MAP kinase family members to drug-sensitive forms by a single amino-acid substitution. Chem Biol 1998; 5:321-8; PMID:9653550; http://dx.doi.org/10.1016/S1074-5521(98)90170-3
- Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, et al. Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 1995; 80:401-11; PMID:7859282; http://dx.doi.org/10.1016/0092-8674(95)90490-5
- Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galán JE, Askenase PW, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 2006; 24:317-27; PMID:16546100; http://dx.doi.org/10.1016/j.immuni.2006.02.004
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/10.1083/jcb.200412022
- Yang CS, Kim JJ, Lee HM, Jin HS, Lee SH, Park JH, Kim SJ, Kim JM, Han YM, Lee MS, et al. The AMPK-PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy 2014; 10:785-802; PMID:24598403; http://dx.doi.org/10.4161/auto.28072
- Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011; 34:213-23; PMID:21349431; http://dx.doi.org/10.1016/j.immuni.2011.02.006
- Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 2012; 122:3476-89; PMID:22945633; http://dx.doi.org/10.1172/JCI60777
- Huang H, Chen HW, Evankovich J, Yan W, Rosborough BR, Nace GW, Ding Q, Loughran P, Beer-Stolz D, Billiar TR, et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol 2013; 191:2665-79.
- Jankovic D, Ganesan J, Bscheider M, Stickel N, Weber FC, Guarda G, Follo M, Pfeifer D, Tardivel A, Ludigs K, et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J Exp Med 2013; 210:1899-910; PMID:23980097; http://dx.doi.org/10.1084/jem.20130084
- Beier JI, Luyendyk JP, Guo L, von Montfort C, Staunton DE, Arteel GE. Fibrin accumulation plays a critical role in the sensitization to lipopolysaccharide-induced liver injury caused by ethanol in mice. Hepatology 2009; 49:1545-53; PMID:19291788; http://dx.doi.org/10.1002/hep.22847
- Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, Tanaka KE, Czaja MJ. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015; 11:271-84; PMID:25650776; http://dx.doi.org/10.1080/15548627.2015.1009787
- Dall'Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small SA, et al. The phospholipase D1 pathway modulates macroautophagy. Nat Commun 2010; 1:142; PMID:21266992; http://dx.doi.org/10.1038/ncomms1144
- Chikh A, Sanzà P, Raimondi C, Akinduro O, Warnes G, Chiorino G, Byrne C, Harwood CA, Bergamaschi D. iASPP is a novel autophagy inhibitor in keratinocytes. J Cell Sci 2014; 127:3079-93; PMID:24777476; http://dx.doi.org/10.1242/jcs.144816
- Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res 2008; 100:35-83; PMID:18620092; http://dx.doi.org/10.1016/S0065-230X(08)00002-X
- Demarchi F, Verardo R, Varnum B, Brancolini C, Schneider C. Gas6 anti-apoptotic signaling requires NF-kappa B activation. J Biol Chem 2001; 276:31738-44; PMID:11425860; http://dx.doi.org/10.1074/jbc.M104457200
- Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest 2009; 119:3502-11; PMID:19955661; http://dx.doi.org/10.1172/JCI40599
- Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140:821-32; PMID:20303873; http://dx.doi.org/10.1016/j.cell.2010.01.040
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 2009; 28:3015-26; PMID:19713938; http://dx.doi.org/10.1038/emboj.2009.242
- Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, Hoffman HM, Feldstein AE. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014; 59:898-910; PMID:23813842; http://dx.doi.org/10.1002/hep.26592
- Fourcot A, Couchie D, Chobert MN, Zafrani ES, Mavier P, Laperche Y, Brouillet A. Gas6 deficiency prevents liver inflammation, steatohepatitis, and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol 2011; 300:G1043-53; PMID:21350191; http://dx.doi.org/10.1152/ajpgi.00311.2010
- Bárcena C, Stefanovic M, Tutusaus A, Joannas L, Menéndez A, García-Ruiz C, Sancho-Bru P, Marí M, Caballeria J, Rothlin CV, et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J Hepatol 2015; 63:670-8.
- Park HJ, Lee SJ, Kim SH, Han J, Bae J, Kim SJ, Park CG, Chun T. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol 2011; 48:720-7; PMID:21095008; http://dx.doi.org/10.1016/j.molimm.2010.10.020
- Pineda-Torra I, Gage M, de Juan A, Pello OM. Isolation, culture, and polarization of murine bone marrow-derived and peritoneal macrophages. Methods Mol Biol 2015; 1339:101-9; PMID:26445783; http://dx.doi.org/10.1007/978-1-4939-2929-0_6
- Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Müller M, Blander JM, Tacke F, Trautwein C. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med 2010; 207:1453-64; PMID:20530204; http://dx.doi.org/10.1084/jem.20091474
- Lodder J, Denaës T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, Lotersztajn S, Teixeira-Clerc F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 2015; 11:1280-92; PMID:26061908; http://dx.doi.org/10.1080/15548627.2015.1058473
- Lu Y, Leung TM, Ward SC, Nieto N. Partial deletion of argininosuccinate synthase protects from pyrazole plus LPS-induced liver injury by decreasing nitrosative stress. Am J Physiol Gastrointest Liver Physiol 2012; 302:G287-95; PMID:22052013; http://dx.doi.org/10.1152/ajpgi.00375.2011
- Xu H, Garver H, Fernandes R, Galligan JJ, Fink GD. Altered L-type Ca2+ channel activity contributes to exacerbated hypoperfusion and mortality in smooth muscle cell BK channel-deficient septic mice. Am J Physiol Regul Integr Comp Physiol 2014; 307:R138-48; PMID:24829499; http://dx.doi.org/10.1152/ajpregu.00117.2014
- Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, He Z, Zeng Y, Hu Y, Sun S, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 2013; 23:201-12; PMID:23318584; http://dx.doi.org/10.1038/cr.2013.6