
ABSTRACT
Proteostasis is important for protecting cells from harmful proteins and is mainly controlled by the HSF1 (heat shock transcription factor 1) stress response pathway. This pathway facilitates protein refolding by molecular chaperones; however, it is unclear whether it functions in autophagy or inclusion formation. The autophagy receptor SQSTM1/p62 is involved in selective autophagic clearance and inclusion formation by harmful proteins, and its phosphorylation at S349, S403, and S407 is required for binding to substrates. Here, we demonstrate that casein kinase 1 phosphorylates the SQSTM1 S349 residue when harmful proteins accumulate. Investigation of upstream factors showed that both SQSTM1 S349 and SQSTM1 S403 residues were phosphorylated in an HSF1 dependent manner. Inhibition of SQSTM1 phosphorylation suppressed inclusion formation by ubiquitinated proteins and prevented colocalization of SQSTM1 with aggregation-prone proteins. Moreover, HSF1 inhibition impaired aggregate-induced autophagosome formation and elimination of protein aggregates. Our findings indicate that HSF1 triggers SQSTM1-mediated proteostasis.
Introduction
The accumulation of harmful proteins, such as misfolded proteins and protein aggregations, leads to cellular dysfunction and cell death. In order to avoid such accumulations, protein quality control is performed via the protein homeostasis (proteostasis) network, a multicompartmental system that coordinates protein synthesis, folding, disaggregation, and degradation.Citation1 HSF1 (heat shock transcription factor 1) is the master regulator of the proteostasis network and is present normally in a negatively regulated state as an inert monomer.Citation2 Upon exposure to a variety of stresses, HSF1 is derepressed; it then trimerizes and translocates to the nucleus. Subsequently, activated HSF1 binds to the heat shock element (HSE), a stress-responsive promoter, resulting in the promotion of expression of heat shock proteins (HSPs) such as the molecular chaperones HSPA1/HSP70 and HSP90AA/HSP90.Citation2
The degradation of harmful proteins is principally mediated by the ubiquitin-proteasome system (UPS) and the autophagy-lysosome pathway.Citation3 Although macroautophagy (the degradation of cellular contents, hereafter referred to as autophagy) has formerly been considered to be a nonselective degradation mechanism, a number of studies have shown that some substrates, such as protein aggregates and impaired organelles, are selectively eliminated by this process.Citation4,5 A range of autophagic receptor proteins are involved in this selective process, e.g., SQSTM1/p62, BNIP3, BNIP3L, FUNDC1, NBR1, CALCOCO2/NDP52, and OPTN.Citation6 SQSTM1, which has an LC3-interacting region, a KEAP1 (kelch like ECH associated protein 1)-interaction region, and a ubiquitin-associated domain,Citation7 contributes to the selective autophagic clearance of protein aggregates, KEAP1, and cytosolic pathogens.Citation8 Recent studies have revealed that SQSTM1 must be phosphorylated at S349, S403, or S407 in order to bind to its substrates.Citation9-12 S349-phosphorylation is mediated by MTORC1 (mechanistic target of rapamycin [serine/threonine kinase] complex 1) and is required for interaction with KEAP1.Citation12 Phosphorylation at S403 by TBK1 (TANK binding kinase 1) and CSNK2/CK2 (casein kinase 2) is necessary for SQSTM1 involvement in the elimination of protein aggregates and mycobacteria.Citation9,11 In addition to its function as an autophagy receptor, SQSTM1 plays an important role in inclusion formation by ubiquitinated proteins. The accumulation of ubiquitinated protein inclusions by impairment of autophagy or proteasomes is suppressed by the loss of SQSTM1.Citation13,14 The formation of protein inclusions and aggresomes is assumed to be a cytoprotective mechanism to sequester harmful proteins.Citation15 Thus, SQSTM1 is one of the key molecules for proteostasis.
Although inclusion formation and autophagic clearance of harmful proteins are essential for proteostasis, it is still unknown how these processes are initiated and regulated. Recently, it was reported that several stress sensor proteins, such as XBP1, HSF1, and STAT3, are involved in the regulation of autophagy-related protein expression. For example, the endoplasmic reticulum stress sensor XBP1 triggers an autophagic response via autophagic vesicle formation and expression of BECN1/Beclin1 and LC3B.Citation16 Moreover, HSF1 cooperatively binds with PSMD10/Gankyrin (26S non-ATPase regulatory proteasome subunit 10) on the ATG7 promoter in cells at an advanced stage of starvation and upregulates ATG7 expression.Citation17 These findings lead us to speculate that stress response pathways are involved in autophagy regulation; thus, it is important to elucidate the process of autophagy regulation in detail.
In this study, we investigated the mechanism of SQSTM1 phosphorylation and the role of phosphorylated SQSTM1 in inclusion formation by ubiquitinated proteins. We found that SQSTM1 phosphorylation was mediated by the HSF1 stress response pathway, and resulted in inclusion formation and autophagic clearance of harmful proteins (aggrephagy). These results suggest that the HSF1 stress response pathway is involved in SQSTM1-associated proteostasis functions.
Results
Colocalization of phosphorylated SQSTM1 with protein aggregates
SQSTM1 is phosphorylated at one site in the KEAP1-interaction region domain (S349) and 2 sites in the ubiquitin associated domain (S403 and S407) (); these phosphorylation events regulate selective autophagic clearance of ubiquitinated proteins, KEAP1, and mycobacteria.Citation9-12 We investigated whether phosphorylation of SQSTM1 was induced by the generation of different types of protein aggregate such as ubiquitinated proteins, nonubiquitinated aggregation-prone protein, and a disease-associated protein. Proteasome inhibition by MG132 led to the accumulation of ubiquitinated proteins and induction of SQSTM1 phosphorylation at both S349 and S403 (). When phosphorylated SQSTM1 was treated with λ protein phosphatase, these bands were not detected by anti-phosphorylated SQSTM1 antibodies (Fig. S1A). Immunocytochemical analysis showed that S349-phosphorylated SQSTM1 and S403-phosphorylated SQSTM1 colocalized with polyubiquitinated protein inclusions (). To examine SQSTM1 phosphorylation further, we used expression of the nonubiquitinated aggregation-prone protein EGFP-STAT5A(E18Δ), a C-terminal frameshift mutant of STAT5A.Citation18 EGFP-STAT5A(E18Δ) was transiently expressed in HeLa cells and phosphorylation of SQSTM1 at S349 and S403 was measured by an immunoblotting analysis. Phosphorylation at these residues was induced by generation of EGFP-STAT5A(E18Δ) aggregates, as also occurs for ubiquitinated inclusions (Fig. S2A), and EGFP-STAT5A(E18Δ) aggregates colocalized with both S349- and S403-phosphorylated SQSTM1 (Fig. S2B).
Figure 1. Phosphorylation of SQSTM1 at S349 and S403 in MG132-treated HeLa cells. (A) Schematic of the SQSTM1 domain structure. SQSTM1 contains a PB1 (Phox and Bem1) domain, a ZZ zinc finger (ZZ) domain, an LC3-interaction region (LIR) motif, a Kelch-like ECH-associated protein 1 (KEAP1)-interaction region (KIR) motif, and a ubiquitin associated (UBA) domain. SQSTM1 is phosphorylated at S349, S403, and S407 (S351, S405, and S409 in mice). (B) HeLa cells were cultured without (−) or with (+) 10 µM MG132 for 12 h, followed by immunoblot analysis of cell lysates. Band intensities were measured, and phosphorylated SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). P values were calculated using the Student t test. *P < 0.01. (C) HeLa cells were cultured with or without 10 µM MG132 for 12 h. Colocalization of SQSTM1 (upper), S349-phosphorylated SQSTM1 (middle, p-SQSTM1 [S349]), and S403-phosphorylated SQSTM1 (lower, p-SQSTM1 [S403]) with ubiquitinated inclusions (Ub) were examined immunohistochemically. Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm.
![Figure 1. Phosphorylation of SQSTM1 at S349 and S403 in MG132-treated HeLa cells. (A) Schematic of the SQSTM1 domain structure. SQSTM1 contains a PB1 (Phox and Bem1) domain, a ZZ zinc finger (ZZ) domain, an LC3-interaction region (LIR) motif, a Kelch-like ECH-associated protein 1 (KEAP1)-interaction region (KIR) motif, and a ubiquitin associated (UBA) domain. SQSTM1 is phosphorylated at S349, S403, and S407 (S351, S405, and S409 in mice). (B) HeLa cells were cultured without (−) or with (+) 10 µM MG132 for 12 h, followed by immunoblot analysis of cell lysates. Band intensities were measured, and phosphorylated SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). P values were calculated using the Student t test. *P < 0.01. (C) HeLa cells were cultured with or without 10 µM MG132 for 12 h. Colocalization of SQSTM1 (upper), S349-phosphorylated SQSTM1 (middle, p-SQSTM1 [S349]), and S403-phosphorylated SQSTM1 (lower, p-SQSTM1 [S403]) with ubiquitinated inclusions (Ub) were examined immunohistochemically. Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm.](/cms/asset/f85a3ecb-4cb0-4727-adb1-15560454f502/kaup_a_1248018_f0001_oc.gif)
We previously showed that SQSTM1 was colocalized with SNCA/α-synuclein aggregates, a common neuropathological hallmark of α-synucleinopathy, using a cell culture model of α-synucleinopathies such as Parkinson disease and dementia with Lewy bodies.Citation19 Here, we examined SQSTM1 phosphorylation states in this model. When SNCA fibrils were introduced into HEK293 cells that exogenously expressed SNCA, Lewy body-like phosphorylated SNCA aggregates were formed intracellularly (). The SNCA aggregates colocalized with SQSTM1 and S349-phosphorylated SQSTM1 (). However, S403-phosphorylated SQSTM1 was not detected at SNCA aggregates (). Similarly, immunoblotting analysis demonstrated that SQSTM1 phosphorylation at S349 was induced not only by the appearance of SNCA aggregates but also by SNCA overexpression, although S403-phosphorylation was not detected in either case (). Since SNCA overexpression induces an increase in its oligomer form,Citation20 S349-phosphorylation presumably resulted from the accumulation of its oligomer.
Figure 2. Generation of SNCA aggregates induces the phosphorylation of SQSTM1 on S349 but not on S403. (A) SNCA fibrils (+Fibril) or PBS-mock (Control) were introduced using a transfection reagent into HEK293 cells exogenously expressing human SNCA. After 24 h, the cells were subjected to immunocytochemical analysis using anti-phosphorylated SNCA (p-SNCA), anti-SQSTM1 (SQSTM1), and anti-phosphorylated SQSTM1 (p-SQSTM1 [S349] and p-SQSTM1 [S403]) antibodies. Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm. Lewy body-like SNCA aggregates are indicated by arrowheads. (B) Lysates of HEK293 (lane 1), HEK293 exogenously expressing human SNCA (lane 2), and HEK293 introduced with SNCA fibrils (lane 3) were analyzed by immunoblotting with anti-SNCA, anti-SQSTM1, anti-phosphorylated SQSTM1, and anti-actin antibodies. Arrowheads indicate molecular species of SNCA including SDS-soluble forms (open arrowhead), SDS-insoluble dimer (closed black arrowhead), and SDS-insoluble aggregates (closed gray arrowhead). Band intensities were measured, and phosphorylated-SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01, **P < 0.05. (C) SNCA fibrils were microinjected into the striatum of mice. After 6 mo, the striatum was immunohistochemically examined using anti-phosphorylated SNCA (p-SNCA), anti-SQSTM1 (SQSTM1), and anti-phosphorylated SQSTM1 (p-SQSTM1 [S351] and p-SQSTM1 [S405]) antibodies. The insets show colocalization images of p-SNCA and SQSTM1. Scales are indicated on the images. Middle panels are high-magnification images of upper panels.
![Figure 2. Generation of SNCA aggregates induces the phosphorylation of SQSTM1 on S349 but not on S403. (A) SNCA fibrils (+Fibril) or PBS-mock (Control) were introduced using a transfection reagent into HEK293 cells exogenously expressing human SNCA. After 24 h, the cells were subjected to immunocytochemical analysis using anti-phosphorylated SNCA (p-SNCA), anti-SQSTM1 (SQSTM1), and anti-phosphorylated SQSTM1 (p-SQSTM1 [S349] and p-SQSTM1 [S403]) antibodies. Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm. Lewy body-like SNCA aggregates are indicated by arrowheads. (B) Lysates of HEK293 (lane 1), HEK293 exogenously expressing human SNCA (lane 2), and HEK293 introduced with SNCA fibrils (lane 3) were analyzed by immunoblotting with anti-SNCA, anti-SQSTM1, anti-phosphorylated SQSTM1, and anti-actin antibodies. Arrowheads indicate molecular species of SNCA including SDS-soluble forms (open arrowhead), SDS-insoluble dimer (closed black arrowhead), and SDS-insoluble aggregates (closed gray arrowhead). Band intensities were measured, and phosphorylated-SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01, **P < 0.05. (C) SNCA fibrils were microinjected into the striatum of mice. After 6 mo, the striatum was immunohistochemically examined using anti-phosphorylated SNCA (p-SNCA), anti-SQSTM1 (SQSTM1), and anti-phosphorylated SQSTM1 (p-SQSTM1 [S351] and p-SQSTM1 [S405]) antibodies. The insets show colocalization images of p-SNCA and SQSTM1. Scales are indicated on the images. Middle panels are high-magnification images of upper panels.](/cms/asset/7b311202-5639-46b6-a83e-0550a716bda3/kaup_a_1248018_f0002_oc.gif)
We further investigated the colocalization of phosphorylated SQSTM1 with SNCA aggregates using an α-synucleinopathy mouse model produced by the microinjection of SNCA fibrils into the striatum.Citation21 At 6 mo after SNCA fibril injection, phosphorylated SNCA-positive inclusions were observed in cell bodies and neurites of the striatum (). Interestingly, SQSTM1 mainly colocalized with the inclusions only at cell bodies (). Likewise, S349 (S351 in mice)-phosphorylated SQSTM1 was predominantly detected in cell bodies rather than neurites (). Phosphorylation of SQSTM1 at S403 (S405 in mice) site did not occur in the in vivo model, similarly to our results using the cell culture model (). These results indicate that SQSTM1 phosphorylation status changes depending on the nature of the protein aggregates.
S349 site of SQSTM1 is phosphorylated by CSNK1 (casein kinase 1)
Although it has been reported that S349 and S403 of SQSTM1 are phosphorylated by MTORC1 and CSNK2-TBK1, respectively,Citation9,11,12 it is also possible that other kinases can mediate phosphorylation of these sites. We explored this possibility using various protein kinase inhibitors. This analysis identified 2 novel pathways for SQSTM1 S349-phosphorylation: one directly mediated by CSNK1/CK1 (casein kinase 1) and the other indirectly by CSNK2. CKI-7, a CSNK1 and SGK (serum/glucocorticoid regulated kinase) inhibitor and TBCA, a CSNK2 inhibitor, inhibited MG132-induced S349-phosphorylation. Both inhibitors had a greater effect than rapamycin, an MTORC1 inhibitor (). Moreover, the combination of rapamycin and CKI-7 or rapamycin and TBCA additively inhibited S349-phosphorylation compared to rapamycin alone (). Phosphorylation was completely blocked by the combination of all 3 inhibitors (). These results suggested that the SQSTM1 (S349)-phosphorylation pathway mediated by CSNK1 and CSNK2 was distinct from that via MTORC1. Similarly, SQSTM1 (S403)-phosphorylation was also examined, but the compounds did not have an inhibitory effect ().
Figure 3. Identification of a novel kinase that phosphorylates SQSTM1 (S349). (A) HeLa cells were treated with rapamycin (MTORC1 inhibitor, 1 µM), CKI-7 (CSNK1 and SGK inhibitor, 100 µM), TBCA (CSNK2 inhibitor, 25 µM), rapamycin and CKI-7, or rapamycin and TBCA, together with MG132 (10 µM). Cell lysates were subjected to immunoblot analysis after 12 h. Band intensities were measured, and phosphorylated-SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01. (B) The dual and triple combination treatments of kinase inhibitors were further examined as described in A. Band intensities of S349-phosphorylated SQSTM1 were measured, and the data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01. (C) Immunoprecipitates of SQSTM1-mycHis (WT, S349A, and S403A) were incubated with CSNK1 or CSNK2 for 1 h, followed by immunoblot analysis using anti-phosphorylated SQSTM1 (p-SQSTM1 [S349] and p-SQSTM1 [S403]) and anti-SQSTM1 antibodies. (D) Colocalization of SQSTM1 with ubiquitinated inclusions was examined when SQSTM1 (S349)-phosphorylation was inhibited by the treatment with kinase inhibitors (1 µM rapamycin, 100 µM CKI-7, and 25 µM TBCA). Immunocytochemical analysis was performed 12 h after treatment. Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm.
![Figure 3. Identification of a novel kinase that phosphorylates SQSTM1 (S349). (A) HeLa cells were treated with rapamycin (MTORC1 inhibitor, 1 µM), CKI-7 (CSNK1 and SGK inhibitor, 100 µM), TBCA (CSNK2 inhibitor, 25 µM), rapamycin and CKI-7, or rapamycin and TBCA, together with MG132 (10 µM). Cell lysates were subjected to immunoblot analysis after 12 h. Band intensities were measured, and phosphorylated-SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01. (B) The dual and triple combination treatments of kinase inhibitors were further examined as described in A. Band intensities of S349-phosphorylated SQSTM1 were measured, and the data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01. (C) Immunoprecipitates of SQSTM1-mycHis (WT, S349A, and S403A) were incubated with CSNK1 or CSNK2 for 1 h, followed by immunoblot analysis using anti-phosphorylated SQSTM1 (p-SQSTM1 [S349] and p-SQSTM1 [S403]) and anti-SQSTM1 antibodies. (D) Colocalization of SQSTM1 with ubiquitinated inclusions was examined when SQSTM1 (S349)-phosphorylation was inhibited by the treatment with kinase inhibitors (1 µM rapamycin, 100 µM CKI-7, and 25 µM TBCA). Immunocytochemical analysis was performed 12 h after treatment. Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm.](/cms/asset/d2c21a0d-f63d-4a6b-bc36-e3eef344dbdf/kaup_a_1248018_f0003_oc.gif)
Next, we performed in vitro phosphorylation assays of SQSTM1. Wild-type SQSTM1 (WT) and S349A and S403A mutants were transiently expressed in SQSTM1 KO HEK293 cells and the proteins were purified by immunoprecipitation. Immunoprecipitation products were incubated with CSNK1 or CSNK2 in the presence of ATP for 1h at 37°C, followed by an immunoblotting analysis. The S349 residue of WT and S403A mutant SQSTM1 was phosphorylated by CSNK1 but not in the S349A mutant (). λ protein phosphatase treatment reduced its phosphorylation (Fig. S1B). For CSNK2, S349-phosphorylated bands were detected at a low level in WT and S403A mutant SQSTM1 (). We did not detect S403-phosphorylation when SQSTM1 WT was incubated with either CSNK1 or CSNK2 (). Our results indicate that SQSTM1 S349 was directly phosphorylated by CSNK1, and that CSNK2 was indirectly involved in this phosphorylation.
We investigated the effect of blocking S349-phosphorylation on SQSTM1 localization with protein aggregates. After a combined treatment with rapamycin, CKI-7 and TBCA, ubiquitinated protein inclusions were no longer formed in MG132-treated HeLa cells; additionally, SQSTM1- and S349-phosphorylated SQSTM1-positive inclusions also disappeared (). An immunoblotting analysis confirmed that ubiquitinated proteins and SQSTM1 in the Triton X-100 insoluble fraction were markedly reduced in kinase inhibitor treated cells (Fig. S3). As shown in , ubiquitinated proteins were normally increased in these cells, proving that the ubiquitination process was not prevented by treatment with the protein kinase inhibitors. We obtained a similar result for the aggregation-prone protein. The inhibition of S349-phosphorylation by kinase inhibitors treatment reduced SQSTM1-colocalization to EGFP-STAT5A(E18Δ) aggregates (Fig. S2C) even though this protein aggregates in a SQSTM1-independent manner.Citation18
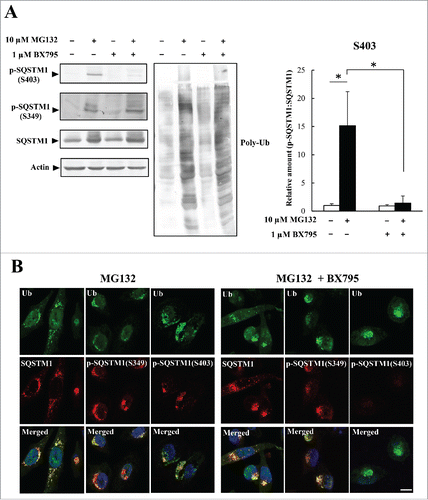
Next, we examined whether S403 phosphorylation was involved in the formation of inclusions by ubiquitinated proteins. HeLa cells were treated with MG132 alone or with a combination of MG132 and the TBK1 inhibitor BX795. In cells treated with MG132 alone, S403-phosphorylated SQSTM1 was detected by immunoblotting analysis (). The combined MG132/BX795 treatment resulted in a 90% reduction in S403 phosphorylation but had no effect on S349 phosphorylation (). Ubiquitin- and SQSTM1-positive inclusions were detected after MG132 treatment; this was also seen in cells treated with BX975 to inhibit S403 phosphorylation (). We confirmed that these inclusions colocalized with S349-phosphorylated SQSTM1 (). Next, we investigated the inclusion formation of ubiquitinated proteins in SQSTM1-mutant-expressing cells. SQSTM1 WT or -mutant (S349A or S403A) was stably expressed in SQSTM1 KO HeLa cells (Fig. S4A). Triton X-100-soluble and -insoluble fractions from control and MG132-treated cells were assessed by immunoblotting with anti- SQSTM1 and anti-poly-ubiquitin antibodies. Triton-insoluble poly-ubiquitinated proteins were significantly decreased in SQSTM1 KO (Vector) and S349A mutant-expressing cells, compared with SQSTM1 WT or -S403 mutant-expressing cells (Fig. S4B and C). Together, our results indicate that S349 phosphorylation is required for inclusion formation by ubiquitinated proteins.
Figure 4. Colocalization of SQSTM1 with ubiquitinated inclusions is not affected by inhibition of SQSTM1 (S403)-phosphorylation. (A) HeLa cells were treated with a BX795 TBK1 inhibitor (1 μM) and MG132 (10 μM) for 12 h. Cell lysates were analyzed by immunoblot analysis. Band intensities were measured, and phosphorylated-SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01. (B) Colocalization of SQSTM1 with ubiquitinated inclusions were immunocytochemically assessed in cells treated with MG132 alone (left) or MG132 and BX795 (right). Cell nuclei counterstained blue with DAPI. Scale bar: 10 μm.
Regulation of SQSTM1 phosphorylation by the HSF1 stress response pathway
When misfolded and unfolded proteins accumulate intracellularly following heat or oxidative stresses, HSF1 is activated and upregulates the expression of proteostasis-related proteins such as HSPs.Citation2,22 In a similar manner, the accumulation of ubiquitinated proteins after inhibition of proteasomes enhanced HSF1 activation. Here, HSF1 activity was measured by an affinity isolation assay using a biotinylated oligonucleotide that included the HSE sequence that is a target sequence for activated HSF1.Citation23 HSF1 activity was induced within 1 h after MG132 treatment and achieved a maximum level after 9 h (Fig. S5). We hypothesized that SQSTM1 phosphorylation was modulated through the HSF1 stress response pathway. To test this hypothesis, HSF1 was inhibited by KRIBB11, an HSF1-specific inhibitor.Citation24 Immunoblotting analysis showed that HSF1 inhibition strongly suppressed both S349- and S403-phosphorylation although ubiquitinated proteins were increased by the MG132 treatment (). This inhibitor did not influence SQSTM1 or MTOR expression (). Immunocytochemical analysis showed that inclusion formation by ubiquitinated proteins did not occur in HeLa cells treated with MG132 and KRIBB11 (), similarly to the effect of kinase inhibitory treatment. Immunoblotting analysis also demonstrated that KRIBB11-treatment markedly reduced Triton X-100-insoluble ubiquitinated proteins and SQSTM1 (Fig. S3). The same result was obtained for the aggregation-prone protein EGFP-STAT5A(E18Δ). Although S349- and S403-phosphorylated SQSTM1 were significantly increased in HeLa cells expressing EGFP-STAT5A(E18Δ), HSF1 inhibition by KRIBB11 reduced their levels (). Immunocytochemical analysis also showed that SQSTM1 was not localized to EGFP-STAT5A(E18Δ) aggregates in KRIBB11-treated cells ().
Figure 5. Phosphorylation of S349 and S403 on SQSTM1 is suppressed by a KRIBB11 HSF1 inhibitor. (A) HeLa cells were treated with 10 µM MG132 and 50 µM KRIBB11 for 12 h. Cell lysates were subjected to immunoblot analysis. Band intensities were measured, and phosphorylated SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01. (B) Colocalization of SQSTM1 with ubiquitinated inclusions were immunocytochemically assessed in cells treated with MG132 alone (left) or MG132 and KRIBB11 (right). Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm. (C) HeLa cells were transiently transfected with mock control or pEGFP-STAT5A(E18Δ) (STAT5A[E18Δ]), and then treated (+) with or without (−) 50 µM KRIBB11. After 12 h, cell lysates were examined by immunoblot analysis. Band intensities of S349- and S403-phosphorylated SQSTM1 were measured, and the data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01, **P < 0.05. (D) Colocalization of SQSTM1 with EGFP-STAT5A(E18Δ) were immunocytochemically assessed in cells treated with DMSO control (left) or KRIBB11 (right). Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm.
![Figure 5. Phosphorylation of S349 and S403 on SQSTM1 is suppressed by a KRIBB11 HSF1 inhibitor. (A) HeLa cells were treated with 10 µM MG132 and 50 µM KRIBB11 for 12 h. Cell lysates were subjected to immunoblot analysis. Band intensities were measured, and phosphorylated SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01. (B) Colocalization of SQSTM1 with ubiquitinated inclusions were immunocytochemically assessed in cells treated with MG132 alone (left) or MG132 and KRIBB11 (right). Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm. (C) HeLa cells were transiently transfected with mock control or pEGFP-STAT5A(E18Δ) (STAT5A[E18Δ]), and then treated (+) with or without (−) 50 µM KRIBB11. After 12 h, cell lysates were examined by immunoblot analysis. Band intensities of S349- and S403-phosphorylated SQSTM1 were measured, and the data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01, **P < 0.05. (D) Colocalization of SQSTM1 with EGFP-STAT5A(E18Δ) were immunocytochemically assessed in cells treated with DMSO control (left) or KRIBB11 (right). Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm.](/cms/asset/69ec0a10-9753-40ec-93ac-b2f88b4169b7/kaup_a_1248018_f0005_oc.gif)
Next, we used HSF1 knockout (HSF1 KO) cells to investigate whether the HSF1 stress response pathway modulates the SQSTM1 phosphorylation pathway. We designed a single guide RNA vector that targeted exon 2 of the HSF1 gene and generated HSF1 KO HeLa cells using the CRISPR/Cas9 system. HSF1 KO HeLa cells were cloned, and gene knockout was validated by an immunoblotting analysis (). In comparison with WT cells, expression of SQSTM1 was unaltered in HSF1 KO HeLa cells although HSPA1 expression was slightly decreased (). When HSF1 KO cells were treated with MG132, phosphorylation of S349 and S403 was reduced to 40% and 3%, respectively, compared with WT cells (). However, exogenous expression of HSF1 in the HSF1 KO cells restored the levels of phosphorylation (). Thus, our observations were not due to off-target effects of the CRISPR/Cas9 system.
Figure 6. Suppression of SQSTM1 S349-phosphorylation in HSF1 KO cells. (A) HSF1 was targeted using the CRISPR/Cas9 system. Cell lysates of HSF1-WT and individual HSF1-targeted clones (#6 and #12) were subjected to immunoblot analysis with anti-HSF1, anti-SQSTM1, anti-HSPA1, and anti-actin. (B) HeLa WT, HeLa HSF1 KO, and HSF1 KO exogenously expressing HSF1 (HeLa HSF1 KO + pEF1-HSF1) were treated with 10 µM MG132 for 12 h. Cell lysates were examined by immunoblot analysis. Band intensities were measured, and phosphorylated-SQSTM1 values were normalized to total SQSTM1. The data are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P < 0.01, **P < 0.05. (C) Colocalization of SQSTM1 with ubiquitinated inclusions were immunocytochemically assessed in cells treated with MG132. Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm.
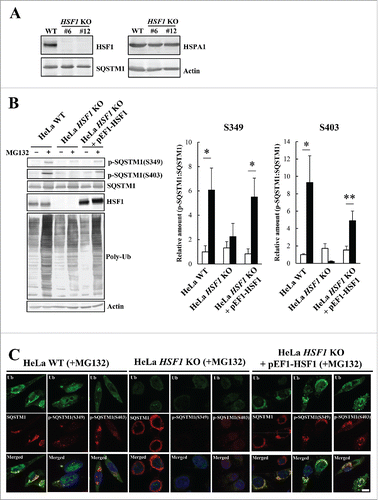
Next, we screened HSF1 KO cells immunocytochemically for inclusion formation by ubiquitinated proteins and for SQSTM1 localization and found no evidence for ubiquitin-positive inclusions (); this finding is consistent with the outcome of the inhibitor experiment. Curiously, SQSTM1 was localized to the perinuclear region when cells were treated with MG132 ( and Fig. S6). Perinuclear SQSTM1 was not detected by either of the anti-phospho SQSTM1 antibodies (), indicating that perinuclear SQSTM1 was unphosphorylated. We then determined whether SQSTM1-perinuclear localization in MG132-treated HSF1 KO cells could be prevented by KRIBB11 treatment (Fig. S6). We found that KRIBB11-treatment suppressed perinuclear localization of SQSTM1 in MG132-treated HSF1 KO cells (Fig. S6). KRIBB11 is known to inhibit HSF1-dependent recruitment of P-TEFb (positive transcription elongation factor b complex) to the HSPA1 promoter,Citation24 but our results here indicate it must have other properties. It has been reported that SQSTM1 shuttles continuously between nuclear and cytosolic compartments at a high rate;Citation25 therefore, SQSTM1 nuclear transport might be impaired in HSF1-deficient cells resulting in the perinuclear accumulation of SQSTM1.
HSF1 stress response pathway regulates autophagic clearance of protein aggregates
It was previously reported that SQSTM1 phosphorylation regulates selective autophagic clearance of ubiquitinated proteins, bacteria, and KEAP1.Citation9-12 We postulated that HSF1 might regulate this process via SQSTM1 phosphorylation. To test this proposal, we performed a metabolic analysis of EGFP-STAT5A(E18Δ) aggregates using the tetracycline (Tet)-On system. It was previously shown that EGFP-STAT5A(E18Δ) undergoes SQSTM1-independent aggresome formation and SQSTM1-dependent autophagic clearance.Citation18 Here, we generated EGFP-STAT5A(E18Δ) aggregates in HeLa TREx-EGFP-STAT5A(E18Δ) cells and placed the cells in tetracycline-free medium for 0, 12, or 24 h. We found that EGFP-STAT5A(E18Δ) aggregates were mostly eliminated within 24 h in control cells (); treatment with kinase inhibitors (rapamycin, CKI-7, and TBCA) or KRIBB11 delayed this elimination (). EGFP intensity was quantified by high-content image analysis using an IN Cell Analyzer 2200 to accurately measure the level of the remaining EGFP-STAT5A(E18Δ) aggregates. Higher EGFP-intensities were present in kinase inhibitor- or KRIBB11-treated cells after 24 h (). The average EGFP intensities in control, kinase inhibitor, and KRIBB11-treated cells after 24 h were 437 ± 424 (arbitrary units), 822 ± 1027, and 1814 ± 1138, respectively (Fig. S7A).
Figure 7. Reduction of EGFP-STAT5A(E18Δ) clearance by inhibition of SQSTM1 phosphorylation. (A) For formation of EGFP-STAT5A(E18Δ) aggregates, HeLa TREx-EGFP-STAT5A(E18Δ) cells were cultured in medium containing tetracycline and 2.5 µM MG132 plus mock, or kinase inhibitors (rapamycin, CKI-7, and TBCA), or KRIBB11. The medium was replaced with normal medium containing mock, or kinase inhibitors, or KRIBB11 and cells were fixed after 0, 12, or 24 h, followed by DAPI staining. The images were obtained by fluorescence microscopy. Scale bar: 100 μm. (B) EGFP-STAT5A(E18Δ) intensities were measured by an IN Cell Analyzer 2200. Values of EGFP intensity in individual cells were plotted on graphs (right), which show means ± SD. Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. *P< 0.01. Cell count (%) was plotted against EGFP-STAT5A(E18Δ) intensity (right). Upper, middle, and lower graphs show 0 h, 12 h, and 24 h, respectively. (C) Expression levels of ATG5 and ATG7 in HeLa_WT (open boxes), KRIBB11-treated HeLa (gray boxes), and HeLa HSF1 KO (closed boxes) cells were measured by real-time PCR. Relative expression values of target genes were normalized against the housekeeping gene GPD/G3PDH (glycerol-3-phosphate dehydrogenase) and are reported as means ± SD (n = 4). Statistical analyses were performed using one-way ANOVA, followed by the Tukey post-hoc test. (D) Autophagosome formation in HeLa_WT, kinase inhibitors (rapamycin, CKI-7, and TBCA) treated HeLa, KRIBB11-treated HeLa, and HeLa HSF1 KO cells was immunocytochemically examined using an anti-LC3 antibody. Cells were cultured in normal conditions (left panels), starvation conditions (middle panels), and with MG132 (right panels) for 5 h. Cell nuclei were counterstained blue with DAPI. Scale bar: 10 μm. a.u., arbitrary units.
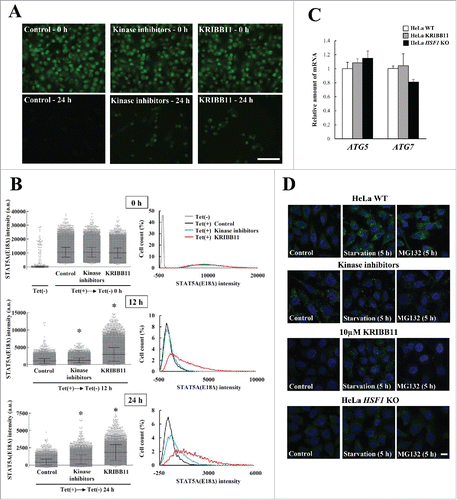
Our data raise the question of whether delayed elimination of protein aggregates is due to inhibition of proteasome activity or autophagy. We therefore examined the effects of impairment of HSF1-mediated stress responses on the proteasome and autophagy. Kinase inhibition and HSF1-deficiency did not affect proteasome activity (Fig. S7B and C). Meanwhile, MG132-induced autophagosome formation was not detected in KRIBB11-treated cells or HSF1 KO cells using immunostaining with an anti-LC3 antibody (). Moreover, LC3 conversion from LC3-I to LC3-II was monitored to assess autophagy flux. However, we could not measure it because combined treatment with MG132-bafilomycin A1 or epoxomicin-bafilomycin A1 to HeLa cells had an undesirable side effect (data not shown). We then performed an autophagy flux assay using an mCherry-EGFP-LC3 reporter system.Citation26 In HeLa-WT cells, MG132 treatment increased mCherry-positive and EGFP-negative LC3-puncta, whereas autophagy induction was inhibited by KRIBB11 treatment (Fig. S8). Similarly, MG132-induced autophagy flux was not detected in HSF1 KO cells (Fig. S8), indicating that the processes of autophagy are impaired in these cells. However, there was a possibility that this inhibition was due to reduced activity in the basic autophagic machinery since it has previously been reported that HSF1 regulates autophagy by directly binding to the ATG7 promoter to upregulate its expression.Citation17,27 We measured ATG7 and ATG5 mRNA expression in these cells and found that there was no significant change in the expression of either gene (). This finding was supported by the outcome of inducing autophagy in starving cells of amino acids: LC3 puncta appeared even in KRIBB11-treated or HSF1 KO cells ( and Fig. S8), indicating that the components of the basic autophagic machinery were not affected by an impairment of HSF1. Therefore, we concluded that HSF1 was specifically required for MG132-induced autophagy. However, triple kinase inhibitor treatment constitutively induced autophagosome formation after MTORC1 inhibition by rapamycin (). Triple kinase inhibitor treatment probably accelerated bulk-degradation by autophagy, and thereby mitigated against the accumulation of EGFP-STAT5A(E18Δ) aggregates compared with KRIBB11-treated cells ( and S7). Overall, HSF1 is a key player in triggering aggrephagy and the impairment of HSF1 causes a reduction in the clearance of protein aggregates.
Discussion
The phosphorylation of SQSTM1 at S349 and S403 is required for stable binding to substrates such as KEAP1 and ubiquitinated proteins.Citation11,12 Proteasome inhibition was found to be induced by phosphorylation of S349 or S403 in agreement with previous reports.Citation11,28 In the present study, we found that CSNK1 could directly phosphorylate SQSTM1 at S349; this novel observation adds to the previous reports that MTORC1, CSNK2, and TBK1 can phosphorylate SQSTM1.Citation9,11,12 There are no canonical consensus sequences for CSNK1 substrates, such as phospho-S/T-X-X-S/T or D/E-X-X-S/T, around the S349 phosphorylation site (342SSKEVDPSTGE352).Citation29 However, CSNK1 phosphorylates noncanonical sequences (S-L-S and K/R-X-K/R-X-X-S/T);Citation29,30 thus, S349 is a noncanonical targeting site for CSNK1. In addition to CSNK1, the specific CSNK2 inhibitor TBCA also suppressed phosphorylation; this effect was achieved indirectly according to the results of the in vitro phosphorylation assay. Importantly, phosphorylation of S349 of SQSTM1 was completely blocked by triple kinase inhibitor treatment (rapamycin, CKI-7, and TBCA), suggesting that these phosphorylation pathways are independent. Phosphorylation of S403 of SQSTM1 was blocked by treatment with the TBK1 inhibitor but not TBCA in HeLa cells, in agreement with a previous report by Pilli et al.Citation9 However, it has previously been reported that CSNK2 phosphorylates S403 of SQSTM1 in MG132-treated Neuro-2A and MEF cells.Citation11 Although we cannot deny the possibility that the S403-phosphorylation process is different among cell lines, it is essential to resolve this discrepancy. In the present study, we examined the phosphorylation status of S349 and S403 simultaneously and found that inhibition of phosphorylation of one site did not interfere with phosphorylation at the other.
We demonstrated here that HSF1 regulates SQSTM1 phosphorylation at both sites upstream of kinase activities. It remains unclear how HSF1 activates MTORC1, CSNK1, CSNK2, and TBK1. A genome-wide analysis shows that genes encoding the kinases are not directly driven by HSF1.Citation22 Also, we found that MTOR expression was not influenced by HSF1 deficiency. Thus, a further downstream factor(s) must be involved in the activation of the kinases. The HSPA1-cochaperone BAG3 gathers misfolded proteins into SQSTM1-containing aggregates, and its expression is mediated by HSF1.Citation31,32 This evidence leads us to speculate that the HSPA1-BAG3 complex may be involved in recruitment of kinases downstream of HSF1. Alternatively, the molecular chaperone HSP90AA may participate in this pathway. Previously, it has been reported that HSP90AA protects CSNK2 from aggregation and dramatically activates CSNK2.Citation33 In addition, TBK1 interacts with HSP90AA, resulting in its protection from proteasome-mediated degradation.Citation34 We speculate that the induction of HSP90AA expression causes an increase in CSNK2 activities and TBK1 levels and subsequent activation of SQSTM1 phosphorylation. Furthermore, pharmacologic inhibition of HSP90AA leads to a decrease in the MTOR-RPTOR/RAPTOR interaction,Citation35 suggesting that HSP90AA might also regulate MTORC1 activity under stress conditions.
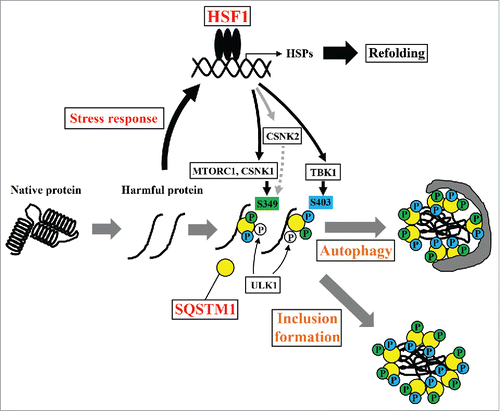
Our study highlighted a novel role for the HSF1 stress response pathway in SQSTM1 function; previously, this pathway was shown to be involved in transcriptional control of proteostasis components such as molecular chaperones.Citation36 SQSTM1 acts as a signaling hub of various transduction pathways and is involved in autophagic degradation and inclusion formation of harmful proteins.Citation7,14,37 Loss of SQSTM1 accelerates cell death that is induced by the overexpression of polyglutamine-expanded huntingtin,Citation38,39 suggesting a key role for SQSTM1 in proteostasis. In the present study, inhibition of SQSTM1 phosphorylation at S349 by kinase inhibitor treatment and loss of HSF1 function suppressed inclusion formation by ubiquitinated proteins and elimination of the aggregation-prone protein; these results indicate the necessity for S349 phosphorylation in SQSTM1 functions and a novel network of HSF1 stress response pathways in proteostasis. We propose multiple functions for HSF1 in proteostasis (). Proteotoxic stresses lead to the activation of HSF1 and the subsequent upregulation of HSP expression, resulting in protein refolding and protection from proteotoxicity. Additionally, HSF1 accelerates inclusion formation and elimination of harmful proteins via SQSTM1 activation that is mediated by several kinases such as MTORC1 and CSNK1. Protein inclusion formation, including the aggresome, is generally believed to be a cytoprotective process by which harmful proteins are compacted and directed toward autophagic clearance.Citation15 Therefore, the HSF1 stress response pathway is required for proteostasis systems. Although phosphorylation of SQSTM1 at S349 increases binding affinity for KEAP1,Citation12 it remains unclear whether this phosphorylation is also required for the direct or indirect interaction with ubiquitinated proteins. It was recently reported that N-terminally arginylated HSPA5/BiP (R-HSPA5), an endoplasmic reticulum chaperone, is a delivery determinant in SQSTM1-mediated autophagic clearance of harmful proteins.Citation40 R-HSPA5 is associated with misfolded proteins in the cytoplasm upon various stresses and binds to the ZZ domain of SQSTM1. Subsequently, this binding induces SQSTM1-oligomerization and increases the interaction between SQSTM1 and LC3.Citation40 This observation leads to speculate that phosphorylation of S349 of SQSTM1 may be involved in the interaction of SQSTM1 with the R-HSPA5-cargo complex. In the future, further structural and functional analyses will be necessary to elucidate the roles of this phosphorylation.
Figure 8. Schematic model of the proteostasis network via the HSF1-SQSTM1 axis. HSF1 is activated by the accumulation of harmful proteins, resulting in the induction of cytoprotective genes including HSPs. Moreover, phosphorylation of SQSTM1 by MTORC1, CSNK1, and TBK1 is also induced via activation of HSF1. Phosphorylated SQSTM1 accelerates inclusion formation and autophagic clearance of harmful proteins.
HSF1 loss does not directly affect the activity of proteasomes and the expression of the autophagy machinery. However, its loss does cause the impairment of the induction of autophagosome formation in proteasome-inhibited cells while starvation-induced autophagy is normal. These observations agree with the results obtained in SQSTM1 deficient cells, indicating that SQSTM1 phosphorylation is specifically required for aggrephagy induction. Itakura et al. report that SQSTM1 oligomerization is essential for its localization to the autophagosome formation site where SQSTM1 associates with upstream autophagy factors such as ULK1 and VMP1.Citation41 Moreover, the interaction of SQSTM1 with LC3 is promoted by SQSTM1 oligomerization.Citation42 Thus, the initiation of aggrephagy might be triggered by SQSTM1 oligomerization and localization to the autophagosome formation site after phosphorylation and substrate capturing of SQSTM1. Other receptor proteins such as OPTN (optineurin) and CALCOCO2/NDP52 have recently been shown to have an important role in the recruitment of the upstream autophagy machinery including ULK1, ZFYVE1/DFCP1, and WIPI1 for selective mitochondrial autophagy.Citation43 It has been reported that ULK1 phosphorylates SQSTM1 at S407.Citation10 Based on these data, we suggest that SQSTM1 oligomerization might be required for the assembly of upstream components for autophagosome nucleation upon aggrephagy induction.
Our analyses showed that the generation of various protein aggregates caused SQSTM1 phosphorylation. Accumulation of ubiquitinated proteins and the aggregation-prone protein induced phosphorylation of SQSTM1 at both S349 and S403, whereas SNCA aggregates and oligomers enhanced its phosphorylation only at S349. This result is in agreement with a pathological analysis of α-synucleinopathies including dementia with Lewy bodies and multiple system atrophy.Citation44 SNCA-positive inclusions are colocalized with SQSTM1 in brains of patients with Parkinson disease, dementia with Lewy bodies, and multiple system atrophy,Citation45 while they are not detected by an anti-phospho-SQSTM1 (S403) antibody.Citation44 Although it is unclear whether this result can be attributed to phosphorylation or dephosphorylation, the phosphorylation status of S403 may be susceptible to the properties of protein aggregates. This puzzling aspect of SQSTM1 phosphorylation might be implicated in the pathogenic mechanism of α-synucleinopathies, and thus further studies will be necessary to determine whether this is the case.
Materials and methods
Animals
C57BL/6J mice were maintained on a 12-h light/dark cycle at 25°C with ad libitum food and water. All experimental designs and procedures were approved by the Committee for Animal Research, Kyoto Prefectural University of Medicine (M23-241), Kyoto Japan, and followed the guidelines of the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Reagents
The following reagents were purchased: MG132 (Peptide Institute, 3175-v), rapamycin (Santa Cruz Biotechnology, sc-3504), and KRIBB11 (Calbiochem, 385570), casein kinase inhibitor CKI-7 (Seikagaku Kogyo; out of production), TBCA (Cayman Chemical, 14831), and BX795 (ChemScene, CS-0259).
Cells
HeLa and HEK293 cells were grown in DMEM medium (Nacalai Tesque, 08458-16) supplemented with 10% fetal bovine serum. Transfection of plasmids DNA was performed using Lipofectamine 2000 (Invitrogen, 11668027) or a CUY21Pro-Vitro electroporator (NEPA GENE, Ichikawa, Japan). Cells were cultured in Hank's balanced salt solution (Nissui, 05905) for observation of amino acid starvation-induced autophagy.
Antibodies
The complete list of antibodies and sources used is given in .
Table 1. Antibodies.
Immunoblotting analysis
Cells were washed with PBS (137 mM NaCl, 8.1 mM Na2HPO4, 2.68 mM KCl, 1.47 mM KH2PO4) and extracted with ice-cold PBS containing 0.5% Triton X-100 (Wako Pure Chemical Industries, 168-11805) and a protease inhibitor cocktail (Nacalai Tesque, 25955-11). Protein concentrations in samples were measured using the Lowry protein assay, followed by addition of Laemmli SDS sample buffer. For immunoblotting analysis, samples were separated on 10% or 15% SDS-polyacrylamide gels using a Tris–glycine buffer system (25 mM Tris, 192 mM glycine, 0.1% SDS). After electrophoresis, the separated proteins were transferred to Immobilon-P membranes (Merck Millipore, IPVH00010). The membranes were blocked with 10% skimmed milk, 5% bovine serum albumin (Nacalai Tesque, 01860-36), or Blocking Solution (Nacalai Tesque, 05151-35) for 1 h, and then incubated with the primary antibodies for 12 h at 25°C. The membranes were then washed with PBS containing 0.1% Tween 20 (Nacalai Tesque, 35624-15) and incubated with alkaline phosphatase-conjugated anti-rabbit IgG (1:4000 dilution; Jackson ImmunoResearch Laboratories Inc., 111-055-144) or anti-mouse IgG (1:2000 dilution; Jackson ImmunoResearch Laboratories Inc., 112-055-167) for 1 h. Immunopositive signals were detected with nitroblue tetrazolium chloride (Nacalai Tesque, 24720-01) and 5-bromo-4-chloro-3-indolylphosphate P-toluidine salt (Nacalai Tesque, 05643-11) reagents.
Drug treatment and immunocytochemistry
HeLa cells were treated with 10 µM MG132 for 12 h for proteasome inhibition. The kinase inhibitors rapamycin (1 µM), CKI-7 (100 µM) and TBCA (25 µM) were added at the same time as MG132 for 12 h. For inhibition of HSF1, HeLa cells were treated with 10 µM KRIBB11 just after pEGFP-STAT5A(E18Δ) transfection or MG132 treatment.Citation46
Immunocytochemical analysis was performed as described previously.Citation18 Cells were fixed with 4% paraformaldehyde (Merck Millipore, 104005) after drug treatment. The fixed cells were permeabilized in PBS containing 0.1% Triton X-100 or 0.01% digitonin (Nacalai Tesque, 123-33) and placed in blocking solution for 30 min. Samples were incubated with the primary antibodies for 12 h at 25°C. After a PBS wash, samples were further incubated with Alexa Fluor 488-labeled goat anti-mouse IgG (Invitrogen, A11029) or Alexa Fluor 594-labeled goat anti-rabbit IgG (Invitrogen, A11037) for 4 h at 25°C. After washing, they were stained with 4′,6-diamidino-2-phenylindole (Dojindo, D523). The cells on cover glasses were mounted on glass slides in an aqueous mounting medium (FluorSave Reagent; Merck Millipore, 345789) and examined under a confocal laser microscope. Confocal laser scanned images were obtained with an LSM 510 META microscope (Carl Zeiss, Oberkochen, Germany). Alexa-Fluor-488 and EGFP were excited using a 488 nm argon laser, and emission was recorded through a 500- to 530-nm band-pass filter. Alexa Fluor 594 was excited with a 543-nm helium-neon laser, and emission was recorded through an LP 560 nm filter. DAPI was excited at 800 nm, with a Mai Tai 2-photon laser (Spectra-Physics, Santa Clara, CA, USA), and fluorescence was recorded through a 390- to 465-nm band-pass filter. Images were obtained through a Plan-Apochromat 1006/1.4 Oil DIC lens (Carl Zeiss). We adjusted the detector gain and laser power settings to avoid oversaturation of SQSTM1-aggegate images. We used lower than usual settings for laser power/gain because the immunofluorescence signal from SQSTM1 aggregates was very bright.
Immunohistochemistry
Under deep anesthesia, mice were intracardially perfused with 0.1 M PBS (0.1 M phosphate buffer, pH 7.4, 0.9% NaCl) followed by 4% paraformaldehyde in 0.1 M PB (0.1 M phosphate buffer, pH 7.4). Brains were dissected into blocks including the cerebral cortex and hippocampus, and post-fixed with the same fixative for 12 h at 4°C. Coronal sections (30 µm) were obtained using a vibratome (DSK, Kyoto, Japan). Sections were permeabilized with 0.1% Triton X-100 in PBS for 30 min, and blocked with 5% normal goat serum in PBS for 1 h. Sections were then treated with primary and secondary antibodies following the immunocytochemical procedures described above. After treatment, the sections were washed with PBS and then with 20 mM Tris-HCl buffer, and mounted in FluorSave. Images were obtained as Z stacks (10–20 z-sections, 1 µm apart, 1024 × 1024 pixels) using an LSM 510 META microscope.
λ protein phosphatase treatment
HeLa cells were treated with 10 µM MG132 for 12 h and harvested in PBS using a cell scraper. Cells were homogenized by sonication, and the insoluble fraction was recovered (20,000 × g, 15 min, 4°C) and resuspended in 1× PMP buffer including 1 mM MnCl2 (New England Biolabs, P0753S). Samples were incubated with 400 units of λ protein phosphatase (New England Biolabs, P0753S) at 37°C for 1 h. SQSTM1 immunoprecipitates were treated with CSNK1 and λ protein phosphatase at 37°C for 1 h in CSNK1 reaction buffer including 1 mM ATP and 1 mM MnCl2.
Cell culture and mouse models of α-synucleinopathy
SNCA fibrils were prepared as described previously.Citation19,47,48 pcDNA-SNCA plasmidCitation49 was transiently transfected into HEK293 cells plated onto 24-well plates, and 4 µg SNCA fibrils were introduced on the following day using Lipofectamine 2000 reagent. Cells were incubated in DMEM medium supplemented with 10% fetal bovine serum for 24 h, and used for immunoblotting analysis and immunocytochemistry.
The α-synucleinopathy mouse model was generated as described previously.Citation21 SNCA fibrils (5 µg/µl) were sonicated and microinjected (1 µl/site) into the striatum using a 30-gauge Hamilton syringe (Hamilton, Reno, NV, USA) at a rate of 0.25 µl/min. Likewise, PBS was microinjected into the striatum of control mice. The coordinates for the striatum were: AP, +0.2 mm from the bregma; ML, ± 2 .0 mm from the midline; and DV, −3 mm below the skull surface. Mice were used for immunohistochemistry 6 mo after injection.
In vitro phosphorylation assay
For the SQSTM1 phosphorylation analysis after CSNK1 and CSNK2 treatment, SQSTM1 WT, SQSTM1S349A, or SQSTM1S403A was expressed in HEK293 SQSTM1 KO cells. The proteins were purified by immunoprecipitation with an anti-SQSTM1 antibody and protein G Mag Sepharose (GE Healthcare, 28951379). A reaction buffer containing 1 mM ATP was added to immunoprecipitation products, and then the mixtures were incubated with or without 1000 units of CSNK1 (New England Biolabs, P6030S) or 500 units of CSNK2 (New England Biolabs, P6010S) for 1 h at 37°C. The kinase reaction was stopped by adding 3 × Laemmli SDS sample buffer. Phosphorylation at S349 or S403 was assessed by immunoblotting analysis using anti-phospho-SQSTM1 (Ser351) and anti-phospho-SQSTM1 (Ser403) antibodies.
Plasmid construction
For construction of the HSF1 expression vector (pEF1-HSF1), human HSF1 cDNA was cloned into pEF1-mycHisB (ThermoFisher, V92120) using reverse transcriptase-polymerase chain reaction (GeneAtlas322; ASTEC, Fukuoka, Japan) with the primers 5′-CGCCACCATGGATCTGCCCGTGGGCCC-3′/5′-GATATCCTAGGAGACAGTGGGGTCC-3′. pEF1-SQSTM1-WT plasmid was constructed using the primers 5′-CCCGCTGATCAGCCTCGAC-3′ /5′-TCACAACGGCGGGGGATGCTTTG-3′ and pEF1-SQSTM1-mycHisCitation18 as the template. The expression vectors for the S349A and S403A mutants of SQSTM1 were generated using the primers 5′-AAGTGGACCCGGCCACAGGTGAACTCC-3′/5′-TCTTTTGAAGACAGATGGGTCC-3′ and 5′-GCCATGGGCTTCTCTGATGA-3′/5′-CAGCATCTGGGAGAGGGACT-3′, respectively, using a KOD-Plus-Mutagenesis kit (Toyobo, SMK-101).
Gene targeting using CRISPR/Cas9 system
We designed target sequences for SQSTM1 and HSF1, and cloned double-strand oligonucleotides for SQSTM1 (5′-AAAGAAGTGGACCCGTCTACGTTTT-3′/5′-GTAGACGGGTCCACTTCTTTCGGTG-3′) and HSF1 (5′-CAGCTTCCACGTGTTCGACCGTTTT-3′/5′-GGTCGAACACGTGGAAGCTGCGGTG-3′) into a GeneArt CRISPR Nuclease vector (ThermoFisher, A21175) according to the manufacturer's instructions. Inserts were confirmed by DNA sequencing analysis. The constructs were transfected into HeLa and HEK293 cells using an electroporator. Targeted clones were obtained by limiting dilution and confirmed by immunoblotting analysis. Target genes of individual clones were amplified by PCR, followed by cloning and sequencing. The SQSTM1 KO_HEK293 clone has 16-base pair (bp) (1039 to 1054 bp) and 5-bp (1046 to 1050 bp) deletions within exon 7 of the SQSTM1 gene. The SQSTM1 KO_HeLa clone has a 1-bp insertion (T) at the 36-bp (1014 to 1049 bp) deletion site within exon 7 of the SQSTM1 gene. The HSF1 KO_HeLa clone has a 10-bp insertion (CCTTGGCAAA) at the 14-bp (131 to 144 bp) deletion site and a 13-bp insertion (TGGAAGTTTGCCA) at the 9-bp (143 to 151 bp) deletion site within exon 2 of the HSF1 gene. Although the copy numbers at the SQSTM1 and HSF1 gene loci in our HeLa cells are unknown, the intact DNA sequence of these genes was never detected.
HSF1 affinity isolation assay
The HSF1 affinity isolation assay was performed as described previously.Citation23 HeLa cells were treated with 10 µM MG132 for 1 to 12 h at 37°C or heat shocked for 1 h at 42°C. Cells were lysed in ice-cold lysis buffer (25 mM HEPES, pH 7.4, 100 mM NaCl, 5 mM EDTA, 0.5% Triton X-100, and protease inhibitor cocktail). Cell extracts were incubated with 1 μg of biotinylated HSE double strand oligonucleotide (5′-biotin-AACGAGAATCTTCGAGAATGGCT-3′ and 5′-AGCCATTCTCGAAGATTCTCGTT-3′) for 30 min at room temperature. The oligonucleotides were precipitated with Streptavidin MagneSphere Paramagnetic Particles (Promega, Z5481) for 1 h at 4°C. HSF1 pulled down with HSE-oligonucleotides was quantified by an immunoblotting analysis using an anti-HSF1 antibody.
High-content analysis using IN Cell Analyzer
We used the Tet-On system in EGFP-STAT5A(E18Δ) cells for analysis of metabolism. The Tet-On construct, pcDNA5-EGFP-STAT5A(E18Δ), was made by inserting a pEGFP-STAT5A(E18Δ) fragment (BglII-SspI) into the BamHI-EcoRV site of pcDNA5 FRT-TO (Invitrogen, V652020). HeLa cells were transfected with pcDNA5-EGFP-STAT5A(E18Δ) and pcDNA6/TR (Invitrogen, V102520), and selected with 40 µg/ml blasticidin (InvivoGen, ant-bl-1) and 200 µg/ml hygromycin B (Calbiochem, 400052) for 2 wk. Single clones (HeLa TREx-EGFP-STAT5A[E18Δ]) were obtained by limiting dilution and confirmed by immunoblotting analysis.
HeLa TREx-EGFP-STAT5A(E18Δ) cells (4 × 104) were plated into 96-well plates and incubated in a CO2 incubator for 18 h (10 wells/group). For formation of EGFP-STAT5A(E18Δ) aggregates, cells were treated with tetracycline and 2.5 µM MG132 for 12 h. Concurrently, cells were treated with 5 µM KRIBB11 or kinase inhibitors (1 µM rapamycin, 100 µM CKI-7, and 25 µM TBCA). Cells were washed 3 times with fresh medium, and then continuously incubated in fresh medium containing 5 µM KRIBB11 or kinase inhibitors (1 µM rapamycin, 100 µM CKI-7, and 25 µM TBCA). After 0, 12, or 24 h, cells were fixed with 4% paraformaldehyde for 15 min and then stained with DAPI. Plates were set in the IN Cell Analyzer 2200 (GE Healthcare, Chicago, IL, USA), and cell images were acquired using a ×10 objective lens (3 fields/well). Excitation and emission wavelengths for DAPI were 390 nm/435 nm, and for EGFP were 475 nm/525 nm. The EGFP intensity in each cell was determined by IN Cell Investigator 1.6 software. For cell segmentation, we chose the collar method and defined a 3 µm collar radius as the cell area. The EGFP intensity per cell was measured and plotted in a graph (n ≥ 5000).
Autophagy flux assay
pBABE-puro mCherry-EGFP-LC3B was a gift from Dr. Jayanta Debnath (Addgene, 22418).Citation50 A DNA region encoding mCherry-EGFP-LC3B was cloned into the pEF1-mycHisB vector (pEF1-mCherry-EGFP-LC3B). HeLa WT or HSF1 KO cells were stably transfected with pEF1-mCherry-EGFP-LC3B, followed by G418 selection. Clones were isolated and used for the autophagy flux assay. Cell images were chosen randomly, and the number of mCherry-only LC3 puncta and mCherry-positive EGFP-positive puncta in individual cells (30 cells) was measured manually.
Statistical analyses
Statistical analyses were conducted using Kaleida Graph 4.1 and GraphPad Prism 5J. Differences between treatments were analyzed for significance using the Student t test or one-way ANOVA with the Tukey post hoc test. Significance was set as ≤0.05. Values in graphs are presented as means ± SD. All experiments were repeated 4 times.
Abbreviations
| CSNK2/CK2 | = | casein kinase 2 |
| CSNK1/CK1 | = | casein kinase 1 |
| GPD/G3PDH | = | glycerol-3-phosphate dehydrogenase |
| HSF1 | = | heat shock transcription factor 1 |
| HSE | = | heat shock element |
| HSPs | = | heat shock proteins |
| KEAP1 | = | Kelch-like ECH associated protein 1 |
| KIR | = | KEAP1-interaction region |
| LIR | = | LC3-interacting region |
| MTORC1 | = | mechanistic target of rapamycin (serine/threonine kinase) complex 1 |
| PB1 | = | (Phox and Bem1) domain |
| proteostasis | = | protein homeostasis |
| SGK | = | serum- and glucocorticoid-inducible kinase |
| TBK1 | = | TANK binding kinase 1 |
| UBA | = | ubiquitin associated |
| UPS | = | ubiquitin-proteasome system |
| ZZ | = | zinc finger |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KAUP_A_1248018_Supplementary_material.zip
Download Zip (1.4 MB)Acknowledgments
We thank Dr. Jayanta Debnath (University of California at San Francisco) for the pBABE-puro mCherry-EGFP-LC3B plasmid.
Funding
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (YW: 15K09320 and 24591272, and MT: 25290014).
Related Research Data
References
- Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem 2015; 84:435-64; PMID:25784053; http://dx.doi.org/10.1146/annurev-biochem-060614-033955
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 1998; 12:3788-96; PMID:9869631; http://dx.doi.org/10.1101/gad.12.24.3788
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780-6; PMID:17051204; http://dx.doi.org/10.1038/nature05291
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID:21189453; http://dx.doi.org/10.4161/auto.7.3.14487
- Knaevelsrud H, Simonsen A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett 2010; 584:2635-45; PMID:20412801; http://dx.doi.org/10.1016/j.febslet.2010.04.041
- Wild P, McEwan DG, Dikic I. The LC3 interactome at a glance. J Cell Sci 2014; 127:3-9; PMID:24345374; http://dx.doi.org/10.1242/jcs.140426
- Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett 2010; 584:1374-8; PMID:20153326; http://dx.doi.org/10.1016/j.febslet.2010.02.017
- Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 2015; 282(24):4672-8; PMID:26432171; http://dx.doi.org/10.1111/febs.13540
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012; 37:223-34; PMID:22921120; http://dx.doi.org/10.1016/j.immuni.2012.04.015
- Lim J, Lachenmayer ML, Wu S, Liu W, Kundu M, Wang R, Komatsu M, Oh YJ, Zhao Y, Yue Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet 2015; 11:e1004987; PMID:25723488; http://dx.doi.org/10.1371/journal.pgen.1004987
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44:279-89; PMID:22017874; http://dx.doi.org/10.1016/j.molcel.2011.07.039
- Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013; 51:618-31; PMID:24011591; http://dx.doi.org/10.1016/j.molcel.2013.08.003
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/10.1016/j.cell.2007.10.035
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517-27; PMID:19250912; http://dx.doi.org/10.1016/j.molcel.2009.01.021
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 2000; 10:524-30; PMID:11121744; http://dx.doi.org/10.1016/S0962-8924(00)01852-3
- Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang Z, et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem 2013; 288:859-72; PMID:23184933; http://dx.doi.org/10.1074/jbc.M112.412783
- Luo T, Fu J, Xu A, Su B, Ren Y, Li N, Zhu J, Zhao X, Dai R, Cao J, et al. PSMD10/Gankyrin induces autophagy to promote tumor progression through cytoplasmic interaction with ATG7 and nuclear transactivation of ATG7 expression. Autophagy 2016; 12:1355-71; PMID:25905985; http://dx.doi.org/10.1080/15548627.2015.1034405
- Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci 2011; 124:2692-701; PMID:21771882; http://dx.doi.org/10.1242/jcs.081232
- Watanabe Y, Tatebe H, Taguchi K, Endo Y, Tokuda T, Mizuno T, Nakagawa M, Tanaka M. p62/SQSTM1-dependent autophagy of Lewy body-like α-synuclein inclusions. PLoS One 2012; 7:e52868; PMID:23300799; http://dx.doi.org/10.1371/journal.pone.0052868
- Yamakawa K, Izumi Y, Takeuchi H, Yamamoto N, Kume T, Akaike A, Takahashi R, Shimohama S, Sawada H. Dopamine facilitates α-synuclein oligomerization in human neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun 2010; 391:129-34; PMID:19900407; http://dx.doi.org/10.1016/j.bbrc.2009.11.015
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338:949-53; PMID:23161999; http://dx.doi.org/10.1126/science.1227157
- Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell 2004; 15:1254-61; PMID:14668476; http://dx.doi.org/10.1091/mbc.E03-10-0738
- Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009; 323:1063-6; PMID:19229036; http://dx.doi.org/10.1126/science.1165946
- Yoon YJ, Kim JA, Shin KD, Shin DS, Han YM, Lee YJ, Lee JS, Kwon BM, Han DC. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J Biol Chem 2011; 286:1737-47; PMID:21078672; http://dx.doi.org/10.1074/jbc.M110.179440
- Pankiv S, Lamark T, Bruun JA, Overvatn A, Bjorkoy G, Johansen T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J Biol Chem 2010; 285:5941-53; PMID:20018885; http://dx.doi.org/10.1074/jbc.M109.039925
- Murrow L, Malhotra R, Debnath J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat Cell Biol 2015; 17:300-10; PMID:25686249; http://dx.doi.org/10.1038/ncb3112
- Desai S, Liu Z, Yao J, Patel N, Chen J, Wu Y, Ahn EE, Fodstad O, Tan M. Heat shock factor 1 (HSF1) controls chemoresistance and autophagy through transcriptional regulation of autophagy-related protein 7 (ATG7). J Biol Chem 2013; 288:9165-76; PMID:23386620; http://dx.doi.org/10.1074/jbc.M112.422071
- Kageyama S, Sou YS, Uemura T, Kametaka S, Saito T, Ishimura R, Kouno T, Bedford L, Mayer RJ, Lee MS, et al. Proteasome dysfunction activates autophagy and the Keap1-Nrf2 pathway. J Biol Chem 2014; 289:24944-55; PMID:25049227; http://dx.doi.org/10.1074/jbc.M114.580357
- Marin O, Bustos VH, Cesaro L, Meggio F, Pagano MA, Antonelli M, Allende CC, Pinna LA, Allende JE. A noncanonical sequence phosphorylated by casein kinase 1 in β-catenin may play a role in casein kinase 1 targeting of important signaling proteins. Proc Natl Acad Sci U S A 2003; 100:10193-200; PMID:12925738; http://dx.doi.org/10.1073/pnas.1733909100
- Kawakami F, Suzuki K, Ohtsuki K. A novel consensus phosphorylation motif in sulfatide- and cholesterol-3-sulfate-binding protein substrates for CK1 in vitro. Biol Pharm Bull 2008; 31:193-200; PMID:18239272; http://dx.doi.org/10.1248/bpb.31.193
- Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ 2013; 20:21-30; PMID:22722335; http://dx.doi.org/10.1038/cdd.2012.72
- Jacobs AT, Marnett LJ. HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J Biol Chem 2009; 284:9176-83; PMID:19179333; http://dx.doi.org/10.1074/jbc.M808656200
- Miyata Y, Yahara I. The 90-kDa heat shock protein, HSP90, binds and protects casein kinase II from self-aggregation and enhances its kinase activity. J Biol Chem 1992; 267:7042-7; PMID:1551911
- Yang K, Shi H, Qi R, Sun S, Tang Y, Zhang B, Wang C. Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol Biol Cell 2006; 17:1461-71; PMID:16394098; http://dx.doi.org/10.1091/mbc.E05-09-0853
- Delgoffe GM, Kole TP, Cotter RJ, Powell JD. Enhanced interaction between Hsp90 and raptor regulates mTOR signaling upon T cell activation. Mol Immunol 2009; 46:2694-8; PMID:19586661; http://dx.doi.org/10.1016/j.molimm.2009.05.185
- Gidalevitz T, Prahlad V, Morimoto RI. The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb Perspect Biol 2011; 3; PMID:21536706; http://dx.doi.org/10.1101/cshperspect.a009704
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkø;y G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/10.1074/jbc.M702824200
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/10.1083/jcb.200507002
- Saitoh Y, Fujikake N, Okamoto Y, Popiel HA, Hatanaka Y, Ueyama M, Suzuki M, Gaumer S, Murata M, Wada K, Nagai Y, et al. p62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies. J Biol Chem 2015; 290:1442-53; PMID:25480790; http://dx.doi.org/10.1074/jbc.M114.590281
- Cha-Molstad H, Sung KS, Hwang J, Kim KA, Yu JE, Yoo YD, Jang JM, Han DH, Molstad M, Kim JG, et al. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol 2015; 17:917-29; PMID:26075355; http://dx.doi.org/10.1038/ncb3177
- Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol 2011; 192:17-27; PMID:21220506; http://dx.doi.org/10.1083/jcb.201009067
- Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, Martens S. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife 2015; 4:e08941; PMID:26413874; http://dx.doi.org/10.7554/eLife.08941
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015; 524:309-14; PMID:26266977; http://dx.doi.org/10.1038/nature14893
- Kurosawa M, Matsumoto G, Sumikura H, Hatsuta H, Murayama S, Sakurai T, Shimogori T, Hattori N, Nukina N. Serine 403-phosphorylated p62/SQSTM1 immunoreactivity in inclusions of neurodegenerative diseases. Neurosci Res 2016; 103:64–70; PMID:26302676; http://dx.doi.org/10.1016/j.neures.2015.08.002
- Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 2001; 12:2085-90; PMID:11447312; http://dx.doi.org/10.1097/00001756-200107200-00009
- Watanabe Y, Ikegawa M, Naruse Y, Tanaka M. A novel splicing variant form suppresses the activity of full-length signal transducer and activator of transcription 5A. FEBS J 2009; 276:6312-23; PMID:19788420; http://dx.doi.org/10.1111/j.1742-4658.2009.07339.x
- Tsujimura A, Taguchi K, Watanabe Y, Tatebe H, Tokuda T, Mizuno T, Tanaka M. Lysosomal enzyme cathepsin B enhances the aggregate forming activity of exogenous α-synuclein fibrils. Neurobiol Dis 2014; 73C:244-53
- Taguchi K, Watanabe Y, Tsujimura A, Tatebe H, Miyata S, Tokuda T, Mizuno T, Tanaka M. Differential expression of α-synuclein in hippocampal neurons. PLoS One 2014; 9:e89327; PMID:24586691; http://dx.doi.org/10.1371/journal.pone.0089327
- Tatebe H, Watanabe Y, Kasai T, Mizuno T, Nakagawa M, Tanaka M, Tokuda T. Extracellular neurosin degrades α-synuclein in cultured cells. Neurosci Res 2010; 67:341-6; PMID:20403393; http://dx.doi.org/10.1016/j.neures.2010.04.008
- N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 2009; 10:173-9; PMID:19148225; http://dx.doi.org/10.1038/embor.2008.238