
ABSTRACT
NRBF2/Atg38 has been identified as the fifth subunit of the macroautophagic/autophagic class III phosphatidylinositol 3-kinase (PtdIns3K) complex, along with ATG14/Barkor, BECN1/Vps30, PIK3R4/p150/Vps15 and PIK3C3/Vps34. However, its functional mechanism and regulation are not fully understood. Here, we report that NRBF2 is a fine tuning regulator of PtdIns3K controlled by phosphorylation. Human NRBF2 is phosphorylated by MTORC1 at S113 and S120. Upon nutrient starvation or MTORC1 inhibition, NRBF2 phosphorylation is diminished. Phosphorylated NRBF2 preferentially interacts with PIK3C3/PIK3R4. Suppression of NRBF2 phosphorylation by MTORC1 inhibition alters its binding preference from PIK3C3/PIK3R4 to ATG14/BECN1, leading to increased autophagic PtdIns3K complex assembly, as well as enhancement of ULK1 protein complex association. Consequently, NRBF2 in its unphosphorylated form promotes PtdIns3K lipid kinase activity and autophagy flux, whereas its phosphorylated form blocks them. This study reveals NRBF2 as a critical molecular switch of PtdIns3K and autophagy activation, and its on/off state is precisely controlled by MTORC1 through phosphorylation.
Introduction
Autophagy is a highly regulated cellular degradation system that engulfs cytosol, organelles, protein aggregates and invading microorganisms into a double-membrane compartment termed the phagophore that matures into an autophagosome; the latter delivers the cargo to lysosomes for degradation.Citation1,2 Dysfunction of autophagy has been implicated in a broad spectrum of human diseases including cancers, neurodegeneration, infectious diseases, metabolic diseases, and aging.Citation3-6
The class III phosphatidylinositol 3-kinase (PtdIns3K) complex plays a key role in autophagy activation by producing phosphatidylinositol-3-phosphate (PtdIns3P) on phagophores.Citation7-9 Several groups have purified the PtdIns3K complex in mammalian cells, which contains at least 7 stoichiometric subunits including PIK3C3, PIK3R4, BECN1, ATG14/ATG14L/Barkor, UVRAG, RUBCN/rubicon and NRBF2.Citation10-21 These 7 subunits form 2 mutually exclusive subcomplexes in which PIK3C3, PIK3R4 and BECN1 are shared. ATG14 forms an autophagy-specific complex with PIK3C3-PIK3R4-BECN1 on phagophore membranes (similar to complex I in yeast),Citation12-15 whereas the UVRAG and RUBCN complex with PIK3C3-PIK3R4-BECN1 is present on endosomal membranes (similar to complex II in yeast).Citation10-12,16 NRBF2, and its yeast counterpart Atg38, was recently identified as the fifth subunit of the autophagic PtdIns3K complex (complex I).Citation13,18-21 Deletion of Nrbf2 in mice or ATG38 in yeast shows a partial autophagy defect, probably due to a function of NRBF2/Atg38 in bridging the interaction between the PIK3C3-PIK3R4 and ATG14-BECN1 subcomplexes.Citation18,20 Interestingly, a suppressive role of NRBF2 in autophagy has also been suggested.Citation21 Therefore, the precise function of NRBF2 remains elusive.
The serine/threonine protein kinase MTOR (mechanistic target of rapamycin) forms a multiprotein complex termed MTORC1 that negatively regulates autophagy.Citation22-25 Amino acid starvation or addition of kinase inhibitors such as rapamycin or Torin 1 suppresses its kinase activity and robustly activates autophagy. To date, only a few MTORC1 substrates in autophagy have been identified. MTORC1 phosphorylates ULK1 (unc51-like autophagy activating kinase 1), an upstream kinase in autophagy,Citation26-29 and this phosphorylation acts counter to ULK1 activation by AMP-activating kinase (AMPK).Citation24 MTORC1 also phosphorylates ATG13,Citation27-30 ATG14,Citation31 and AMBRA132 to regulate autophagy activation. In addition, MTORC1 phosphorylates UVRAG to negatively regulate the late stage of autophagosome-endosome maturation.Citation33 Although a link between MTORC1 and the autophagy essential PtdIns3K complex has been proposed previously,Citation7 the molecular connection between these 2 key enzymatic activities has not been firmly established. Here, we report the identification of NRBF2 as a substrate of MTORC1. MTORC1-mediated NRBF2 phosphorylation functions as a critical molecular switch for PtdIns3K and autophagy activation.
Results
Identification of NRBF2 as a stoichiometric component of PtdIns3K
NRBF2, and its yeast counterpart Atg38, was reported as the fifth subunit of the autophagic PtdIns3K complex (complex I in yeast).Citation18-21 Consistent with this notion, we found that NRBF2 predominately associates with PIK3C3, PIK3R4, BECN1 and ATG14 in the cellular complex isolated by tandem affinity purification from human osteosarcoma U2OS cells (). We have further analyzed binding regions mediating the interaction between NRBF2 and ATG14 by deletion mapping. The minimum binding regions has been narrowed down to amino acids (aa) 343–392 in ATG14 and aa 52–107 in NRBF2 (), which is different from a previous report.Citation18,20
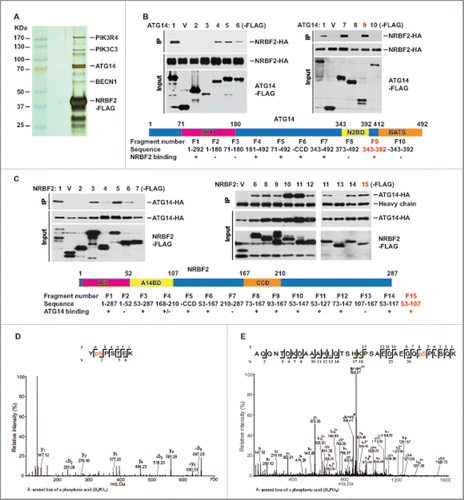
Figure 1. NRBF2 interacts with ATG14 as a stoichiometric component of the PtdIns3K complex. (A) NRBF2 is a stoichiometric component of the PtdIns3K complex. Silver staining of the tandem affinity-purified NRBF2 complex or vector alone in U2OS cells. All the marked bands were identified by mass spectrometry. (B-C) Mapping of the NRBF2-ATG14 binding. Different truncated fragments of Flag-NRBF2 or Flag-ATG14 were expressed with either HA-ATG14 or HA-NRBF2. Immunoprecipitation was performed using the anti-Flag resin. The minimum binding regions were marked in red. V, vector alone. (D-E) Identification of NRBF2 phosphorylation sites at S113 (D) and S120 (E) by mass spectrometry analysis.
NRBF2 is phosphorylated at S113 and S120
Mass spectrometry analysis identified human NRBF2 as a phospho-protein, mainly at serine 113 and serine 120 (). S113 is evolutionarily conserved among vertebrates and S120 is less conserved (). In nutrient-rich wide-type (WT) human (U2OS) cells or mouse embryonic fibroblasts (MEFs), endogenous NRBF2 exists as a doublet on western blots. The slower migrating band shifted down upon calf-intestinal phosphatase (CIP) treatment (), confirming that either human or mouse NRBF2 is a phospho-protein. Both human and mouse endogenous NRBF2 phosphorylation were inhibited by amino acid starvation (Earle's balanced salt solution; EBSS) or MTORC1 inhibition (rapamycin treatment) (). We generated a rabbit polyclonal antibody against the phospho S120 peptide; this phospho-specific antibody specifically recognizes the phosphorylated form of human NRBF2 (). Nrbf2 KO MEFs were complemented with human or mouse WT NRBF2. In these cells, stably expressed NRBF2 also existed as a doublet on western blots (). Nrbf2 KO MEFs were also complemented with either human NRBF2 S113A or S120A mutant or mouse NRBF2S112A (equivalent to S113 in human) mutant. Mutation of human S120 to alanine significantly reduced NRBF2 phosphorylation, whereas the human S113A mutation has a minor but noticeable reduction on NRBF2 phosphorylation (), indicating that S120 is the major phosphorylation site and S113 is a minor phosphorylation site in human cells; S112 is the only phosphorylation site in mouse NRBF2 (). These data indicate that human NRBF2 is heavily phosphorylated at S113 and S120 in unstressed cells.
Figure 2. NRBF2 phosphorylation is negatively regulated by autophagic stress. (A) Alignment of NRBF2 sequences in vertebrates. S113 and S120 are marked in red. (B) Phosphorylation of endogenous NRBF2 in mouse and human cells upon autophagic stress. Mouse embryonic fibroblasts (mouse, upper) and osteosarcoma U2OS cells (human, lower) were treated with EBSS (2 h) or rapamycin (Rapa, 50 nM, 2 h), then endogenous NRBF2 was detected using anti-NRBF2 antibody in western blotting. The phosphorylation of NRBF2 in human U2OS cells was detected by a phospho-specific antibody against human NRBF2 S120. Cell lysate from unstressed cells was also treated with CIP for 1 h as a control. (C) Phosphorylation sites in human and mouse NRBF2. NRBF2 knockout (KO) MEFs were transiently transfected with Flag-tagged human wild-type (WT), S113A or S120A NRBF2, as well as mouse WT or S112A NRBF2 mutants. NRBF2 was detected with anti-Flag antibody. (D) Generation of human NRBF2S113A,120A double mutant that eliminates NRBF2 phosphorylation. NRBF2 KO cells were transiently transfected with Flag-tagged wild-type (WT) NRBF2 (treated with or without CIP), NRBF2S113,120A (AA), and NRBF2S113,120D (DD) mutants. NRBF2 was detected using an anti-Flag antibody. (E) Characterization of the phospho-specific antibody against human NRBF2 S120 using overexpressed NRBF2. NRBF2 KO MEFs were transiently transfected with Flag-tagged WT NRBF2, NRBF2S113,120A (AA), or NRBF2S113,120D (DD) mutants. NRBF2 phosphorylation was detected using anti-p-S120 antibody, and NRBF2 were detected with anti-Flag antibody. (F) Human NRBF2 phosphorylation is negatively regulated by autophagic stress. Nrbf2 KO MEFs were transfected with a plasmid encoding Flag-tagged WT NRBF2 and treated with EBSS (2 h), rapamycin (50 nM, 2 h) or Torin 1 (100 nM, 2 h). NRBF2 phosphorylation was detected using the phospho-specific antibody against human NRBF2 S120 or mobility shift. NRBF2, SQSTM1 and LC3 were detected by western blotting.

Given that both phosphorylation sites S113 and S120 contribute to stress regulated phosphorylation of human NRBF2, we generated an NRBF2 phosphorylation dead mutant with both S120 and S113 changed to alanine (S113A S120A; AA mutant), and an NRBF2 phosphorylation constitutively active mutant with both S120 and S113 changed to aspartic acids (S113D S120D; DD mutant). These mutants, as well as human Flag-tagged NRBF2 WT, were stably expressed in nrbf2−/− MEFs with no NRBF2 expression.Citation20 The NRBF2 AA mutant completely abolished its phosphorylation, and the NRBF2 DD mutant mimicked the phosphorylated form of NRBF2 (). Because MTORC1-mediated NRBF2 phosphorylation is only completely eliminated by the double rather than single mutation, and later on our functional analysis also suggested that single mutants have similar but weaker phenotypes than double mutants (data not shown), we focused on human NRBF2 S113 and S120 double mutants (AA and DD mutants) in the subsequent functional studies.
NRBF2 phosphorylation is negatively regulated by starvation and MTORC1 inhibition
The rabbit polyclonal phospho-specific antibody against the phosphorylated S120 peptide, generated in our lab, specifically recognized NRBF2 WT but not AA or DD mutants (). The nrbf2−/− MEFs complemented with NRBF2 WT were treated with EBSS for amino acid starvation or rapamycin and Torin 1 for MTORC1 inhibition. NRBF2 phosphorylation detected by anti-phospho-NRBF2 antibody or mobility shift was reduced upon EBSS, rapamycin or Torin 1 treatment compared with that in untreated cells (). Thus, this result further confirmed that NRBF2 phosphorylation at S120 is inhibited by autophagic stress.
NRBF2 is a substrate of MTORC1 in vitro
To identify the protein kinases responsible for NRBF2 phosphorylation, we performed an in vitro kinase assay using purified recombinant NRBF2 with immunoprecipitated AMPK, MTORC1 or ULK1 kinases crucial for autophagy regulation. Recombinant NRBF2 was phosphorylated by immunoprecipitated MTORC1-RPTOR and ULK1 but not AMPK (). TSC2, a known substrate of AMPK;Citation34 Atg13, a known substrate of MTORC1;Citation27-30 and BECN1, a known substrate of ULK1,Citation35 were used as positive controls (). The kinase dead mutants of AMPK, MTORC1 and ULK1 were used as negative controls (). The NRBF2S113,120A double mutant and S120A single mutant nearly completely abolished MTORC1-mediated phosphorylation (), confirming that these serines are the major sites for MTORC1-mediated phosphorylation. Interestingly, the NRBF2S113,120A double mutant could still be phosphorylated by ULK1 (), suggesting that ULK1 might phosphorylate NRBF2 at a different site(s) ().
Figure 3. NRBF2 is a substrate of MTORC1. (A-C) Immunoprecipitated AMPK (A), MTORC1-RPTOR (B) and ULK1 (C) were incubated with the indicated recombinant proteins in an in vitro kinase assay. Phosphorylation is detected by autoradiogram and input proteins were Coomassie Blue stained. TSC2 (aa 1300–1367), ATG13, and BECN1 (aa 1–85) served as positive controls for AMPK, MTORC1 and ULK1 kinase activities, respectively. WT, wild type; KD, kinase dead mutant. (D) Recombinant GST-tagged WT, S120A and S113A S120A NRBF2 proteins were used in the in vitro kinase assay with immunoprecipitated MTORC1. (E) Recombinant GST-tagged WT and S113A S120A NRBF2 proteins were used in the in vitro kinase assay with immunoprecipitated ULK1. (F) A schematic diagram to show that NRBF2 is phosphorylated by MTORC1.
NRBF2 S113 and S120 phosphorylation dictates its interaction with the PtdIns3K subcomplex
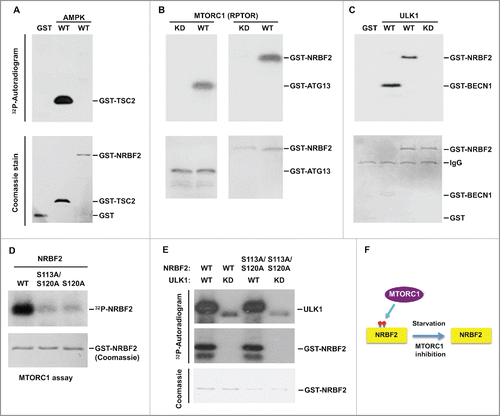
NRBF2 is proposed to bridge the interaction between PIK3C3-PIK3R4 and ATG14-BECN1.Citation18,20 We tested the binding of NRBF2 WT, AA (S113A S120A) and DD (S113D S120D) mutants with PtdIns3K components in a co-immunoprecipitation assay. Surprisingly, the NRBF2 AA mutant differs from the NRBF2 DD mutant in binding either PIK3C3-PIK3R4 or ATG14-BECN1. The phosphorylation-mimicking NRBF2 DD mutant pulled down significantly more PIK3C3 and PIK3R4 than the NRBF2 AA mutant, whereas the phosphorylation dead NRBF2 AA mutant preferentially interacted with ATG14-BECN1, under both nutrient-rich and starved conditions (). NRBF2 WT, heavily phosphorylated under untreated conditions, strongly interacted with PIK3C3 and PIK3R4, at a level similar to the NRBF2 DD mutant. NRBF2 WT, lightly phosphorylated upon amino acid starvation and rapamycin treatment, promoted the interaction with ATG14-BECN1, at a level similar to the NRBF2 AA mutant (). These data suggest that the NRBF2 phosphorylation state dictates its interaction with different PtdIns3K subcomplexes, and MTORC1 can regulate the binding preference of WT NRBF2 to PIK3C3-PIK3R4 or ATG14-BECN1 via modulating NRBF2 phosphorylation ().
Figure 4. NRBF2 S113 S120 phosphorylation differentiates its interaction with PtdIns3K subcomplexes. (A-H) Interactions between NRBF2 (unphosphorylated or phosphorylated mutants) and individual PtdIns3K complex components. HEK293T cells were transfected with MYC-PIK3C3 (A), HA-PIK3R4 (C), HA-ATG14 (E), or HA-BECN1 (G) together with Flag-tagged NRBF2 wild-type (WT), AA, or DD mutant, and treated with EBSS (2 h), rapamycin (50 nM, 2 h), or left untreated. Flag-NRBF2 and its associated proteins were immunoprecipitated uisng anti-Flag M2 resin and probed by individual antibodies in western blotting. The relative ratios of MYC-PIK3C3 (B), HA-PIK3R4 (D), HA-ATG14 (F), or HA-BECN1 (H) to NRBF2 were quantified and standardized. The error bars represent the standard error of the mean from 3 independent experiments within the same treatment group. *, P < 0.05. (I) A schematic showing that phosphorylated NRBF2 preferentially interacts with PIK3C3 and PIK3R4, while unphosphorylated NRBF2 preferentially interacts with ATG14 and BECN1.
NRBF2 S113 and S120 phosphorylation regulates its interaction with the ULK1 protein complex
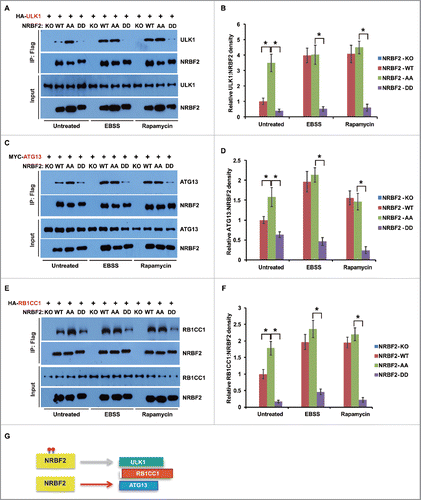
A key event in autophagy inhibition is that MTORC1 phosphorylates the Atg1/ULK1 protein kinase complex upstream of PtdIns3K.Citation26-30 We tested if NRBF2 phosphorylation affects its association with the ULK1 complex in a co-immunoprecipitation assay. The NRBF2 AA mutant promoted its association with the ULK1 complex components ULK1, RB1CC1 and ATG13, whereas the NRBF2 DD mutant reduced its interaction with ULK1, RB1CC1 and ATG13 (). Similar to the NRBF2 DD mutant, NRBF2 WT under untreated conditions had a significantly reduced interaction with the ULK1 complex components ULK1, RB1CC1 and ATG13, compared with the NRBF2 AA mutant or NRBF2 WT upon amino acid starvation and rapamycin treatment (). These findings suggest that MTORC1-mediated NRBF2 phosphorylation critically regulated its interaction with ULK1 protein kinase ().
Figure 5. NRBF2 S113 S120 phosphorylation regulates its interaction with the ULK1 protein complex. (A-F) Interaction between NRBF2 (unphosphorylated or phosphorylated forms) and individual ULK1 complex components. HEK293T cells were transfected with HA-ULK1 (A), MYC-ATG13 (C), or HA-RB1CC1 (E), respectively, together with NRBF2 wild-type (WT), AA, or DD mutant, and treated with EBSS (2 h), rapamycin (50 nM, 2 h) or left untreated. Flag-NRBF2 and its associated proteins were immunoprecipitated using anti-Flag M2 resin and probed with individual antibodies in western blotting. The relative ratios of HA-ULK1 (B), MYC-ATG13 (D), or HA-RB1CC1 (F) to NRBF2 were quantified and standardized. The error bars represent the standard error of the mean from 3 independent experiments within the same treatment group. *, P < 0.05. (G) A schematic showing the interaction between different forms of NRBF2 with the ULK1 protein complex ULK1, RB1CC1 and ATG13. Gray arrow means seldom interact, and red arrow means preferentially interact.
NRBF2 S113 and S120 phosphorylation functions as a switch to control autophagic PtdIns3K complex assembly and ULK1 complex association
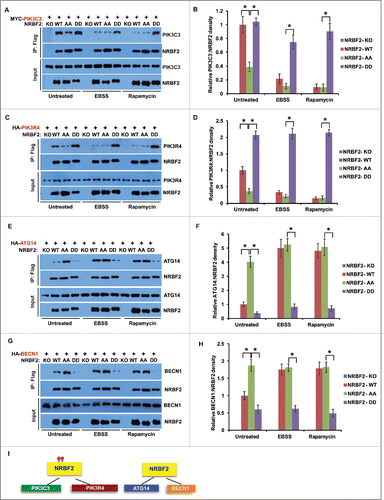
To investigate the function of NRBF2 S113 and S120 phosphorylation in PtdIns3K regulation, nrbf2−/− MEFs were stably complemented with human NRBF2 WT, AA, or DD mutants (). Immunoprecipitations were performed using anti-Flag, anti-BECN1 or anti-ATG14 antibodies. This experiment was designed to address if PtdIns3K complex assembly or ULK1 complex association was regulated by NRBF2 phosphorylation (). The results of anti-Flag immunoprecipitation indicated that the NRBF2 AA mutant preferentially interacted with ATG14-BECN1, as well as ULK1-RB1CC1, whereas the DD mutant preferentially interacted with PIK3C3-PIK3R4 (). WT NRBF2 behaved similarly to the NRBF2 DD mutant under untreated conditions, while WT NRBF2 behaved similarly to the NRBF2 AA mutant upon amino acid starvation or MTORC1 inhibition, which was consistent with co-immunoprecipitation results with the overexpressed NRBF2 ( and ). Meanwhile, significantly more autophagic PtdIns3K complex was assembled in cells expressing the NRBF2 AA mutant detected by both ATG14 () and BECN1 () endogenous immunoprecipitations, whereas much less autophagic PtdIns3K complex assembly was detected in the cells expressing the NRBF2 DD mutant (). In the cells expressing WT NRBF2, less autophagic PtdIns3K complex assembly was detected under untreated conditions, whereas autophagic PtdIns3K complex assembly was significantly increased in cells treated with Torin 1. In cells expressing WT NRBF2 under untreated conditions, less ULK1 complex assembly was detected, which was similar to that in cells expressing the NRBF2 DD mutant. In contrast, in cells expressing WT NRBF2 upon Torin 1 treatment, more ULK1 complex association was promoted, which was similar to that in cells expressing the NRBF2 AA mutant (). Furthermore, endogenous immunoprecipitation using NRBF2, ATG14 and BECN1 antibodies indicated that the MTORC1 activity regulated the assembly of the PIK3C3 complex and ULK1 complex in WT U2OS cells, but not in NRBF2 KD U2OS cells (). These data strongly suggest that NRBF2 S113 and S120 phosphorylation functions as a critical switch to control autophagic PtdIns3K complex assembly and ULK1 complex association.
Figure 6. NRBF2 S113 S120 phosphorylation differentially regulates PtdIns3K complex and ULK1 complex assembly. (A) Generation of stable cell lines in Nrbf2 knockout (KO) mouse embryonic fibroblasts (MEFs) complemented with Flag-tagged NRBF2 wild-type (WT), AA or DD mutants. The expression of Flag-NRBF2 was detected with an anti-Flag antibody in western blotting. (B) A schematic showing how phosphorylation of NRBF2 affects PtdIns3K complex assembly. (C-F) NRBF2 S113 S120 phosphorylation negatively regulates PtdIns3K complex and ULK1 complex assembly. Nrbf2 KO MEFs stably complemented with Flag-tagged NRBF2 WT, AA, or DD mutant were treated with Torin 1 (50 nM, 2 h) or left untreated. Endogenous PtdIns3K complex and ULK1 complex were detected (C). (D-F) Endogenous immunoprecipitations were performed using anti-Flag antibody (D), mouse anti-ATG14 (E) or mouse anti-BECN1 primary antibodies (F), and analyzed by western blotting in the upper panels and quantitative analyses in the bottom panels. The error bars represent the standard error of the mean from 3 independent experiments within the same group. *, P < 0.05.
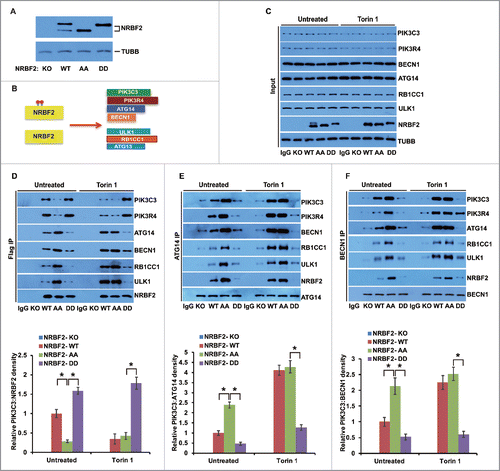
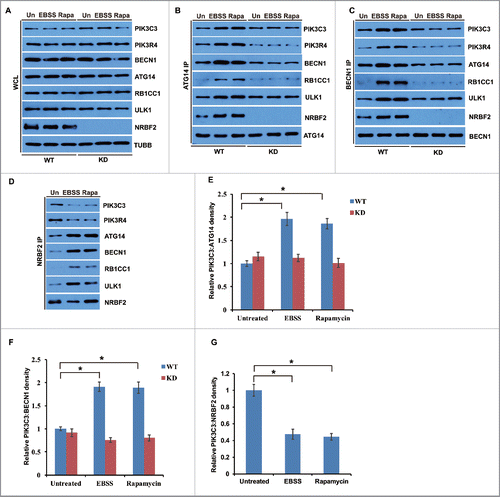
Figure 7. MTORC1-mediated NRBF2 phosphorylation regulates the assembly of the PIK3C3 complex and ULK1 complex in the endogenous co-immunoprecipitation. (A-D) WT and NRBF2 KD U2OS cells were treated with EBSS (2 h), rapamycin (Rapa, 50 nM, 2 h) or left untreated. Endogenous PtdIns3K complex and ULK1 complex were detected (A). (B-D) Endogenous immunoprecipitations were performed using mouse anti-ATG14 (B), mouse anti-BECN1 (C) or rabbit anti-NRBF2 primary antibodies (D). Endogenous NRBF2 and its associated proteins were probed with individual antibodies in western blotting. (E-G) Quantitative analysis of (B-D). The error bars represent the standard error of the mean from 3 independent experiments within the same treatment group. *, P < 0.05.
NRBF2 S113 and S120 phosphorylation affects PtdIns3K lipid kinase activity
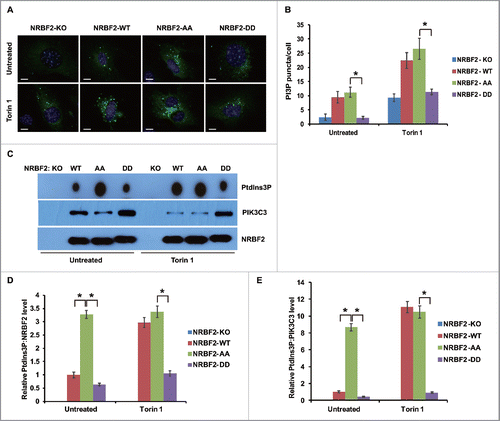
We further investigated if NRBF2 phosphorylation regulates PtdIns3K lipid kinase activity. PtdIns3K phosphorylates phosphatidylinositol to generate PtdIns3P.Citation7-9 The autophagic PtdIns3P binding protein ZFYVE1/DFCP1Citation36 (GFP tagged) was used as a probe to detect PtdIns3P production in cells expressing human WT NRBF2 and phosphorylation mutants. PtdIns3P production shown as GFP-ZFYVE1 puncta was significantly reduced in cells expressing the NRBF2 DD mutant compared with those in cells expressing NRBF2 WT or the AA mutant (). Furthermore, we tested the PtdIns3K lipid kinase activity associated with NRBF2 WT or phosphorylation mutants in an in vitro assay. Lipid kinase activity of immunoprecipitated PtdIns3K complex associated with NRBF2 WT in unstressed cells was similar to that associated with the NRBF2 DD mutant, and both were much lower than the NRBF2 AA mutant (). Torin 1 treatment stimulated NRBF2 WT-associated PtdIns3K lipid kinase activity, to a level similar to the NRBF2 AA mutant. However, NRBF2 DD-associated PtdIns3K lipid kinase activity remained unchanged in cells treated with Torin 1 (). These data suggest that MTORC1-mediated NRBF2 phosphorylation negatively regulates PtdIns3K lipid kinase activity.
Figure 8. NRBF2 S113 S120 phosphorylation regulates PtdIns3K lipid kinase activity. (A) NRBF2 S113 S120 phosphorylation negatively regulates autophagic PtdIns3P production. Autophagic PtdIns3P binding protein GFP-ZFYVE1 was expressed and detected in Nrbf2 knockout (KO) mouse embryonic fibroblasts (MEFs) stably complemented with Flag-tagged NRBF2 WT, AA, or DD mutant. Scale bar, 10 µm. (B) Quantitative analysis of (A). The error bars represent the standard error of the mean from 3 independent experiments within the same group. *, P < 0.05. (C) NRBF2 S113 S120 phosphorylation regulates PtdIns3P production in vitro. Nrbf2 KO MEFs stably complemented with Flag-tagged NRBF2 WT, AA, or DD mutant were treated with Torin 1 (50 nM, 2 h) or left untreated, and then subjected to PtdIns3K lipid kinase assay. (D-E) Quantitative analysis of (C). The relative ratios of PtdIns3P production to NRBF2 (D) and PIK3C3 (E) were quantified and standardized. The error bars represent the standard error of the mean from 3 independent experiments within the same group. *, P < 0.05.
NRBF2 S113 and S120 phosphorylation negatively regulates autophagy
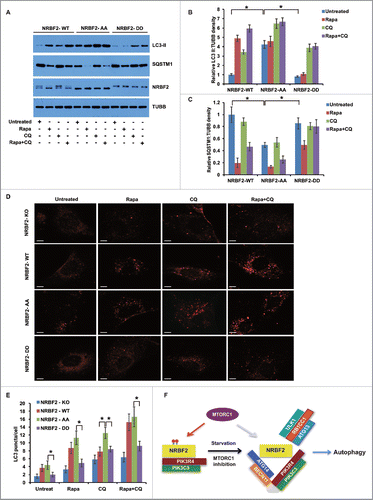
PtdIns3K-ULK1 complex assembly and lipid kinase activity are critical for autophagy activation.Citation7-9 Therefore, we investigated if NRBF2 S113 and S120 phosphorylation modulates autophagy. Autophagy flux was evaluated by western blot and immunostaining. Autophagy was activated and autophagy flux was accelerated in cells expressing the NRBF2 AA mutant, because more LC3 lipidation (LC3-II form) and SQSTM1/p62 degradation were detected in cells expressing the NRBF2 AA mutant compared with those in cells expressing WT NRBF2 (). Meanwhile, the NRBF2 DD mutant behaved similarly to the NRBF2 WT in the unstressed conditions, but failed to activate autophagy as seen with NRBF2 WT upon rapamycin treatment (). Immunostaining of LC3 puncta also supported the activating role of the NRBF2 AA mutant and the inhibitory role of the NRBF2 DD mutant in autophagy (). Taken together, we conclude that MTORC1-mediated NRBF2 phosphorylation was required to fine-tune autophagy activity ().
Figure 9. NRBF2 S113 S120 phosphorylation regulates autophagy. (A) NRBF2 S113 S120 phosphorylation regulates autophagy flux. Nrbf2 knockout (KO) mouse embryonic fibroblasts (MEFs) stably complemented with NRBF2 wild-type (WT), AA, or DD mutant were treated with rapamycin (Rapa, 50 nM, 2 h), CQ (20 µM, 2h) and rapamycin + CQ (50 nM rapamycin and 20 µM CQ, 2 h) or left untreated. Autophagy flux was analyzed by probing LC3-II and SQSTM1 in western blotting. The protein levels of NRBF2 and TUBB were also monitored. (B-C) Quantitative analysis of (A). The relative ratios of LC3-II (B) or SQSTM1 (C) to TUBB were standardized. The error bars represent the standard error of the mean from 3 independent experiments within the same group. *, P < 0.05. (D) Representative LC3 staining images of Nrbf2 KO MEFs stably complemented with NRBF2 WT, AA, or DD mutant, treated with rapamycin (Rapa, 50 nM, 2 h), CQ (20 µM, 2 h) and rapamycin + CQ (50 nM rapamycin and 20 µM CQ, 2 h) or left untreated. Endogenous LC3 staining was performed using an anti-LC3 antibody, and representative images were taken by 40× magnification. Scale bar: 10 µm. (E) Quantitative analysis of (D). LC3 puncta per cell were quantified in 100 cells for each panel. The error bars represent the standard error of the mean from 3 independent experiments within the same group. *, P < 0.05. (F) A working model of MTORC1-mediated NRBF2 S113 S120 phosphorylation as a switch to regulate PtdIns3K complex assembly, lipid kinase activity, ULK1 association and autophagy.
Discussion
In this study, we demonstrate that NRBF2 functions as a fine-tuning autophagy regulator. Under fed conditions, MTORC1 is active and NRBF2 is phosphorylated by MTORC1. Phosphorylated NRBF2 inhibits autophagy, preferentially binds a nonautophagic form of the PtdIns3K complex consisting of PIK3C3-PIK3R4 only, and this NRBF2-associated PtdIns3K complex has low lipid kinase activity. When MTORC1 is inhibited and autophagy is activated, NRBF2 becomes unphosphorylated. This form of NRBF2 binds ATG14-BECN1, promotes autophagic PtdIns3K complex assembly, stimulates PtdIns3P production, promotes ULK1 association and activates autophagy.
The fine-tuning function might help to explain the weak or even contradicting autophagy phenotype in NRBF2-deficient cells.Citation18-21 NRBF2 could function as an activator or inhibitor depending on its phosphorylation status that relies on the expression levels and activity of MTORC1. ATG38 KO yeast maintain 50% of autophagy activity,Citation18 whereas Nrbf2 knockout mice survive to adulthood without neurodegeneration phenotypes.Citation20 NRBF2/Atg38-deficient cells still retain partial autophagic activity.Citation18-21 NRBF2 is probably not essential for PtdIns3K activity and autophagy, but functions as a critical regulator to more readily activate autophagy when challenged with stresses. MTORC1-mediated phosphorylation at S120 is more prominent in human NRBF2. This site is not evolutionarily conserved in mouse NRBF2. Another site (S113 in human and S112 in mouse) is evolutionarily conserved. Phosphorylation at both sites is regulated by autophagic stresses. It is currently not clear if Atg38 is also regulated by TOR-mediated phosphorylation in yeast.
It has been shown that PIK3C3 exists in several different autophagic or nonautophagic subcomplexes.Citation37 ATG14 might help to differentiate the regulation (inhibition or activation) of different PIK3C3 complexes by the upstream energy sensing kinase AMPK in response to starvation.Citation37 Our results support this model. NRBF2 might be critical for the switch from the nonautophagic PIK3C3-PIK3R4 complex to the autophagic PIK3C3-PIK3R4-BECN1- ATG14 complex. A crystal structure of the endosomal PIK3C3 complexCitation38 and an EM structure of the autophagic PIK3C3 complexCitation39-40 were recently revealed. These studies suggest that PtdIns3K subunits form a V/Y-shaped complex, with Vps15/PIK3R4-Vps34/PIK3C3 at one arm, BECN1-ATG14 (complex I) or Vps30/BECN1-Vps38/UVRAG (complex II) at the other arm, and NRBF2 at the base. It will be interesting to determine the conformational change of NRBF2 by phosphorylation in the autophagic PIK3C3 complex (complex I), as an understanding of the conformational change within the complex under both fed and stressed conditions might help to explain how it functions as a critical switch for the assembly and activation of the autophagic PtdIns3K complex. Identification of NRBF2 as an MTORC1 substrate in autophagy will further facilitate our understanding of how MTORC1-mediated phosphorylation events coordinate and specify different steps in autophagy. In addition, NRBF2 is also phosphorylated by ULK1, however, probably through a different site(s), and its function and mechanism in this regard remains to be determined.
Materials and methods
Reagents, antibodies and plasmids
Mouse anti-Flag (F3165), mouse anti-HA (H3663) and rabbit anti-LC3 (L7543) antibodies were purchased from Sigma Aldrich Co. Rabbit anti-NRBF2 (A301–851A), rabbit anti-PIK3R4 (A302–571A) and rabbit anti-RB1CC1 (A301–574A) antibodies were purchased from Bethyl Laboratories. Rabbit anti-PIK3C3 (38–2100) antibody was purchased from Thermo fisher Scientific. Mouse anti-BECN1 (E-8; sc-48341) and mouse anti-MYC (9E10; sc-40) antibodies were purchased from Santa Cruz Biotechnology. Mouse anti-ATG14 (M184–3) antibody was purchased from MBL International Corporation. Mouse anti-SQSTM1 (H00008878-M01) antibody was purchased from Novus Biologicals. Rabbit anti-ULK1 (8054T) antibody was obtained from Cell Signaling Technology. Mouse anti-TUBB/β-tubulin (E7) was purchased from DSHB (Developmental Studies Hybridoma Bank). Anti-phospho-NRBF2 S120 antibodies were generated by immunizing rabbits with the corresponding phospho-peptide, LSQKYS*PSTEK. The phospho-specific antibodies were affinity purified (Cell Signaling Technology).
pCDNA4/TO (Invitrogen, V102020) was modified by introducing the coding sequence of 3× FLAG tag (between NotI and XhoI sites) and designated as pCDNA4-FLAG. The WT or mutant NRBF2 coding sequences were inserted between EcoRV and NotI sites to generate pCDNA4-FLAG-NRBF2, and these plasmids were used to establish stable cell lines. The coding sequence of the HA tag was introduced into the N terminus of the ATG14 coding sequence after the initiation codon ATG by PCR, which was cloned into pCDNA5/FRT/TO (Invitrogen, V601020) between BamHI and Not I sites.
Human NRBF2 plasmids (single-mutant S113 or S120) were constructed with site-directed mutagenesis method using pCDNA4-WT NRBF2 as template. The primer sequences are available in . The PCR amplification were done with heating at 95°C for 30 sec, and followed by 18 cycles: 95°C for 30 sec, 53°C for 30 sec and 68°C for 4 min. PCR fragments were digested with 10 U DpnI enzyme at 37°C for 1 h and gel-purified by QIAquick Gel Extraction Kit (QIAGEN, 28706) according to the instructions of the manufacturer. The extracted products were transformed and verified by DNA sequencing. Double-mutant S113 S120 NRBF2 plasmids AA and DD were constructed using pCDNA4-S113A or S113D NRBF2 as templates, with S120A and S120D primers, respectively.
Table 1. The primers for the NRBF2 mutants.
Cell culture, cell transfection and generation of stable cell line
nrbf2−/− MEF cells were generated as described previously.Citation20 HEK293T, U2OS and MEF cells were cultured in DMEM (Sigma Aldrich Co., D5796) supplemented with 10% HyClone™ fetal bovine serum (GE Healthcare Life Sciences, SH30088.03) and 1% Gibco™ penicillin-streptomycin solution (Thermo Fisher Scientific, 15140122). Cell transfection was performed using Lipofectamine® 2000 (Thermo Fisher Scientific, 11668019; for MEF cells) or polyethylenimine (PEI; Polysciences Inc., 23966–2; for HEK293T cells) according to protocols provided by the manufacturers. To establish the NRBF2-complemented stable MEF cell lines, recombinant pCDNA4 plasmids containing the sequence of NRBF2 WT, AA or DD mutants were transfected into nrbf2−/− MEF cells. Stable pools were obtained in the presence of 100 μg/ml hygromycin B (Thermo Fisher Scientific, 10687010). Single colonies were picked and screened for the expression of Flag-NRBF2 using mouse anti-Flag antibody, and confirmed using rabbit anti-NRBF2 antibody by western blotting ().
The inducible shRNA NRBF2 knockdown U2OS cell lines were established according to the manufacturer. The shRNA sequences were designed with siRNA Target Finder (Ambion) and cloned into BglII and HindIII sites of pSuperior.puro vector (Oligoengine, 38029). The shRNA coding sequence for human NRBF2 knockdown is GATCCCCGAGGCTATTTCTTGTCACATTCAAGAGAGGTTGTTCTGTTCCACTGCTTTTTA, with the targeted sequence of NRBF2 in italics. The inducible knockdown stable cell lines were generated from U2OSTetR cells.Citation13 To measure the knockdown efficiency, cells were treated with 500 ng/mL doxycycline (DOX; Sigma Aldrich Co., D9891) for 3 d, followed by western blotting against NRBF2.
Tandem affinity purification of NRBF2 cellular complex
The tandem affinity purification strategy to fractionate the NRBF2 complexes from human cells was performed as described previously with some modification.Citation11,41-43 Briefly, the stable cell line capable of expressing ZZ-NRBF2–3× Flag upon DOX induction was obtained. The lowest dose of DOX to induce expression of exogenous NRBF2 close to the endogenous level was chosen for complex purification. The cells were grown in DMEM with 10% fetal bovine serum and harvested near confluence. The cell pellet was washed with chilled phosphate-buffered saline (PBS; Sigma Aldrich Co., D8662) 3 times and then suspended in TAP buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% NP-40 [Sigma Aldrich Co., I8896], 1 mM NaF, 1 mM Na3VO4, 1 mM EDTA and Protease inhibitor cocktail [Roche Life Science, 5056489001]). The resuspended cell pellets were gently vortexed for 1 min after a 30-min incubation on ice. The homogenate was centrifuged for 20 min at 10,000 × g. The supernatant was transferred to a fresh tube. Then 0.8 mL of packed IgG beads (GE Healthcare Life Sciences, 17–0969–01) was added to the supernatant, followed by gentle rotation overnight at 4°C. The bound protein was eluted by TEV protease (Sigma Aldrich Co., T4455) cleavage and further purified by anti-Flag antibody-conjugated beads (Sigma Aldrich Co., A2220). The final eluates from the Flag-M2 beads using 3× Flag peptide were resolved by SDS-PAGE on a 4–12% gradient gel and visualized by silver staining. Specific bands were cut off and subjected to mass spectrometry (MS) analysis.
Mass spectrometry analysis
Protein bands on the SDS-PAGE gel were destained, and then reduced in 10 mM DTT at 56°C for 30 min followed by alkylation in 55 mM iodoacetamide at dark for 1 h. After that the protein bands were in-gel digested with sequencing grade trypsin (10 ng/μL trypsin, 50 mM ammonium bicarbonate, pH 8.0) overnight at 37°C. Peptides were extracted with 5% formic acid-50% acetonitrile and 0.1% formic acid-75% acetonitrile sequentially and then concentrated to ∼20 μl. The extracted peptides were separated by an analytical capillary column (50 μm × 10 cm) packed with 5-μm spherical C18 reversed phase material (YMC, 063691). An Agilent 1100 binary pump was used to generate an HPLC gradient as follows: 0–5% B in 5 min, 5–40% B in 25 min, 40–100% B in 15 min (B = 0.1 M acetic acid-70% acetonitrile). The eluted peptides were sprayed into a QSTAR XL mass spectrometer (AB Sciex, Foster City, CA, USA) equipped with a nano-ESI ion source. The mass spectrometer was operated in information-dependent mode with 1 MS scan followed by 3 MS/MS scans for each cycle. Database searches were performed on an in-house Mascot server (Matrix Science Ltd., London, UK) and the following variable modifications were included: oxidation on methionine, carbamidomethylation on cysteine, phosphorylation on serine, threonine, and tyrosine. The tandem mass spectra of matched phosphorylated peptides were manually checked for their validity.
AMP-activated protein kinase complex purification and the kinase assay
The kinase substrate protein was prepared from either E. coli or the transfected HEK293 cells. The proteins were eluted from the affinity beads, followed by dialysis against 50 mM Tris, pH 8.0, 10% glycerol overnight. AMPK complexes were prepared from the HEK293 cells, in which MYC-PRKAA1, MYC-PRKAG1, and Flag-PRKAB1 were transfected using PEI. The cells were lysed with Mild Lysis Buffer (MLB) containing 10 mM Tris, pH 7.5, 2 mM EDTA, 100 mM NaCl, 0.5% NP-40, 50 mM NaF, 1 mM Na3VO4, and Roche Protease inhibitor cocktail. AMPK complexes were immuno-purified using Flag-M2 beads, which were pre-equilibrated with the MLB. The AMPK immune-complex was extensively washed with the MLB, followed once by buffer E containing 50 mM Tris, pH 8.0, and 0.05% NP-40. The AMPK complex was eluted with buffer E containing 200 μg/ml 3× Flag peptide. The resulting AMPK complexes were further purified using a desalting column with the buffer containing 50 mM Tris, pH 8.0, 10% glycerol. For the kinase assay, 50–100 ng of AMPK complexes were incubated with 0.5 μg of the substrate protein (or the proteins of interest) in the presence of kinase buffer containing 20 mM HEPES, pH 7.4, 1 mM EGTA, 0.4 mM EDTA, 5 mM MgCl2, 0.05 mM DTT, 0.2 mM AMP, 0.1 mM ATP. For the radioisotope assay, 2 μCi 32P-γ-ATP (perkinElmer, NEG035C010MC) was included in the reaction. The reaction was carried out at 37°C for 30 min and terminated by adding SDS-PAGE sample buffer.
MTORC1 assay
MTORC1 complex was prepared from the HEK293 cells, in which MYC-MTOR, and HA-RPTOR were transfected using PEI. MTORD2338A was used as the kinase inactive mutant. To obtain MTORC1, the cells were lysed with CHAPS buffer containing 40 mM HEPES, pH 7.4, 2 mM EDTA, 10 mM pyrophosphate, 10 mM gylcerophosphate, 0.3% CHAPS (Amresco, 0465). This zwitterionic detergent buffer was used as a base solution for the following immunoprecipitation, and washing. MTORC1 (MYC-MTOR-HA-RPTOR) complex was immuno-purified using HA-beads, which were pre-equilibrated with the CHAPS buffer, and then the immune complex was extensively washed with the CHAPS buffer several times. The resulting beads containing MTORC1 complex was washed with kinase assay buffer containing 25 mM HEPES, pH 7.4, 50 mM KCl, 5% glycerol, 10 mM MgCl2, 5 mM MnCl2, 0.1 mM DTT once. For the kinase assay, the MTORC1 immune complex was incubated with 0.5 μg of the substrate protein (or the proteins of interest) in the kinase buffer containing 50 μM ATP, and 2 μCi 32P-γ-ATP for the radioisotope assay. The reaction was carried out at 37°C for 30 min on the vibrating incubator, and terminated by adding SDS-PAGE sample buffer. Phosphorylation of the substrate proteins was determined by 32P-autoradiogram.
ULK1 kinase assay
ULK1 kinase was prepared from HEK293 cells, in which Flag-ULK1 (wild-type or kinase-dead K46R) was transfected using PEI. The ULK1 kinase was immuno-purified using Flag-M2 beads as described for the AMPK preparation. The purified ULK1 kinase from Flag-M2 beads was desalted using a micro-desalting spin column with buffer containing 50 mM Tris, pH 8.0, 10% glycerol. For the kinase assay, 50–100 ng of ULK1 kinase was incubated with 0.5 μg of the substrate protein (or the proteins of interest) in the presence of kinase buffer containing 20 mM HEPES, pH 7.4, 1 mM EGTA, 0.4 mM EDTA, 5 mM MgCl2, 0.05 mM DTT, 0.05 mM ATP, 2 μCi 32P-γ-ATP for the radioisotope assay. The reaction was carried out at 37°C for 30 min and terminated by adding SDS-PAGE sample buffer. In parallel, the same kinase reaction was carried out using ULK1K46R as the negative control. Phosphorylation of the substrate proteins was determined by 32P-autoradiogram.
Immunoprecipitation assays
For Flag tag immunoprecipitation assay, whole-cell lysates lysed with TAP buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% NP-40, 1 mM NaF, 1 mM Na3VO4, 1 mM EDTA and Protease inhibitor cocktail) were collected after pelleting cellular debris using centrifugation at 1,5000xg, 10 min. Lysate was then incubated with 20 μl anti-Flag M2 Affinity Gel (Sigma Aldrich Co., A2220) for 16 h at 4°C. Beads were washed 3 times with TAP buffer and then eluted with a 1:10 ratio of 200 μg/ml 3× Flag peptide to lysate. The eluate were subjected to western blotting. For endogenous immunoprecipitation assay, whole-cell lysates lysed using TAP buffer were collected after pelleting cellular debris using centrifugation. Lysate was pre-cleaned by incubating with 20 μl protein A/G agarose (Santa Cruz Biotechnology, sc-2003) and goat anti-mouse IgG (Santa Cruz Biotechnology, sc-2005) for 30 min at 4°C. After centrifugation, the supernatant was incubated with mouse anti-BECN1 antibody (1:500 dilution) or mouse anti-ATG14 antibody (1:500 dilution) for 1 h at 4°C, followed by the addition of 20 μl protein A/G agarose and incubation overnight at 4°C. Beads were washed 3 times with TAP buffer and then mixed with 60 μl SDS loading buffer. The supernatant fractions were subjected to western blotting according to standard protocols. The immunoprecipitation efficiency was evaluated by the amount of the immunoprecipitated proteins normalized with the bait proteins.
Immunostaining assay
For LC3 immunostaining assay, NRBF2-KO, NRBF2-WT, NRBF2-AA and NRBF2-DD stable cell lines were trypsinized and transferred to 6-well dishes containing coverslips. After 24 h, cells were cultured in the following conditions: untreated, rapamycin (50 nM), CQ (20 µM) and rapamycin + CQ (50 nM rapamycin and 20 µM CQ) for 2 h. Then cells were fixed using ice-cold methanol for 4 min at 4°C. Cells were then washed 3 times with PBS and blocked with blocking buffer (2.5% BSA [Sigma Aldrich Co., V900933] + 0.1% Triton X-100 [Sigma Aldrich Co., T8787] in PBS) at room temperature for 2 h. Cells were incubated with rabbit anti-LC3 antibodies (1:10000 dilution) at 4°C overnight, washed with PBS buffer and then incubated with goat anti-rabbit secondary antibodies (1:10,000 dilution; Santa Cruz Biotechnology, sc-2004) for 2 h at room temperature. For the ZFYVE1 assay, NRBF2-KO, NRBF2-WT, NRBF2-AA and NRBF2-DD stable cell lines were transfected with pEGFP-ZFYVE1 according to the protocol described above. 24 h after transfection, these cells were trypsinized and transferred to 6-well dishes containing coverslips. After another 24 h, cells were treated with 50 nm Torin 1 for 2 h. Then the cells were fixed using 4.0% paraformaldehyde for 10 min at 4°C. Cells were then washed 3 times with PBS and stained with DAPI buffer (1:10,000 dilution) at room temperature for 5 min. Slides were examined by using a laser scanning microscope (Zeiss LSM Z2 META ultraviolet-visible).
In vitro PtdIns3K lipid kinase assay
Nrbf2 KO MEFs and NRBF2-WT, NRBF2-AA and NRBF2-DD stable cell lines were lysed in lysis buffer containing 20 mM HEPES (pH 7.0), 150 mM NaCl, 1 mM EDTA, 1.0% Triton X-100, Proteinase Inhibitor Cocktail and Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, 78420). Immunoprecipitation was performed with Flag-M2 beads. Immune complexes were washed 3 times in lysis buffer, followed by an additional wash in TNE buffer (10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 100 uM Na3VO4). A fraction (1/5) of the beads of immununoprecipitates was aliquoted for western blot. Residual beads were resuspended in 60 μl TNE buffer containing 20 μg sonicated phosphatidylinositol (Avanti Polar Lipids, 840042C), and incubated with 50 μM cold ATP, 10 μCi 32P-ATP, and 5 mM MgCl2 at room temperature for 30 min. The reaction was terminated by adding 20 μl 8 M HCl, and the organic phase was extracted with 160 μl chloroform:methanol (1:1). Extracted phospholipid products were resolved by TLC using a silica-coated gel 60 F254 (EMD Millipore, S07875) and a solvent composed of chloroform:methanol:H2O:ammonium hydroxide (v:v:v:v, 9:7:1.7:0.3). 32P-PtdIns3P was subjected to autoradiography on an X-ray film.
Image quantification and statistical analysis
Results of western blots and lipid kinase assay were quantified by densitometry analysis using ImageJ software (NIH, USA) with same parameters. The data were analyzed by ANOVA and the pairwise comparisons were done by Bonferroni post-hoc test. Experiments were repeated at least 3 times for quantification. Immunostaining results were quantified by counting puncta formation and the statistical significance of differences between mean values (P < 0.05) was evaluated using the unpaired Student t test. At least 3 different visual fields containing at least 100 cells were counted for each condition. All data were expressed as the mean ± standard deviation.
Abbreviations
| AMPK | = | AMP-activating kinase |
| ATG14 | = | autophagy-related 14 |
| KD | = | knockdown |
| KO | = | knockout |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MEFs | = | mouse embryonic fibroblasts |
| MTOR | = | mechanistic target of rapamycin |
| NR | = | nutrient rich |
| NRBF2 | = | nuclear receptor binding factor 2 |
| PIK3C3/Vps34 | = | phosphatidylinositol 3-kinase, catalytic subunit type 3 |
| PIK3R4/p150/Vps15 | = | phosphoinositide-3-kinase, regulatory subunit 4 |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| PtdIns3P | = | phosphatidylinositol-3-phosphate |
| RB1CC1/FIP200 | = | RB1 inducible coiled-coil 1 |
| SQSTM1/p62 | = | sequestosome 1 |
| TAP | = | tandem affinity purification |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| WT | = | wild type |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dandan Chen, Jianxu Li, and Muqing Cao for technical assistance.
Funding
The work was supported by grants to Q.Z. from the Welch Foundation (I-1864), the Cancer Prevention & Research Institute of Texas (RP140320 and RP170382), an American Cancer Society Research Scholar Grant (RSG-11–274–01-CCG) and NIH R01 (GM116908), and the National Basic Research Program of China (973 Program, 2013CB117301), the National Natural Science Foundation of China (31528018 and 31472101), the 111 Project (B16044) and Beijing Nova program (xx2013055) to X. M.. The work was partly supported by China Scholarship Council to X. M. and S.Z.
Related Research Data
References
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463-77; PMID:15068787; http://dx.doi.org/10.1016/S1534-5807(04)00099-1
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931-7; PMID:17712358; http://dx.doi.org/10.1038/nrm2245
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961-7; PMID:17972889; http://dx.doi.org/10.1038/nrc2254
- Zhi X, Zhong Q. Autophagy in cancer. F1000Prime Rep 2015; 7:18; PMID:25750736; http://dx.doi.org/10.12703/P7-18
- Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J 2008; 410:1-17; PMID:18215151; http://dx.doi.org/10.1042/BJ20071427
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol 2010; 20:355-62; PMID:20356743; http://dx.doi.org/10.1016/j.tcb.2010.03.002
- Levine B, Liu R, Dong X, Zhong Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol 2015; 25:533-44; PMID:26071895; http://dx.doi.org/10.1016/j.tcb.2015.05.004
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8:688-99; PMID:16799551; http://dx.doi.org/10.1038/ncb1426
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 2008; 10(7):776-87; PMID:18552835; http://dx.doi.org/10.1038/ncb1740
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms 2 distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360-72; PMID:18843052; http://dx.doi.org/10.1091/mbc.E08-01-0080
- Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A 2008; 105:19211-6; PMID:19050071; http://dx.doi.org/10.1073/pnas.0810452105
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385-96; PMID:19270696; http://dx.doi.org/10.1038/ncb1846
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11:468-76; PMID:19270693; http://dx.doi.org/10.1038/ncb1854
- Sun Q, Westphal W, Wong KN, Tan I, Zhong Q. Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A 2010; 107:19338-43; PMID:20974968; http://dx.doi.org/10.1073/pnas.1010554107
- He S, Ni D, Ma B, Lee JH, Zhang T, Ghozalli I, Pirooz SD, Zhao Z, Bharatham N, Li B, et al. PtdIns(3)P-bound UVRAG coordinates Golgi-ER retrograde and Atg9 transport by differential interactions with the ER tether and the beclin 1 complex. Nat Cell Biol 2013; 15:1206-19; PMID:24056303; http://dx.doi.org/10.1038/ncb2848
- Araki Y, Ku WC, Akioka M, May AI, Hayashi Y, Arisaka F, Ishihama Y, Ohsumi Y. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J Cell Biol 2013; 203:299-313; PMID:24165940; http://dx.doi.org/10.1083/jcb.201304123
- Cao Y, Wang Y, Abi Saab WF, Yang F, Pessin JE, Backer JM. NRBF2 regulates macroautophagy as a component of Vps34 Complex I. Biochem J 2014; 461:315-22; PMID:24785657; http://dx.doi.org/10.1042/BJ20140515
- Lu J, He L, Behrends C, Araki M, Araki K, Jun Wang Q, Catanzaro JM, Friedman SL, Zong WX, Fiel MI, Li M, Yue Z. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat Commun 2014; 5:3920; PMID:24849286
- Zhong Y, Morris DH, Jin L, Patel MS, Karunakaran SK, Fu YJ, Matuszak EA, Weiss HL, Chait BT, Wang QJ. Nrbf2 protein suppresses autophagy by modulating Atg14L protein-containing Beclin 1-Vps34 complex architecture and reducing intracellular phosphatidylinositol-3 phosphate levels. J Biol Chem 2014; 289:26021-37; PMID:25086043; http://dx.doi.org/10.1074/jbc.M114.561134
- Arsham AM, Neufeld TP. Thinking globally and acting locally with TOR. Curr Opin Cell Biol 2006; 18:589-97; PMID:17046229; http://dx.doi.org/10.1016/j.ceb.2006.09.005
- Munson MJ, Ganley IG. MTOR, PIK3C3 and autophagy: Signaling the beginning from the end. Autophagy 2015; 11:2375-6; PMID:26565689; http://dx.doi.org/10.1080/15548627.2015.1106668
- Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 2015; 125:25-32; PMID:25654547; http://dx.doi.org/10.1172/JCI73939
- Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature 2015; 517:302-10; PMID:25592535; http://dx.doi.org/10.1038/nature14190
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; http://dx.doi.org/10.1091/mbc.E08-12-1249
- Chang YY, Neufeld TP. 2009. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell 2009; 20:2004-14; PMID:19225150; http://dx.doi.org/10.1091/mbc.E08-12-1250
- Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318; http://dx.doi.org/10.1074/jbc.M900573200
- Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009; 5:973-9; PMID:19597335; http://dx.doi.org/10.4161/auto.5.7.9296
- Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy 2013; 9:1983-95; PMID:24013218; http://dx.doi.org/10.4161/auto.26058
- Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, De Zio D, Nazio F, Antonioli M, D'Orazio M, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol 2015; 17:20-30; PMID:25438055; http://dx.doi.org/10.1038/ncb3072
- Kim YM, Jung CH, Seo M, Kim EK, Park JM, Bae SS, Kim DH. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell 2015; 57:207-18; PMID:25533187; http://dx.doi.org/10.1016/j.molcel.2014.11.013
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577-90; PMID:14651849; http://dx.doi.org/10.1016/S0092-8674(03)00929-2
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741-50; PMID:23685627; http://dx.doi.org/10.1038/ncb2757
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685-701; PMID:18725538; http://dx.doi.org/10.1083/jcb.200803137
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013; 152:290-303; PMID:23332761; http://dx.doi.org/10.1016/j.cell.2012.12.016
- Rostislavleva K, Soler N, Ohashi Y, Zhang L, Pardon E, Burke JE, Masson GR, Johnson C, Steyaert J, Ktistakis NT, et al. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science 2015; 350:aac7365; PMID:26450213; http://dx.doi.org/10.1126/science.aac7365
- Baskaran S, Carlson LA, Stjepanovic G, Young LN, Kim do J, Grob P, Stanley RE, Nogales E, Hurley JH. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. eLife 2014; 3:e05115; PMID:25490155; http://dx.doi.org/10.7554/eLife.05115
- Young LN, Cho K, Lawrence R, Zoncu R, Hurley JH. Dynamics and architecture of the NRBF2-containing phosphatidylinositol 3-kinase complex I of autophagy. Proc Natl Acad Sci U S A 2016; 113:8224-9; PMID:27385829; http://dx.doi.org/10.1073/pnas.1603650113
- Fan W, Tang Z, Chen D, Moughon D, Ding X, Chen S, Zhu M, Zhong Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 2010; 6:614-21; PMID:20495340; http://dx.doi.org/10.4161/auto.6.5.12189
- Chen D, Fan W, Lu Y, Ding X, Chen S, Zhong Q. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol Cell 2012; 45:629-41; PMID:22342342; http://dx.doi.org/10.1016/j.molcel.2011.12.036
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013; 502:254-7; PMID:24089205; http://dx.doi.org/10.1038/nature12606