
ABSTRACT
Macroautophagy/autophagy is a tightly regulated intracellular catabolic pathway involving the lysosomal degradation of cytoplasmic organelles and proteins to be recycled into metabolic precursors. AMBRA1 (autophagy and Beclin 1 regulator 1) has a central role in the autophagy signaling network; it acts upstream of MTORC1-dependent autophagy by stabilizing the kinase ULK1 (unc-51 like autophagy activating kinase 1) and by favoring autophagosome core complex formation. AMBRA1 also regulates the cell cycle by modulating the activity of the phosphatase PPP2/PP2A (protein phosphatase 2) and degradation of MYC. Of note, post-transcriptional regulation mediated by noncoding microRNAs (MIRNAs) contributes significantly to control autophagy. Here we describe a new role for the microRNA MIR7–3HG/MIR-7 as a potent autophagy inhibitor. Indeed, MIR7–3HG targets the 3′ untranslated region (UTR) of AMBRA1 mRNA, inducing a decrease of both AMBRA1 mRNA and protein levels, and thus causing a block in autophagy. Furthermore, MIR7–3HG, through AMBRA1 downregulation, prevents MYC dephosphorylation, establishing a positive feedback for its own transcription. These data suggest a new and interesting role of MIR7–3HG as an anti-autophagic MIRNA that may affect oncogenesis through the regulation of the tumor suppressor AMBRA1.
KEYWORDS:
Introduction
Macroautophagy (hereafter referred to as autophagy) is a highly conserved catabolic pathway that is involved in cellular degradation of long-lived proteins or dysfunctional cellular components, through the action of lysosomes.Citation1 Central to this process is the formation of the autophagosome, a double-membraned vesicle, which is responsible for the delivery of cytosolic cargoes to the lysosome.Citation2 In addition to its housekeeping and homeostatic function, autophagy constitutes a key prosurvival response that allows adaptation to unfavorable conditions.Citation3,4,5 Autophagy is essential for normal development,Citation6,7 and defects in this process are linked to numerous human diseases, including neurodegeneration and cancer; this fact underlines why tight control of autophagy is essential.Citation8,9 In a tumor microenvironment, autophagy promotes cancer cell survival in response to metabolic stress.Citation10,11 However, hyperactivation of autophagy can lead to cell death, and human cancers often display inactivating mutations in autophagy-promoting genes.Citation12,13 Thus, in relation to tumorigenesis, the role of autophagy is complex and likely depends on the genetic background of the cell as well as on the environmental cues the cell is exposed to.Citation14
MicroRNAs (MIRNAs), a class of small noncoding RNAs, can bind the 3′ untranslated region (UTR) of target messenger RNAs (mRNAs), causing translational inhibition or mRNA destabilization.Citation15 In recent years, MIRNAs have been closely linked to the post-translational regulation of almost all fundamental biological pathways, including autophagy.Citation16,17,18,19 Conversely, autophagy is also important to maintain MIRNA homeostasis.Citation20 Recent data provide evidence that under stress conditions, a number of MIRNAs including MIR30A, MIR101, MIR130A, and MIR196 are capable of modulating autophagy activity by changing intracellular levels of key autophagy proteins.Citation21,22
This bidirectional crosstalk has been defined as the autophagoMIR pathway,Citation23 and its deregulation is obviously central to cancer oncogenesis and progression. Here we describe a new member of this pathway. In fact, MIR7–3HG turns out to be an anti-autophagic MIRNA that acts through the downregulation of AMBRA1, a key protein in autophagy. AMBRA1 has been identified as a pivotal factor in regulating autophagy in vertebrates,Citation24,25 which promotes the interaction of BECN1/Beclin 1 with its target lipid kinase, PI3K3C3/Vps34, thereby mediating the early phase of autophagy in autophagosome nucleation.Citation26 Several AMBRA1 interaction partners have been identified so far,Citation27 highlighting the pivotal role of this protein as an efficient scaffold-molecule that is able to coordinate many intracellular processes with autophagy.Citation28,29 Also, a number of AMBRA1 post-translational modifications were characterized, while very little is known about its transcriptional regulation. In this study we have characterized MIR7–3HG and AMBRA1 regulatory interaction in a number of cell lines, and subsequently focused on a model of lung cancer (the A549 cell line) exhibiting low levels of AMBRA1 and high levels of the MIR7–3HG transcriptional regulator MYC.Citation28,30 Of note, even though MIR7–3HG is generally described to have a tumor-suppressive role,Citation31 in some instances the opposite effect has also been reported. In particular, in lung carcinomas, a poor prognosis is associated with MIR7–3HG overexpression.Citation30 Furthermore, inhibition of MIR7–3HG causes reduced proliferation and increased apoptosis in HeLa and lung carcinoma cell lines, respectively,Citation32 suggesting that high MIR7–3HG expression is not necessarily beneficial in terms of inhibiting carcinogenesis. In the present study, we also highlight MIR7–3HG as an oncogenic MIRNA promoting its own MYC-mediated transcription through the downregulation of the tumor suppressor AMBRA1.
Results
In silico analysis of most significant MIRNA candidates to target the AMBRA1 3′ UTR
In order to identify and study which MIRNAs could potentially regulate autophagy through targeting the AMBRA1 3′ UTR, we performed a global analysis using 4 independent target prediction algorithms, DIANA-microT v3.032, TargetScan 5.2,Citation33,34 microrna.orgCitation35 and PicTar.Citation36 We thus obtained a list of the MIRNA families broadly conserved among vertebrates (Fig. S1A). The majority of predicted MIRNAs were unknown regulators of autophagy, with the exception of MIR23B. In fact, MIR23B regulates autophagy, associated with radio-resistance in pancreatic cancer cells, through the targeting of ATG12 and HMGB2.Citation37 By contrast, the other predicted MIRNAs (MIR7–3HG, MIR9 and MIR200B) we identified could be potentially novel regulators of autophagy. To determine whether these MIRNAs' binding sites were functional on the full-length AMBRA1 3′-UTR, HeLa cells were cotransfected with MIRNA overexpressing plasmids and a reporter plasmid, containing Renilla luciferase fused to the AMBRA1 full-length 3′-UTR sequence. Indeed, MIR7–3HG overexpression suppressed significantly (47%) the activity of Renilla luciferase compared to the control, whereas MIR200B and MIR23B over-dosage had no effects on the reporter activity (Fig S1B). MIR9 caused, as well, a mild decrease (27%) in Renilla expression. As a consequence, we decided to focus our attention on MIR7–3HG whose overexpression led to a remarkable downregulation of Renilla activity (Fig. S1B).
AMBRA1 mRNA is a direct target of MIR7–3HG
A stable interaction based on partial base pairing between mature MIRNA and the 3′-UTR region of an mRNA (the “MIRNA responsive element”, MRE) is crucial for MIRNA-dependent regulation of target mRNA and protein levels. A seed sequence is essential for the MIRNA::mRNA interaction and it is located in the MIRNA 5′-end, at position 2 to 7. In the seed region occurs the perfect match between a MIRNA and its mRNA target, while additional residues of the mature MIRNAs stabilize the contact.Citation38 To validate the effect of MIR7–3HG on AMBRA1, its wild-type 3′-UTR region was cloned downstream of the cDNA encoding Renilla luciferase in the psiCHECK-2 vector (). In parallel, we also created a mutant version of this construct by introducing 2 nonconsecutive base changes in the seed region of the AMBRA1-specific MRE for MIR7–3HG (). Co-transfection of MIR7–3HG, together with the wild-type luciferase vector in HEK293 cells, resulted in a significant decrease in the luciferase activity, compared with control levels (). By contrast, MIR7–3HG had no significant effects on the luciferase levels expressed by the mutant construct (). These results indicate that MIR7–3HG can downregulate AMBRA1 levels by directly targeting the above described MRE present in its 3′-UTR region.
Figure 1. MIR7–3HG (MIR-7) targets AMBRA1. (A) Schematics representing sequences of AMBRA1 MRE (top sequence) or its artificially mutated form (bottom sequence) cloned within the 3′-UTR of the reporter Renilla luciferase in the vector psiCHECK-2. Mutations are marked in lower case letters. MIR7–3HG seed sequence is in bold. (B) Normalized luciferase activity in lysates from HEK293 cells, co-transfected with the wild-type or mutant AMBRA1 luciferase constructs and negative control (Empty Vector) or MIR7–3HG plasmids (indicated as MIR7; mean ± SD of 3 independent experiments, *p < 0.05.) N.S., not significant. (C) Western blot analysis of cytoplasmic AMBRA1 levels 24 h after transfection with MIR7–3HG-encoding plasmids (indicated as MIR7) or negative control (Empty Vector). ACTB was used as an endogenous control. One representative western blot of 3 independent experiments is shown. The right panel shows ImageJ densitometry analysis of 3 independent experiments (mean ± SD of independent experiments,**p < 0.01). (D) AMBRA1 mRNA expression was analyzed by quantitative RT-qPCR 24 h after transfection with MIR7–3HG-encoding plasmids (indicated as MIR7) or negative control (Empty Vector). Relative quantification was measured by means of the comparative cycle threshold (ΔΔCt) method (mean ± SD of 3 independent experiments, *p < 0.05).
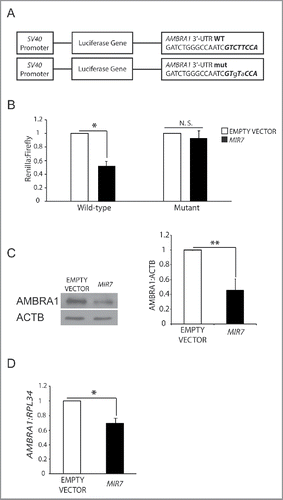
Effect of MIR7–3HG overexpression on cellular AMBRA1 levels
To assess whether MIR7–3HG affected the translation of the endogenous AMBRA1, we analyzed its protein levels upon transfection with MIR7–3HG-overexpressing plasmid in HeLa cells (Fig. S1C). Western blot analysis indicates that MIR7–3HG reduces the endogenous levels of AMBRA1, coherently with the luciferase assay results (). In addition, we also checked AMBRA1 mRNA levels, and demonstrated that they were, indeed, affected by MIR7–3HG overexpression (). We could thus conclude that MIR7–3HG mediates the direct regulation of AMBRA1 expression.
Overexpression of MIR7–3HG results in an autophagy decrease in HeLa cells
Given the effect that MIR7–3HG has on AMBRA1, and considering the role of AMBRA1 in autophagy, we analyzed the role of this MIRNA in autophagy signaling and progression. To date, MAP1LC3/LC3 is the most reliable marker of autophagosomes, whose formation upon autophagy induction can be monitored by LC3-I to LC3-II conversion and by detecting LC3-positive cytosolic puncta. We found that, in basal conditions (DMEM with 10% serum), the overexpression of MIR7–3HG in HeLa cells reduced LC3 conversion compared to the control (). Also, since autophagosome accumulation can result either from increased de novo autophagosome biosynthesis or inhibition of the autophagy flux, we also measured the on-rate/off-rate of autophagy by using the lysosomal inhibitor chloroquine. When we overexpressed MIR7–3HG, we observed a decrease in the accumulation of LC3-II, with respect to the control (). To further confirm our data, we monitored the autophagy flux and autophagosome formation by measuring LC3-positive puncta in immunofluorescence analyses. In cells transfected with the control vector, we detected a significant number of LC3-II puncta and we observed accumulation of dots after chloroquine treatment (). Conversely, MIR7–3HG did not increase LC3 puncta formation after chloroquine treatment (), supporting our finding that MIR7–3HG induces a block in the autophagic flux (autophagy on-rate). These observations were also strengthened by the analysis of LC3 conversion and dot accumulation upon autophagy induction by starvation (). In line with what was observed in basal conditions, after MIR7–3HG overexpression LC3 puncta did not further increase upon starvation. To further assess the effect of MIR7–3HG on autophagy, we quantified the levels of the autophagy receptor SQSTM1/p62, a protein incorporated into phagophores (autophagosome precursors) and degraded in autolysosomesCitation39 in basal and starvation conditions. Upon MIR7–3HG overexpression, there is a clear reduction in SQSTM1 degradation in both conditions and in comparison with the control vector ().
Figure 2. MIR7–3HG (MIR7) overexpression downregulates basal autophagy. (A) Analysis of cytoplasmic LC3-I and LC3-II levels, by using anti-LC3A/B antibody, on extracts from cells untreated or treated with chloroquine (CHQ; 20 μM, 30 min). All samples were analyzed 24 h after transfection with MIR7–3HG-encoding plasmids (indicated as MIR7) or negative control (Empty Vector). ACTB was used as a loading control. One representative western blot of 3 independent experiments is shown. The right graphs show the quantification of autophagy flux as the ratio between LC3-II in chloroquine-treated and LC3-II in untreated cells, the rate of LC3-I in LC3-II conversion and the quantification of LC3-II in the untreated samples. ImageJ densitometry analysis of 3 independent experiments (mean ± SD of independent experiments, *p < 0.05, ***p < 0.001). (B) MIR7–3HG blocked LC3 dot accumulation in samples treated with chloroquine. Cells were transfected with MIR7–3HG (indicated as MIR7) or Empty Vector and untreated (upper panels) or treated with chloroquine (20 μM, 30 min, lower panels). Scale bar: 5 μm. GFP fluorescence is shown in the red channel. The right graph shows the quantification of LC3 dots per transfected cell as the ratio between chloroquine-treated and -untreated cells (mean ± SD of 3 independent experiments, *p < 0.05). Statistical analysis was performed using the Student t test. (C) Overexpression of MIR7–3HG downregulates starvation-induced autophagy. Starvation-induced and autophagy related LC3-I to LC3-II conversion, by using anti-LC3A/B antibody, after MIR7–3HG overexpression (indicated as MIR7) is shown by immunoblots of control or MIR7–3HG-transfected cells that were nonstarved (STV-) or starved for 2 h (STV+). ACTB was used as loading control. The right graph shows ImageJ densitometry analysis of 3 independent experiments (mean ± SD of independent experiments, *p < 0.05, **p < 0.01). (D) Analysis of LC3 dot formation in HeLa cells, transfected with MIR7–3HG (indicated as MIR7) or empty vector. Autophagy was assessed under starvation conditions (2 h, lower panels). Scale bar: 5 μm. GFP fluorescence is shown in the red channel. The right graph shows the quantification of LC3 dots per transfected cell as the ratio between starved and nonstarved cells (mean ± SD of 3 independent experiments, **p < 0.01). Statistical analysis was performed using the Student t test. (E) Analysis of the starvation-induced and autophagy-related SQSTM1 degradation in MIR7–3HG transfected HeLa cells (indicated as MIR7). The right graph shows ImageJ densitometry analysis of 3 independent experiments (mean ± SD of independent experiments, ***p < 0.001, **p < 0.01).
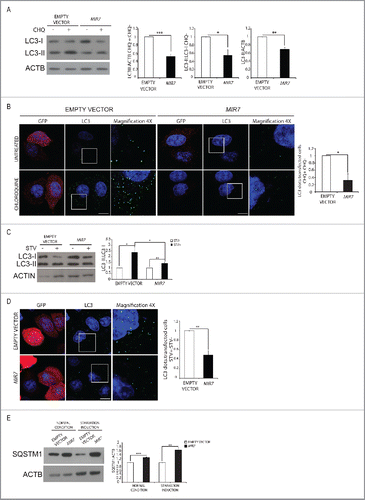
Further, in order to verify whether or not the observed autophagy modulation was achieved by MIR7–3HG by downregulating other autophagy-related key molecules besides AMBRA1, we also analyzed the effect of MIR7–3HG on other autophagy proteins that play a central role in the regulation of autophagy; among them ULK1 and BECN1, both involved in autophagy inductionCitation40 and a few crucial ATG genes (ATG3, ATG5, ATG7 and ATG12) in the elongation and closure of phagophores. Interestingly, we did not observe any changes in the protein levels of the analyzed autophagy markers after MIR7–3HG overexpression (Fig. S2A, B).
Last, we checked whether the observed regulation was specific for tumor cell lines, such as HeLa cells, or common to other nontumor cells, such as HEK293 (immortalized human kidney epithelium). We obtained, after MIR7–3HG overexpression, a decrease in AMBRA1 but also in autophagy flux and autophagosome formation level as seen with HeLa cells (Fig. S3). Moreover, we investigated the effect of MIR7–3HG also in 2 other nontumor cell lines, NIH3T3 (immortalized murine fibroblasts) or RPE1 (immortalized human retinal epithelium). In all cases MIR7–3HG donwregulated AMBRA1 and inhibited autophagy flux (Fig. S4).
In summary, MIR7–3HG overexpression downregulates autophagy in both normal and starvation conditions.
MIR7–3HG affects autophagy by downregulating AMBRA1
To demonstrate that MIR7–3HG was acting on autophagy via AMBRA1, we investigated whether MIR7-insensitive AMBRA1 mRNA could rescue MIR7–3HG-dependent autophagy regulation. To this aim, we generated 2 new constructs carrying the AMBRA1 coding sequence with a wild-type or mutated AMBRA1 3′-UTR tail (AMBRA1[3′-UTR-WT] and AMBRA1[3′-UTR-MUT], respectively). The mutation is hypothesized to disrupt the specific MIR7–3HG binding to AMBRA1 mRNA (). We then overexpressed these constructs simultaneously with MIR7–3HG in HEK293 cells and verified that, at variance with the wild-type form, the mutant construct was indeed unresponsive to MIR7–3HG (). Of the greatest importance, the AMBRA1[3′-UTR-MUT] construct was able to completely rescue autophagy in the given conditions to control levels, as monitored by LC3 and SQSTM1/p62 in an autophagy flux assay ().
Figure 3. Overexpression of MIR7–3HG-insensible AMBRA1 rescues the autophagy phenotype. (A) Scheme representing sequences of AMBRA1 mRNA with wild-type 3′-UTR (top sequence) or its artificially mutated form in the MIR7–3HG responsive element (bottom sequence). Mutations are marked in lower case letters. MIR7–3HG seed sequence is in bold. (B) Western blot analysis of AMBRA1 wild-type or mutated 24 h after cotransfection with MIR7–3HG-encoding plasmids (indicated as MIR7) or negative control (Empty Vector). Different bands are detected upon AMBRA1 overexpression, with the band indicated by the arrow corresponding to the expected size-range for full-length AMBRA1. The other bands, with lower molecular weights, correspond to AMBRA1 full-length cleavage products.Citation28 ACTB was used as endogenous control. One representative western blot of 3 independent experiments is shown. The right panel shows ImageJ densitometry analysis of the band of 3 independent experiments (mean ± SD of independent experiments,**p <0.01). (C) Analysis of autophagy flux following overexpression of AMBRA1 wild type or mutated, 24 h after cotransfection with MIR7–3HG-encoding plasmids (indicated as MIR7) or negative control (Empty Vector). Western blot analysis of LC3-II and SQSTM1 accumulation, using LC3B and SQSTM1 antibody, in extracts from cells untreated or treated with chloroquine (CHQ; 20 μM, 30 min). ACTB was used as loading control. One representative western blot of 3 independent experiments is shown. The right graphs show the quantification of autophagy flux measures as the ratio between LC3-II and SQSTM1 in chloroquine-treated and untreated cells (upper graphs), the rate of LC3-I to LC3-II conversion and the quantification of LC3-II (lower graphs) in the untreated samples. The data show ImageJ densitometry analysis of 3 independent experiment (mean ± SD of independent experiments, *p < 0.05,**p < 0.01, ***p < 0.001).
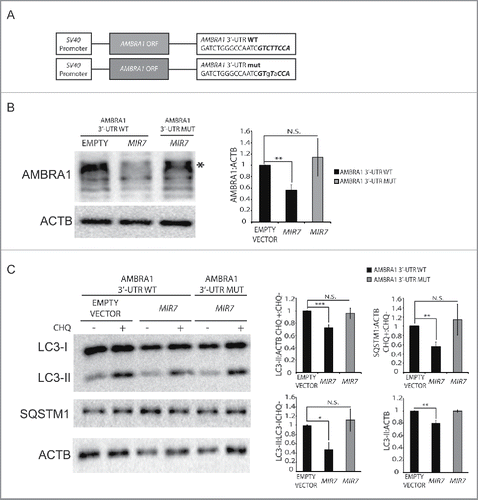
Altogether, these results suggest the existence of a clear epistatic relationship between MIR7 and AMBRA1 in autophagy downregulation, irrespective of the cell type analyzed.
AntagoMIR against MIR7–3HG induces an increase of AMBRA1 protein levels and positively regulates autophagy
In the previous set of experiments, we observed that overexpression of MIR7–3HG caused a dramatic decrease in AMBRA1 protein levels, leading to an efficient block in the autophagy process. Based on these results, we next moved to another cellular system of tumor origin (A549 cells), exhibiting a very low level of AMBRA1.Citation28 siMIRNA are chemically engineered oligonucleotides conceived to block specific endogenous MIRNAs. To test whether silencing endogenous MIR7–3HG could have an impact on target mRNA and protein levels, we obtained MIR7–3HG-specific siMIRNA (siMIR7–3HG, Fig. S5). We next transfected A549 cells with siMIR7–3HG or nonspecific control siMIRNA (Ctrl siRNA), and then monitored AMBRA1 protein and mRNA levels. We observed that transfection with siMIR7–3HG led to an increase in AMBRA1 protein () and a milder but significant increase in AMBRA1 mRNA levels () compared to the control antagoMIRs, confirming the MIR7–3HG-dependent AMBRA1 regulation.
Figure 4. Inhibition of endogenous MIR7–3HG (MIR7) leads to AMBRA1 upregulation and increased autophagy. (A) Western blot analysis in the A549 cell line of cytoplasmic AMBRA1 and SQSTM1 levels, 72 h after transfection with siMIR7 (siRNA anti-MIR7–3HG) or negative control (Ctrl siRNA, control siRNA). ACTB was used as an endogenous control. One representative western blot of 3 independent experiments is shown. The right panels show ImageJ densitometry analysis of 3 independent experiments (mean ± SD of independent experiments, **p < 0.01). (B) AMBRA1 mRNA expression was analyzed by quantitative RT-PCR, 72 h after overexpression of siMIR7–3HG (indicated as siMIR7). Relative quantification was measured by means of the comparative cycle threshold (ΔΔCt) method (mean ± SD of 3 independent experiments, *p < 0.05). (C) Analysis of cytoplasmic LC3-I and LC3-II levels, by using anti-LC3A/B antibody, on extracts from cells untreated or treated with chloroquine (20 μM, 30 min). All samples were analyzed 72 h after transfection with siMIR7 (siRNA anti-MIR7–3HG) or negative control (Ctrl siRNA, control siRNA). ACTB was used as a loading control. One representative western blot of 3 independent experiments is shown. The right graphs show the quantification of autophagy flux as the ratio between LC3-II in chloroquine-treated and untreated cells, the rate of LC3-I to LC3-II conversion and the quantification of LC3-II in the untreated samples. ImageJ densitometry analysis of 3 independent experiments (mean ± SD of independent experiments, *p < 0.05).
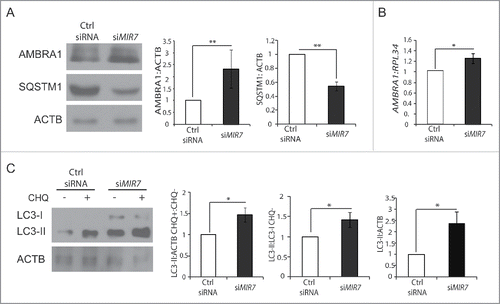
Next, we analyzed autophagy upon biologically relevant inhibition of MIR7–3HG activity. In line with the previous experiments, interference of MIR7–3HG in the lung carcinoma cell line A549 by siMIR7–3HG caused an increase in the rate of LC3-I to LC3-II conversion and SQSTM1 degradation, when compared with control cells grown in basal conditions ( and ). The second step was to analyze the autophagic flux after MIR7–3HG silencing using the siMIR7–3HG antagoMIR. As shown in , after chloroquine treatment, we could appreciate a higher autophagosome accumulation in samples interfered for MIR7–3HG, respective to the control.
AntagoMIR against MIR7–3HG induces an increase of MYC phosphorylation at serine 62
Recently, we have demonstrated that AMBRA1, through a direct binding with the catalytic subunit of the serine/threonine-protein phosphatase 2A (PPP2/PP2A), is able to mediate the dephosphorylation of phospho-MYC on serine 62 (p-MYC [S62]) and, consequently, its proteasomal degradation.Citation28,6
Primed by this evidence, we analyzed the level of p-MYC (S62) upon transfection of A549 cells with siMIR7–3HG. Of note, this cell line exhibits a clear inverse correlation between AMBRA1 (low) and p-MYC (S62) (high) protein levels.Citation28 When we transfected the A549 cell line with siMIR7–3HG, besides an AMBRA1 increase, we observed a decrease of p-MYC (S62) (). As previously shown,Citation28,6 also in this system, the downregulation of p-MYC (S62) induced MYC degradation ().
Figure 5. Inhibition of endogenous MIR7–3HG (MIR7) leads to an increase of MYC dephosphorylation and to a decrease in cell proliferation. (A) Western blot analysis of AMBRA1, p-MYC (S62) and MYC levels, 72 h after A549 cells transfection with siMIR7 (siRNA anti-MIR7–3HG) or negative siRNA control (Ctrl siRNA, control siRNA). ACTB was used as a loading control. One representative western blot of 3 independent experiments is shown. The right graph shows the ratio between p-MYC (S62) and MYC proteins (ImageJ densitometry analysis of 3 independent experiments [mean ± SD of independent experiments, ***p < 0.001]). (B) Cells transfected with siMIR7–3HG or negative siRNA control (Ctrl siRNA, control siRNA) were treated with 50 μg ml−1 cycloheximide (CHX) and collected at the indicated time points. Protein extracts of cells were analyzed by western blot, using antibodies against MYC. ACTB was used as a loading control. The right panel shows ImageJ densitometry analysis of the band of 3 independent experiments (mean ± SD of independent experiments,**p < 0.01, *p < 0.05). (C) Western blot analysis in the A549 cell line of cytoplasmic p-MYC (S62) 72 h after transfection with a plasmid encoding AMBRA1WT, AMBRA1PXP or with GLB1/ß-galactosidase, as a negative control. The right graph shows the relative quantification of endogenous MIR7–3HG in A549 cells after transfection. The level of endogenous MIR7–3HG was analyzed by quantitative RT-PCR. Relative quantification was measured using the comparative cycle threshold (ΔΔCt) method (mean ± SD of 3 independent experiments, *p < 0.05).
![Figure 5. Inhibition of endogenous MIR7–3HG (MIR7) leads to an increase of MYC dephosphorylation and to a decrease in cell proliferation. (A) Western blot analysis of AMBRA1, p-MYC (S62) and MYC levels, 72 h after A549 cells transfection with siMIR7 (siRNA anti-MIR7–3HG) or negative siRNA control (Ctrl siRNA, control siRNA). ACTB was used as a loading control. One representative western blot of 3 independent experiments is shown. The right graph shows the ratio between p-MYC (S62) and MYC proteins (ImageJ densitometry analysis of 3 independent experiments [mean ± SD of independent experiments, ***p < 0.001]). (B) Cells transfected with siMIR7–3HG or negative siRNA control (Ctrl siRNA, control siRNA) were treated with 50 μg ml−1 cycloheximide (CHX) and collected at the indicated time points. Protein extracts of cells were analyzed by western blot, using antibodies against MYC. ACTB was used as a loading control. The right panel shows ImageJ densitometry analysis of the band of 3 independent experiments (mean ± SD of independent experiments,**p < 0.01, *p < 0.05). (C) Western blot analysis in the A549 cell line of cytoplasmic p-MYC (S62) 72 h after transfection with a plasmid encoding AMBRA1WT, AMBRA1PXP or with GLB1/ß-galactosidase, as a negative control. The right graph shows the relative quantification of endogenous MIR7–3HG in A549 cells after transfection. The level of endogenous MIR7–3HG was analyzed by quantitative RT-PCR. Relative quantification was measured using the comparative cycle threshold (ΔΔCt) method (mean ± SD of 3 independent experiments, *p < 0.05).](/cms/asset/7245afd0-93da-4403-9386-f0e3502a47ce/kaup_a_1269989_f0005_b.gif)
Given the reported activity of p-MYC (S62) on the MIR7–3HG promoter and MIR7–3HG transcription,Citation30 we sought to investigate whether an AMBRA1 unable to bind the p-MYC (S62) phosphatase PPP2/PP2A (AMBRA1PXP)Citation28 decreased the impact of MYC on MIR7–3HG transcription. Interestingly, overexpression of wild-type AMBRA1, but not of its mutant form could regulate MIR7–3HG expression in lung cancer cells ().
These data strongly suggest that AMBRA1-mediated regulation of p-MYC affects MIR7–3HG expression, thus establishing a feedback loop.
MIR7–3HG modulation in A549 cells has an impact on cell proliferation
Given the role of AMBRA1 in cell cycle regulationCitation28 and in order to assess if AMBRA1 modulation mediated by MIR7–3HG could have an effect on cell proliferation, we analyzed the proliferation rate of A549 cells after MIR7–3HG inhibition by a cell viability assay. Indeed, MIR7–3HG inhibition led to a decrease of proliferation when compared with control cells that was rescued upon AMBRA1 overexpression (). In line with this finding, overexpression of MIR7–3HG in HeLa cells, that express moderate levels of MIR7, led to a cell number increase (). Also in this case, AMBRA1 overexpression compensated for the effect of MIR7–3HG overexpression, priming the cells to proliferate similar to the control.
Figure 6. MIR7–3HG (MIR7) affects proliferation in an AMBRA1-dependent manner. (A) The graph shows the MIR7–3HG overexpression effect on cell proliferation in HeLa cells. (B) The left graph shows the siMIR7–3HG effect on cell proliferation in A549 cells. In (A) and (B) the proliferation rate was measured by the AlamarBlue® Cell Viability assay. All samples were analyzed 48 h after transfection with siMIR7–3HG or the negative control siRCtrl (mean ± SD of independent experiments, *p < 0,05; ***p < 0.001). (C) Inverse correlation between MIR7–3HG levels and AMBRA1 expression in lung cancer. Expression levels of AMBRA1 (left graphs) and MIR7–3HG (right graphs) in LUAD (upper graphs) and LUSC (lower graphs) datasets from TCGA comparing normal (NT) to tumor primary (TP) samples. Center lines in the boxplot show the medians; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, outliers are represented by dots.
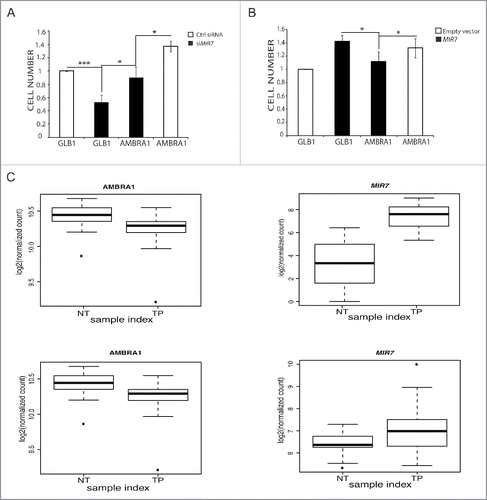
MIR7–3HG and AMBRA1 expression inversely correlate in human lung tumor samples compared to normal tissues
Primed by our findings in the lung tumor cell line A549, we set out to investigate the reciprocal AMBRA1-MIR7–3HG regulation in silico in human lung tumor samples. To this aim, RNA-Seq data for 58 and 51 paired samples have been analyzed for LUAD (LUng ADenocarcinoma) and LUSC (LUng Small Cells cancer) data sets deposited in TCGA (The Cancer Genome Atlas database), respectively. We decided to focus only on the paired samples since the expression levels of AMBRA1 vary remarkable from sample to sample. It is thus important to be able to compare the tumor samples to normal samples from the same patient as a reference to identify cases in which the gene is up- or downregulated.
We found that more than 40% and 60% of the LUAD and LUSC samples, respectively, have a negative log2FC for the AMBRA1 gene, indicating that in these tumor samples AMBRA1 is downregulated with respect to the normal samples. The other cluster of patient data in which the log2FC is positive have not been further investigated in this study, since other mechanisms such as the absence of other regulators of its expression levels could account for expression changes, considering the high heterogeneous spectrum of cancer-related mechanisms. We thus focused on the samples characterized by log2FC, and we analyzed the levels of MIR7–3HG in tumor and normal samples. We observed that MIR7–3HG was always overexpressed in the given tumor cases for LUAD and in 90% of the LUSC samples where AMBRA1 was downregulated (), supporting the existence of a tight correlation between MIR7–3HG and AMBRA1 expression levels in cancer patient data.
Discussion
Autophagy plays essential roles in different cellular processes, and many studies have recently demonstrated the involvement of MIRNA-mediated regulation of proteins in the autophagy pathway. Very recently a correlation was described between MIR23A and AMBRA1 in autophagy regulation during UV-stress-induced premature senescence.Citation41 However, only few of these studies have investigated in depth the effect of MIRNAs on autophagic flux or attempted to identify the physiological context in which the MIRNA may be linked to autophagy. In this study, we introduce MIR7–3HG as a new autophagy-regulating MIRNA that acts as an oncogene in lung cancer cells, by blocking autophagy in favor of cell proliferation.
Data obtained from the in silico analysis of AMBRA1, a key protein in autophagy regulation, identified MIR7–3HG as a strong candidate for AMBRA1 regulation. Indeed, we observed that MIR7–3HG overexpression causes a significant decrease in AMBRA1 levels; we also demonstrated that this regulation occurs trough the binding of MIR7–3HG to a canonical MIR7–3HG responsive element present in the 3′-UTR region of AMBRA1 mRNA, a domain that is broadly conserved among vertebrates. In fact, taking advantage of a luciferase assay, we validated AMBRA1 mRNA as a direct target of MIR7–3HG.
To investigate the AMBRA1-MIR7–3HG axis in autophagy regulation we next decided to study LC3-II levels upon AMBRA1 downregulation in both basal and starvation conditions or in a number of tumor and nontumor cell lines. We demonstrated by several approaches that MIR7–3HG overexpression decreases the conversion rate of LC3-I into LC3-II, positively modulates SQSTM1/p62 and plays an important role in blocking the autophagy flux. Also, we have proven the AMBRA1 binding by MIR7–3HG to be functional to this regulation. Altogether, these results demonstrate that MIR7–3HG has a role in autophagy regulation via AMBRA1. It should be mentioned that an opposite and specific role for MIR7–3HG overexpression in human lung cancer cells has been previously reported,Citation42 although based on a different protocol of transduction of MIR7–3HG, on a fluorescent-reporter detection method to monitor autophagy and, most important, at a very much later time point. Indeed, we cannot exclude, based on our data, that in a later phase of cell growth, autophagy gets primed as a secondary feedback response, resulting in decreased cell survival.
Recent data strongly support a physiological and pathological relevance for the crosstalk between autophagy and other pathways.Citation43,44 As an example, autophagy-related proteins also affect cell proliferation.Citation26,28,45,46 Indeed, to adapt to nutrient deprivation or other cell stresses, a simultaneous regulation of autophagy and cell growth occurs in eukaryotic organisms. First, a plethora of signaling molecules and pathways have opposite effects on cell growth and autophagy, supporting the idea that these processes might represent mutually exclusive cell fates.Citation47 Such a reciprocal inhibition between autophagy and cell growth can occur either through direct regulatory mechanisms, most of which remain to be determined to date, or through the same signaling pathways that act independently and simultaneously on autophagy and cell growth.
AMBRA1, a well-characterized positive regulator of the autophagy pathway, affects cell proliferation both in vitro and in vivo.Citation24,26,28 In more detail, embryos homozygous for the gene-trap mutation in the Ambra1 locus (Ambra1gt/gt embryos) are affected by a dramatic hyperproliferative phenotype of the neuroepithelium, appearing at the onset of neurulation.Citation26 Moreover, AMBRA1 downregulation or its overexpression in human cell lines result in a significant increase or decrease of the cell proliferation rate, respectively.Citation26,28 By taking these lines of evidence into account, the relevance of AMBRA1 in controlling cell proliferation and guaranteeing cell survival upon autophagic stress is evident.
Primed by these findings, we thus investigated whether ovexpression of MIR7–3HG, in our cellular model, could affect at the same time autophagy and cell proliferation. Overexpression of MIR7–3HG in HeLa and A549 cell lines resulted in a decrease of the autophagy flux, as previously described, and, at the same time, in a parallel increase in cell proliferation. By contrast, we obtained an opposite effect when we used an antagoMIR construct that decreases MIR7–3HG expression, leading to a decrease in cell proliferation and an increase in autophagy.
In recent years, AMBRA1 has emerged as a scaffold molecule that serves as a platform for autophagy-related complexes and as an early autophagy regulator, linking this process to a number of other cellular activities.Citation27 In this work, we have demonstrated that MIR7–3HG, via AMBRA1 downregulation, could be the circuit breaker between autophagy and cell proliferation. Indeed, through MIR7–3HG inhibition or activation, cells could be able to control both autophagy and proliferation.
Of note, we have previously demonstrated that AMBRA1, through its interaction with the phosphatase PPP2/PP2A, dephosphorylates the oncogene MYC, thereby enhancing its proteasomal degradation.Citation28 In this way, AMBRA1 is able to control MYC cellular levels, thus acting as a tumor suppressor. In various lung cancer cell lines, we observed a low expression level of AMBRA1 and high expression levels of phosphorylated MYC.Citation28 In lung cancer, MIR7–3HG expression is regulated by MYC, which promotes MIR7–3HG transcription.Citation48 Here, based on the interference of MIR7–3HG in A549 lung cancer cells, we suggest that MIR7–3HG maintains the balance of this inverse correlation between AMBRA1 and phospho-MYC. In fact, by blocking MIR7–3HG expression in A549 cells, we obtained an increase in AMBRA1; this finding correlates with a parallel enhancement in autophagy levels and a decrease in phosphorylated MYC levels. Through our experiments, we thus found that MIR7–3HG, through AMBRA1 downregulation, can regulate not only autophagy, but also the levels of MYC, a transcription factor regulating MIR7–3HG's own activity. This would establish a positive feedback for MIR7–3HG transcription.
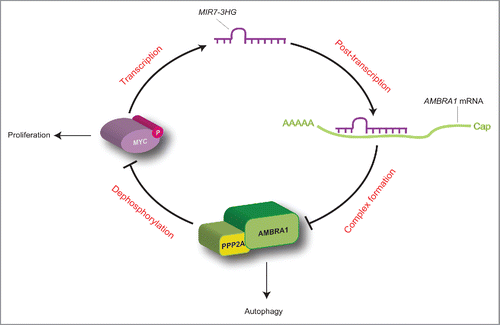
In sum, we identified a regulatory loop, in which there is a mutual regulation between MIR7–3HG and AMBRA1. On the one hand, there is a direct post-transcriptional downregulation of MIR7–3HG on AMBRA1 expression; on the other hand, AMBRA1 is able to downregulate MIR7–3HG transcription by promoting proteasomal degradation of its transcriptional regulator MYC (). Such evidence, together with the observation that MIR7–3HG is deregulated in lung cancer cells, in which autophagy and AMBRA1 play a central role, leads us to hypothesize that MIR7–3HG and AMBRA1 interaction could be critical for the onset and progression of lung carcinoma. Of the highest importance, both LUAD and LUSC lung cancer in humans (with the A549 cell lined being derived from a LUAD tumor), show a striking inverse correlation between AMBRA1 mRNA and MIR7–3HG levels, which is lost in normal tumor from the same patients.
Figure 7. Proposed working model for the MYC-MIR7–3HG-AMBRA1 axis. In this regulatory loop, MIR7–3HG downregulates AMBRA1 expression, and promotes its own transcription mediated by MYC. This model favors cell proliferation to the detriment of autophagy (for details see the text).
The importance of both autophagy and MIRNAs in cancer is emerging in recent years. It is widely demonstrated that altered autophagy is associated with tumor formation and progression, and with an altered response to several types of cancer treatment. The results reported here suggest that MIRNAs have the potential of modulating autophagy through regulation of expression of key autophagy genes, such as AMBRA1, providing evidence for a new role of MIRNAs highly correlated with cancer biology.
The finding that MIR7–3HG modulates the expression of the tumor suppressor AMBRA1, thereby promoting its expression, supports indeed the idea that MIR7–3HG is a promising prognostic biomarker and a therapeutic target for lung cancer. Our findings imply that by modulating MIR7–3HG in lung cancer cells, it could be possible to switch a cell phenotype towards a more autophagic profile and a less proliferative state.
In the future, it could be interesting to investigate, in nonmalignant cells, whether there is a correlation between the presence of polymorphisms or genetic rearrangements of the AMBRA1 3′-UTR or if that could have a protective role in cancer insurgence and progression.
Material and methods
Cell culture and reagents
HEK293, NIH3T3, RPE1 and HeLa cells were cultured in Dulbecco's modified Eagle's medium (LONZA, BE12–604F) supplemented with 10% fetal bovine serum (GIBCO, 10270–106), in a CO2-humidified incubator at 37°C. A549 cell lines were similarly cultured in RPMI 1640 (LONZA, BE12–702F) supplemented with 10% fetal bovine serum. For autophagy induction cells were washed with phosphate-buffered saline (UCS Diagnostic, PBS1199) and cultured for 2 h in Earle's balanced salt solution (Sigma-Aldrich, E2888). All the cell lines were transiently transfected with expression vectors using Lipofectamine 2000 (Invitrogen, 11668–019) as indicated by the supplier. Viral infection of A549 cells with the retroviral AMBRA1-coding constructs was performed as previously described.Citation25,28 When indicated, cells were incubated in the presence of 50 μg ml−1 cycloheximide (Sigma Aldrich, 200–636–0) and 20 μM chloroquine (Sigma Aldrich, 50–63–5).
Plasmid constructs
A construct coding for MIR7–3HG overexpression was produced by cloning the pri-MIRNA sequence, with 100 nucleotides upstream and downstream of the pre-MIRNA, in the pSP65 plasmid into the U1snRNA expression cassette (according to DentiCitation49). The full-length human 3′-UTR sequence of AMBRA1 was amplified by PCR and then directionally cloned in NotI and XhoI unique restriction site of the psicheck2 plasmid (Promega, C8021), downstream of the Renilla luciferase gene. This kind of vector also contains the firefly luciferase to normalize transfection efficiency. AMBRA1–3′-UTR mutated in the MIR7–3HG recognition element was created by site-directed mutagenesis (Agilent Technologies, 200518), using AMBRA1 3′ UTR as a template. The linker primers used during the study were; AMBRA1 5′- CATCTCGAGAGACAAACGTTGCACTGGTG-3′; 5′- CATGCGGCCGCGAGGGGCATGTCATCATTTT-3′; AMBRA1 mutant primers 5′- AGCCTCCAGAGAGTGAACAGTGTACCAGATCTGGGCCAATCATCCT-3′ and the exact reverse complement.
Target prediction
MIRNA targets were predicted using publicly available bioinformatics tools. Target scan (http://www.targetscan.org/); Pictar (http://pictar.mdc-berlin.de/cgi-bin/new_PicTar_vertebrate.cgi?species = vertebrate), MIRanda (http://www.microrna.org/microrna/getGeneForm.do), diana TOOLS (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r = microT_CDS/index)
Immunoblotting and antibodies
Cells were lysed at the indicated time points in RIPA buffer (50 mM TRIS-HCl, pH 7.4, 150 mM NaCl, 1% NP40 [ThermoFhisher Scientific, FNN0021], 0.25% Na-deoxycholate [ThermoFisher Scientific, 89904]) supplemented with complete protease inhibitor cocktail (Roche, 04–693–131–001) and 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich, P7626). Protein extracts (20 mg per well) were analyzed using 7% or 12% SDS-polyacrylamide gels and then transferred onto nitrocellulose membranes (Millipore, IPVH00010). Membranes were blocked in 5% nonfat milk in TBST (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.1% Tween 20 [Sigma-Aldrich, P 9416], pH 7.4) for 1 h and then incubated with a 5% milk-containing TBST solution containing the following primary antibodies: Anti-ACTB (Sigma Aldrich, A2066; 1:2,000), anti-p-MYC (S62) (Abcam, ab78318), anti-SQSTM1 (Santa Cruz Biotechnology, sc-28359), anti-BECN1 (Santa Cruz Biotechnology, sc-11427), anti-AMBRA1 (Santa Cruz Biotechnology, sc-398204), and anti-LC3 (Cell Signaling Technolog,y 27755 and 12741). Following incubation with horseradish peroxidase-coupled secondary anti-mouse (Bio-Rad, 170–6516) and anti-rabbit antibody (Bio-Rad, 170–6515), bands were revealed by chemiluminescence. Band intensities were quantified using ImageJ software.
Immunocytochemistry
Cells were washed in PBS and fixed with 4% paraformaldehyde in PBS for 30 min. After permeabilization with 0.4% Triton X-100 (Sigma-Aldrich, X100) in PBS for 5 min, cells were blocked in 3% normal goat serum (Sigma-Aldrich, G9023) in PBS and incubated overnight at 4°C with primary antibodies; we used the antibodies directed against LC3. Cells were then washed in blocking buffer and incubated for 1 h with labeled anti-mouse (Molecular Probes, Alexa Fluor 488, A11017 or Alexa Fluor 555, A20187) or anti-rabbit (Jackson Immuno Research, FITC, 111–095–003 or Cy3, 115–165–166) secondary antibodies. Nuclei were stained with 1 mg/ml DAPI and examined under a Zeiss LSM 700 100_ oil-immersion objective (Zeiss, Oberkoechen, Germany). We used ‘ZEN 2009 Light edition’ software for image analysis. All measurements in this work were performed by a blind approach. All analyses were performed in nonsaturated single z-confocal planes.
RNA isolation and real-time RT-PCR for AMBRA1 mRNA quantification
RNA was extracted from HeLa and A549 cells using QIAGEN MIRNeasy kit (217004) following the manufacturer's instructions. Extracted RNA (800 ng) was used for reverse transcription and 500 ng of random primers for each μg of RNA was added to each tube. Samples were heated to 70°C for 5 min followed by immediate incubation on ice. M-MLV reaction buffer (1X), dNTPs, RNase inhibitor (1 U/μl), and M-MLV reverse transcriptase (8U/reaction) (Promega, 9PIM170) were added to the mix and were incubated for 1 h at 37°C. Real-time PCR was performed using LightCycler 480 SYBR Green I Master and the LightCycler 480 System (Roche) following the manufacturer's instructions. mRNA change was quantified with the 2−ΔΔCT method using RPL34 as mRNA control. The following primers where used for quantitative real-time PCR: Ambra1 (F: AACCCTCCACTGCGAGTTGA, R: TCTACCTGTTCCGTGGTTCTCC), RPL34 (F: GTCCCGAACCCCTGGTAATAGA, R: GGCCCTGCTGACATGTTTCTT).
Real-time RT-PCR for endogenus MIRNA quantification
RNA was extracted from A549 cells. Extracted RNA (800 ng) was used for reverse transcription following the manufacturer's instructions using miScript Reverse Transcriptase Mix, miScript RT Buffer, RNase-Free Water (Quiagen, 218061). qPCR reactions were performed following manufacturer's instructions for QuantiTect SYBR Green PCR Master Mix, miScript Univerthe sal Primer (Quiagen, 218073). MIRNA change was quantified with the 2−ΔΔCT method using RNU6–1 (RNA, U6 small nuclear 1) as control.
Luciferase assay
Firefly and Renilla luciferase activities were measured 48 h post-transfection using the dual-luciferase reporter assay system (Promega, E1910) according to the manufacturer's instructions. Results were expressed as Renilla luciferase activity normalized to firefly luciferase activity.
siRNA MIRNA
The A549 cells were transfected with miRCURY LNA™, against MIR7–3HG (MIMAT0000252) and as control we utilized microRNA inhibitor negative control A purchase from Exigon.
Statistical analyses
Statistical analyses were performed using the Student 2-tailed t test. Data are shown as means ± SD of n independent experiments. Values of p < 0.05 were considered significant.
Analyses of lung cancer data sets from The Cancer Genome Atlas database
RNA-Seq data for LUAD and LUSC paired samples have been downloaded from The Cancer Genome Atlas (TCGA) and pre-processed with the TCGAbiolinks R package.Citation50 In particular, we downloaded the data for 58 and 51 patient samples for lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), respectively. At first, we searched for possible outliers using the TCGAnalyse Preprocessing function of TCGAbiolinks, which performs an Array Array Intensity correlation analyses, i.e. a square symmetric matrix of Pearson correlation among all samples. According to this analysis we did not find samples with correlation lower than 0.6 in LUAD so all the samples have been retained for further analyses, whereas one of the samples was discarded for LUSC. Afterwards, using the TCGAnalyze_Normalization function of TCGAbiolinks, we normalized the mRNA transcripts using the EDASeq R package. In this step, we used normalization procedures to adjust for GC-content and gene-length effects on read counts and full quintile normalization.Citation51 Fold change (FC) of tumor samples with respect to normal has been calculated and log2 (log2FC) transformed and used to classify the patient samples according to changes in AMBRA1 expression levels of tumor versus normal samples. The MIR7–3HG microRNA expression levels corresponding to the same samples for which RNA-Seq data were available have been also analyzed as log2 transformed normalized RSEM counts (for a total of 53 and 48 samples, for LUAD and LUSC, respectively). The data were produced using the Illumina HiSeq 2000 mRNA/MIRNA sequencing platform.
Abbreviations
| A549 | = | human lung carcinoma |
| AMBRA1 | = | autophagy and Beclin 1 regulator 1 |
| AMBRAPXP | = | AMBRA1 mutated in the PPP2/PP2A binding site |
| AMBRA13′-UTR-WT | = | AMBRA1 3′ untranslated region - wild type |
| AMBRA13′-UTR-MUT | = | AMBRA1 3′ untranslated region mutated in MIR7–3HG binding region |
| CHQ | = | chloroquine |
| HEK293 | = | human embryonic kidney 293 |
| MAP1LC3/LC3 | = | microtubule associated protein 1 light chain 3 |
| MIR | = | microRNAs |
| MRE | = | miRNA responsive element |
| MYC | = | v-myc avian myelocytomatosis viral oncogene homolog |
| PPP2/PP2A | = | protein phosphatase 2 |
| UTR | = | untranslated region |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
KAUP_A_1269989_SUPPLEMENTARY.zip
Download Zip (5.9 MB)Acknowledgements
We thank Mrs. M. Acuña Villa for secretarial work and Dr. Vanda Turcanova for the excellent technical support. We are indebted with Dr. Ubaldo Gioia, (Rome, Italy) for kindly providing us with the MIR7–3HG, MIR-9 and Dr. Francesca De Vito (Rome, Italy) for kindly providing us with the MIR-200b. We are grateful to Dr. Juliane Becher, Antonio Colaprico (Bruxelles, Belgium), and Richa Batra (Copenhagen, Denmark) for the useful advice and critical discussion of this work, and to Dr. Sandrine Humbert (Grenoble, France) to grant the execution of a number of the revision experiments in her laboratory.
Funding
This research was supported in part by grants from the Telethon Foundation (GGP14202), AIRC (IG2016-18906), FISM (2013), the Italian Ministry of Health (Progetto Giovani Ricercatori GR2011–2012 to FS) and the Danish National Supercomputer for Life Science Computerome. VC is supported by the Lundbeck Foundation (R209–2015–3505) and the KBVU (R146-A9471). This work has been also supported by a KBVU grant from the Danish Cancer Society (R146-A9364) to FC. We are also grateful to the Bjarne Saxhof Foundation, the Lundbeck Foundation (R167–2013–16100), the NovoNordisk Foundation (7559) and the European Union (Horizon 2020 MEL-PLEX, grant agreement 642295). Further, FC and EP labs in Copenhagen are part of the Center of Excellence in Autophagy, Recycling and Disease (CARD), funded by the Danish National Research Foundation.
Related Research Data
References
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861-73; PMID:18006683; http://dx.doi.org/10.1101/gad.1599207
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961-7; PMID:17972889; http://dx.doi.org/10.1038/nrc2254
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451(7182):1069-75.
- Abada A, Elazar Z. Getting ready for building: Signaling and autophagosome biogenesis. EMBO Rep 2014; 15:839-52; PMID:25027988; http://dx.doi.org/10.15252/embr.201439076
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. A signalling pathway controlling MYC degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 2004; 6:308-18; PMID:15048125; http://dx.doi.org/10.1038/ncb1110
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432(7020):1032-6. Epub 2004 Nov 3; PMID:15525940; http://dx.doi.org/10.1038/nature03029
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Chen N, Debnath J. Autophagy and tumorigenesis. FEBS Lett 2010; 584:1427-35; PMID:20035753; http://dx.doi.org/10.1016/j.febslet.2009.12.034
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51-64; PMID:16843265; http://dx.doi.org/10.1016/j.ccr.2006.06.001
- Karantza-Wadsworth V, White E. Role of autophagy in breast cancer. Autophagy 2007; 3:610-3; PMID:17786023; http://dx.doi.org/10.4161/auto.4867
- Corcelle EA, Puustinen P, Jäättelä M. Apoptosis and autophagy: Targeting autophagy signalling in cancer cells -‘trick or treats’?. FEBS J 2009; 276:6084-96; PMID:19788415; http://dx.doi.org/10.1111/j.1742-4658.2009.07332.x
- Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biol Ther 2011; 11:157-68; PMID:21228626; http://dx.doi.org/10.4161/cbt.11.2.14622
- Eisenberg-Lerner A, Kimchi A. The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis 2009; 14(4):376-91. Review; PMID:19172397; http://dx.doi.org/10.1007/s10495-008-0307-5
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215-33; PMID:19167326; http://dx.doi.org/10.1016/j.cell.2009.01.002
- Gupta SK, Thum T. Non-coding RNAs as orchestrators of autophagic processes. J Mol Cell Cardiol 2015; 95:26-30; pii: S0022-2828(15)30116-4. [Epub ahead of print] Review.
- Jing Z, Han W, Sui X, Xie J, Pan H. Interaction of autophagy with microRNAs and their potential therapeutic implications in human cancers. Cancer Lett 2015; 356(2 Pt B):332-8. Epub 2014 Oct 7. Review; PMID:25304373; http://dx.doi.org/10.1016/j.canlet.2014.09.039
- Zhai H, Fesler A, Ju J. MicroRNA: A third dimension in autophagy. Cell Cycle 2013; 12(2):246-50. Epub 2012 Jan 15. Review; PMID:23255136; http://dx.doi.org/10.4161/cc.23273
- Frankel LB, Lund AH. MicroRNA regulation of autophagy. Carcinogenesis 2012; 33:2018-25; PMID:22902544; http://dx.doi.org/10.1093/carcin/bgs266
- Gibbings D, Mostowy S, Voinnet O. Autophagy selectively regulates MIRNA homeostasis. Autophagy 2013; 9(5):781-3. Epub 2013 Feb 19; PMID:23422216; http://dx.doi.org/10.4161/auto.23694
- Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, Liu CG, Yang JM. Regulation of autophagy by a beclin 1-targeted microRNA, MIR-30a, in cancer cells. Autophagy 2009; 5:816-23; PMID:19535919; http://dx.doi.org/10.4161/auto.9064
- Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF, Hébuterne X, et al. A synonymous variant in IRGM alters a binding site for MIR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat Genet 2011; 43:242-5; PMID:21278745; http://dx.doi.org/10.1038/ng.762
- Gundara JS, Robinson BG, Sidhu SB. Evolution of the “autophagaMIR”. Autophagy 2011; 7(12):1553-4; PMID:22024754; http://dx.doi.org/10.4161/auto.7.12.17762
- Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 2010; 191:155-68; PMID:20921139; http://dx.doi.org/10.1083/jcb.201002100
- Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol 2013; 15(4):406-16. Epub 2013 Mar 24; PMID:23524951; http://dx.doi.org/10.1038/ncb2708
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447:1121-5; PMID:17589504
- Cianfanelli V, Nazio F, Cecconi F. Connecting autophagy: AMBRA1 and its network of regulation. Mol Cell Oncol 2015; 2(1):e970059; PMID:27308402; http://dx.doi.org/10.4161/23723548.2014.970059
- Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, De Zio D, Nazio F, Antonioli M, D'Orazio M, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol 2015; 17(5):706; PMID:25925585; http://dx.doi.org/10.1038/ncb3171
- Mei Y, Su M, Soni G, Salem S, Colbert CL, Sinha SC. Intrinsically disordered regions in autophagy proteins. Proteins 2014; 82:565-78; PMID:24115198; http://dx.doi.org/10.1002/prot.24424
- Chou YT, Lin HH, Lien YC, Wang YH, Hong CF, Kao YR, Lin SC, Chang YC, Lin SY, Chen SJ, et al. EGFR promotes lung tumorigenesis by activating MIR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res 2010; 70:8822-31; PMID:20978205; http://dx.doi.org/10.1158/0008-5472.CAN-10-0638
- Kalinowski FC, Brown RA, Ganda C, Giles KM, Epis MR, Horsham J, Leedman PJ. microRNA-7: a tumor suppressor miRNA with therapeutic potential. Int J Biochem Cell Biol 2014; 54:312-7. Epub 2014 Jun 5. Review; PMID:24907395; http://dx.doi.org/10.1016/j.biocel.2014.05.040
- Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human MIRNAs and indications for an involvement of MIRNA in cell growth and apoptosis. Nucleic Acids Res 2005; 33:1290-7; PMID:15741182; http://dx.doi.org/10.1093/nar/gki200
- Maragkakis M, Reczko M, Simossis VA, Alexiou P, Papadopoulos GL, Dalamagas T, Giannopoulos G, Goumas G, Koukis E, et al. DIANA-microT web server: elucidating microRNA functions through target prediction. Nucleic Acids Res 2009; 37:W273-6; PMID:19406924; http://dx.doi.org/10.1093/nar/gkp292
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 2007; 27:91-105; PMID:17612493; http://dx.doi.org/10.1016/j.molcel.2007.06.017
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009; 19:92-105; PMID:18955434; http://dx.doi.org/10.1101/gr.082701.108
- Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: Targets and expression. Nucleic Acids Res 2008; 36:D149-53; PMID:18158296; http://dx.doi.org/10.1093/nar/gkm995
- Wang P, Zhang J, Zhang L, Zhu Z, Fan J, Chen L, Zhuang L, Luo J, Chen H, Liu L, et al. MicroRNA 23b regulates autophagy associated with radioresistance of pancreatic cancer cells. Gastroenterology 2013; 145(5):1133-1143.e12. Epub 2013 Aug 2; PMID:23916944; http://dx.doi.org/10.1053/j.gastro.2013.07.048
- Le Sage C, Agami R. Immense promises for tiny molecules: uncovering MIRNA functions. Cell Cycle 2006; 5:1415-21; PMID:16775415; http://dx.doi.org/10.4161/cc.5.13.2890
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen HF, Øvervatn A, Bjørkøy G, Johansen T. SQSTM1/p62 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/10.1074/jbc.M702824200
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672-6; PMID:10604474; http://dx.doi.org/10.1038/45257
- Zhang JA, Zhou BR, Xu Y, Chen X, Liu J, Gozali M, Wu D, Yin ZQ, Luo D. MiR-23a-depressed autophagy is a participant in PUVA- and UVB-induced premature senescence. Oncotarget 2016; 7(25):37420-37435
- Tazawa H, Yano S, Yoshida R, Yamasaki Y, Sasaki T, Hashimoto Y, Kuroda S, Ouchi M, Onishi T, Uno F, et al. Genetically engineered oncolytic adenovirus induces autophagic cell death through an E2F1-microRNA-7-epidermal growth factor receptor axis. Int J Cancer 2012; 131(12):2939-50; PMID:22492316; http://dx.doi.org/10.1002/ijc.27589
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15(7):713-20. Review. Erratum in:Nat Cell Biol. 2013 Aug;15(8):1017; PMID:23817233; http://dx.doi.org/10.1038/ncb2788
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40(2):280-93. Review; PMID:20965422; http://dx.doi.org/10.1016/j.molcel.2010.09.023
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013; 154(6):1269-84; PMID:24034250; http://dx.doi.org/10.1016/j.cell.2013.08.015
- Puustinen P, Rytter A, Mortensen M, Kohonen P, Moreira JM, Jäättelä M. CIP2A oncoprotein controls cell growth and autophagy through mTORC1 activation. J Cell Biol 2014; 204(5):713-27; PMID:24590173; http://dx.doi.org/10.1083/jcb.201304012
- Neufeld TP. Autophagy and cell growth–the yin and yang of nutrient responses. J Cell Sci 2012; 125(Pt 10):2359-68. Epub 2012 May 30; PMID:22649254; http://dx.doi.org/10.1242/jcs.103333
- Chou YT, Lin HH, Lien YC, Wang YH, Hong CF, Kao YR, Lin SC, Chang YC, Lin SY, Chen SJ, et al. EGFR promotes lung tumorigenesis by activating MIR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res 2010; 70:8822-31; PMID:20978205; http://dx.doi.org/10.1158/0008-5472.CAN-10-0638
- Denti MA, Rosa A, Sthandier O, De Angelis FG, Bozzoni I. A new vector, based on the PolII promoter of the U1 snRNA gene, for the expression of siRNAs in mammalian cells. Mol Ther 2004; 10(1):191-9; PMID:15272480; http://dx.doi.org/10.1016/j.ymthe.2004.04.008
- Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 2016; 44(8):e71; PMID:26704973; http://dx.doi.org/10.1093/nar/gkv1507
- Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinformatics 2011; 12:480; PMID:22177264; http://dx.doi.org/10.1186/1471-2105-12-480