
ABSTRACT
Mitophagy is a highly specialized process to remove dysfunctional or superfluous mitochondria through the macroautophagy/autophagy pathway, aimed at protecting cells from the damage of disordered mitochondrial metabolism and apoptosis induction. PINK1, a neuroprotective protein mutated in autosomal recessive Parkinson disease, has been implicated in the activation of mitophagy by selectively accumulating on depolarized mitochondria, and promoting PARK2/Parkin translocation to them. While these steps have been characterized in depth, less is known about the process and site of autophagosome formation upon mitophagic stimuli. A previous study reported that, in starvation-induced autophagy, the proautophagic protein BECN1/Beclin1 (which we previously showed to interact with PINK1) relocalizes at specific regions of contact between the endoplasmic reticulum (ER) and mitochondria called mitochondria-associated membranes (MAM), from which the autophagosome originates. Here we show that, following mitophagic stimuli, autophagosomes also form at MAM; moreover, endogenous PINK1 and BECN1 were both found to relocalize at MAM, where they promoted the enhancement of ER-mitochondria contact sites and the formation of omegasomes, that represent autophagosome precursors. PARK2 was also enhanced at MAM following mitophagy induction. However, PINK1 silencing impaired BECN1 enrichment at MAM independently of PARK2, suggesting a novel role for PINK1 in regulating mitophagy. MAM have been recently implicated in many key cellular events. In this light, the observed prevalent localization of PINK1 at MAM may well explain other neuroprotective activities of this protein, such as modulation of mitochondrial calcium levels, mitochondrial dynamics, and apoptosis.
Introduction
Mutations in the PINK1 gene represent the second most frequent cause of autosomal recessive early onset Parkinson disease (PD).Citation1 PINK1 encodes a mitochondrial protein kinase that protects mitochondrial integrity at different levels, by regulating mitochondrial morphology and transport, calcium buffering, complex I activity and ATP production.Citation2-4 Besides these functions, PINK1 plays a key role in the activation of mitophagy, a selective, timely and tightly regulated process aimed at eliminating aged and dysfunctional mitochondria through the autophagy machinery. An efficient mitophagy is essential to protect neuronal cells from the harm of disordered mitochondrial metabolism and the release of proapoptotic proteins, which would ineluctably trigger neuronal cell death.Citation5 Upon conditions of massive mitochondrial depolarization (such as that induced by the mitochondrial uncoupler CCCP), PINK1 selectively accumulates on the surface of damaged mitochondria, where it phosphorylates and recruits both ubiquitin and PARK2, another protein mutated in recessive PD.Citation6,7 A recent study showed that PARK2 could also be recruited by BECN1,Citation8 a pro-autophagic protein with ubiquitous cellular localization.Citation9 Once relocalized, PARK2 induces the ubiquitination and proteasomal degradation of proteins of the outer mitochondrial membrane (OMM),Citation10 leading to inhibition of fusion and trafficking of dysfunctional mitochondria.Citation11-15 Next, these organelles are associated to the forming autophagosome membranes by specific ubiquitin-binding receptor proteins such as SQSTM1/p62,Citation16,17 and subsequently incorporated within autophagosomes. Finally, the autophagosomes fuse with lysosomes to complete the mitophagic process (for a review see refs. Citation18,19).
While these phases have been well characterized, the process of autophagosome biogenesis during mitophagy has only recently started to be elucidated. In a recent study on mitochondrial depolarization, PINK1 was found to recruit the autophagy receptors CALCOCO2/NDP52 and OPTN (optineurin) to mitochondria in a PARK2-independent manner. In turn, these receptors recruit the early autophagy factors ULK1, ZFYVE1/DFCP1 and WIPI1 to focal spots proximal to mitochondria, promoting mitophagy.Citation20 However, another study showed that OPTN stabilization does require PARK2, and that recruitment to damaged mitochondria of ZFYVE1/DFCP1, a key component of the so-called “omegasome,” that represents the autophagosome precursor, is independent of OPTN.Citation21 Moreover, the role of BECN1 in autophagosome formation during mitophagy has not yet been explored.
BECN1 is a key component of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex (including BECN1, ATG14, PIK3C3/Vps34 and PIK3R4/Vps15),Citation22 that is required for the nucleation of the phagophore and omegasome generation.Citation23 Recently it has been shown that, in starvation-induced autophagy, the PtdIns3K complex relocalizes to specific regions of apposition between the ER and mitochondria termed “mitochondria-associated membranes” (MAM), that represent the site of formation of autophagosomes.Citation24 As we previously demonstrated that PINK1 interacts directly with BECN1, and is able to enhance basal and starvation-induced autophagy,Citation9 the aim of this study was to explore whether, in conditions of activated mitophagy, PINK1 and BECN1 could also interplay in the process of autophagosome formation.
Results
BECN1 is implicated in later stages of mitophagy
As we confirmed that, in our experimental model, BECN1 interacts with PARK2 both in basal conditions and upon CCCP exposure (Fig. S1), we sought to verify whether BECN1 was required for PARK2 recruitment to damaged mitochondria, as previously reported.Citation8 To this aim, we silenced BECN1 in PINK1- or PARK2-overexpressing SH-SY5Y cells and treated them with CCCP. After short times of treatment (30 min to 3 h), we evaluated several markers of early mitophagy, including PINK1 accumulation ( and ), and PARK2 recruitment ( and , Fig. S2), as well as ubiquitination and degradation of the mitochondrial proteins MFN2, OPA1 and VDAC1 ().Citation25 Surprisingly, in contrast to what was previously reported, PARK2 was normally recruited to depolarized mitochondria in the absence of BECN1; moreover, no differences could be observed in any of the other markers in scramble versus BECN1-silenced cells. Treatment of cells with 10 µM CCCP (as adopted by Choubey and collaborators)Citation8 resulted in a lower accumulation of PINK1 and recruitment of PARK2 to mitochondria, again with no significant differences between control and BECN1-silenced cells (data not shown). These findings show that, at least in our experimental system, BECN1 is not required to trigger the early steps of the mitophagic process.
Figure 1. BECN1 is not required to trigger the early steps of mitophagy. (A and B) Confocal analysis of PINK1 accumulation and PARK2 recruitment at mitochondria upon CCCP treatment in shSCR and shBECN1 SH-SY5Y cells, overexpressing PINK1-GFP (A) or YFP-PARK2 (B). Cells were treated with DMSO alone or with 25 μM CCCP for 3 h and mitochondria were immunostained using the mitochondrial marker TOMM20 (red). Scale bar: 10 μm. (C and D) Statistical analysis of data from A-B (mean ± SD of n = 3, 30 cells per experiment). Histograms report the percentage of cells showing colocalization of PINK1 (C) or PARK2 (D) with mitochondria. **p < 0.001. (E) WB of PINK1 and early mitophagic markers in shSCR and shBECN1 SH-SY5Y cells overexpressing HA-PARK2. Degradation (shorter exposure) and ubiquitination (longer exposure, upper band) of the mitochondrial proteins MFN2, OPA1 and VDAC1 were assessed at the indicated early time points of treatment with 25 μM CCCP. Values were normalized against TUBA/tubulin. Separated blots indicate that we joined together distant parts from the same gel.
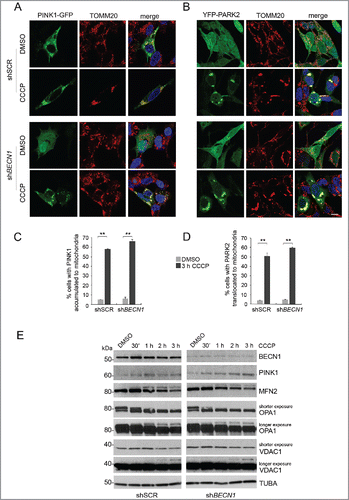
We next evaluated whether BECN1 could be implicated in the later phases of mitophagy, by exposing cells to 25 µM CCCP for longer times (up to 12 h). To this aim, we monitored the degradation of mitochondrial proteins residing in the inner membrane and the matrix (TIMM23 and COX4I1, respectively) (), the levels of mitochondrial DNA (mtDNA) (), and the proportion of cells in which mitochondria had disappeared ( and ), all representing markers of advanced mitophagy.Citation25 In BECN1-silenced cells, we could consistently observe a significant impairment of mitochondrial clearance at longer treatment times, as shown by delayed degradation of both TIMM23 and COX4I1, accumulation of mtDNA and a reduced number of cells with no mitochondria. These findings were paralleled by a significantly higher amount of cleaved PARP and proportion of TUNEL-positive cells, indicative of increased CCCP-induced apoptosis in cells lacking BECN1 (Fig. S3). Thus, it appears that BECN1 is essential for a successful completion of mitophagy, which in its absence cannot proceed efficiently, regardless of PARK2 recruitment and proteasome-mediated degradation of the OMM proteins. No differences between control and BECN1-silenced cells could be observed in the reduction of PARK2 levels after 12 h of CCCP treatment, which is caused by proteasomal-mediated degradation of PARK2.Citation26,27 Similarly, LC3 cleavage was not affected by BECN1 silencing, as previously reported ().Citation28
Figure 2. BECN1 downregulation significantly impairs the advanced stages of CCCP-induced mitophagy. shSCR and shBECN1 SH-SY5Y cells stably expressing HA-PARK2 (A-D) or YFP-PARK2 (E and F) were treated with 25 µM CCCP up to 12 h. (A) Western blot of PARK2, LC3 and late mitophagic markers (TIMM23 and COX4I1). Separated lines indicate that we joined together distant parts from the same gel. (B and C) Densitometric analysis of data from (A). Values were normalized against TUBA/tubulin. (D) Analysis of mtDNA levels by RT-PCR. (E) Confocal analysis of shSCR and shBECN1 SH-SY5Y cells transfected with plasmids encoding YFP-PARK2 and DsRed-Mito after 12 h, 25 µM CCCP exposure. Scale bar: 10 µm. (F) Histogram reporting the statistical analysis of data from (E), expressed as mean ± SD of the percentage of cells with no mitochondria. *p<0.05; **p<0.001.
PINK1 and BECN1 are enhanced in the MAM compartment
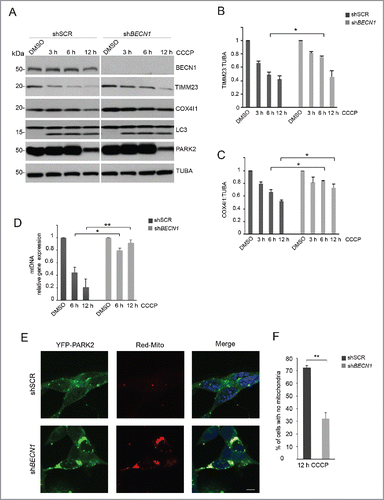
We and others described previously that PINK1 and BECN1 are found in various subcellular compartments.Citation9,29,30 To characterize the subcellular domain(s) in which PINK1-BECN1 binding takes place, both in basal conditions and upon mitophagic stress, we performed co-immunoprecipitation (co-IP) experiments in cytoplasmic and mitochondria-enriched fractions from both wild-type SH-SY5Y cells or cells stably expressing PINK1 (), and from HEK293 cells transiently overexpressing the 2 constructs (Fig. S4). These experiments showed a strong interaction between PINK1 and BECN1 in the mitochondria-enriched fraction, which increased after CCCP treatment, whereas only a mild binding was found in the cytoplasmic fraction.
Figure 3. PINK1 and BECN1 increase in the MAM fraction upon CCCP treatment. (A and B) co-IP analysis of PINK1-BECN1 interaction in cytoplasmic and crude mitochondrial fractions from SH-SY5Y cells, both in basal conditions and upon 25 µM CCCP treatment of 6 h. Cross-contaminations of cytoplasmic and mitochondrial-enriched fractions were assessed by evaluating TIMM23 and TUBA/tubulin as specific markers. (A) Endogenous BECN1 was immunoprecipitated using mouse anti-BECN1 antibody, followed by immunoblotting to detect co-immunoprecipitated endogenous PINK1 with rabbit anti PINK1. shBECN1 SH-SY5Y cells were used as control (CTR). (B) Overexpressed PINK1-HA was immunoprecipitated using mouse anti-HA antibody covalently attached to crosslinked agarose bead particles, and co-immunoprecipitated endogenous BECN1 was detected using mouse anti-BECN1. Preimmune IgG were used as negative control. (C-F) Confocal analysis of BECN1 and PINK1 colocalization with ER and mitochondrial markers upon treatment with 25 µM CCCP for 6 h. SH-SY5Y cells expressing BECN1-mCherry (left panels) or PINK1-mCherry (right panels) were co-transfected with ER-GFP (C and D) or stained with the mitochondrial marker TOMM20 (green) (E and F). Scale bar: 10 µm. (G) Histogram reporting Mander's overlap coefficients relative to BECN1 and PINK1 colocalization with ER and mitochondria (mean ± SD of n = 3, 10 cells per experiments). *p<0.05; **p<0.001. (H) Western blot of PINK1 and BECN1 distribution in Percoll-purified subcellular fractions isolated from SH-SY5Y cells exposed to DMSO or 25 µM CCCP for 6 h. Fractions were separated by SDS-PAGE and immunoblotted with specific antibodies for each fraction. VDAC1 and TUBA/tubulin were used as markers for pure mitochondrial and cytosolic fractions, respectively; CANX (calnexin) was considered as a marker of both ER and MAM, whereas ACSL4 was adopted as a MAM-specific marker. PNS, post-nuclear supernatant; Micro, microsome fraction; MitoP, pure mitochondria; MAM, mitochondria-associated membranes. Separated lines indicate that we joined together distant parts from the same gel.
To further substantiate this observation, we assessed by confocal microscopy how these 2 proteins would shift among different subcellular compartments upon CCCP exposure. However, whereas the colocalization of PINK1 with TOMM20 significantly increased as expected, the mitochondrial localization of BECN1 was only minimally affected. Conversely, we observed a markedly enhanced overlay of BECN1 with an ER-selective dye (ER-GFP), whereas PINK1-ER colocalization increased only slightly ().
This finding was intriguing, as it was apparently in contrast with our previous subfractionation data. However, it should be noted that the adopted fractionation protocol does not generate pure mitochondria, but only a crude mitochondrial fraction still containing portions of ER membranes attached to the OMM (the so called MAM). These contact sites between ER and mitochondrial membranes are so tight and strong that they cannot be separated by simple subcellular fractionation protocols,Citation31 as confirmed by the expression of the typical ER marker CANX (calnexin) also in the mitochondrial-enriched fraction ( and S4).Citation32
To investigate whether the interaction between PINK1 and BECN1 could involve MAM, gradient centrifugation experiments were performed to obtain a separation of the MAM from pure mitochondria. Interestingly, both endogenous PINK1 and BECN1 were already present in the MAM fraction in basal conditions, and their localization to this fraction markedly increased after CCCP treatment. PINK1 levels were also enhanced in the pure mitochondrial fraction (although to a much lesser extent), while BECN1 was found to increase in the microsomal fraction containing the ER, in line with the confocal imaging data ().
We were able to confirm these findings both in a distinct cellular model (HEK293 cells) (Fig. S5), and in SH-SY5Y cells treated with 1 µM valinomycin, a selective mitochondrial uncoupler that induces mitophagy (Fig. S6), indicating that the accumulation of both PINK1 and BECN1 at MAM is a consistent event in conditions of triggered mitophagy. CCCP-induced MAM translocation was also observed for PINK1G309D, a pathogenic mutant with impaired kinase activity (Fig. S7).
PINK1 enhances BECN1 relocalization at MAM
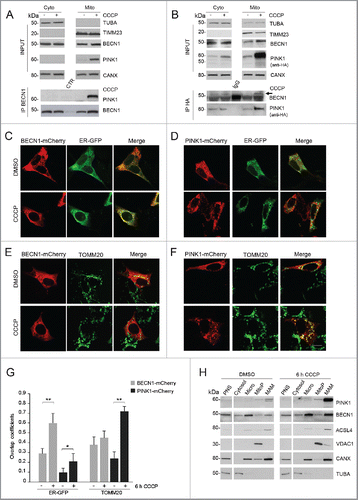
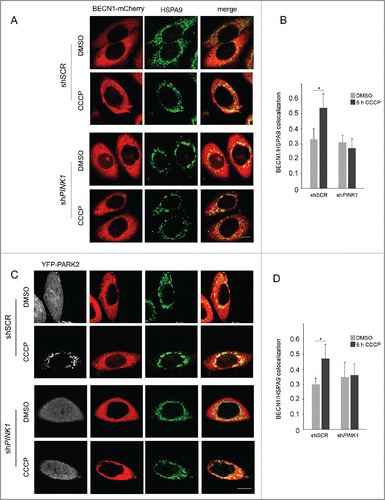
The previous findings suggest that, as already observed for starvation-induced autophagy, MAM could represent the site where autophagosomes are shaped in conditions of mitophagic stimuli, and that PINK1 may play a role in this process, likely through its interaction with BECN1. To further explore this hypothesis, we tested whether PINK1 could influence BECN1 relocalization to the MAM compartment after CCCP treatment. To this aim, we monitored BECN1 levels after gradient centrifugation of PINK1-silenced SH-SY5Y cells. Interestingly, upon CCCP exposure, BECN1 levels failed to increase in the MAM fraction, while the enhancement in the microsomal fraction containing the ER was comparable to that observed in the presence of PINK1 ().
Figure 4. PINK1 influences CCCP-induced BECN1 relocalization at MAM. (A) Quantitative real-time PCR showing PINK1 cDNA levels in SH-SY5Y cells infected with scramble (shSCR) vs PINK1 shRNA (shPINK1). (B) WB of endogenous BECN1 distribution in Percoll-purified subcellular fractions isolated from shPINK1 SH-SY5Y cells exposed to either DMSO or 25 µM CCCP for 6 h. Fractions were separated by SDS-PAGE and immunoblotted with specific antibodies for each fraction (details as in ). Separated lines indicate that we joined together distant parts from the same gel. (C) Confocal analysis of BECN1 and HSPA9 colocalization. shSCR and shPINK1 SH-SY5Y cells, overexpressing BECN1-mCherry, were treated with DMSO or 25 µM CCCP for 6 h and stained for the MAM-localized protein HSPA9. The insets show enlarged views of colocalized areas. (D) Histogram reporting Mander's overlap coefficients relative to BECN1 and HSPA9 colocalization shown in (C) (mean ± SD of n = 3, 10 cells per experiments). Scale bar: 10 µm. *p≤ 0.05.
To further confirm this observation, we monitored by confocal microscopy the colocalization of BECN1 with HSPA9/GRP75, a protein that selectively localizes at MAM.Citation33 In line with previous data, CCCP treatment significantly enhanced BECN1-HSPA9 colocalization in control cells, whereas this enhancement was lost in PINK1-silenced cells (). Taken together, these findings show that, upon a mitophagic stimulus, PINK1 is required to promote BECN1 relocalization to the MAM compartment.
PARK2 also localizes at MAM but is not implicated in BECN1 relocalization upon CCCP
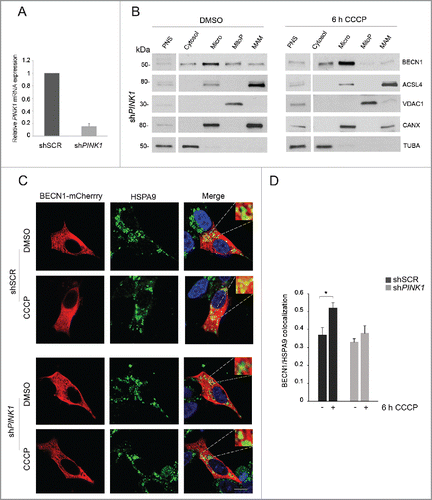
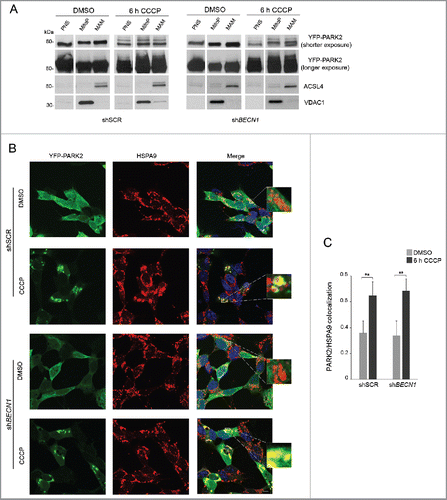
Previous studies have shown that PARK2 accumulates at MAM following excitotoxic stimuli and that it is able to modulate ER-mitochondria tethering.Citation34-36 While it is well known that PARK2 markedly relocalizes to mitochondria in conditions of activated mitophagy and only in the presence of PINK1,Citation37 we sought to verify whether PARK2 would also accumulate at MAM in these conditions, and whether this would be mediated by its interaction with BECN1. To this aim, we performed gradient centrifugation and confocal microscopy experiments in SH-SY5Y cells overexpressing YFP-PARK2. By western blotting, we detected PARK2 in pure mitochondrial and MAM fractions in basal conditions, with enhancement after 6 h CCCP treatment (). In line with these results, colocalization of PARK2 with the MAM-specific marker HSPA9 was already evident in basal conditions and significantly increased upon mitochondrial depolarization (). In both experiments, PARK2 relocalization to MAM remained unaffected by silencing of BECN1.
Figure 5. PARK2 localization at MAM. (A-C) shSCR and shBECN1 SH-SY5Y cells, overexpressing YFP-PARK2, were treated with DMSO or 25 μM CCCP for 6 h. (A) Immunoblots of subcellular fractions subjected to gradient centrifugation, resolved in SDS-PAGE and revealed with specific antibodies. VDAC1 was used as a marker of pure mitochondria, whereas ACSL4 was adopted as a MAM-specific marker. PNS, post-nuclear supernatant; MitoP, pure mitochondria; MAM, mitochondria-associated membranes. PARK2 accumulation after CCCP treatment is more appreciable at higher exposure; the upper bands evident at lower exposure reflect PARK2 ubiquitination. Separated lines indicate that we joined together distant parts from the same gel. (B) Confocal analysis of YFP-PARK2 and the MAM-localized protein HSPA9. The insets display enlarged views of colocalized areas. Scale bar: 10 μm. (C) Histogram reporting Mander's overlap coefficients relative to YFP-PARK2 and HSPA9 colocalization shown in (B) (mean ± SD of n = 3, 10 cells per experiments). **p < 0.001.
To further clarify the interaction of PARK2 with BECN1, we explored whether PARK2 is required for BECN1 accumulation at MAM in conditions of mitophagy. To this aim, we made use of HeLa cells, that are devoid of PARK2 and are therefore largely used as a PARK2-free cellular model,Citation21 and monitored by confocal microscopy the colocalization of BECN1 and HSPA9. Similar to what we observed in SH-SY5Y cells, BECN1-HSPA9 overlay significantly increased upon CCCP treatment even in the absence of PARK2, whereas this enhancement was completely abolished by PINK1 silencing (). Overexpression of PARK2 in this experimental system did not affect BECN1-HSPA9 colocalization, and was not able to rescue the effects induced by lack of PINK1 (). Taken together, these results suggest that PARK2 and BECN1 do not need each other to relocalize to MAM upon mitochondrial depolarization, whereas both require PINK1.
Figure 6. BECN1- HSPA9 overlay significantly increased upon CCCP treatment even in the absence of PARK2.shSCR and shPINK1 HeLa cells overexpressing only BECN1-mCherry (red) (A and B) or BECN1-mCherry (red) and YFP-PARK2 (gray) (C and D) were treated with DMSO or 25 μM CCCP for 6 h and then immunostained with the MAM-localized protein HSPA9 (green). Scale bar: 10 μm. (B and D) Histograms reporting Mander's Overlap Coefficients relative to BECN1-mCherry and HSPA9 colocalization showed in A and C (mean ± SD of n = 3, 10 cells per experiments). *p < 0.05.
To evaluate whether PARK2 could ubiquitinate BECN1, we performed a ubiquitination assay in HeLa cells either overexpressing PARK2 or empty vector, but failed to observe ubiquitination of BECN1 in basal conditions or after 3 h of CCCP treatment (data not shown).
BECN1 and PINK1 enhance the formation of ER-mitochondria contact sites after mitophagic stimuli
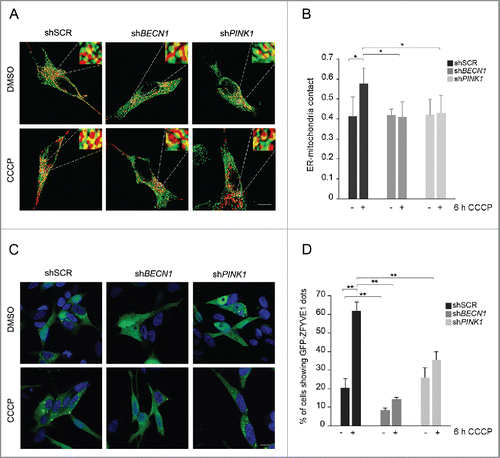
The sites of association between ER and mitochondria are known to dynamically increase upon specific conditions such as starvation and apoptotic stimuli,Citation38 and PARK2 was already reported to modulate this process.Citation34,36 Since we showed that both PINK1 and BECN1 accumulate in the MAM fraction after mitochondrial depolarization, we sought to explore whether ER-mitochondria contact sites would be enhanced by CCCP, and whether BECN1 and/or PINK1 would be required for this process. Accordingly, we transfected SH-SY5Y cells with Red-Mito and ER-GFP constructs, labeling mitochondria and ER with fluorescent proteins, and quantified by confocal microscopy the contact points of the 2 fluorescent signals. Treatment of cells with CCCP resulted in a significantly higher mitochondria-ER colocalization, indicating increased tethering between these 2 organelles ( and S8). However, this was abolished by either BECN1 or PINK1 silencing, showing that both proteins were essential for this process ().
Figure 7. BECN1 and PINK1 influence ER-mitochondria contact sites and omegasome formation upon CCCP treatment. (A) Representative 3D-reconstructions of ER (green) and mitochondria (red) in stably expressing ER-GFP and Red-Mito shSCR, shBECN1 and shPINK1 SH-SY5Y cells, treated with DMSO or 25 µM CCCP for 6 h. Insets show higher magnification details and yellow areas indicate regions of close apposition between the 2 organelles. (B) Histogram reporting Manders' coefficients relative to data from (A), expressed as mean ± SD (n = 3, 10 cells per experiment). (C) Confocal analysis of shSCR, shBECN1 and shPINK1 cells stably expressing GFP-ZFYVE1, under basal conditions or upon treatment with 25 µM CCCP for 12 h. (D) Histogram displays the percentage of cells showing GFP-ZFYVE1-positive dots, relative to data from (C) (mean ± SD of n = 3, 10 cells per experiment). *p < 0.05; **p < 0.001. Scale bar: 10 µm.
BECN1 and PINK1 are required for the process of autophagosome formation during mitophagy
In line with previous observations of the central role of MAM in starvation-induced autophagy,Citation24 we hypothesized that these regions would also be centrally implicated in the mitophagy process. To examine this we performed gradient centrifugation and confocal microscopy experiments following the protein ZFYVE1/DFCP1 that, upon autophagy activation, translocates to the ER and binds membranes rich in phosphatidylinositol-3-phosphate (PtdIns3P) to form the omegasomes.Citation21,39 After 6-h CCCP treatment, GFP-ZFYVE1 levels increased in the MAM fraction (Fig. S9A), as seen in starvation,Citation24 and this was confirmed also in the presence of a distinct mitophagic stimulus such as valinomycin (Fig. S6). Moreover, colocalization of GFP-ZFYVE1 with HSPA9 was similar upon CCCP treatment as in starvation (Fig. S9B), confirming that autophagosomes originate at MAM sites also in conditions of CCCP-induced mitophagy.
We next evaluated whether PINK1 and BECN1 would be implicated in the process of omegasome formation during mitophagy. To monitor this process, we assessed by confocal microscopy the expression pattern of GFP-ZFYVE1, which in basal conditions is diffusely fluorescent, whereas after autophagy induction it forms discrete dots that correspond to the omegasomes. CCCP treatment resulted in a marked increase of cells showing GFP-ZFYVE1 dots, confirming activation of the process of autophagosome biogenesis. BECN1 silencing resulted in a markedly reduced proportion of GFP-ZFYVE1 punctate cells also in basal conditions, which failed to increase as expected upon CCCP treatment. No differences could be observed between control and PINK1-silenced untreated cells; however, after CCCP treatment, the proportion of PINK1-silenced cells showing GFP-ZFYVE1 dots was about half that of the controls, indicating that PINK1 was also required for a correct assembly of the omegasome during mitophagy ().
To explore the mechanism through which PINK1 could affect BECN1-mediated autophagosome formation, we explored whether BECN1 would be phosphorylated by PINK1 in the presence of CCCP, using starvation-induced BECN1 phosphorylation as a positive control.Citation40 After 3 h and 6 h CCCP treatment, we observed the appearance of BECN1 phosphorylated bands, which, however, remained unmodified after PINK1 silencing, indicating that BECN1 is not directly phosphorylated by PINK1 (Fig. S10). To further substantiate this observation, we replicated the key findings of this study (ER-mito contact sites, recruitment of BECN1 at MAM and omegasome formation) in PINK1-silenced SH-SY5Y cells overexpressing either wild-type PINK1 or PINK1-KDD, an artificial triple kinase-dead mutant construct.Citation41 Upon CCCP treatment, cells overexpressing PINK1-KDD showed a comparable increase of ER-mito tethering, BECN1 localization to MAM and omegasome formation as those overexpressing wild-type PINK1 (Fig. S11–S13), confirming that the kinase activity of PINK1 is not required for these processes.
Discussion
In recent years, mitophagy has clearly emerged as a critical process to selectively remove dysfunctional mitochondria before they can make irreversible damage, especially in long-living cells such as neurons. Two PD-related proteins, PINK1 and PARK2, play a central role in the activation of the mitophagic cascade, with PINK1 selectively accumulating on depolarized mitochondria and recruiting PARK2.Citation18
We recently showed that PINK1 directly interacts with BECN1, a key pro-autophagic protein, to promote autophagy.Citation9 However, the role of BECN1 in CCCP-induced mitophagy is still controversial. In a recent study on PC12 cells, BECN1 was found to interact with PARK2 mainly in the cytosol, and to induce its translocation to depolarized mitochondria, enhancing the early steps of mitophagy.Citation8 However, another study on HeLa cells reported that mitochondrial clearance after 24-h CCCP treatment was independent from BECN1.Citation42 In our SH-SY5Y experimental system, silencing of BECN1 was not able to affect PARK2 recruitment or any other early step of CCCP-induced mitophagy. Yet, we showed that several markers of late mitophagy were all consistently impaired in BECN1-silenced cells at 6–12 h of CCCP treatment, clearly demonstrating that BECN1 is required for a timely accomplishment of the mitophagic process. Cells lacking BECN1 also showed a significantly increased rate of apoptotic cell death, confirming the neuroprotective role of mitophagy, whose impairment in depolarized mitochondria shifts the balance toward the activation of pro-apoptotic pathways.Citation43-45 The discrepancies between our findings and previously published data are possibly explained by the distinct experimental conditions adopted. For instance, significant differences between PC12 and SH-SY5Y cells in response to cellular stressors have been already demonstrated.Citation46 Similarly, while BECN1 is able to significantly expedite mitochondrial clearance at short CCCP exposure times, alternative mitophagy pathways (such as those regulated by MAPK/ERK) are likely to be activated by prolonged mitochondrial damage,Citation47 possibly also explaining the apparent paradox that activated mitophagy has been observed in conditions of stable PINK1 knockdown.Citation48
In our experimental setting, CCCP induced an evident LC3 lipidation, widely adopted as a general marker of activated autophagy, that, however, was unaffected by either PINK1 or BECN1 silencing. The latter finding confirms a previous study showing that CCCP-related LC3 lipidation is independent of BECN1 but requires ATG9, a membrane protein implicated in transporting vesicles to the expanding phagophore.Citation28 While the authors concluded that CCCP-induced mitophagy does not critically depend on BECN1, our findings of impaired mitophagy in BECN1-silenced cells argue against this conclusion. In fact, LC3 lipidation alone cannot be considered a universal indicator of mitophagy, as it explores only a distinct step of autophagosome biogenesis arising in the ER-Golgi intermediate compartment, independent of the process of omegasome formation.Citation23 Moreover, recent studies demonstrate the existence of at least 2 distinct pathways of macroautophagy, the canonical LC3-dependent pathway and an LC3-and ATG9-independent, but BECN1-dependent, pathway. This alternative process was implicated in mitochondrial clearance during erythrocyte maturation, suggesting it could play a role in mitophagy.Citation49
The issue of which cellular membranes give origin to the omegasome has represented a long-debated subject, with distinct evidence pointing to either the ER or the OMM as candidate sites. A recent study has elegantly reconciled these hypotheses showing that, upon starvation-induced autophagy, autophagosomes form at specific regions of close contact between the ER and mitochondria (MAM).Citation24 These regions serve the important function of tethering the 2 organelles together by proteins located on the opposing membranes, allowing their close communication.Citation50 Several early markers of autophagosome formation, including BECN1 and ZFYVE1/DFCP1, translocate to MAM under starved conditions, and inhibition of this translocation prevents the initiation of the process.Citation24
To date, the mechanism of autophagosome formation during mitophagy has not been explored in detail. Here we show for the first time that omegasomes form at MAM also in conditions of CCCP-induced mitophagy, and that both BECN1 and PINK1 are required for this process. Indeed, we found that endogenous PINK1 and BECN1 localize at MAM, and their localization, as well as interaction, are strongly enhanced in the presence of mitophagic stimuli. After BECN1 silencing, omegasome formation is already significantly impaired in basal conditions, confirming the role of BECN1 in maintaining normal levels of constitutive autophagy,Citation29 and this process completely fails to enhance after mitophagy activation. CCCP-induced omegasome formation is also dramatically decreased in PINK1-silenced cells compared with controls, suggesting a novel intriguing function of PINK1 in promoting mitophagy.
Interestingly, we showed that BECN1 is not directly phosphorylated by PINK1 and that the PINK1-KDD construct, that is devoid of kinase activity,Citation41 still maintains the same ability to promote ER-mito tethering, BECN1 recruitment at MAM and omegasome formation as its wild-type counterpart. While these findings argue against the involvement of PINK1 kinase activity in this pathway, our data indicate that PINK1 and BECN1 are respectively placed on the opposed mitochondrial and ER sides of MAM, and that PINK1 silencing significantly impairs the relocalization of BECN1 at MAM. Collectively, these findings support the hypothesis that, in conditions of activated mitophagy, the direct interaction between PINK1 and BECN1 may serve to “attract” ER-residing BECN1 toward mitochondria, similarly to what has previously been shown for the SNARE protein STX17 (syntaxin 17) upon starvation-induced autophagy.Citation24 This also results in an increased tethering between the 2 organelles, as we show that CCCP treatment induces a significant enrichment of the ER-mitochondria contact sites, which is abolished by silencing either of the 2 proteins.
Upon mitochondrial depolarization, PARK2 translocates to mitochondria in a PINK1-dependent fashion.Citation51 Here we show that, in these conditions, PARK2 levels increase both in pure mitochondrial and MAM fractions, confirming previous observations that PARK2 accumulates both at mitochondria and ER-mitochondria junctions in conditions of excitotoxicity,Citation35 and modulates ER-mitochondrial crosstalk.Citation34,36 The finding that PINK1, PARK2 and BECN1 all localize at MAM after CCCP treatment is intriguing. However, while PARK2 and BECN1 are dispensable for each other's translocation at MAM, both proteins require PINK1. The recent report that, upon mitochondria depolarization, PINK1 is also able to activate a PARK2-independent cascade leading to the recruitment of several early autophagy factors, including ZFYVE1/DFCP1, to focal spots proximal to mitochondria, clearly suggests that PINK1 can activate mitophagy through a number of distinct and parallel pathways.Citation20
Indeed, it is tempting to speculate that this eclectic protein could represent a master regulator of MAM functions, that include not only autophagosome biogenesis but also other key cellular events, such as calcium transport from the ER to mitochondria, mitochondrial dynamics, and apoptosis control.Citation50 For instance, we recently found that PINK1-mediated phosphorylation of BCL2L1/Bcl−xL impairs its N terminus cleavage.Citation52 This impairment is expected to maintain the inhibitory activity of BCL2L1 on the MAM-residing protein VDAC1, thus preventing CYCS/cytochrome c release and apoptosis.Citation53 Similarly, we and others have previously shown that PINK1 mutations result in mitochondrial accumulation of calcium, which could be rescued by Ruthenium Red, a mitochondrial calcium influx blocker.Citation54,55 Since MAM represent the core sites for the direct and selective exchange of calcium signals between ER and mitochondria, it will be worth investigating whether PINK1 could be directly implicated in this mechanism.Citation56
Besides PINK1 and PARK2, MAM have been recently studied also in connection with other PD-related proteins. In fact, SNCA/α-synuclein localizes at MAM, and its pathogenic mutations impair the formation of contact sites between ER and mitochondria.Citation57 Moreover, PARK7/DJ-1 was also found to favor ER-mitochondrial crosstalk and enhance calcium transfer.Citation58 The localization of so many disease-related proteins in the same subcellular compartment further strengthens the existence of common biological pathways, which are likely of relevance also for other neurodegenerative diseases. For instance, using different in vitro and in vivo models, BECN1 was shown to ameliorate the neuronal pathology as well as promote the autophagic clearance of mutant proteins such as SNCA, HTT (huntingtin) and ATXN3/ataxin-3.Citation59-61 In murine models of Alzheimer disease, BECN1 was also found to directly interact with PARK2, and to promote β-amyloid degradation.Citation62-64 Similarly, a recent study demonstrated that PINK1 was able to counteract mitophagy impairment and neurotoxicity in a Drosophila model of Huntington disease.Citation65 In light of these findings, the localization of PINK1, PARK2 and BECN1 in the MAM compartment opens a novel intriguing scenario to further explore the biological functions of these pivotal neuroprotective proteins.
Materials and methods
Reagents and media
Reagents were purchased from the following manufacturers: 100-mm cell culture dishes (Falcon, 3–353003), penicillin-streptomycin-glutamine (Life Techonologies,10378–016), Dulbecco's modified Eagle's medium (DMEM; Life Technologies, 31966–021), fetal bovine serum (Euroclone, ECS0180L), G-418 (Life Technologies 11811–023), puromycin (Sigma-Aldrich, P9620), Lipofectamine 2000 (Invitrogen, 11668–019), polybrene (Sigma-Aldrich, H9268), DMSO (Sigma-Aldrich, 472301), CCCP (Sigma-Aldrich, C2759), valinomycin (Sigma-Aldrich, V0627), protease inhibitor cocktail (Sigma-Aldrich, P8340), phosphatase inhibitors (Thermo Scientific, 78420), protein G-agarose beads (Santa Cruz Biotechnology, SC2002), protein A-agarose beads (Santa Cruz Biotechnology, sc-2001), normal mouse IgG (Santa Cruz Biotechnology, sc-2025), normal rabbit IgG (Santa Cruz Biotechnology, sc-2027), EZview™Red Anti-HA affinity gel (Sigma- Aldrich, E6779), Bradford Assay reagent (Bio-Rad, 5000205), Laemmli sample buffer (Bio-Rad, 1610737), Novex Sharp pre-stained protein standard (Invitrogen, LC5800), 4–20% Mini-PROTEAN TGX gel (BioRad, 456-1094), RIPA buffer (Cell Signaling Technology, 9806S), Pierce ECL western blotting substrate (PIERCE, 32106), HEPES (pH 7.4; Sigma-Aldrich, H3375), D-mannitol (Sigma-Aldrich, M4125), sucrose (Merck Millipore, 100892.9050), Percoll (Sigma-Aldrich, P1644), EGTA (Sigma-Aldrich, E8145), Tris-HCl (pH 7.4; Sigma-Aldrich, T2663), Dulbecco's phosphate-buffered saline (PBS; Euroclone, ECB4004L), paraformaldehyde (Sigma-Aldrich, P6148), Triton X-100 (Sigma-Aldrich, T8787), normal goat serum (Life Technologies, PCN5000).
Antibodies
The following antibodies were used for western blotting, immunoprecipitation and immunofluorescence assays: rabbit anti-PINK1 (Novus Biologicals, BC100–494), mouse anti-HA (Sigma-Aldrich, H3663), rat anti-HA (Roche Applied Science, 10744700), mouse anti-BECN1 (BD Biosciences, BD612113), rabbit anti-BECN1 (Novus Biologicals,NB500–249), goat anti-MYC (Novus Biologicals, NB600–335), mouse anti-MYC (Santa Cruz Biotechnology, sc-40), mouse anti-MFN2 (Abcam, ab56889), mouse anti-OPA1 (BD Biosciences, BD612607), rabbit anti-VDAC (Cell Signaling Technology, 4661), mouse anti-TIMM23/TIM23 (BD Biosciences, BD611222), mouse anti-TOMM20/TOM20 (BD Biosciences, BD612278), mouse anti-COX4I1 (Abcam, ab14744), rabbit anti-PARK2 (Abcam, ab15954),rabbit anti-LC3B (Sigma-Aldrich, L7543), rabbit anti-cleaved PARP (Cell Signaling Technology, 95416), rabbit anti-CANX/calnexin (Sigma-Aldrich, C4731), rabbit anti-ACSL4/FACL4 (Abcam, ab137525), rabbit anti-HSPA9/GRP75 (Cell Signaling Technology, 3593), rabbit anti-GFP (Cell Signaling Technology, 2555) and mouse anti-TUBA/α-tubulin (Sigma-Aldrich, T9026).
Horseradish-peroxidase-conjugated secondary antibodies were: goat anti-mouse (Chemicon International, AP124P), goat anti-rabbit (Bio-Rad 1706515), goat anti-rat (Cell Signaling Technology, 7077), and rabbit anti-goat (Bio-Rad, 1721034).
Secondary antibodies used in immunofluorescence experiments were: goat anti-mouse Alexa Fluor 488 (Life Technologies, A21121), goat anti-mouse Alexa Fluor 555 (Life Technologies, A21127), and goat anti-rabbit Alexa Fluor 488 (Life Technologies, A11008).
Plasmids
BECN1-MYC, PINK1-HA and PINK1-KDD-HA constructs have been described previously.Citation9,52 Other constructs were: ER-GFP (Invitrogen, O36212), Red-Mito (Clontech, 632421), pRK5-HA-Parkin (Addgene, 17613; deposited by Ted Dawson), YFP-PARK2 (Addgene, 23955; deposited by Richard Youle). GFP-ZFYVE1/DFCP1 was kindly provided by N. T. Ktistakis (Babraham Institute, Cambridge, UK). The BECN1-mCherry and PINK1-mCherry vectors used in immunofluorescence experiments were generated by subcloning mCherry sequence at the 3′ end of full-length BECN1 and PINK1 cDNAs.
Cell cultures and treatments
HEK293, 293T, HeLa and SH-SY5Y cells were maintained in DMEM, supplemented with 2 mM L-glutamine, 200 U/ml penicillin, 200 mg/ml streptomycin and 10% heat inactivated fetal bovine serum at 37°C in 95% humidifier air and 5% CO2. For experiments of mitochondrial depolarization, cells were exposed to 25 µM CCCP or the vehicle DMSO at the indicated times.
Transfections, infections and RNA interference
SH-SY5Y and HeLa cells were transfected with Lipofectamine 2000 reagent. SH-SY5Y cells stably expressing PINK1-HA, GFP-ZFYVE1, ER-GFP and Red-Mito cDNAs were generated maintaining the transfected cells in the presence of selection media with G418 for at least 2 wk.
HEK293 and 293T cells were transfected with the calcium phosphate method using the ProFection Mammalian Transfection System (Promega, E1200). To knock down PINK1 and BECN1 expression in SH-SY5Y cells (shPINK1 and shBECN1), pMISSION validated shRNA bacterial glycerol stocks were purchased from Sigma-Aldrich (PINK1 clone ID: NM_032409.1–548s1c1; BECN1 clone ID: NM_003766.2–1117s1c1). The MISSION pLKO.1-puro scrambled shRNA was used as a negative control (shSCR). Lentiviral packaging vectors pMISSION-gagpol and pMISSION-vsvg (Sigma-Aldrich SHP001) were used as packaging constructs. The corresponding plasmids were purified from the JM109 strain and used to generate lentiviral particles in 293T packaging cells (ATCC, Number CRL-11268). The SH-SY5Y cells were infected overnight with the viral supernatant, in the presence of 8 mg/ml polybrene. Stable transductants were obtained by adding 2 mg/ml puromycin up to 10 d. Stable knockdown was confirmed by immunoblot and RT-PCR.
RT-PCR
Total RNA was extracted using the High Pure RNA Isolation Kit (Roche, 11828665001) and then reverse transcribed with SuperScript II Reverse Transcriptase (Invitrogen, 18064–014). For mtDNA quantification, total DNA was extracted using DNeasy Blood & Tissue Kit (Qiagen, 69581). Resulting cDNAs or DNAs were quantified by real-time PCR using SYBR green master mix (Life Technologies, 4368577) on the HT-7900 platform (Life Technologies), using the following primers: PINK1-Fw: 5′-CAAGAGGCTCAGCTACCTGCAC-3′ and PINK1-Rev: 5′-TGTCTCACGTCTGGAGGCACT-3′; BECN1-Fw: 5′-AGGAACTCACAGCTCCATTAC-3′ and BECN1-Rev: 5′-AATGGCTCCTCTCCTGAGTT-3′; mtDNA-Fw: 5′-AGGACAAGAGAAATAAGGCC-3′ and mtDNA-Rev: 5′-TAAGAAGAGGAATTGAACCTCTGACTGTAA-3′. The relative expression was calculated using the ΔΔCT method.
Immunoblotting
Cells were lysed in RIPA buffer containing protease and phosphatase inhibitors and protein extracts were quantified by Bradford assay. Equivalent amounts of lysates were resolved by electrophoresis through a 4–20% Mini-PROTEAN TGX gel and probed with the primary and secondary antibodies listed above. Detection was performed by using Pierce ECL western blotting substrate. All experiments were normalized by TUBA/α-tubulin expression. Image contrast and brightness were optimized using Adobe Photoshop CS6 (Adobe Systems Inc.). Densitometry measurements were calculated using ImageQuant TL Software (GE Healthcare). In all panels, values are plotted as fold-change relative to control, which has been set to a value of 1.
Coimmunoprecipitation
For immunoprecipitation experiments of mitochondrial and cytosolic fractions, HEK293 and SH-SY5Y cells, exposed or not to the indicated stress, were fractionated in advance as described below. IPs were performed by incubating 500 mg of each fraction with the indicated antibodies on a rotary shaker overnight at 4°C. Mouse or rabbit IgG were used as controls of nonspecific co-IP. The following day, 50 μl (50% suspension in Isolation Buffer) of protein A- or G-beads were added and further incubated for 6 h on a rotary shaker at 4°C. Beads were then washed 3 times with isolation buffer, immunocomplexes were eluted in Laemmli sample buffer at 95°C for 5 min and finally processed for western blotting.
Subcellular fractionation
The protocol for isolation of mitochondrial and cytoplasmic fractions was adapted from Wieckowski et al. Citation31 Briefly, cells were resuspended in isolation buffer (225 mM D-Mannitol, 75 mM sucrose, 0.1 mM EGTA, 30 mM Tris-HCl, pH 7.4) and disrupted by passing 25 times through a 25 g needle, followed by centrifugation for 8 min at 800 g at 4°C to discard nuclei and cell debris. The resulting supernatant was further centrifuged for 20 min at 10,000 g at 4°C. Pellets contained the crude mitochondrial-enriched fractions, whereas cytoplasmic/microsomal fractions remained in the supernatants. For experiments using cytoplasmic and crude mitochondrial fractions, equal amounts of proteins were analyzed by western blotting and probed with the indicated antibodies. Crude mitochondria were ultracentrifuged on a 30% Percoll-gradient at 100,000 g for 1 h at 4°C to further purify MAM and pure mitochondria. Cytoplasmic fractions were ultracentrifuged at 100,000 g for 1 h at 4°C to isolate cytosolic and microsomal fractions.
Mitophagy assessment
To evaluate mitophagy induction, PINK1 accumulation and PARK2 recruitment at mitochondria were assessed in confocal microscopy by colocalization experiments with the mitochondrial marker TOMM20 or with overexpressing Red-Mito; data were presented as the percentage of cells with PINK1 or PARK2 localized at mitochondria. Mitophagy progression was calculated by monitoring the disappearance of TOMM20 or Red-Mito signal and counting cells devoid of mitochondria, as well as by RT-PCR of mtDNA content. Omegasome formation was assessed in SH-SY5Y cells stably expressing GFP-ZFYVE1, by counting the percentage of cells exhibiting the accumulation of GFP-ZFYVE1 dots (≥ 5 dots/cell).
Modulation of the OPA1, MFN2 and VDAC1 levels in western blotting was used as a measure of early mitophagy, whereas the degradation of the inner mitochondrial membrane protein TIMM23 and the matrix protein COX4I1 was followed to monitor advanced mitophagy.Citation66
Immunofluorescence and confocal imaging
SH-SY5Y cells were grown for 24 h on glass cover slips before immunofluorescence. Where indicated, cells were transfected with the indicated plasmids. For immunofluorescence experiments, cells were washed with PBS and fixed in 4% paraformaldehyde in PBS for 20 min at room temperature and permeabilized by incubation in PBS containing 0.2% Triton X-100 and 10% goat serum for 1 h. Then, cells were incubated for 2 h at room temperature with primary antibodies resuspended in PBS plus 0.1% Triton X-100 and 1% goat serum. After PBS washing, primary antibodies were visualized using the appropriate secondary antibodies conjugated with either Alexa Fluor 488 or Alexa Fluor 555. After 3 washes with PBS, cells were mounted on glass slides using the ProLong Gold antifade reagent with DAPI (Molecular Probes, P36935), visualized with laser scanning confocal microscopy (C2 Confocal Microscopy System), by using the laser lines 488 nm (green) or 561 nm (red) and a 60×1.4 NA Plan Apo objective (Nikon Corporation), controlled by NIS Element AR 4.13.04 software. Image manipulation and merging were performed by using the appropriate tools of ImageJ 1.49i software. Quantification of colocalization, expressed in terms of Mander's Overlap Coefficient, was calculated using the JacoP plugin of ImageJ software.
Analysis of ER-mitochondria contact sites
To evaluate ER-mitochondria contact sites, cells expressing ER-GFP and Red-Mito were excited with the laser lines 488 nm or 561 nm, and 20–40 images, separated by 0.2 μm along the z-axis, were acquired. Deconvolution, 3D-reconstruction and surface rendering were realized using the VolumeJ plugin of ImageJ. Mander's colocalization coefficient, calculated by the JacoP plugin, was used to quantitatively assess the fraction of ER juxtaposing to mitochondria.
To measure the mitochondrial perimeter in contact with ER, single-plane confocal images of SH-SY5Y cells stably expressing ER-GFP and Red-Mito were used. The mean intensity of mitochondrial and ER fluorescence was calculated, and a threshold corresponding to 75% of the mean Red-Mito intensity was used to obtain binary images of mitochondria. To trace the profile of the mitochondrial perimeter, the ImageJ iso-photcontour2 plugin was applied to binary images of mitochondria. To evaluate the amount of mitochondrial perimeter in contact with the ER, we followed the method reported by Filadi et al.Citation67 ER-mitochondria colocalization was calculated as the ratio (R) between mitochondrial perimeter pixels colocalized with the ER and total mitochondrial perimeter pixels.
Phos-tag gel
To check BECN1 phosphorylation in the presence and absence of PINK1 shRNA, Phos-tag SDS-PAGE was performed using Phos-tag™ acrylamide (Wako Pure Chemical Industries, 304-93521), as described by the manufacturer. Briefly, shSCR and shPINK1 SH-SY5Y cells were treated with DMSO or CCCP, total cell lysates were prepared in lysis buffer (20 mM HEPES, 120 mM NaCl, 1% Triton X-100) containing protease inhibitor cocktail and resolved by 7% SDS-PAGE using a gel containing 50 μM Phos-tag acrylamide and MnCl2. Immunoblotting was conducted normally after soaking the gels in 1 mM EDTA for 10 min to remove the Mn2+. All other steps in this analysis were identical to normal SDS-PAGE and immunoblotting protocols. Transfer was conducted onto PVDF membrane.
FACS analysis
Cells were washed in PBS and fixed in 70% ethanol overnight at 4°C. The next day, samples were washed in PBS, 1% BSA and then processed according to the TUNEL-based In Situ Cell Death Detection Kit Fluorescein (Roche Diagnostic, 1684795), in conjunction with propidium iodide staining. Single-cell suspensions were analyzed by Cyan ADP (Beckman Coulter). Apoptosis was scored by quantifying the population of TUNEL- and propidium iodide-positive cells. Flow-cytofluorimetric data were plotted and analyzed by using the Summit 4.3 software (Beckman Coulter).
Statistical analysis
Densitometric results, counts at confocal microscopy and cytofluorimetric data were represented as histograms; values were obtained from at least 3 independent experiments and expressed as means ± SD, calculated with GraphPad Prism 5. Unpaired Student t test was used to assess the differences between 2 groups. A P-value of less than 0.05 was considered as significant.
Abbreviations
| ACSL4/FACL4 | = | acyl-CoA synthetase long-chain family member 4 |
| AMBRA1 | = | autophagy and Beclin 1 regulator 1 |
| ATG | = | autophagy related |
| BECN1 | = | Beclin 1 |
| CCCP | = | carbonyl cyanide m-chlorophenylhydrazone |
| co-IP | = | co-immunoprecipitation |
| COX4I1 | = | cytochrome c oxidase subunit 4I1 |
| ER-GFP | = | endoplasmic reticulum-targeted green fluorescent protein |
| FACS | = | fluorescence-activated cell sorting |
| GFP | = | green fluorescent protein |
| HSPA9/GRP75 | = | heat shock protein family A (Hsp70) member 9 |
| KDD | = | kinase dead domain |
| MAM | = | mitochondria-associated membranes |
| MAP1LC3/LC3 | = | microtubule- associated protein light chain 3 |
| Red-Mito | = | mitochondria-targeted red fluorescent protein |
| MFN2 | = | mitofusin 2 |
| mtDNA | = | mitochondrial DNA |
| OMM | = | outer mitochondrial membrane |
| OPA1 | = | OPA1, mitochondrial dynamin like GTPase |
| PARK2 | = | parkin RBR E3 ubiquitin protein ligase |
| PARP | = | poly(ADP-ribose) polymerase |
| PD | = | Parkinson disease |
| PINK1 | = | PTEN induced putative kinase 1 |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| PtdIns3P | = | phosphatidylinositol-3-phosphate |
| TIMM23 | = | translocase of inner mitochondrial membrane 23 |
| TOMM20 | = | translocase of outer mitochondrial membrane 20 |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| VDAC1 | = | voltage dependent anion channel 1 |
| VPS | = | vacuolar protein sorting |
| WIPI1 | = | WD repeat domain, phosphoinositide interacting 1 |
| YFP | = | yellow fluorescent protein |
| ZFYVE1/DFCP1 | = | zinc finger FYVE-type containing 1 |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KAUP_A_1277309_Supplementary_material.zip
Download Zip (6.6 MB)Acknowledgments
We are grateful to Dr. R. Filadi (Department of Biomedical Sciences, University of Padua, Italy) for his help with mitochondrial and ER morphology analysis.
Funding
This work was supported by grants from the Italian Ministry of Health (Bando Giovani Ricercatori 2009 and 2011), Italian Ministry of University and Research (FIRB Accordi di Programma 2010) and Telethon Foundation Italy (grant nr. GGP10140).
Related Research Data
References
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004; 304:1158-60; PMID:15087508; http://dx.doi.org/10.1126/science.1096284
- Deas E, Plun-Favreau H, Wood NW. PINK1 function in health and disease. EMBO Mol Med 2009; 1:152-65; PMID:20049715; http://dx.doi.org/10.1002/emmm.200900024
- Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci 2014; 37:315-24; PMID:24735649; http://dx.doi.org/10.1016/j.tins.2014.03.004
- Valente EM, Michiorri S, Arena G, Gelmetti V. PINK1: one protein, multiple neuroprotective functions. Future Neurol 2009; 4:575-90; http://dx.doi.org/10.2217/fnl.09.39
- Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet 2012; 3:297; PMID:23267366
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014; 510:162-6; PMID:24784582
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 2014; 460:127-39; PMID:24660806; http://dx.doi.org/10.1042/BJ20140334
- Choubey V, Cagalinec M, Liiv J, Safiulina D, Hickey MA, Kuum M, Liiv M, Anwar T, Eskelinen EL, Kaasik A. BECN1 is involved in the initiation of mitophagy: it facilitates PARK2 translocation to mitochondria. Autophagy 2014; 10:1105-19; PMID:24879156; http://dx.doi.org/10.4161/auto.28615
- Michiorri S, Gelmetti V, Giarda E, Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R, Arena G, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ 2010; 17:962-74; PMID:20057503; http://dx.doi.org/10.1038/cdd.2009.200
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 2011; 286:19630-40; PMID:21454557; http://dx.doi.org/10.1074/jbc.M110.209338
- Chen Y, Dorn GW. 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013; 340:471-5; PMID:23620051; http://dx.doi.org/10.1126/science.1231031
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 2010; 19:4861-70; PMID:20871098; http://dx.doi.org/10.1093/hmg/ddq419
- Glauser L, Sonnay S, Stafa K, Moore DJ. Parkin promotes the ubiquitination and degradation of the mitochondrial fusion factor mitofusin 1. J Neurochem 2011; 118:636-45; PMID:21615408; http://dx.doi.org/10.1111/j.1471-4159.2011.07318.x
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 2010; 5:e10054; PMID:20383334; http://dx.doi.org/10.1371/journal.pone.0010054
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011; 147:893-906; PMID:22078885; http://dx.doi.org/10.1016/j.cell.2011.10.018
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010; 6:1090-106; PMID:20890124; http://dx.doi.org/10.4161/auto.6.8.13426
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119-31; PMID:20098416; http://dx.doi.org/10.1038/ncb2012
- Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 2012; 125:795-9; PMID:22448035; http://dx.doi.org/10.1242/jcs.093849
- Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 2015; 85:257-73; PMID:25611507; http://dx.doi.org/10.1016/j.neuron.2014.12.007
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015; 524:309-14; PMID:26266977; http://dx.doi.org/10.1038/nature14893
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A 2014; 111:E4439-48; PMID:25294927; http://dx.doi.org/10.1073/pnas.1405752111
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol 2010; 20:355-62; PMID:20356743; http://dx.doi.org/10.1016/j.tcb.2010.03.002
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 2013; 14:759-74; PMID:24201109; http://dx.doi.org/10.1038/nrm3696
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013; 495:389-93; PMID:23455425; http://dx.doi.org/10.1038/nature11910
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A 2000; 97:13354-9; PMID:11078524; http://dx.doi.org/10.1073/pnas.240347797
- Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2012; 2:1002; PMID:23256036; http://dx.doi.org/10.1038/srep01002
- Chen D, Chen X, Li M, Zhang H, Ding WX, Yin XM. CCCP-Induced LC3 lipidation depends on Atg9 whereas FIP200/Atg13 and Beclin 1/Atg14 are dispensable. Biochem Biophys Res Commun 2013; 432:226-30; PMID:23402761; http://dx.doi.org/10.1016/j.bbrc.2013.02.010
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/10.1016/j.cell.2005.07.002
- Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, Piacentini M, Levine B, Cecconi F. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J 2011; 30:1195-208; PMID:21358617; http://dx.doi.org/10.1038/emboj.2011.49
- Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc 2009; 4:1582-90; PMID:19816421; http://dx.doi.org/10.1038/nprot.2009.151
- Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, Cooper TJ, Thomas G, Simmen T. The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell 2008; 19:2777-88; PMID:18417615; http://dx.doi.org/10.1091/mbc.E07-10-0995
- Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta 2013; 1833:213-24; PMID:22575682; http://dx.doi.org/10.1016/j.bbamcr.2012.04.013
- Cali T, Ottolini D, Negro A, Brini M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim Biophys Acta 2013; 1832:495-508; PMID:23313576; http://dx.doi.org/10.1016/j.bbadis.2013.01.004
- Van Laar VS, Roy N, Liu A, Rajprohat S, Arnold B, Dukes AA, Holbein CD, Berman SB. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol Dis 2015; 74:180-93; PMID:25478815; http://dx.doi.org/10.1016/j.nbd.2014.11.015
- Gautier CA, Erpapazoglou Z, Mouton-Liger F, Muriel MP, Cormier F, Bigou S, Duffaure S, Girard M, Foret B, Iannielli A, et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum Mol Genet 2016; 25(14):2972-84 [Epub ahead of print]; PMID:27206984
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 2010; 107:378-83; PMID:19966284; http://dx.doi.org/10.1073/pnas.0911187107
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 2006; 174:915-21; PMID:16982799; http://dx.doi.org/10.1083/jcb.200604016
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685-701; PMID:18725538; http://dx.doi.org/10.1083/jcb.200803137
- Fogel AI, Dlouhy BJ, Wang C, Ryu SW, Neutzner A, Hasson SA, Sideris DP, Abeliovich H, Youle RJ. Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol Cell Biol 2013; 33:3675-88; PMID:23878393; http://dx.doi.org/10.1128/MCB.00079-13
- Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci U S A 2005; 102:5703-8; PMID:15824318; http://dx.doi.org/10.1073/pnas.0500617102
- Hollville E, Carroll RG, Cullen SP, Martin SJ. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol Cell 2014; 55:451-66; PMID:24999239; http://dx.doi.org/10.1016/j.molcel.2014.06.001
- Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 2012; 111:1208-21; PMID:23065344;http://dx.doi.org/10.1161/CIRCRESAHA.112.265819
- Carroll RG, Hollville E, Martin SJ. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep 2014; 9:1538-53; PMID:25456142; http://dx.doi.org/10.1016/j.celrep.2014.10.046
- Rambold AS, Lippincott-Schwartz J. Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle 2011; 10:4032-8; PMID:22101267; http://dx.doi.org/10.4161/cc.10.23.18384
- Klintworth H, Newhouse K, Li T, Choi WS, Faigle R, Xia Z. Activation of c-Jun N-terminal protein kinase is a common mechanism underlying paraquat- and rotenone-induced dopaminergic cell apoptosis. Toxicol Sci 2007; 97:149-62; PMID:17324951; http://dx.doi.org/10.1093/toxsci/kfm029
- Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy 2007;3:663-6; PMID:17622797; http://dx.doi.org/10.4161/auto.4625
- Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009; 284:13843-55; PMID:19279012; http://dx.doi.org/10.1074/jbc.M808515200
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654-8; PMID:19794493; http://dx.doi.org/10.1038/nature08455
- Naon D, Scorrano L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim Biophys Acta 2014; 1843:2184-94;PMID:24875902;http://dx.doi.org/10.1016/j.bbamcr.2014.05.011
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795-803; PMID:19029340; http://dx.doi.org/10.1083/jcb.200809125
- Arena G, Gelmetti V, Torosantucci L, Vignone D, Lamorte G, De Rosa P, Cilia E, Jonas EA, Valente EM. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ 2013; 20:920-30; PMID:23519076; http://dx.doi.org/10.1038/cdd.2013.19
- Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A 2000; 97:3100-5; PMID:10737788; http://dx.doi.org/10.1073/pnas.97.7.3100
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 2009; 33:627-38; PMID:19285945; http://dx.doi.org/10.1016/j.molcel.2009.02.013
- Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, Dallapiccola B, Valente EM, Masliah E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson's disease by disturbing calcium flux. J Neurochem 2009; 108:1561-74; PMID:19166511; http://dx.doi.org/10.1111/j.1471-4159.2009.05932.x
- Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S, Poletti F, et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun Signal 2011; 9:19; PMID:21939514; http://dx.doi.org/10.1186/1478-811X-9-19
- Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, Voos W, Schon EA, Przedborski S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci 2014; 34:249-59;PMID:24381286;http://dx.doi.org/10.1523/JNEUROSCI.2507-13.2014
- Ottolini D, Cali T, Negro A, Brini M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum Mol Genet 2013; 22:2152-68; PMID:23418303; http://dx.doi.org/10.1093/hmg/ddt068
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci 2009; 29:13578-88; PMID:19864570; http://dx.doi.org/10.1523/JNEUROSCI.4390-09.2009
- Shibata M, Lu T, Furuya T, Degterev A, Mizushima N, Yoshimori T, MacDonald M, Yankner B, Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J Biol Chem 2006; 281:14474-85;PMID:16522639;http://dx.doi.org/10.1074/jbc.M600364200
- Nascimento-Ferreira I, Nobrega C, Vasconcelos-Ferreira A, Onofre I, Albuquerque D, Aveleira C, Hirai H, Deglon N, Pereira de Almeida L. Beclin 1 mitigates motor and neuropathological deficits in genetic mouse models of Machado-Joseph disease. Brain 2013; 136:2173-88; PMID:23801739; http://dx.doi.org/10.1093/brain/awt144
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008; 118:2190-9; PMID:18497889
- Lonskaya I, Hebron ML, Desforges NM, Franjie A, Moussa CE. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol Med 2013; 5:1247-62; PMID:23737459; http://dx.doi.org/10.1002/emmm.201302771
- Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet 2011; 20:2091-102; PMID:21378096;http://dx.doi.org/10.1093/hmg/ddr091
- Khalil B, El Fissi N, Aouane A, Cabirol-Pol MJ, Rival T, Lievens JC. PINK1-induced mitophagy promotes neuroprotection in Huntington's disease. Cell Death Dis 2015; 6:e1617; PMID:25611391; http://dx.doi.org/10.1038/cddis.2014.581
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/10.1080/15548627.2015.1100356
- Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci U S A 2015; 112:E2174-81; PMID:25870285; http://dx.doi.org/10.1073/pnas.1504880112