
ABSTRACT
Autophagy and apoptosis are 2 fundamental biological mechanisms that may cooperate or be antagonistic, although both are involved in deciding the fate of cells in physiological or pathological conditions. These 2 mechanisms coexist simultaneously in cells and share common upstream signals and stimuli. Autophagy and apoptosis play pivotal roles in cancer development. Autophagy plays a key function in maintaining tumor cell survival by providing energy during unfavorable metabolic conditions through its recycling mechanism, and supporting the high energy requirement for metabolism and growth. This review focuses on gastrointestinal stromal tumors and cell death through autophagy and apoptosis, taking into account the involvement of both of these processes in tumor development and growth and as mechanisms of drug resistance. We also focus on the crosstalk between autophagy and apoptosis as an emerging field with major implications for the development of novel therapeutic options.
Introduction
Autophagy, or ‘self-eating’, and apoptosis or ‘self-killing’ are 2 distinct mechanisms that may cooperate or be antagonist in cells, although both are involved in deciding cell fate. Autophagy plays a dual role in cells, acting either as a prosurvival or prodeath mechanism, whereas apoptosis is the best described form of programmed cell death, determining exclusively cell death.Citation1 Autophagy is a dynamic, evolutionarily conserved and tightly regulated strategy developed by cells to degrade long-lived or damaged cytosolic proteins and organelles, providing energy and nutrients.Citation2,3 Autophagy is typically a stress adaptation mechanism in response to a wide variety of stimuli, including starvation, hypoxia, endoplasmic reticulum stress, and oxidative stress, prompting a prosurvival pathway. Indeed, stress above a certain threshold may promote nonapoptotic autophagic forms of cell death. Autophagy is principally cytoprotective, defending cells from dysfunctional organelles and protein aggregates or functioning as a recycling process. However, excessive or persistent autophagy can also lead to cytotoxicity, promoting cell death rather than survival.Citation4 In some instances, the 2 mechanisms are observed to coexist simultaneously in cells as the result of shared common upstream signals and stimuli.
The term ‘autophagy’ was first used in 1963 by de Duve and Wattiaux to describe a process in mammals that leads to the engulfment of cytoplasmic material that is subsequently degraded.Citation5,6 To date, 3 distinct primary mechanisms of autophagy have been distinguished according to the route of delivery of the cytoplasmic material to the lysosomes and the substrates: microautophagy, chaperone-mediated autophagy and macroautophagy. Chaperone-mediated autophagy and microautophagy involve the direct uptake into lysosomes of unfolded individual soluble proteins, and the nonspecific engulfment of cytoplasm, which may include cell organelles, respectively.Citation7 In macroautophagy, hereafter referred to as autophagy, portions of cytoplasm and intracellular organelles are sequestered in double-membrane bound compartments, named phagophores, that expand and mature into autophagosomes, which ultimately are delivered to lysosomes for degradation.
Apoptosis is an evolutionarily conserved process, which is essential for organism development, growth and tissue homeostasis.Citation8 Apoptosis leads to the prompt destruction of cellular structures and organelles and culminates with cellular shrinkage and apoptotic body formation that avoids leakage of cellular contents, preventing inflammation and tissue destruction. The apoptotic cascade may be activated through 2 main pathways: the extrinsic pathway, involving stimulation of members of the tumor necrosis factor receptor superfamily, and the intrinsic pathway, which involves mitochondria.
Apoptosis and autophagy in cancer
Apoptosis and autophagy are complex and interconnected cellular processes that play a major role in determining cellular fate. These 2 processes are important in maintaining cellular homeostasis and play critical functions in cancer development and progression. Evasion from apoptosis is one of the essential changes in a cell that drives its malignant transformation. Apoptosis was originally thought to be the only form of programmed cell death that, once perturbed, promotes tumorigenesis and impairs chemotherapy.Citation9,10 Autophagy was initially considered a housekeeping process for degrading and recycling long-lived proteins, cellular aggregates and damaged organelles, and has now emerged as having a paradoxical role in cancer, acting as both tumor promoter and tumor-suppressor, by blocking inflammation and necrosis and maintaining genomic stability.Citation11-13 Currently, this dual role is not fully understood and the underlying mechanism has not been clarified. However, the consensus appears to be that autophagy suppresses tumor initiation and reduces genomic instability, but promotes the survival of established tumors.Citation13,14 Furthermore, although the exact mechanisms are not totally known, it is thought that the dual role of autophagy in cancer is context-dependent.Citation11 Autophagy is regulated approximately by more than 30 autophagy-related (ATG) genes in mammals and is finely regulated by epigenetic processes, including methylation and miRNA deregulation.Citation15,16 This process serves as an alternative energy source to maintain cell homeostasis and viability during metabolic stress or stimuli. Thus, autophagy is thought to sustain the cell's energetic needs during hypoxia, and in this context has emerged as a new player in the progression and expression of a more aggressive phenotype in hypoxic solid tumors.Citation13,17 In general, autophagy plays a key role in maintaining tumor cell survival by providing energy during unfavorable metabolic conditions through its recycling mechanism, and supporting the high requirement of cancer cells for metabolism and growth. This mechanism is particularly advantageous for cells located far from blood vessels and experiencing hypoxia and paucity of nutrients.Citation18
Apoptosis guarantees the integrity of many processes, including tissue development and appropriate growth. During cancer development, apoptosis is one of the major barriers that have to be overcome. Cancer cells, in addition to their limitless replicative potential, are further strengthened by evading apoptosis, which facilitates their survival, proliferation and drug resistance.Citation19-21 Several mechanisms have been identified for cancer cell evasion of apoptosis. One of the most common mechanisms is the loss of pro-apoptotic regulators, for example through mutations in tumor suppressor genes, including TP53. Another mechanism involves aberrant expression of survival proteins, including BCL2, heat shock proteins, and inhibitor of apoptosis (IAP) family of proteins. It is likely that each tumor develops its own specific strategies to escape from cell death and promote resistance to anticancer therapies.Citation20
Autophagy and apoptosis are linked by a complex interrelationship that is critical in dictating cell fate. Autophagy occurs in response to stress or damage; however, when cellular damage is too extensive, apoptosis is initiated in order to eliminate cells. In cells with flawed apoptosis, autophagy may serve as a backup strategy to stop cell processes or a strategy to promote other cell death mechanisms, possibly including necrosis. The regulators of both apoptosis and autophagy thus interact with each other to determine the appropriate cellular consequence resulting from stress and damage.
The autophagic self-cannibalization mechanism currently represents a hot topic in cancer research, given its double-edged sword role both in cancer and cell death, and is an issue of great debate. Studies are accumulating on the importance of autophagy and apoptosis in cancer development; in particular, major components of the pathways regulating both processes are constitutively overexpressed or mutated in several solid tumors.Citation22 Most human cancers have either mutations in TP53 or defects in other genes of the TP53 pathway, and trp53-null mice are highly prone to tumorigenesis.Citation23 In addition, most anticancer treatments work by triggering the cell to undergo apoptosis via a mechanism often mediated by TP53. Thus, it has been postulated that cells resistant to apoptosis, through such means as overexpression of the anti-apoptotic protein BCL2 or TP53 mutation, will also be resistant to chemotherapy and radiotherapy.Citation24-26 However, whereas apoptosis is the primary mechanism for cell killing by DNA-damaging anticancer agents in some lymphomas, this is not the case in most epithelial tumors, and changing the sensitivity to apoptosis does not alter the overall level of cell death.Citation27-29 Autophagy can also be considered a tumor-inhibiting process, as activation of many oncoproteins, including MTOR, AKT1, and BCL2, results in autophagy inhibition.Citation30,31 Conversely, with an upregulation of autophagy, cells have increased metabolism and stress tolerance, thus allowing their survival.Citation32 For example, a number of studies have suggested the catabolic process of autophagy in breast cancer as a mechanism of resistance to chemotherapy and targeted inhibitors.Citation33 Therefore, strategies targeting cancer-type-specific cell death regulators may become an essential option to sensitize tumor cells to anticancer drugs used alone or most likely as a combination of target-specific compounds, together with standard chemotherapeutic agents.Citation34
Autophagy and apoptosis in gastrointestinal stromal tumors
This review aims to focus on the role of autophagy and apoptosis in gastrointestinal stromal tumors (GIST) as mechanisms of treatment resistance and novel therapeutic options in GIST.
Gastrointestinal stromal tumors
GIST are the most common sarcoma of the gastrointestinal tract arising from the interstitial cells of Cajal. Nevertheless, they are considered rare, with an incidence in Europe of 1.5 new cases per million per year.Citation35 Approximately 70-80% of GIST cases harbor gain-of-function mutations in KIT, whereas approximately 5-7% of cases display activating mutations in PDGFRA (platelet derived growth factor receptor α.36,37 Oncogenic activation of the 2 tyrosine kinase receptors KIT and PDGFRA, the main molecular drivers of GIST, results in auto-phosphorylation of tyrosine residues, with consequent activation of crucial downstream signaling cascades involved in apoptosis, differentiations, neoplastic transformation, and malignant progression. The remaining 10-15% of GIST cases do not present any alteration of these genes and are usually referred to as KIT-PDGFRA wild-type (WT) GIST, representing a heterogeneous family of patients with distinct molecular hallmarks ().Citation38,39 WT GIST cases exhibit distinctive characteristics with respect to the KIT-PDGFRA mutant cases. The majority of the WT GIST cases present mutations in genes for SDH subunits A, B, C or D, BRAF, KRAS or NF1 and may show IGF1R overexpression.Citation40,41 Identification of these distinctive molecular events allowed the introduction of tyrosine kinase inhibitors (TKIs) in the medical treatment of advanced disease. The introduction of TKIs, with imatinib as the forefather, led to a drastic improvement of both progression-free survival and overall survival.Citation42 The response of GIST to TKIs is strictly correlated with KIT and PDGFRA genotypes.Citation43-46 Patients with KIT exon 11 mutations have a response rate to imatinib of 83.5%, compared with 48% for patients with exon 9 mutations, whereas patients with the exon 18 PDGRFAD842V mutation are primarily resistant to imatinib.Citation46-49 KIT-PDGFRA WT GIST cases are also resistant to imatinib, but the percentage of response or stable disease is variable according to different studies. Evidence of a potential role of mutational status as a prognostic factor has progressively emerged. The presence of KIT exon 11 insertion/deletion involving one or both of the Trp557 and Lys558 amino acid residues correlates with a poor clinical outcome, whereas patients harboring a PDGFRA mutation, particularly D842V, show a better prognosis than patients with other mutations.Citation50 Despite this knowledge, imatinib remains the only first-line treatment available for GIST management, and after a median time of 20-24 mo, patients experience disease progression. The most common investigated mechanism of imatinib resistance involves acquisition of KIT-PDGFRA secondary mutations.Citation51,52 Alternative mechanisms, extensively investigated in oncology but still poorly explored in GIST, include pharmacogeneticCitation44,45,54-59 and epigeneticCitation60,61 mechanisms of resistance to imatinib. Presently, when GIST progression occurs, sunitinib and regorafenib represent standard second- and third-line treatments, respectively.Citation62,63 Nonetheless, GIST progression after treatment failure remains a problem, as no new options exist, representing an area of unmet clinical need. It is likely that autophagy is enhanced when cancer cells face environmental stress, including ionizing radiation, chemotherapy, TKI therapy, and nutritional deficiency.Citation64,65
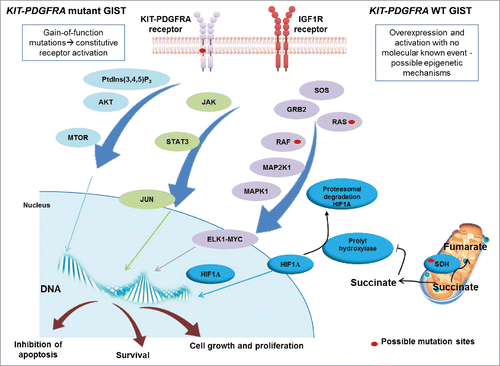
Figure 1. Schematic representation of the signaling in GIST. KIT and PDGFRA mutant GIST cases are characterized by gain-of-function mutations that activate the tyrosine kinase receptor in a constitutive and ligand-independent manner. KIT and PDGFRA WT GIST cases do not present gain-of-function alterations but may overexpress IGF1R. KIT, PDGFRA and IGF1R are tyrosine kinase receptors and their activation results in the promotion of downstream cascades, including PtdIns(3,4,5)P3-MTOR, JAK-JUN and RAS-MAPK/ERK, which lead to uncontrolled cell proliferation and growth, survival and inhibition of apoptosis. KIT and PDGFRA WT GIST, in addition, can harbor mutations in 1 of the 4 subunits of the SDH gene, which cause loss of function. This leads to cytoplasmatic accumulation of succinate, which downregulates prolyl hydroxylase, responsible for promoting HIF1A degradation. Succinate accumulation results in increased levels of HIF1A, which enters the nucleus and activates transcription factors.
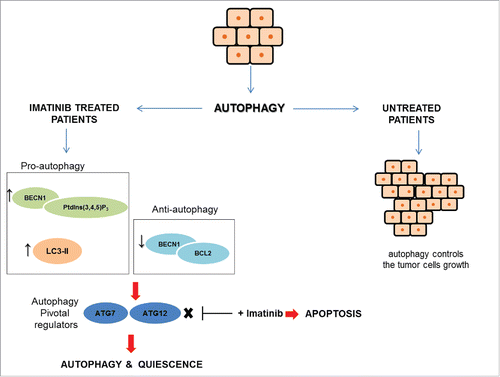
Autophagy in GIST
The idea that autophagy might play a pivotal role in GIST progression has become increasingly popular over recent years (). In 2008, Miselli and collaborators reported the first strong indirect evidence that imatinib response in GIST patients is sustained by autophagy rather than apoptosis.Citation66 The morphological, biochemical and immuno-phenotypical profiles, related to autophagy and apoptosis proteins, were analyzed in 13 surgically resected samples of GIST patients, 11 of which were treated with imatinib and 2 of which were untreated. Western blot analyses revealed high levels of the pro-autophagy BECN1 (Beclin 1)-phosphatidylinositol 3-kinase complex and LC3-II protein, and low levels of the anti-autophagy complex BECN1-BCL2, while no apoptosis-related protein expression was found.Citation66 Interestingly, given the presence of autophagy-related markers in both untreated and imatinib-treated patients, the authors hypothesized that autophagy was part of the molecular profile of GIST. Based on these data, the authors suggested 2 different mechanisms: autophagy-controlled tumor cell growth in untreated cases, and autophagy as a stress response in imatinib-induced starvation in treated patients (). Unfortunately, the lack of suitable material prevented a direct demonstration of autophagy, but these findings paved the way to further in-depth investigations. Gupta and coworkers later reported that a significant proportion of cells were able to survive under imatinib therapy, entering in a state of reversible quiescence and activating an autophagy-dependent survival mechanism.Citation67 The rationale that led to investigate and prove that GIST cells survive imatinib and undergo quiescence is based on the stable tumor burden, which is usually observed in GIST patients. Less than 2% of imatinib-treated patients experience complete radiographic regression whereas more often stable disease (indicated by viable nonproliferating tumor cells) is observed.Citation67 Furthermore, using RNAi-mediated silencing against ATG7 and ATG12, 2 key autophagy regulators, the authors demonstrated that ATG knockdown synergizes with imatinib in killing GIST cells. This study represented the first experimental evidence clearly supporting the involvement of autophagy in the survival of imatinib-treated GIST cells. These studies led to the idea that the inability of imatinib to kill GIST cells may promote selection and growth of resistant subclones harboring secondary KIT mutations. Further studies showed that autophagy inhibition by lysosomotropic agents, which act by inhibiting lysosomal acidification and consequent block of the terminal stages of autophagic proteolysis, sensitize GIST cell death following imatinib treatment, both in vitro and in vivo.Citation67,68 Overall, these studies suggest imatinib and lysosomotropic agents as an interesting therapeutic option with the potential to tremendously advance clinical outcome in GIST patients. Regrettably, no further studies have been conducted or so far reported, leaving this intriguing opportunity unexplored and with remaining uncertainty. More recently, Burger and coworkers proposed lysosomal sequestration as the main mechanism responsible for affecting intracellular levels of imatinib.Citation69 Using a GIST cell line model (GIST-T1), the authors demonstrated that inhibition of imatinib lysosomal sequestration significantly increased apoptosis in imatinib-sensitive GIST-T1 cells. Together these studies suggest that lysosomal targeting represents a promising novel strategy to eradicate imatinib-resistant GIST. However, the exact mechanism, autophagy block or lysosomal sequestration inhibition, that leads to this anticancer activity remains to be determined.
Figure 2. Autophagy as part of the molecular profile in both untreated and imatinib treated GIST cells. In untreated GIST cells (A), autophagy controls tumor cell growth. In treated GIST cells, imatinib binds KIT-PDGFRA receptors, blocking their downstream signaling cascade (B); however, a portion of the cells undergoes quiescence and activates autophagy-related survival mechanisms, which, in turn, may promote growth of resistant subclones, characterized by additional, imatinib-resistant mutations (C).
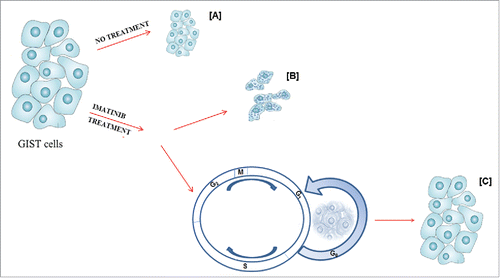
Figure 3. GIST and autophagy signaling. GIST cells can take advantage of autophagy by 2 different mechanisms. In untreated cells, autophagy controls tumor cells growth, whereas, in imatinib-treated cells autophagy is a stress response to starvation. In treated GIST cells, the pro-apoptotic BECN1 (Beclin 1)-PtdIns(3,4,5)P3 complex and LC3-II are expressed at high levels, while the anti-autophagy BECN1-BCL2 complex is at low levels. Autophagy signaling involves 2 key regulators, ATG7 and ATG12, that drive the cells toward autophagy and quiescence. ATG7 and ATG12 inhibition in association with imatinib promotes death cell through apoptosis.
To date, as noted above, imatinib is the only first-line treatment available for GIST management. Unfortunately, the majority of patients undergo imatinib dose escalation or switch to sunitinib due to the emergence of secondary resistance.Citation51 Regrettably, most patients, in particular those harboring KIT exon 17 secondary mutations, are less sensitive to sunitinib.Citation51,70 Regorafenib has demonstrated clinical benefits in imatinib/sunitinib-resistant GIST patients and is currently the only approved standard third-line option.Citation63 Overall, management of imatinib-resistant GIST is problematic, as sunitinib and regorafenib are only moderately well tolerated and median progression free survival remains fairly short.Citation71 In view of this critical scenario, research is focusing on identifying new compounds that can address the disease progression. A study by Bauer and colleagues reported KIT ubiquitination followed by proteasome-mediated degradation and GIST cell apoptosis as a consequence of HSP90AA1 inhibition.Citation72 Subsequently, Hsueh and collaborators reported autophagy as one of the molecular mechanisms partially involved in AUY922-induced KIT degradation in GIST cell lines.Citation73,74 In particular, they demonstrated that AUY922, an inhibitor of the chaperone HSP90AA1, of which KIT is a client, can effectively downregulate the expression of both phosphorylated- and total KIT protein levels via autophagy, inhibiting GIST cell line growth. Although proteasome and autophagy pathways have long been considered as 2 independent degradation machineries, these data, together with a growing body of literature, suggest the existence of a cross-talk between these 2 protein degradation pathways.Citation73-75 Based on this molecular evidence, a phase II trial with AUY922 in patients with advanced GIST who previously failed treatment with imatinib and sunitinib (NCT01389583), is ongoing.Citation76
Apoptosis in GIST
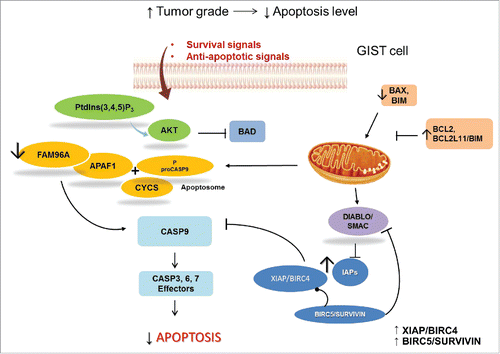
Apoptosis is tightly related to tumor development and as such it has also been widely investigated in GIST (). Indeed, one of the first studies linking apoptosis to GIST prognosis dates back to almost 10 y ago, when Wang and collaborators showed a significant negative correlation between apoptosis and degree of GIST differentiation.Citation77 The authors stratified patients according to tumor status and observed that the apoptotic index (evaluated by TUNEL assay) is gradually decreased in specimens from benign GIST, potential malignant and malignant GIST patients. Overall, the study implicated programmed cell death evasion in GIST pathogenesis. The finding was not so obvious, considering that the KIT downstream cascade has variable biological effects and the signaling pathway data in different cancer cell models may not predict which are critical for oncogenic behavior in GIST.Citation78 More recently, Ma and coworkers corroborated the involvement of apoptosis in GIST pathogenesis, correlating KIT expression level (as determined by immunohistochemistry) with GIST development through apoptosis inhibition and proliferation promotion.Citation79 Specifically, the authors found KIT-positive staining in 50 out of 68 screened cases and most of these patients (88%) presented KIT mutations. They later showed that KIT signaling plays a critical role in the tumor-growth rate by its relationship with proliferation and apoptosis. KIT expression shows a positive correlation with the cell proliferation marker MKI67/Ki-67 and a negative correlation with the pro-apoptotic protein APAF1. APAF1, participating in the intrinsic (mitochondrial) apoptosis pathway, associates with CYCS/cytochrome c and CASP9 to form the apoptosome, constituting a critical scaffold for CASP9 activation, and leading to apoptosis.Citation80 Recently, Schwamb and collaborators suggested a pro-apoptotic-tumor suppressor role for FAM96A, by interacting with APAF1.Citation81 The authors revealed that FAM96A protein expression is profoundly reduced or missing in 2 independent cohorts of GIST samples. In addition, they observed FAM96A reduction in tumorigenic interstitial cell of Cajal stem cells, indicating that loss of FAM96A pro-apoptotic activity occurs early in GIST oncogenesis.Citation81 Interestingly, re-establishment of FAM96A expression in GIST cell lines increases tumor sensitivity to apoptosis. Thus, identifying novel pro-apoptotic molecules such as FAM96A and elucidating their mechanisms may represent an innovative tool to fully restore mitochondrial apoptosis in GIST patients. The ability of GIST cells to escape apoptosis may involve multiple pathways. Baykara and colleagues reported a high expression of the anti-apoptotic protein BIRC5/survivin in tumor tissue from GIST patients.Citation82 A recent paper by Zhang and coworkers corroborated the diminished apoptotic activity in GIST, as they found expression of anti-apoptotic BCL2 and under-expression of the pro-apoptotic BAX in the majority of GIST patients.Citation83 Another research group investigated the effect of Peginterferon alfa-2b (pegylated interferon alfa-2b), alone or in combination with imatinib on imatinib-resistant GIST cell lines.Citation84 The combination of imatinib and Peginterferon alfa-2b promotes significant inhibition of cell proliferation, specifically in imatinib-resistant GIST cell lines. This effect might be due to perturbation of the PI3K-AKT-MTOR pathway, in particular the downregulation of the expression level of MTOR protein, and the induction of apoptosis. Therefore, the authors concluded that Peginterferon alfa-2b and imatinib combination may be an attractive therapeutic option for imatinib-resistant GIST.Citation84 According to the studies thus far described, the ability to escape apoptosis in response to a variety of stimuli is clearly confirmed as a hallmark of GIST pathogenesis. In addition, apoptosis also plays an important role in imatinib response.
Figure 4. GIST and apoptosis signaling. Survival signals can activate the PtdIns(3,4,5)P3-AKT cascade, which phosphorylates and inactivates the pro-apoptotic BCL2-family member BAD. In GIST, pro-apoptotic proteins, such as BAX, are downregulated and anti-apoptotic regulators are expressed. Anti-apoptotic proteins, including the anti-apoptotic BCL2 family members and inhibitor of apoptosis (IAP) proteins, are regulated by DIABLO/SMAC, which is mutated in GIST samples. IAPs are upregulated in GIST. XIAP/BIRC4, directly inhibits effector caspases, whereas BIRC5/survivin has indirect anti-apoptotic effects by stabilizing XIAP and inhibiting DIABLO. Apoptosis is also regulated through the mitochondrial (intrinsic) pathway. Pro-apoptotic BCL2 family proteins, including, BAX and BCL2L11/BIM, are important mediators of these signals. Activation of mitochondria promotes the release of CYCS/cytochrome C that binds APAF1 to form the apoptosome and subsequent activation of the initiator proCASP9. APAF1 interacts with the pro-apoptotic tumor suppressor FAM96A, which is downregulated in GIST.
Cancer cells commonly exhibit a high proliferation rate, with an increased energy consumption and augmented glucose uptake up to 200-fold higher compared with normal cells.Citation85 The glucose over-consumption is exploited in certain positron emission tomography (PET) methods using fluorodeoxyglucose (FDG, 18F-labeled) as a glucose analog. A study by Van den Abbeele and collaborators showed that untreated GIST cases are highly FDG-avid tumors, regulated by the constitutively active KIT-PI3K-AKT pathway, and display a dramatic loss of glucose uptake within 24 h of imatinib treatment. Overall, this indicates a high KIT-dependent glucose turnover, allowing early prediction of tumor response to imatinib by PET.Citation86,87 Tarn and colleagues observed a significant reduction in glucose uptake in imatinib-treated GIST cells, mediated by a decrease of the glucose transporter SLC2A4/GLUT4.Citation78 The authors also showed that glucose uptake in GIST cells, evaluated via FDG-PET, occurs in an AKT-signaling-dependent manner in both imatinib-sensitive and resistant cell models, as constitutively activated AKT1 or AKT2 isoforms are capable of reversing the imatinib inhibitory effect on glucose uptake.Citation78 Interestingly, glucose uptake is prognostic of tumor response to imatinib, but does not correlate with decreased cell growth and tumor burden reduction, achieved through apoptosis, in an AKT-independent manner. Indeed, constitutively activated AKT1 or AKT2 isoforms do not rescue GIST cells from imatinib-mediated apoptosis, despite the observation that glucose uptake is not shut down. Overall, this work suggests that after imatinib treatment, the 2 mechanisms of cell growth and glucose uptake are mutually exclusive events in GIST, and the therapeutic effect observed with imatinib is independent from AKT-signaling and glucose deprivation.Citation78 Mühlenberg and coworkers also investigated the disruption of glucose metabolism as a therapeutic approach in GIST.Citation86 The authors used 2-deoxyglucose (2DG), a non-metabolizable glucose analog, to show inhibition of KIT glycosylation, which abrogates KIT signaling. Additionally, the combination of 2DG with imatinib and ABT263, an antagonist of the anti-apoptotic BCL2 protein, may enhance the apoptotic response. Sustained cell growth is one of the fundamental hallmarks of cancer. Besides augmented glucose uptake many cancers also show increased need for fatty acids as an energy source.Citation88-91 AMACR (α-methylacyl-CoA racemase) is indispensable in the catabolism of branched fatty acids and its overexpression, leading to a growth advantage, has been reported in several carcinomas.Citation90 Recently, Li and colleagues reported AMACR overexpression in a large series of GIST tumor samples and cell lines. AMACR overexpression is in part sustained by gene amplification, also reflected at the protein level. However, increased gene copies were not observed in most of the samples, suggesting that alternative regulatory mechanisms, including miRNA downregulation, are likely to contribute to AMACR overexpression.Citation91 Interestingly, the authors found that AMACR amplification/overexpression confers aggressiveness through enhanced cell proliferation rather than apoptosis.Citation91 Indeed, AMACR-silenced GIST cell lines were found in G1-phase arrest without apoptosis. Nevertheless, a pro-apoptotic signal cannot be excluded, as a recent study reported a G2/M-phase arrest and a reduced expression of the apoptosis-inhibitor BIRC5 in prostatic trifluoroibuprofen-treated cancer cells.Citation92 As a confirmation, imatinib-resistant AMACR-amplified GIST cells are susceptible to the AMACR-inactivator ebselen oxide through decline in cell viability, G1-phase arrest, and induction of apoptosis.Citation91
A further study by Liu and collaborators demonstrated that one of the mechanisms contributing to the pro-apoptotic activity of imatinib in GIST cells is the upregulation of soluble histone H2AFX.Citation92 The authors proposed a model in which excessive levels of free histone H2AFX cause chromatin aggregation, which promotes impaired transcription, abrogating ongoing gene transcription, and thereby sensitizing tumor cells to undergo apoptosis.
Returning to , the achievement of a disease long-term stabilization in the presence of residual tumor, together with in vitro studies,Citation93 undoubtedly suggests that GIST cell quiescence plays an important role in imatinib therapy. The major regulators of tumor cell quiescence have not been clearly identified; however, a number of studies suggest the critical involvement of the cyclin-dependent kinase inhibitor CDKN1B/p27Kip1, which is normally expressed at a low level in cells that are in the S or G2 phase of the cell cycle.Citation94 Liu and colleagues reported that GIST cells escaping from apoptosis after imatinib treatment overexpress nuclear CDKN1B/p27Kip1, indicating that the cells have withdrawn from the cell cycle and are quiescent.Citation95 Mechanistically this event is associated with an imatinib-induced modulation of the anaphase promoting complex (ANAPC)-FZR1/CDH1 signaling axis. The ANAPC-FZR1/CDH1 complex is essential for reinforcing G0-G1 arrest and hence removing cells from the proliferative pool, and keeping them in a quiescent state. The work by Liu and collaborators provided evidence of an imatinib-mediated downregulation of the SKP2 protein in vitro, which is associated with increased nuclear FZR1/CDH1; the ANAPC-FZR1/CDH1 complex promotes CDKN1B/p27Kip1 accumulation and suppression of CDK in G0-G1.Citation94-98 Thereafter, Boichuk and coworkers corroborated the involvement of the ANAPC-FZR1/CDH1-SKP2-CDKN1B/p27Kip1 signaling axis in imatinib-induced quiescence in vitro, and provided the same evidence in cell line and patient-derived mouse xenografts in vivo.Citation99 The authors also demonstrated the involvement of the DREAM complex, a multi-subunit complex recently identified as an additional key regulator of quiescence. DREAM complex formation occurs in G0 phase, negatively regulates the cell cycle, and includes RB1/pRB (RB transcriptional corepressor 1) and the related “pocket protein” family members, RBL1/p107 and RBL2/p130. The RBL2 protein, whose expression is induced by imatinib treatment, accumulates in G0 and interacts with E2F4 to repress E2F-dependent gene transcription.Citation98,99 Interestingly, RBL2 protein levels are also regulated by SKP2,Citation100,101 emphasizing that quiescence involves multiple interrelated pathways.
Overall, autophagy and cell quiescence safeguard GIST cells from apoptosis.Citation67,74,94 However, the role of IAPs has only recently been investigated in GIST by Falkenhorst and coworkers.Citation102 The authors showed that 2 IAPs, XIAP/BIRC4 and BIRC5/survivin, are highly expressed both at the mRNA and protein level in primary GIST and cell line models. Interestingly, the authors also showed that IAP inhibitors may improve the apoptotic response to KIT inhibitors. Additional functional studies are required to draw a definitive conclusion on the constitutive overexpression of these IAPs in apoptotic-resistant GIST. Nevertheless, the authors showed that IAPs, particularly XIAP and BIRC5, may co-mediate the prosurvival signaling of oncogenic KIT.
Autophagy, apoptosis and miRNA in GIST
Nannini and collaborators recently highlighted the involvement of miRNA, short noncoding RNA, in GIST pathogenesis and imatinib response.Citation60 To date, no studies have associated deregulated miRNA to altered expression of autophagy genes in GIST, despite the fact that the functions of some essential autophagy genes are regulated by miRNA. Unlike autophagy, a different fate is reserved for apoptosis, as 4 different studies have associated miRNA deregulation with this pathway.Citation103-104 Kim and colleagues reported an inverse relationship between MIR494 and KIT expression in 31 GIST patients and observed a perturbation of the KIT downstream molecular pathway in GIST cells after MIR494 treatment.Citation103 Additionally, MIR494 overexpression results in both cell cycle arrest and apoptosis. Gits and coworkers showed that MIR17HG/MIR17–92 and MIR221-MIR222 cluster members are important onco-miRNA contributing to GIST development.Citation104 In support of this finding, overexpression of MIR17, MIR20A, and MIR222 in 2 different GIST cell lines promote cell growth inhibition and apoptosis induction. In a further study, Ihle and colleagues reported that both MIR222 and MIR221 reduce viability and induce apoptosis in vitro in 3 different GIST cell lines via the KIT, AKT, and BCL2 signaling cascade.Citation105 Recently, Shi and collaborators reported a correlation between MIR518A-5p and apoptosis.Citation106 The authors found MIR518A-5p downregulation in imatinib-resistant GIST patients by miRNA microarray and provided evidence of a functional interaction with the 3′ UTR of the PIK3C2A gene. The PIK3C2A gene belongs to the phosphatidylinositol (PtdIns) 3-kinase family, which regulates various cellular processes, including cell growth, proliferation and apoptosis. The authors further documented that transfection of the imatinib-resistant GIST882 cell line with MIR518A-5p reduces cell proliferation and promotes apoptosis. The authors showed PIK3C2A downregulation, both at mRNA and protein levels, after MIR518A-5p transfection, indicating that low expression of MIR518A-5p is likely to cause PIK3C2A upregulation, promoting cancer survival, and prompting resistance to imatinib in GIST.Citation106
Concluding remarks and future perspectives
Despite the available data, many questions still remain regarding both autophagy and apoptosis and these warrant more detailed investigation. Autophagy inhibition in combination with imatinib represents a valuable strategy to improve GIST treatment (). Future studies should also examine the precise mechanism that determines whether GIST cells undergo apoptosis or quiescence.
Table 1. Novel therapeutic targets and imatinib combination treatments for GIST.
CRISPR/Cas9 is a relatively new system that has emerged as a powerful genome editing tool, facilitating and expanding our capacity to engineer mammalian genomes. Such a system would be very helpful in GIST, considering that the cell models, which are not commercially available and accessible only upon request to the laboratory that developed the line, are not representative of the complexity of GIST. In particular, there are no established PDGFRA mutated, KIT WT or PDGFRA WT GIST lines. The possibility to introduce specific and clinically relevant mutations has the potential to create powerful in vitro models of the disease, possibly introducing a new era of drug discovery and validation. This aspect is of particular interest in GIST, as KIT and PDGFRA WT GIST may be regarded as therapeutic orphans, and considering that the only available treatment option involves TKIs.
The studies conducted so far have identified many promising therapeutic targets as new treatment options and suggested new combination strategies to improve the effectiveness of imatinib treatment and improve patient response. Another significant application of the CRISPR/Cas9 system will be the knockout of specific genes, such as the IAPs XIAP/BIRC4 and BIRC5 and the autophagic regulators AGT7 and AGT12. In view of the extensive crosstalk between autophagy and apoptosis, it is likely that proteins involved in the intersections of these pathways, including BECN1-BCL2, AGT12-ATG3 and ATG12-MCL1, may have particular importance in GIST. Therefore, a further interesting strategy is to exploit gene editing to generate knockin and knockout models of these genes. These finding have the potential to be critical for the successful development of novel therapeutic strategies in GIST.
Abbreviations
| 2DG | = | 2-deoxyglucose |
| AKT1 | = | AKT serine/threonine kinase 1 |
| AKT2 | = | AKT serine/threonine kinase 2 |
| AMACR | = | α-methylacyl-CoA racemase |
| APAF1 | = | apoptotic peptidase activating factor 1 |
| ANAPC | = | anaphase promoting complex |
| ATG7 | = | autophagy-related 7 |
| ATG12 | = | autophagy-related 12 |
| BAD | = | BCL2 associated agonist of cell death |
| BAX | = | BCL2 associated X, apoptosis regulator |
| BCL2L1 | = | BCL2 like 1 |
| BCL2, BCL2 | = | apoptosis regulator |
| BECN1 | = | Beclin 1 |
| BIM | = | BCL2 like 11 |
| BIRC5/survivin | = | baculoviral IAP repeat containing 5 |
| BRAF | = | B-Raf proto-oncogene, serine/threonine kinase |
| Cas9 | = | CRISPR associated protein 9 |
| FZR1/CDH1 | = | fizzy/cell division cycle 20 related 1 |
| CDK | = | cyclin-dependent kinase |
| CDKN1B/p27Kip1 | = | cyclin-dependent kinase inhibitor 1B |
| CRISPR | = | clustered regularly-interspaced short palindromic repeats |
| CYCS | = | cytochrome c, somatic |
| DIABLO/SMAC | = | diablo IAP-binding mitochondrial protein |
| DREAM, DP, RB-like | = | E2F4 and MuvB multisubunit protein complex |
| ELK1, ELK1 | = | ETS transcription factor |
| E2F4, E2F | = | transcription factor 4 |
| FAM96A | = | family with sequence similarity 96 member A |
| FDG | = | fluorodeoxyglucose |
| FZR1 | = | fizzy/cell division cycle 20 related 1 |
| GIST | = | gastrointestinal stromal tumor |
| GRB2 | = | growth factor receptor bound protein 2 |
| HIF1A/HIF1α | = | hypoxia inducible factor 1 α subunit |
| HSP90AA1 | = | heat shock protein 90 α family class A member 1 |
| H2AFX/H2AX | = | H2A histone family member X |
| IAP | = | inhibitor of apoptosis |
| IGF1R | = | Insulin like growth factor 1 receptor |
| JAK | = | Janus Kinase |
| JUN | = | Jun proto-oncogene, AP-1 transcription factor subunit |
| KIT, KIT | = | proto-oncogene receptor tyrosine kinase |
| KRAS | = | KRAS proto-oncogene, GTPase |
| MAP1LC3-II/LC3-II | = | microtubule associated protein 1 light chain 3 |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| MYC | = | v-myc avian myelocytomatosis viral oncogene homolog |
| NF1 | = | neurofibromin 1 |
| PDGFRA | = | platelet derived growth factor receptor α |
| Peginterferon alfa-2b | = | pegylated interferon alfa-2b |
| PET | = | positron emission tomography |
| PIK3C2A | = | phosphatidylinositol-4-phosphate 3-kinase catalytic subunit type 2 α |
| PtdIns3P | = | phosphatidylinositol-3-phosphate |
| RAF | = | RAF family |
| RAS | = | RAS type GTPase family |
| SDH | = | succinate dehydrogenase |
| SKP2 | = | S-phase kinase associated protein 2 |
| SLC2A4/GLUT4 | = | solute carrier family 2 member 4 |
| SOS | = | SOS Ras/Rac guanine nucleotide exchange factor |
| STAT3 | = | signal transducer and activator of transcription 3 |
| TKI | = | tyrosine kinase inhibitor |
| TKR | = | tyrosine kinase receptor |
| TP53 | = | tumor protein p53 |
| TUNEL | = | terminal nucleotidyl transferase-mediated nick end labelling |
| WT | = | wild type |
| XIAP/BIRC4 | = | X-linked inhibitor of apoptosis |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 2013; 1833:3448-59; PMID:23770045; http://dx.doi.org/10.1016/j.bbamcr.2013.06.001
- Lapaquette P, Guzzo J, Bretillon L, Bringer MA. Cellular and Molecular Connections between Autophagy and Inflammation. Mediators Inflamm 2015; 2015:398483; PMID:26221063; http://dx.doi.org/10.1155/2015/398483
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726-34; PMID:16148885; http://dx.doi.org/10.1038/nrc1692
- Martin SJ. Oncogene-induced autophagy and the Goldilocks principle. Autophagy 2011; 7:922-3; PMID:21552010; http://dx.doi.org/10.4161/auto.7.8.15821
- Editorial, Focusing on autophagy. Nat Cell Biol 2010; 12:813; PMID:20811352; http://dx.doi.org/10.1038/ncb0910-813
- De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol 1966; 28:435-92; PMID:5322983; http://dx.doi.org/10.1146/annurev.ph.28.030166.002251
- Todde V, Veenhuis M, van der Klei IJ. Autophagy: principles and significance in health and disease. Biochim Biophys Acta 2009; 1792:3-13; PMID:19022377; http://dx.doi.org/10.1016/j.bbadis.2008.10.016
- Du Toit A. Cell death: balance through a bivalent regulator. Nat Rev Mol Cell Biol 2013; 14:546; PMID:23900393; http://dx.doi.org/10.1038/nrm3637
- Qian HR, Yang Y. Functional role of autophagy in gastric cancer. Oncotarget 2016; 7(14):17641-51; PMID:26910278
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51-64; PMID:16843265; http://dx.doi.org/10.1016/j.ccr.2006.06.001
- Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev 2011; 21:113-9; PMID:21255998; http://dx.doi.org/10.1016/j.gde.2010.12.008
- Long JS, Ryan KM. New frontiers in promoting tumour cell death: targeting apoptosis, necroptosis and autophagy. Oncogene 2012; 31:5045-60; PMID:22310284; http://dx.doi.org/10.1038/onc.2012.7
- Janji B, Viry E, Moussay E, Paggetti J, Arakelian T, Mgrditchian T, Messai Y, Noman MZ, Van Moer K, Hasmim M, et al. The multifaceted role of autophagy in tumor evasion from immune surveillance. Oncotarget 2016; 7(14):17591-607; PMID:26910842
- Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol 2011; 99:287-92; PMID:21722986; http://dx.doi.org/10.1016/j.radonc.2011.06.002
- Sui X, Zhu J, Zhou J, Wang X, Li D, Han W, Fang Y, Pan H. Epigenetic modifications as regulatory elements of autophagy in cancer. Cancer Lett 2015; 360:106-13; PMID:25687886; http://dx.doi.org/10.1016/j.canlet.2015.02.009
- Fulda S, Kogel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015; 34:5105-13; PMID:25619832; http://dx.doi.org/10.1038/onc.2014.458
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717-29; PMID:21406549; http://dx.doi.org/10.1101/gad.2016111
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8:528-39; PMID:21587219; http://dx.doi.org/10.1038/nrclinonc.2011.71
- Indovina P, Pentimalli F, Casini N, Vocca I, Giordano A. RB1 dual role in proliferation and apoptosis: cell fate control and implications for cancer therapy. Oncotarget 2015; 6:17873-90; PMID:26160835; http://dx.doi.org/10.18632/oncotarget.4286
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57-70; PMID:10647931; http://dx.doi.org/10.1016/S0092-8674(00)81683-9
- Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis 2000; 21:485-95; PMID:10688869; http://dx.doi.org/10.1093/carcin/21.3.485
- Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 2004; 4:592-603; PMID:15286739; http://dx.doi.org/10.1038/nrc1412
- Attardi LD. The role of p53-mediated apoptosis as a crucial anti-tumor response to genomic instability: lessons from mouse models. Mutat Res 2005; 569:145-57; PMID:15603759; http://dx.doi.org/10.1016/j.mrfmmm.2004.04.019
- Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. p53 status and the efficacy of cancer therapy in vivo. Science 1994; 266:807-10; PMID:7973635; http://dx.doi.org/10.1126/science.7973635
- Amundson SA, Myers TG, Scudiero D, Kitada S, Reed JC, Fornace AJ, Jr. An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res 2000; 60:6101-10; PMID:11085534
- Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer 2003; 3:117-29; PMID:12563311; http://dx.doi.org/10.1038/nrc992
- Lock RB, Stribinskiene L. Dual modes of death induced by etoposide in human epithelial tumor cells allow Bcl-2 to inhibit apoptosis without affecting clonogenic survival. Cancer Res 1996; 56:4006-12; PMID:8752171
- Han JW, Dionne CA, Kedersha NL, Goldmacher VS. p53 status affects the rate of the onset but not the overall extent of doxorubicin-induced cell death in rat-1 fibroblasts constitutively expressing c-Myc. Cancer Res 1997; 57:176-82; PMID:8988061
- Kemp CJ, Sun S, Gurley KE. p53 induction and apoptosis in response to radio-and chemotherapy in vivo is tumor-type-dependent. Cancer Res 2001; 61:327-32; PMID:11196181
- Zarzynska JM. The importance of autophagy regulation in breast cancer development and treatment. Biomed Res Int 2014; 2014:710345; PMID:25317422; http://dx.doi.org/10.1155/2014/710345
- Ozpolat B, Benbrook DM. Targeting autophagy in cancer management - strategies and developments. Cancer Manag Res 2015; 7:291-9; PMID:26392787; http://dx.doi.org/10.2147/CMAR.S34859
- White E. The role for autophagy in cancer. J Clin Invest 2015; 125:42-6; PMID:25654549; http://dx.doi.org/10.1172/JCI73941
- Zambrano J, Yeh ES. Autophagy and Apoptotic Crosstalk: Mechanism of Therapeutic Resistance in HER2-Positive Breast Cancer. Breast Cancer (Auckl) 2016; 10:13-23; PMID:26997868
- Radogna F, Dicato M, Diederich M. Cancer-type-specific crosstalk between autophagy, necroptosis and apoptosis as a pharmacological target. Biochem Pharmacol 2015; 94:1-11; PMID:25562745; http://dx.doi.org/10.1016/j.bcp.2014.12.018
- Valsangkar N, Sehdev A, Misra S, Zimmers TA, O'Neil BH, Koniaris LG. Current management of gastrointestinal stromal tumors: Surgery, current biomarkers, mutations, and therapy. Surgery 2015; 158:1149-64; PMID:26243346; http://dx.doi.org/10.1016/j.surg.2015.06.027
- Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998; 279:577-80; PMID:9438854; http://dx.doi.org/10.1126/science.279.5350.577
- Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003; 299:708-10; PMID:12522257; http://dx.doi.org/10.1126/science.1079666
- Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol 2004; 22:3813-25; PMID:15365079; http://dx.doi.org/10.1200/JCO.2004.05.140
- Nannini M, Biasco G, Astolfi A, Pantaleo MA. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST). J Med Genet 2013; 50:653-61; PMID:23833252; http://dx.doi.org/10.1136/jmedgenet-2013-101695
- Pantaleo MA, Nannini M, Corless CL, Heinrich MC. Quadruple wild-type (WT) GIST: defining the subset of GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling pathways. Cancer Med 2015; 4:101-3; PMID:25165019; http://dx.doi.org/10.1002/cam4.325
- Nannini M, Astolfi A, Paterini P, Urbini M, Santini D, Catena F, Indio V, Casadio R, Pinna AD, Biasco G, et al. Expression of IGF-1 receptor in KIT/PDGF receptor α wild-type gastrointestinal stromal tumors with succinate dehydrogenase complex dysfunction. Future Oncol 2013; 9:121-6; PMID:23252569; http://dx.doi.org/10.2217/fon.12.170
- Belinsky MG, Rink L, Flieder DB, Jahromid MS,Schiffman JD, Godwin AK, Mehren Mv. Overexpression of insuline-like growth factor 1 receptor and frequent mutational inactivation of SDH in wild-type SDHB-negative gastrointestinal stromal tumors. Genes Chromosomes Cancer 2013; 52:214-24; PMID:23109135; http://dx.doi.org/10.1002/gcc.22023
- Barrios CH, Blackstein ME, Blay JY, Casali PG, Chacon M, Gu J, Kang YK, Nishida T, Purkayastha D, Woodman RC, et al. The GOLD ReGISTry: a Global, Prospective, Observational Registry collecting longitudinal data on patients with advanced and Localised Gastrointestinal Stromal Tumours. Eur J Cancer 2015; 51:2423-33; PMID:26248685; http://dx.doi.org/10.1016/j.ejca.2015.07.010
- Angelini S, Ravegnini G, Fletcher JA, Maffei F, Hrelia P. Clinical relevance of pharmacogenetics in gastrointestinal stromal tumor treatment in the era of personalized therapy. Pharmacogenomics 2013; 14:941-56; PMID:23746188; http://dx.doi.org/10.2217/pgs.13.63
- Ravegnini G, Sammarini G, Angelini S, Hrelia P. Pharmacogenetics of tyrosine kinase inhibitors in gastrointestinal stromal tumor and chronic myeloid leukemia. Expert Opin Drug Metab Toxicol 2016; 12:733-42; PMID:27149004; http://dx.doi.org/10.1080/17425255.2016.1184649
- Ravegnini G, Nannini M, Sammarini G, Astolfi A, Biasco G, Pantaleo MA, Hrelia P, Angelini S. Personalized Medicine in Gastrointestinal Stromal Tumor (GIST): Clinical Implications of the Somatic and Germline DNA Analysis. Int J Mol Sci 2015; 16:15592-608; PMID:26184165; http://dx.doi.org/10.3390/ijms160715592
- Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumors. J Clin Oncol 2003; 21:4342-9; PMID:14645423; http://dx.doi.org/10.1200/JCO.2003.04.190
- Debiec-Rychter M, Dumez H, Judson I, Wasag B, Verweij J, Brown M, Dimitrijevic S, Sciot R, Stul M, Vranck H, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 2004; 40:689-95; PMID:15010069; http://dx.doi.org/10.1016/j.ejca.2003.11.025
- Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, Shiraga S, Bainbridge T, Morich J, Heinrich MC. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005; 23:5357-64; PMID:15928335; http://dx.doi.org/10.1200/JCO.2005.14.068
- Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 2006; 24:4764-74; PMID:16954519; http://dx.doi.org/10.1200/JCO.2006.06.2265
- Maleddu A, Pantaleo MA, Nannini M, Biasco G. The role of mutational analysis of KIT and PDGFRA in gastrointestinal stromal tumors in a clinical setting. J Transl Med 2011; 9:75; PMID:21605429; http://dx.doi.org/10.1186/1479-5876-9-75
- Songdej N, von Mehren M. GIST treatment options after tyrosine kinase inhibitors. Curr Treat Options Oncol 2014; 15:493-506; PMID:24952730; http://dx.doi.org/10.1007/s11864-014-0295-3
- Maleddu A, Pantaleo MA, Nannini M, Di Battista M, Saponara M, Lolli C, Biasco G. Mechanisms of secondary resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumours (Review). Oncol Rep 2009; 21:1359-66; PMID:19424610; http://dx.doi.org/10.3892/or_00000361
- Angelini S, Pantaleo MA, Ravegnini G, Zenesini C, Cavrini G, Nannini M, Fumagalli E, Palassini E, Saponara M, Di Battista M, et al. Polymorphisms in OCTN1 and OCTN2 transporters genes are associated with prolonged time to progression in unresectable gastrointestinal stromal tumours treated with imatinib therapy. Pharmacol Res 2013; 68:1-6; PMID:23127916; http://dx.doi.org/10.1016/j.phrs.2012.10.015
- Angelini S, Ravegnini G, Nannini M, Bermejo JL, Musti M, Pantaleo MA, Fumagalli E, Venturoli N, Palassini E, Consolini N, et al. Folate-related polymorphisms in gastrointestinal stromal tumours: susceptibility and correlation with tumour characteristics and clinical outcome. Eur J Hum Genet 2015; 23:817-23; PMID:25227144; http://dx.doi.org/10.1038/ejhg.2014.198
- Koo DH, Ryu MH, Ryoo BY, Beck MY, Na YS, Shin JG, Lee SS, Kim EY, Kang YK. Association of ABCG2 polymorphism with clinical efficacy of imatinib in patients with gastrointestinal stromal tumor. Cancer Chemother Pharmacol 2015; 75:173-82; PMID:25417047; http://dx.doi.org/10.1007/s00280-014-2630-6
- Brahmi M, Alberti L, Dufresne A, Ray-Coquard I, Cassier P, Meeus P, Decouvelaere AV, Ranchère-Vince D, Blay JY, et al. KIT exon 10 variant (c.1621 A >C) single nucleotide polymorphism as predictor of GIST patient outcome. BMC Cancer 2015; 15:780; PMID:26498480; http://dx.doi.org/10.1186/s12885-015-1817-5
- Rutkowski P, Bylina E, Klimczak A, Switaj T, Falkowski S, Kroc J, Lugowska I, Brzeskwiniewicz M, Melerowicz W, Osuch C, et al. The outcome and predictive factors of sunitinib therapy in advanced gastrointestinal stromal tumors (GIST) after imatinib failure - one institution study. BMC Cancer 2012; 12:107; PMID:22439647; http://dx.doi.org/10.1186/1471-2407-12-107
- Ravegnini G, Nannini M, Simeon V, Musti M, Sammarini G, Saponara M, et al. Polymorphisms in DNA repair genes in gastrointestinal stromal tumors: susceptibility and correlation with tumor characteristic and clinical outcome. Tumor Biol 2016; Epub ahead of print
- Nannini M, Ravegnini G, Angelini S, Astolfi A, Biasco G, Pantaleo MA. miRNA profiling in gastrointestinal stromal tumors: implication as diagnostic and prognostic markers. Epigenomics 2015; 7:1033-49; PMID:26447534; http://dx.doi.org/10.2217/epi.15.52
- Pantaleo MA, Ravegnini G, Astolfi A, Simeon V, Nannini M, Saponara M, Urbini M, Gatto L, Indio V, Sammarini G, et al. Integrating miRNA and gene expression profiling analysis revealed regulatory network in GIST. Epigenomics 2016; 8(10):1347-1366; Epub ahead of print; PMID:27625077; http://dx.doi.org/10.2217/epi-2016-0030
- Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006; 368:1329-38; PMID:17046465; http://dx.doi.org/10.1016/S0140-6736(06)69446-4
- Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M, Joensuu H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013; 381:295-302; PMID:23177515; http://dx.doi.org/10.1016/S0140-6736(12)61857-1
- Patel S. Managing progressive disease in patients with GIST: factors to consider besides acquired secondary tyrosine kinase inhibitor resistance. Cancer Treat Rev 2012; 38:467-72; PMID:22035971; http://dx.doi.org/10.1016/j.ctrv.2011.10.001
- Ertmer A, Huber V, Gilch S, Yoshimori T, Erfle V, Duyster J, Elsässer HP, Schätzl HM. The anticancer drug imatinib induces cellular autophagy. Leukemia 2007; 21:936-42; PMID:17330103
- Miselli F, Negri T, Gronchi A, Losa M, Conca E, Brich S, Fumagalli E, Fiore M, Casali PG, Pierotti MA, et al. Is autophagy rather than apoptosis the regression driver in imatinib-treated gastrointestinal stromal tumors? Transl Oncol 2008; 1:177-86; PMID:19043528; http://dx.doi.org/10.1593/tlo.08157
- Gupta A, Roy S, Lazar AJ, Wang WL, McAuliffe JC, Reynoso D, McMahon J, Taguchi T, Floris G, Debiec-Rychter M, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci U S A 2010; 107:14333-8; PMID:20660757; http://dx.doi.org/10.1073/pnas.1000248107
- Rubin BP, Debnath J. Therapeutic implications of autophagy-mediated cell survival in gastrointestinal stromal tumor after treatment with imatinib mesylate. Autophagy 2010; 6:1190-1; PMID:20930540; http://dx.doi.org/10.4161/auto.6.8.13430
- Burger H, den Dekker AT, Segeletz S, Boersma AWM, d Bruijn P, Debiec-Rychter M, Taguchi T, Sleijfer S, Sparreboom A, Mathijssen RH, et al. Lysosomal sequestration determines intracellular imatinib levels. Mol Pharmacol 2015; 88:477-87; PMID:26108972; http://dx.doi.org/10.1124/mol.114.097451
- Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB, Fletcher JA, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008; 26:5352-9; PMID:18955458; http://dx.doi.org/10.1200/JCO.2007.15.7461
- Bauer S, Joensuu H. Emerging agents for the treatment of advanced, imatinib-resistant gastrointestinal stromal tumors: Current status and future directions. Drugs 2015; 75:1323-34; PMID:26187774; http://dx.doi.org/10.1007/s40265-015-0440-8
- Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res 2006; 66:9153-61; PMID:16982758; http://dx.doi.org/10.1158/0008-5472.CAN-06-0165
- Hsueh YS, Yen CC, Shih NY, Chiang NJ, Li CF, Chen LT. Autophagy is involved in endogenous and NVP-AUY922-induced KIT degradation in gastrointestinal stromal tumors. Autophagy 2013; 9:220-33; PMID:23196876; http://dx.doi.org/10.4161/auto.22802
- Hsueh YS, Chang HH, Chiang NJ, Yen CC, Li CF, Chen LT. MTOR inhibition enhances NVP-AUY922-induced autophagy-mediated KIT degradation and cytotoxicity in imatinib-resistant gastrointestinal stromal tumors. Oncotarget 2014; 5:11723-36; PMID:25375091; http://dx.doi.org/10.18632/oncotarget.2607
- Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12:836-41; PMID:20811356; http://dx.doi.org/10.1038/ncb0910-836
- https://clinicaltrials.gov/ct2/show/NCT01389583
- Wang Q, Kou YW. Study of the expressions of p53 and bcl-2 genes, the telomerase activity and apoptosis in GIST patients. World J Gastroenterol 2007; 13:2626-8; PMID:17552015; http://dx.doi.org/10.3748/wjg.v13.i18.2626
- Tarn C, Skorobogatko YV, Taguchi T, Eisenberg B, von Mehren M, Godwin AK. Therapeutic effect of imatinib in gastrointestinal stromal tumors: AKT signaling dependent and independent mechanisms. Cancer Res 2006; 66:5477-86; PMID:16707477; http://dx.doi.org/10.1158/0008-5472.CAN-05-3906
- Ma YY, Yu S, He XJ, Xu Y, Wu F, Xia YJ, Guo K, Wang HJ, Ye ZY, Zhang W, et al. Involvement of c-KIT mutation in the development of gastrointestinal stromal tumors through proliferation promotion and apoptosis inhibition. Onco Targets Ther 2014; 7:637-43; PMID:24833907
- Brenner D, Mak TW. Mitochondrial cell death effectors. Curr Opin Cell Biol 2009; 21:871-7; PMID:19822411; http://dx.doi.org/10.1016/j.ceb.2009.09.004
- Schwamb B, Pick R, Fernández SB, Völp K, Heering J, Dötsch V, Bösser S, Jung J, Beinoraviciute-Kellner R, Wesely J, et al. FAM96A is a novel pro-apoptotic tumor suppressor in gastrointestinal stromal tumors. Int J Cancer 2015; 137:1318-29; PMID:25716227; http://dx.doi.org/10.1002/ijc.29498
- Baykara M, Akkus M, Yildiz R, Gonul II , Dursun A, Coskun U, Benekli M, Sevinc A, Dane F, Buyukberber S. Survivin expression and its potential clinical significance in gastrointestinal stromal sarcoma. Int Immunopharmacol 2011; 11:2227-31; PMID:22020290; http://dx.doi.org/10.1016/j.intimp.2011.10.005
- Zhang Y, Gu ML, Zhou XX, Ma H, Yao HP, Ji F. Altered expression of ETV1 and its contribution to tumorigenic phenotypes in gastrointestinal stromal tumors. Oncol Rep 2014; 32:927-34; PMID:24970355
- Zhang LY, Huang JS, Pi ZM, Yu MY. Inhibition effect of Peg-IFNalpha-2b and Imatinib alone or combination on imatinib-resistant gastrointestinal stromal tumors cell lines. Int J Clin Exp Pathol 2015; 8:11340-7; PMID:26617858
- Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab 2016; 23:27-47; PMID:26771115; http://dx.doi.org/10.1016/j.cmet.2015.12.006
- Muhlenberg T, Grunewald S, Treckmann J, Podleska L, Schuler M, Fletcher JA, Bauer S. Inhibition of KIT-glycosylation by 2-deoxyglucose abrogates KIT-signaling and combination with ABT-263 synergistically induces apoptosis in gastrointestinal stromal tumor. PLoS One 2015; 10:e0120531; PMID:25781619; http://dx.doi.org/10.1371/journal.pone.0120531
- Van den Abbeele AD, Gatsonis C, de Vries DJ, Melenevsky Y, Szot-Barnes A, Yap JT, Godwin AK, Rink L, Huang M, Blevins M, et al. ACRIN 6665/RTOG 0132 phase II trial of neoadjuvant imatinib mesylate for operable malignant gastrointestinal stromal tumor: monitoring with 18F-FDG PET and correlation with genotype and GLUT4 expression. J Nucl Med 2012; 53:567-74; PMID:22381410; http://dx.doi.org/10.2967/jnumed.111.094425
- Ferdinandusse S, Denis S, IJlst L, Dacremont G, Waterham HR, Wanders RJ. Subcellular localization and physiological role of alpha-methylacyl-CoA racemase. J Lipid Res 2000; 41:1890-6; PMID:11060359
- Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG, Pienta KJ, Ghosh D, Chinnaiyan AM. alpha-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA 2002; 287:1662-70; PMID:11926890; http://dx.doi.org/10.1001/jama.287.13.1662
- Zhou M, Chinnaiyan AM, Kleer CG, Lucas PC, Rubin MA. Alpha-Methylacyl-CoA racemase: a novel tumor marker over-expressed in several human cancers and their precursor lesions. Am J Surg Pathol 2002; 26:926-31; PMID:12131161; http://dx.doi.org/10.1097/00000478-200207000-00012
- Li CF, Chen LT, Lan J, Chou FF, Lin CY, Chen YY, Chen TJ, Li SH, Yu SC, Fang FM, et al. AMACR amplification and overexpression in primary imatinib-naïve gastrointestinal stromal tumors: a driver of cell proliferation indicating adverse prognosis. Oncotarget 2014; 5:11588-603; PMID:25473890; http://dx.doi.org/10.18632/oncotarget.2597
- Festuccia C, Gravina GL, Mancini A, Muzi P, Cesare ED, Kirk R, Smith M, Hughes S, Gibson R, Lian LY, et al. Trifluoroibuprofen inhibits α-methylacyl coenzyme A racemase (AMACR/P504S), reduces cancer cell proliferation and inhibits in vivo tumor growth in aggressive prostate cancer models. Anticancer Agents Med Chem 2014; 14:1031-41; PMID:24712326; http://dx.doi.org/10.2174/1871520614666140327152607
- Liu Y, Tseng M, Perdreau SA, Rossi F, Antonescu C, Besmer P, Fletcher JA, Duensing S, Duensing A. Histone H2AX is a mediator of gastrointestinal stromal tumor cell apoptosis following treatment with imatinib mesylate. Cancer Res 2007; 67:2685-92; PMID:17363589; http://dx.doi.org/10.1158/0008-5472.CAN-06-3497
- Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD, Fletcher JA. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 2004; 23:3999-4006; PMID:15007386; http://dx.doi.org/10.1038/sj.onc.1207525
- Liu Y, Perdreau SA, Chatterjee P, Wang L, Kuan SF, Duensing A. Imatinib mesylate induced quiescence in gastrointestinal stromal tumor cells through the CDH1-SKP2-p27Kip1 signaling axis. Cancer Res 2008; 68:9015-23; PMID:18974147; http://dx.doi.org/10.1158/0008-5472.CAN-08-1935
- Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol 2003; 13:41-7; PMID:12507555; http://dx.doi.org/10.1016/S1044-579X(02)00098-6
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 2004; 428:190-3; PMID:15014502; http://dx.doi.org/10.1038/nature02330
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG, Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004; 428:194-8; PMID:15014503; http://dx.doi.org/10.1038/nature02381
- Boichuk S, Parry JA, Makielski KR, Litovchick L, Baron JL, Zewe JP, Wozniak A, Mehalek KR, Korzeniewski N, Seneviratne DS, et al. The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res 2013; 73:5120-9; PMID:23786773; http://dx.doi.org/10.1158/0008-5472.CAN-13-0579
- Smith EJ, Leone G, DeGregori J, Jakoi L, Nevins JR. The accumulation of an E2F-p130 transcriptional repressor distinguishes a G0 cell state from a G1 cell state. Mol Cell Biol 1996; 16:6965-76; PMID:8943352; http://dx.doi.org/10.1128/MCB.16.12.6965
- Tedesco D, Lukas J, Reed SI. The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev 2002; 16:2946-57; PMID:12435635; http://dx.doi.org/10.1101/gad.1011202
- Falkenhorst J, Grunewald S, Mühlenberg T, Marino-Enriquez A, Reis AC, Corless C, Heinrich M, Treckmann J, Podleska LE, Schuler M, et al. Inhibitor of apoptosis Proteins (IAPs) are commonly dysregulatd in GIST and can be pharmacologically targeted to enhance the pro-apoptotic activity of imatinib. Oncotarget 2016; 7:41390-403; PMID:27167336
- Kim WK, Park M, Kim YK, Tae YK, Yang HK, Lee JM, Kim H. MicroRNA-494 downregulates KIT and inhibits gastrointestinal stromal tumor cell proliferation. Clin Cancer Res 2011; 17:7584-94; PMID:22042971; http://dx.doi.org/10.1158/1078-0432.CCR-11-0166
- Gits CM, van Kuijk PF, Jonkers MB, Boersma AW, van Ijcken WF, Wozniak A, Sciot R, Rutkowski P, Schöffski P, Taguchi T, et al. MiR-17-92 and miR-221/222 cluster members target KIT and ETV1 in human gastrointestinal stromal tumours. Br J Cancer 2013; 109:1625-35; PMID:23969726; http://dx.doi.org/10.1038/bjc.2013.483
- Ihle MA, Trautmann M, Kuenstlinger H, Huss S, Heydt C, Fassunke J, Wardelmann E, Bauer S, Schildhaus HU, Buettner R, et al. miRNA-221 and miRNA-222 induce apoptosis via the KIT/AKT signalling pathway in gastrointestinal stromal tumours. Mol Oncol 2015; 9:1421-33; PMID:25898773; http://dx.doi.org/10.1016/j.molonc.2015.03.013
- Shi Y, Gao X, Hu Q, Li X, Xu J, Lu S, Liu Y, Xu C, Jiang D, Lin J, et al. PIK3C2A is a gene-specific target of microRNA-518a-5p in imatinib mesylate-resistant gastrointestinal stromal tumor. Lab Invest 2016; 96(6):652-60; PMID:26950487; http://dx.doi.org/10.1038/labinvest.2015.157