
ABSTRACT
Pancreatic ductal adenocarcinoma (PDAC) is the most aggressive and lethal cancer. The role of autophagy in the pathobiology of PDAC is intricate, with opposing functions manifested in different cellular contexts. MIR506 functions as a tumor suppressor in many cancer types through the regulation of multiple pathways. In this study, we hypothesized that MIR506 exerted a tumor suppression function in PDAC by inducing autophagy-related cell death. Our results provided evidence that downregulation of MIR506 expression was associated with disease progression in human PDAC. MIR506 triggered autophagic flux in PDAC cells, which led to autophagy-related cell death through direct targeting of the STAT3 (signal transducer and activator of transcription 3)-BCL2-BECN1 axis. Silencing and inhibiting STAT3 recapitulated the effects of MIR506, whereas forced expression of STAT3 abrogated the effects of MIR506. We propose that the apoptosis-inhibitory protein BCL2, which also inhibits induction of autophagy by blocking BECN1, was inhibited by MIR506 through targeting STAT3, thus augmenting BECN1 and promoting autophagy-related cell death. Silencing BECN1 and overexpression of BCL2 abrogated the effects of MIR506. These findings expand the known mechanisms of MIR506-mediated tumor suppression to activation of autophagy-related cell death and suggest a strategy for using MIR506 as an anti-STAT3 approach to PDAC treatment.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is considered the most lethal and aggressive of all neoplasms, with a dismal 5-y survival rate hovering around 6% and a median survival duration of less than 6 mo.Citation1,2 Less than 15% of patients with PDAC are suitable for surgical resection because most have locally advanced or distant metastatic disease at the time of diagnosis.Citation3,4 Even the most advanced chemotherapeutics and targeted therapies have brought only modest survival benefits.Citation5 Thus, effective prognostic markers and novel treatment strategies and agents are urgently needed to improve the therapeutic outcome of PDAC.
Macroautophagy (referred to as autophagy) is an evolutionarily conserved cellular process that plays an important role in maintaining cellular homeostasis and overcoming metabolic stress and nutrient deprivation.Citation6,7 Autophagy is observed in a wide range of cancer cells and its function varies significantly depending on cancer subtype and distinct cellular context. Under appropriate conditions, autophagy has cytoprotective roles and leads to therapeutic resistance by removing or mitigating harmful stimuli. Conversely, excessive or prolonged autophagy functions as a tumor-suppressive mechanism by inducing autophagy-related cell death (type II programmed cell death or PCD) independent of, or in parallel with, apoptosis and necrosis in certain cellular conditions.Citation8-11 Autophagy-related cell death can be induced by inhibition of anti-autophagic proteins in some apoptosis-resistant cancers, including PDAC.Citation12-14 The role of autophagy in the pathobiology of PDAC is intricate, with opposing functions, but the mechanisms have not been well documented.Citation15
STAT3 (signal transducer and activator of transcription 3) is a potent transcription factor that regulates the expression of more than 1,000 genes involved in multiple aspects of cellular processes.Citation16 The prominent roles of STAT3 in regulating autophagy are supported by growing evidence.Citation17-22 STAT3 functions as the main transcription regulator of several autophagy-related genes, such as BCL2, BECN1, PIK3C3, PIK3R1, BNIP3, and HIF1A. Because these genes function in a context-dependent manner, the crosstalk between autophagy and STAT3 signaling may decide the survival or death of a cell.Citation23-25
MIR506, a component of an X chromosome-linked miRNA cluster, has been reported to function as an important tumor suppressor in many human cancers. Previous studies have identified MIR506 as a key gatekeeper for the network regulating epithelial-to-mesenchymal transition by regulating SNAI2, VIM, and CDH2, and its expression is associated with good prognosis in ovarian and gastric cancers.Citation26,27 MIR506 suppresses proliferation and induces senescence by directly targeting the CDK4-CDK6-FOXM1 axis.Citation28 The antiproliferative function of MIR506 has also been established in esophageal, gastric, colorectal, and cervical cancers as well as glioblastoma and hepatocellular carcinoma.Citation29 MIR506 is involved in the responses of ovarian cancers and pancreatic cancer to chemotherapy through regulation of RAD51 and SPHK1.Citation30,31 The role of MIR506 in autophagic regulation, however, has not been reported.
Recent reports have shown that expression of MIR506 is downregulated in PDAC tissues.Citation31,32 Furthermore, STAT3 was recently identified as a direct target of MIR506.Citation33 We hypothesized that MIR506 exerted a tumor suppression function in PDAC by inducing autophagy-related cell death through targeting the STAT3 pathway. In the current study, we investigated the expression of MIR506 in different PDAC tissues and its relationship with the prognosis of the patients. Further experiments with patient-derived xenograft (PDX) cell systems revealed that MIR506 functions as a tumor suppressor by inducing autophagy-related cell death through STAT3 signaling pathways. Our findings have unveiled a previously unrecognized mechanism underlying the anticancer effects of MIR506 against human PDAC.
Results
MIR506 downregulation is associated with disease progression in human PDAC
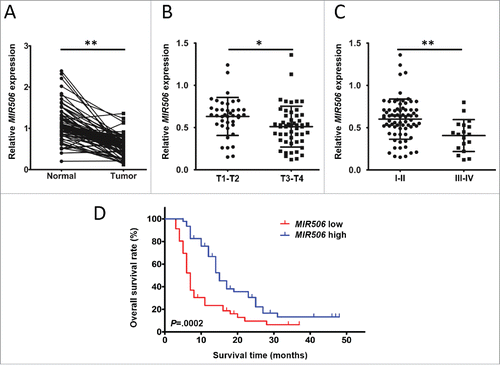
Quantification of MIR506 expression in 92 matched pairs of human PDAC and adjacent normal tissues by quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) showed that the relative MIR506 expression levels were significantly lower in PDAC tissues than in the adjacent normal tissues (P < 0.01; ). Clinicopathological analyses showed that lower MIR506 expression in the tumors was significantly correlated with T status and TNM stage (TNM Classification of Malignant Tumors) (Table S1). The relative MIR506 expression levels were significantly lower in the advanced tumor than early stage tumor (lower in the III-IV group and T3-T4 group than in the I-II group and T1-T2 group) (P < 0.05 and P < 0.01, respectively; and ). Kaplan-Meier survival analysis indicated that patients whose tumor expressed a lower level of MIR506 had a significantly lower overall survival rate ().
Figure 1. MIR506 downregulation correlates with disease progression in human PDAC. (A) Comparison of MIR506 expression in 92 matched pairs of PDAC tissues and corresponding nontumor tissues via qRT-PCR. RNU6 was used as an internal control. (B) Comparison of MIR506 expression in stage T1-T2 and stage T3-T4 PDAC tissues. (C) Comparison of MIR506 expression in stage I-II and stage III-IV PDAC tissues. *P < 0.05; **P < 0.01. (D) Kaplan-Meier curves compared the overall survival rates of PDAC patients whose tumor expressed a low or high level of MIR506. The median value was used as the cut-off point for definition of low and high MIR506 expression groups.
MIR506 induces cell death in human PDAC cells
The effect of MIR506 on proliferation of PDAC cells was evaluated by immunofluorescent staining for MKI67/Ki-67 in PDX lines MDA-PATC53, MDA-PATC124, and MDA-PATC148. MIR506 dramatically decreased the MKI67-positive rate in PDAC cells (Fig. S1A). Consistent with this finding was the observation that MIR506 significantly impaired the colony-formation capacity of PDAC cells (Fig. S1B). Assessment of the cell cycle distribution of these cells by propidium iodide (PI) staining and bromodeoxyuridine (BrdU) assay demonstrated that all MIR506-transfected cells had a higher fraction in G1 phase and a corresponding lower fraction in S phase than cells transfected with control miRNA (MIRctrl) (Fig. S2). Luciferase reporter assay showed that MIR506 suppressed proliferation and induced G1 arrest by directly targeting CDK4-CDK6 (Fig. S3) in PDAC cells, which was similar to what we reported in ovarian cancer cells.
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed to evaluate the impact of MIR506 on PDAC cell viability. Surprisingly, cell viability significantly decreased in a time-dependent manner in MIR506-transfected PDAC cells (), which contrasted with previous findings that MIR506 resulted only in blockage of cell viability relative to that of control miRNA-treated cells.
Figure 2. MIR506 induces cell death in human PDAC cells. (A) MDA-PATC53, MDA-PATC124, and MDA-PATC148 cells were transfected with MIRctrl or MIR506 for 5 d. Cell viability was measured by the MTT assay every day. (B) After transfection with MIRctrl or MIR506, cells were trypsinized and collected in PBS, and cell death was quantified by trypan blue exclusion assay every day. Data represent mean ± SD of at least of 3 independent experiments. **P < 0.01.
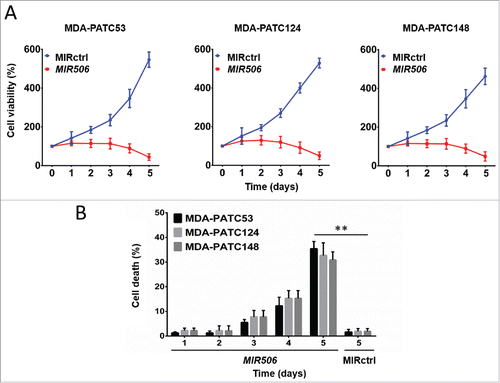
We performed a trypan blue exclusion assay to confirm whether the MIR506-mediated decrease of PDAC cell viability was associated with cell death. The results show that MIR506 induced cell death in a time-dependent manner (). These results reveal that MIR506 not only inhibited the proliferation of PDAC cells but also induced their death.
Cell death caused by MIR506 is dependent on autophagy
To determine the mode of cell death caused by MIR506, we first investigated whether the cytotoxicity was related to apoptosis. PDAC cells treated with MIR506 or the apoptosis inducer 5-fluorouracil (5-FU) were exposed to the selective inhibitor of apoptosis N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (z-VAD-FMK). Cell death was analyzed by ANXA5-PI staining followed by flow cytometry. Z-VAD-FMK dramatically decreased the ANXA5-positive cell population induced by 5-FU but showed no effect on that induced by MIR506 (). The results of the trypan blue exclusion assay also showed that z-VAD-FMK significantly suppressed the apoptotic cell death caused by 5-FU but failed to protect the cells from death caused by MIR506 (). To confirm these results, we performed Apo-BrdU assays (to detect apoptotic cells) and immunoblotting. As shown in , MIR506 failed to induce a significant increase in the Apo-BrdU-positive cell population. Consistent with this finding was the absence of cleaved CASP3 (caspase 3) and cleaved CASP9 in lysates of MIR506-transfected cells (). These data show that MIR506, unlike 5-FU, induced predominantly non-apoptotic cell death.
Figure 3. Cell death caused by MIR506 is dependent on autophagy. (A) MDA-PATC53 cells were transfected with MIRctrl or MIR506 or pretreated with apoptosis-inducer 5-FU in the absence (DMSO) or presence of the selective apoptosis inhibitor z-VAD-FMK (z-VAD; 20 mM) for 96 h; (B) MDA-PATC53 cells were transfected with MIRctrl or MIR506 or pretreated with the necrosis promoter t-BHP in the absence (DMSO) or presence of necrosis inhibitor IM-54 (10 mM) for 96 h; (C) MDA-PATC53 cells were pretreated with the autophagy inhibitor 3-MA (5 mM) or CQ (50 μM) or transfected with shRNAs to knock down ATG5 or ATG7 first, and then transfected with MIRctrl or MIR506 for 96 h. Cells were trypsinized and analyzed by flow cytometry after being stained with FITC-labeled ANXA5-PI. (D-F) After pretreatment as described in (A-C), cell death was also quantified by trypan blue exclusion assay. (G) MDA-PATC53 cells were transfected with MIRctrl or MIR506 or pretreated with 5-FU in the absence (DMSO) or presence of z-VAD-FMK (20 mM) for 96 h. Cells were labeled with Apo-BrdU and analyzed by flow cytometry. (H) After pretreatment as described in (G), whole-cell extracts of MDA-PATC53 cells were subjected to immunoblotting with anti-CASP3 and -CASP9 antibodies. (I) MDA-PATC53 cells were pretreated with or without the LMP inducer LLOMe (2 mM) or transfected with MIRctrl or MIR506 for 96 h. Cells were fixed for immunofluorescent staining with anti-LGALS1 or -LGALS3 antibody and visualized with a fluorescence microscope. Data represent mean ± SD of at least of 3 independent experiments. NS, not significant; *P < 0.05. Scale bar: 10 μm.
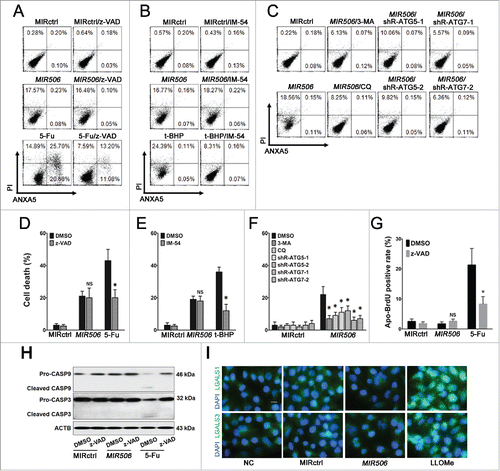
To determine the role of necrosis in PDAC cell death, we pretreated cells with a selective inhibitor of necrosis, 2-(1H-Indol-3-yl)-3-pentylamino-maleimide (IM-54), to antagonize the cell death induced by MIR506 or the known necrosis inducer tert-butyl hydroperoxide (t-BHP). IM-54 significantly reduced the ANXA5-negative, PI-positive cell population () and abrogated t-BHP-induced cell death (), but it did not protect the cells from death and did not decrease the ANXA5-negative, PI-positive cell population induced by MIR506. These results show that necrosis was not the mode of MIR506-induced cell death.
To determine whether MIR506-induced cell death was associated with autophagy, we examined the effect of the autophagy inhibitors 3-methyladenine (3-MA) and chloroquine (CQ) in PDAC cells. Genetic inhibition of autophagy was also used to demonstrate the requirement of autophagy in the MIR506-induced cell death (Fig. S4). Pretreatment with 3-MA or CQ, or knockdown of the essential autophagy genes ATG5 or ATG7 by shRNA significantly prevented MIR506-induced cell death and caused a dramatic decrease in the ANXA5-negative, PI-positive cell population in MIR506-treated cells ( and ). These results suggest that autophagy is the major mechanism of MIR506-induced cell death in PDAC cells.
Lysosomal membrane permeabilization (LMP) could be induced by a broad array of stimuli and leads to lysosomal cell death, which can present with apoptotic or necrotic features according to the extent of lysosomal hydrolase leakage. Because lysosomal cell death is often confused with autophagic cell death due to the accumulation of autophagosomes, we performed immunofluorescence by staining with LMP-specific markers LGALS1 and LGALS3 to rule out this possibility.Citation34 The LMP inducer L-leucyl-L-leucine methyl ester (LLOMe) was used as a positive control. The results showed that MIR506 could not induce LMP ().
MIR506 induces autophagic flux in PDAC cells
To confirm that MIR506 can induce autophagy in PDAC cells, we performed an immunofluorescence analysis for LC3, a marker of autophagosome formation. The formation of LC3-labeled vacuoles was markedly increased in the cytoplasm after transfection with MIR506 and could be prevented with 3-MA ( and ). We evaluated the formation of autolysosomes by flow cytometry after staining with acridine orange (AO). As shown in Fig. S5, MIR506 significantly induced the formation of autolysosomes in PDAC cells, and the AO-positive cell population was reduced by the autophagy inhibitor 3-MA.
Figure 4. MIR506 induces autophagic flux in PDAC cells. (A) MDA-PATC53 cells were transfected with MIRctrl or MIR506 in the absence or presence of the autophagy inhibitor 3-MA (5 mM) for 72 h. Cells were fixed for immunofluorescent staining with anti-LC3 antibody and visualized with a fluorescence microscope. (B) The LC3 puncta in each cell were counted. n = 100 cells/sample, **P<0.01. (C) MDA-PATC53 cells were transfected with MIRctrl or MIR506 for 48 h and infected with Autophagy Tandem Sensor RFP-GFP-LC3 in the absence or presence of 3-MA (5 mM) for 24 h. Then, cells were visualized with a fluorescence microscope. (D) The autophagosomes (yellow dots) and autolysosomes (red-only dots) in each cell were counted. n = 100 cells/sample. (E) MDA-PATC53 cells were transfected with MIRctrl or MIR506 in the absence or presence of the vacuolar-type H+-ATPase inhibitor Baf-A (10 nM). Whole-cell extracts were subjected to immunoblotting with anti-LC3 and -SQSTM1 antibodies. Scale bar: 10 μm.
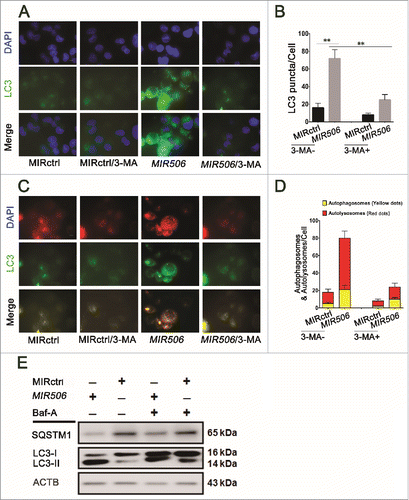
Autophagic flux was also monitored with the Premo Autophagy Tandem Sensor RFP-GFP-LC3. RFP fluorescence was detectable in both autophagosomes and autolysosomes, but GFP fluorescence was quenched in autolysosomes by the low-pH environment. MIR506 significantly increased the formation of autophagosomes and autolysosomes, indicating that it enhanced autophagic flux ( and ). Conversion of non-lipidated soluble LC3 (LC3-I) to phosphatidylethanolamine-conjugated LC3 (LC3-II) serves as a sensitive indicator of autophagy, and immunoblotting analysis revealed a dramatic increase in the ratios of LC3-II to LC3-I and LC3-II to actin in response to MIR506 transfection. Our findings on the clearance of SQSTM1, another distinct feature of autophagy, were consistent with these results. To determine whether the observed high levels of autophagic markers were due to increased autophagic flux, we treated cells with the vacuolar-type H+-ATPase inhibitor bafilomycin A1 (Baf-A), which prevents fusion between autophagosomes and lysosomes, thus inhibiting LC3 degradation.Citation35 Treatment with Baf-A caused significant increases of LC3-II accumulation in MIR506-transfected cells indicating that MIR506 can enhance autophagic flux ( and S6). Taken together, these results support the idea that MIR506 induced autophagic flux in PDAC cells.
MIR506 directly targets STAT3 and induces autophagy-related cell death in a STAT3-dependent manner
The miRNA target analysis algorithm TargetScan 6.0 predicted that the 3′-untranslated region (UTR) of STAT3 mRNA contains a potential binding site of MIR506 and this site is highly conserved among different species (). We chose STAT3 as a validation target because it is involved in multiple aspects of the autophagic process and was shown to be a direct target of MIR506 in glioma cells in a recent report by Peng et al.Citation33 To examine the regulation of STAT3 by MIR506 we performed a luciferase reporter assay in MDA-PATC53 cells. Using a dual-luciferase reporter system, we showed that MIR506 significantly inhibited the luciferase reporter activity of the wild-type but not the mutant STAT3 3′-UTR, indicating that STAT3 is a direct target of MIR506 in PDAC cells ().
Figure 5. MIR506 directly targets STAT3 and induces autophagy-related cell death in a STAT3-dependent manner. (A) A MIR506 binding site was predicted in the 3′-UTR of STAT3 by TargetScan. (B) A luciferase reporter assay showed that MIR506 directly targeted the STAT3 3′-UTR. MDA-PATC53 cells were cotransfected with wild-type or mutant STAT3 3′-UTR-luciferase reporter and MIRctrl or MIR506 mimic for 72 h before analysis. The firefly and renilla luciferase activities were measured using a Dual Luciferase Reporter Assay System. (C-D) MDA-PATC53, MDA-PATC124, MDA-PATC148, MDA-PATC50, and MDA-PATC69 cells were transfected with MIRctrl or MIR506. Whole-cell extracts were subjected to immunoblotting with anti-STAT3, anti-p-STAT3, and anti-LC3 antibodies. (E) The expression level of STAT3 was detected by qRT-PCR and normalized to the endogenous control GAPDH. (F) Cell death was quantified by trypan blue exclusion assay. Data represent the mean ± SD of at least 3 independent experiments. NS, not significant; **P < 0.01.
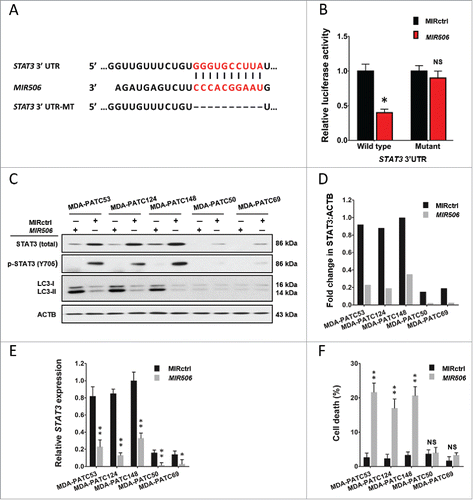
Immunoblotting and qRT-PCR analyses showed that MIR506 significantly reduced the expression of STAT3 at both protein and mRNA levels in all 5 PDAC cell lines tested (). Marked conversion of LC3-I to LC3-II was detected only in cell lines with a high baseline level of STAT3, including MDA-PATC53, MDA-PATC124, and MDA-PATC148 (). We also performed flow cytometry with AO staining to confirm the induction of autophagy. The results revealed that the AO-positive cell population increased significantly in response to MIR506 only in cell lines with a high baseline STAT3 level (Fig. S7). Consistent with these results, autophagy-related cell death was observed in cells with a high STAT3 level but not in those with a low STAT3 level (). These results indicate that MIR506 triggered autophagy-related cell death and that this effect was dependent on STAT3.
MIR506 induces autophagy-related cell death through the STAT3 signaling pathway
The STAT3 signaling pathway, a prominent stress-responsive pathway, is involved in multiple aspects of the autophagic process and modulates autophagy via both transcription-dependent and transcription-independent pathways. Immunoblotting analysis for STAT3-regulated autophagy-related genes revealed a dramatic increase in BECN1 and decrease in BCL2 after MIR506 transfection in PDAC cells (). Because BECN1 is essential for autophagy upregulation in response to stress and BCL2 is a central inhibitor of this process by directly binding BECN1 via its BH3 domain, we hypothesized that MIR506 induces autophagy-related cell death through the STAT3-BCL2-BECN1 signaling pathway.
Figure 6. MIR506 induces autophagy-related cell death through the STAT3 signaling pathway. (A) MDA-PATC53, MDA-PATC124, and MDA-PATC148 cells were transfected with MIRctrl or MIR506 for 72 h. Whole-cell extracts were subjected to immunoblotting with anti-STAT3, anti-p-STAT3, anti-BECN1, -BCL2, and -LC3 antibodies. (B-C) MDA-PATC53, MDA-PATC124, and MDA-PATC148 cells were treated with the indicated concentrations of the STAT3 inhibitor Stattic for the indicated times. (D-E) Cell death was quantified by trypan blue exclusion assay. (F) MDA-PATC53, MDA-PATC124, and MDA-PATC148 cells were transfected with control siRNA (siR-ctrl) or 1 of 2 siRNAs against STAT3 (siR-STAT3–1 or siR-STAT3–2). Whole-cell extracts were subjected to immunoblotting with anti-STAT3, anti-p-STAT3, anti-BECN1, -BCL2, and -LC3 antibodies. (G) Cell death was quantified by trypan blue exclusion assay. (H-I) MDA-PATC53 cells overexpressing wild-type or Y705-mutated STAT3 were transfected with MIRctrl or MIR506 and analyzed by immunoblotting and trypan blue exclusion assay as described above. (J-K) MDA-PATC53 cells were transfected with control shRNA (shR-ctrl) or 1 of 2 shRNAs against BECN1 (siR-BECN1–1 or siR-BECN1–2) and analyzed by immunoblotting and trypan blue exclusion assay as described above. (L-M) MDA-PATC53 cells overexpressing BCL2 were transfected with MIRctrl or MIR506 and analyzed by immunoblotting and trypan blue exclusion assay as described above. Data represent the mean ± SD of at least of 3 independent experiments. NS, not significant; *P < 0.05; **P < 0.01.
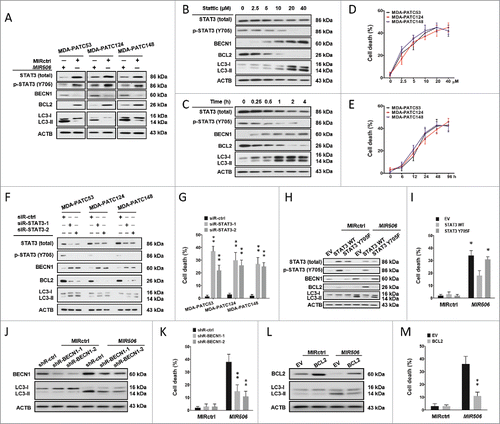
To confirm that autophagy-related cell death triggered by MIR506 was mediated by repression of STAT3 in PDAC cells, gain-of-function and loss-of-function studies were carried out. First, we treated PDAC cells with a selective STAT3 inhibitor, Stattic, which inhibits activation, dimerization, and nuclear translocation of STAT3 by interacting with the SH2 domain to imitate the suppressive effect of MIR506. Following the decline of phosphorylated STAT3 (p-STAT3; Y705) expression, BECN1 increased and BCL2 decreased in both dose-dependent and time-dependent manners in total proteins isolated from the treated cells. The LC3 conversion and AO staining showed dramatic enhancement of autophagic flux (, and S8). The trypan blue exclusion assay also revealed a gradual increase of cell death ( and ). Silencing of STAT3 by small-interfering RNA (siRNA) knockdown (si-STAT3–1 and si-STAT3–2), similar to MIR506, significantly increased autophagic flux and cell death ( and S9).
To determine whether ectopic expression of STAT3 could rescue the effects of MIR506, we established MDA-PATC53 cells that stably overexpressed wild-type or Y705-mutant STAT3 and subsequently transfected them with MIR506. Overexpression of wild-type STAT3 partially abrogated the MIR506-induced downregulation of both total STAT3 and p-STAT3, while overexpression of mutated STAT3 rescued only the downregulation of total STAT3. The forced expression of wild-type STAT3 significantly counteracted the autophagic flux and cell death triggered by MIR506, suggesting that STAT3 mediated the autophagy-related cell death-inducing function of MIR506 in PDAC ( and S10).
To further confirm the major role of BECN1 and BCL2 in autophagy-related cell death induced by MIR506, we first silenced BECN1 by siRNA (si-BECN1–1 and si-BECN1–2) in MDA-PATC53. Western blot showed that knockdown of BECN1 abrogated the conversion of LC3 caused by MIR506 (). Knockdown of BECN1 also reduced the AO-positive cell ratio and cell death caused by MIR506 ( and S11). Next, we overexpressed BCL2 by transfecting MDA-PATC53 cells with a BCL2 expression plasmid. Overexpression of BCL2 inhibited the conversion of LC3, reduced the AO-positive cell ratio ( and S12) and rescued the cell death caused by MIR506 ().
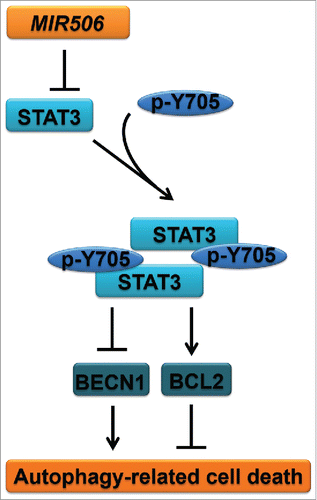
Together, these results suggest that MIR506-mediated regulation of the STAT3-BCL2-BECN1 signaling pathway is one of the major mechanisms underlying induction of autophagy-related cell death. The proposed mechanism is summarized in .
Figure 7. Schematic of the proposed mechanism of MIR506 induction of autophagy-related cell death in PDAC cells through the STAT3 signaling pathway.
Discussion
In this study, we showed that decreased expression of MIR506 accompanied disease progression in human PDAC. The normal pancreatic tissue is a mixture of acinar, ductal and endocrine tissue. The relative contributions of each component may vary in different samples, which may explain the heterogeneity observed in normal tissues. Future studies are needed to discern the relative expression levels of MIR506 in different cell components in normal pancreas and understand its role in cellular differentiation. The most important contribution of this study to the understanding of MIR506 as a tumor suppressor is that we showed for the first time that MIR506 triggered autophagic flux and autophagy-related cell death in PDAC cells. We further demonstrated that MIR506 regulated autophagy-related cell death through direct targeting of STAT3. Silencing and inhibiting STAT3 recapitulated the effects of MIR506, whereas forced expression of wild-type STAT3, but not the kinase-inactive mutant form, abrogated the effects of MIR506. The regulatory effect of MIR506 on STAT3 led to inhibition of BCL2 and activation of BECN1. These findings demonstrate that autophagy-related cell death is an unrecognized mechanism underlying the anticancer effects of MIR506 against human PDAC cells.
Autophagy is an evolutionarily conserved process that recycles nonessential cytosolic materials for degradation by lysosomes to maintain cellular homeostasis and overcome metabolic stress and nutrient deprivation.Citation36-37 The roles of autophagy in PDAC are divergent and the molecular mechanisms have not been fully explained.Citation38 In PDAC cells, autophagy usually functions as a cytoprotective strategy in response to hypoxic and acidic microenvironments with starved and stressful metabolic conditions.Citation39-44 Fujii et al. first revealed that high LC3 expression levels were significantly correlated with poor outcome and shorter disease-free intervals in PDAC.Citation45 Yang et al. demonstrated that the growth of PDAC had a distinct dependence on autophagy in vivo and in vitro.Citation46 Although autophagy often contributes to therapeutic resistance, excessive or prolonged autophagy causes autophagy-related cell death as a tumor-suppressive mechanism in response to some treatments, leading to tumor regression.Citation47 Gemcitabine, a standard chemotherapeutic agent for the treatment of PDAC, induces autophagy-related cell death by activating the expression of VMP1, which could trigger autophagy by interacting with BECN1.Citation48 Capecitabine induces excessive autophagy that leads to autophagy-related cell death by inhibiting SRC kinase, which plays a role in regulating autophagy.Citation49 Cannabinoids show a synergistic effect with gemcitabine in inhibiting cell growth by inducing reactive oxygen species-mediated autophagy-related cell death that sensitizes chemoresistant cells to gemcitabine.Citation50 Proton pump inhibitors show antiproliferative activities in cancer therapy by inhibiting the vacuolar ATPase, which plays a crucial role in tumorigenesis. In PDAC, omeprazole modulated chemoresistance and induced tumor cell death directly by enhancing autophagic activity in combination with either gemcitabine or 5-FU.Citation51
BCL2 is a crucial regulator of PCD by preventing cells from undergoing apoptosis. Overexpression of BCL2 contributes to the development of resistance to chemotherapy, radiation, and hormone therapy. Recent evidence indicates that BCL2 is also involved in regulation of autophagy-related cell death (type II PCD). BCL2 inhibits induction of autophagy by binding and blocking BECN1, a central autophagy-promoting tumor suppressor. Therapeutic targeting of BCL2 in cancer cells with BCL2 overexpression-induced chemoresistance, either by small molecule inhibitors or antisense methodology, is emerging as an effective anticancer strategy. Along these lines, silencing of BCL2 induces autophagy-related cell death in BCL2-overexpressing breast cancer cells. In human leukemic cells, downregulation of BCL2 triggers autophagy-related cell death.Citation52 Sorafenib induces cell death in vitro and suppresses tumor growth in vivo through an autophagy-dependent mechanism mediated by BECN1 in hepatocellular carcinoma.Citation53 Overexpression of BCL2 is common in PDAC and results in an extreme resistance to apoptotic PCD. In the present study, MIR506 showed a prominent autophagy-related cell death-inducing function mediated through the STAT3-BCL2-BECN1 pathway.
Taken together, our findings provide evidence that MIR506 triggered autophagy-related cell death in human PDAC cells and that this function was mediated through downregulation of STAT3, which in turn inhibited BCL2 and promoted BECN1 function. These results provided a mechanistic characterization of MIR506-induced autophagy-related cell death and suggest that further exploration of this approach, as a potentially effective therapeutic option against human PDAC, is warranted.
Materials and methods
Clinical specimens
Paired human PDAC and adjacent nontumor pancreatic tissues were collected from archived surgical specimens from 92 patients with biopsy-proven PDAC who underwent operation at Tianjin Medical University General Hospital between 2009 and 2013. Samples were immediately snap-frozen in liquid nitrogen and stored in liquid nitrogen until RNA extraction. Each patient's clinical and biological information was registered and follow-up data were recorded after informed consent was obtained. This study was approved by the institutional research ethics committee at Tianjin Medical University General Hospital. The patients' demographic and clinical characteristics are summarized in Table S1.
Patient-derived xenograft cell lines and cell culture
Human PDAC PDX cell lines MDA-PATC50, MDA-PATC53, MDA-PATC69, MDA-PATC124, and MDA-PATC148 were established as previously reported and cultured in Dulbecco modified essential/F12 50:50 medium supplemented with 10% fetal bovine serum in an incubator with 5% CO2 atmosphere at 37°C.Citation54
RNA isolation and qRT-PCR
The total RNA, including miRNA, was extracted from cells and tissues by using the mirVana Isolation Kit (Ambion, AM1560). Reverse-transcription was performed by using TaqMan miRNA Reverse Transcription kit (Applied Biosystems, 4366596) or SuperScript II Reverse Transcriptase (Invitrogen, 18064014). TaqMan MicroRNA Assay and TaqMan Gene Expression Assays (Applied Biosciences, A25576 and 4331182, respectively) were used to detect and quantify MIR506 and STAT3. Relative expression was normalized to the endogenous control GAPDH or RNU6 using the 2−ΔΔCt method. Experiments were carried out in triplicate.
Reagents and antibodies
The following antibodies were used: anti-LGALS1 (Abcam, ab25138) 1:1,000; anti-LGALS3 (Abcam, ab53082) 1:1,000; anti-LC3 (Cell Signaling Technology, 3868) 1:500; anti-STAT3 (Cell Signaling Technology, 12640) 1:500; anti-phospho-STAT3 (Tyr705; Cell Signaling Technology, 9145) 1:250; anti-CASP3/caspase-3 (Santa Cruz Biotechnology, sc-7148) 1:500; anti-CASP9/caspase-9 (Santa Cruz Biotechnology, sc-70505) 1:500; anti-BECN1/Beclin-1 (Santa Cruz Biotechnology, sc-11427) 1:500; anti-BCL2/Bcl-2 (Santa Cruz Biotechnology, sc-7382) 1:250; anti-SQSTM1 (Santa Cruz Biotechnology, sc-28359) 1:500; and anti-ACTB/β-Actin (Santa Cruz Biotechnology, sc-47778) 1:1,000. See Table S2 for a complete list of antibodies used. Other chemicals used included IM-54 (Sigma, SML0412), 5-FU (Sigma, 03738), z-VAD-FMK (Sigma, V116), t-BHP (Sigma, 416665), 3-MA (Sigma, M9281), AO (Sigma, A8097), Baf-A (Sigma, B1793), Stattic (Sigma, S7947), CQ (Sigma, C6628), and LLOMe (Santa Cruz Biotechnology, sc-285992).
Transfection
To generate the Y705-mutated STAT3 sequence, nucleotide sequences coding amino acid residues Y705 of STAT3 were mutated to F705 using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, 210519). Introduction of the mutation was verified by sequencing. Human wild-type BCL2, and wild-type and mutated STAT3 cDNAs were cloned into the pcDNA3.1 plasmid (Invitrogen, V79020) and verified by sequencing. ShRNA plasmids for BECN1 (TRCN0000299789 and TRCN0000299790), ATG5 (TRCN0000151474 and TRCN0000330392), and ATG7 (TRCN0000007586 and TRCN0000007587) were purchased from Sigma. Cells were transfected with 4 μg of plasmids by FuGENE HD (Promega, E2311) for 48 h, followed by G418 selection for 3 wk.
The miRNA mimics of MIR506 and control miRNA (MIRctrl) were purchased from Dharmacon (c-300846–05 and CN-001000–01). SiRNAs for STAT3 (SASI_Hs01_00121206 and 00121207) were obtained from Sigma. Cells were seeded in 6-well plates at 2× 105/well and allowed to attach overnight. They were then transfected with MIR506 mimic, MIRctrl, or siRNA for STAT3 by using Lipofectamine RNAiMAX (Invitrogen, 13778150). Total RNA and protein were collected 48 h after transfection.
Luciferase reporter assay
The 3′-UTR sequences of STAT3 containing the wild-type or mutant predicted binding site of MIR506 were cloned into the pGL3 vector (Promega, E1751). MDA-PATC53 cells transfected with MIRctrl or MIR506 mimic were seeded and co-transfected with pGL3 and pRL-TK vector (Promega, E1751 and E2241) using FuGENE HD. The firefly and renilla luciferase activities were measured using a Dual Luciferase Reporter Assay System according to the manufacturer's protocol (Promega, E1910).
Cell viability assay
Cell viability was assessed by MTT assay. MIRctrl- or MIR506-transfected cells were seeded into 96-well plates and incubated for 24, 48, 72, 96, or 120 h at 37°C. MTT (Sigma, M2128) was added to each well at a final concentration of 5 mg/L and incubated for a further 4 h. After removal of the medium, 150 μL of dimethyl sulfoxide (DMSO) was added to each well to dissolve the crystal formazan dye. The absorbance at a wavelength of 540 nm (A540) was determined for each well using a microplate reader.
Cell death assay
Cell death was first quantified by trypan blue exclusion assay. Both floating and adherent cells were trypsinized, collected, and resuspended in 1 mL of phosphate-buffered saline solution (PBS; Gibco, 10010023). Cell death was determined as the percentage of stained cells (blue) over the total cells. Cell death was also assessed by using the Apoptosis Detection Kit (BD PharMingen, 556547). The treated cells were incubated with fluorescein isothiocyanate (FITC)-conjugated anti-ANXA5 antibody and PI in binding buffer and analyzed by flow cytometry.
Apo-BrdU apoptosis assays
Cell apoptosis was assessed with an Apo-BrdU kit (BD PharMingen, 556405). Cells were fixed and treated with terminal deoxynucleotidyl transferase to catalyze an addition of Br-dUTP to the 3′-hydroxyl termini of fragmented DNA, which were then stained with FITC-conjugated anti-BrdU antibody. DNA break sites were identified and analyzed by flow cytometry to determine apoptotic fractions.
Western blotting
Cells were harvested and subjected to lysis in RIPA Buffer (Thermo Fisher Scientific, 89901) with 1:100 Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, 78426). The whole-cell lysate from each sample was separated by electrophoresis on a 10% polyacrylamide gel. The membrane was blocked in 5% nonfat milk in Tris Buffered Saline (TBS; BIO-RAD, 1706435, pH7.6) with 0.1% Tween-20 (Sigma, P2287) and incubated with primary antibodies at 4°C overnight. The secondary antibodies (Santa Cruz Biotechnology, sc-2077 or sc-2375) were used at a concentration of 1:10,000. The proteins were visualized using the SuperSignal West Pico chemiluminescent substrate or Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, 34080 or 34095).
Immunofluorescence microscopy imaging
Cells were seeded into Lab-Tek II Chamber Slides (Thermo Fisher Scientific, 154526) and cultured in complete medium. After treatment, cells were fixed for 15 min with 4% paraformaldehyde, permeated and blocked in blocking solution (1× PBS with 10% normal goat serum (Sigma, G9023) and 0.5% NP40 (Abcam, ab142227) for 4 h, and incubated with anti-human LC3 antibody (1:500 dilution), anti-human LGALS1 (1:1,000 dilution), or anti-human LGALS3 (1:1,000 dilution) at 4°C overnight and then with Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, A-11034) at 1:1,000 dilution at ambient temperature for 1 h. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific, P36931). The fluorescence images were captured with a ZEISS Axioplan 2 microscope.
Autophagic flux measurement
Autophagic flux was measured in cells transfected with Autophagy Tandem Sensor RFP-GFP-LC3 (Life Technologies, P36239). After 24 h, the fluorescence images were captured using a ZEISS Axioplan 2 microscope and the autophagosomes (yellow dots) and autolysosomes (only red dots) were counted.
Acridine orange staining
After treatment, the cells were incubated with 1 mg/mL AO solution (Sigma, A8097) for 15 min in complete medium. The cells were then collected and analyzed by flow cytometry. In AO-stained cells, the acidic compartments (autolysosomes) appeared red. The intensity of the red fluorescence indicated the degree of autophagy.
Statistical analysis
Data are expressed as the mean ± SD of at least 3 separate experiments performed in triplicate. A Student t test was performed to compare the differences between groups. P values < 0.05 were considered statistically significant. Statistically significant differences are indicated in figures by asterisks (*P < 0.05, **P < 0.01). SPSS 17.0 software (SPSS Inc., Chicago, IL) was used for all statistical analyses.
Abbreviations
| 3-MA | = | 3-methyladenine |
| 5-FU | = | 5-fluorouracil |
| AO | = | acridine orange |
| ATG5 | = | autophagy-related 5 |
| ATG7 | = | autophagy-related 7 |
| Baf-A | = | bafilomycin A1 |
| BCL2 | = | BCL2, apoptosis regulator |
| BECN1 | = | Beclin 1 |
| BrdU | = | bromodeoxyuridine |
| CASP3 | = | caspase 3 |
| CASP9 | = | caspase 9 |
| CQ | = | chloroquine |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| FITC | = | fluorescein isothiocyanate |
| IM-54 | = | 2-(1H-Indol-3-yl)-3-pentylamino-maleimide |
| LLOMe | = | L-leucyl-L-leucine methyl ester |
| LMP | = | lysosomal membrane permeabilization |
| MTT | = | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PBS | = | phosphate-buffered saline |
| PCD | = | programmed cell death |
| PDAC | = | pancreatic ductal adenocarcinoma |
| PDX | = | patient-derived xenograft |
| PI | = | propidium iodide |
| qRT-PCR | = | quantitative reverse-transcriptase polymerase chain reaction |
| shRNA | = | short-hairpin RNA |
| siRNA | = | small-interfering RNA |
| SQSTM1 | = | sequestosome 1 |
| STAT3 | = | signal transducer and activator of transcription 3 |
| t-BHP | = | tert-butyl hydroperoxide |
| z-VAD-FMK | = | N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone |
Disclosure of potential conflicts of interest
The authors have no potential conflicts of interest to disclose.
Supplementary files
Download MS Word (4.7 MB)Acknowledgments
We thank Kathryn L. Hale, Department of Scientific Publications at The University of Texas MD Anderson Cancer Center, for editing this manuscript. We thank Drs. Gregory A. Hawkins, Peiqing Sun, and Peter A. Antinozzi for valuable discussion on the results and figures.
Funding
This study was partially supported by the U.S. National Institutes of Health/National Cancer Institute under awards P30CA016672 to MD Anderson Cancer Center and P30CA012197 to Wake Forest Baptist Comprehensive Cancer Center. This work was also supported by a grant from the National Foundation for Cancer Research (to W.Z.) and the Skip Viragh Family Foundation (to J.F.). W.Z. was supported by the Hanes and Willis Family Endowed Professorship in Cancer. L.S. was supported by a Postdoctoral Fellowship from the Tianjin Medical University General Hospital and Comprehensive Cancer Center of Wake Forest Baptist Medical Center.
Related Research Data
References
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014; 371:1039-49; PMID:25207767; http://dx.doi.org/10.1056/NEJMra1404198
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5-29; http://dx.doi.org/10.3322/caac.21254
- Neoptolemos JP. Adjuvant treatment of pancreatic cancer. Eur J Cancer 2011; 47 Suppl 3:S378-80; PMID:21944022; http://dx.doi.org/10.1016/S0959-8049(11)70210-6
- Barugola G, Partelli S, Marcucci S, Sartori N, Capelli P, Bassi C, Pederzoli P, Falconi M. Resectable pancreatic cancer: who really benefits from resection? Ann Surg Oncol 2009; 16:3316-22; PMID:19707831; http://dx.doi.org/10.1245/s10434-009-0670-7
- Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011; 364:1817-25; PMID:21561347; http://dx.doi.org/10.1056/NEJMoa1011923
- Hale AN, Ledbetter DJ, Gawriluk TR, Rucker EB 3rd. Autophagy: regulation and role in development. Autophagy 2013; 9:951-72; PMID:24121596; http://dx.doi.org/10.4161/auto.24273
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344-8; PMID:21127245; http://dx.doi.org/10.1126/science.1193497
- Ito H, Aoki H, Kühnel F, Kondo Y, Kubicka S, Wirth T, Iwado E, Iwamaru A, Fujiwara K, Hess KR, et al. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J Natl Cancer Inst 2006; 98:625-36; PMID:16670388; http://dx.doi.org/10.1093/jnci/djj161
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004; 304:1500-2; PMID:15131264; http://dx.doi.org/10.1126/science.1096645
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004-10; PMID:18971948; http://dx.doi.org/10.1038/nrm2529
- Berry DL, Baehrecke EH. Autophagy functions in programmed cell death. Autophagy 2008; 4:359-60; PMID:18212526; http://dx.doi.org/10.4161/auto.5575
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/10.1016/j.cell.2005.07.002
- Akar U, Chaves-Reyez A, Barria M, Tari A, Sanguino A, Kondo Y, Kondo S, Arun B, Lopez-Berestein G, Ozpolat B. Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 2008; 4:669-79; PMID:18424910; http://dx.doi.org/10.4161/auto.6083
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 2004; 6:1221-8; PMID:15558033; http://dx.doi.org/10.1038/ncb1192
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, Kimmelman AC. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 2014; 4:905-13; PMID:24875860; http://dx.doi.org/10.1158/2159-8290.CD-14-0362
- Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 2005; 65:939-47; PMID:15705894
- Kang R, Tang D, Lotze MT, Zeh HJ 3rd. AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and bioenergetics through the IL6-pSTAT3 pathway. Autophagy 2012; 8:989-91; PMID:22722139; http://dx.doi.org/10.4161/auto.20258
- Gong J, Muñoz AR, Chan D, Ghosh R, Kumar AP. STAT3 down regulates LC3 to inhibit autophagy and pancreatic cancer cell growth. Oncotarget 2014; 5:2529-41; PMID:24796733; http://dx.doi.org/10.18632/oncotarget.1810
- Niso-Santano M, Shen S, Adjemian S, Malik SA, Mariño G, Lachkar S, Senovilla L, Kepp O, Galluzzi L, Maiuri MC, et al. Direct interaction between STAT3 and EIF2AK2 controls fatty acid-induced autophagy. Autophagy 2013; 9:415-7; PMID:23221979; http://dx.doi.org/10.4161/auto.22910
- Huang YH, Yang PM, Chuah QY, Lee YJ, Hsieh YF, Peng CW, Chiu SJ. Autophagy promotes radiation-induced senescence but inhibits bystander effects in human breast cancer cells. Autophagy 2014; 10:1212-28; PMID:24813621; http://dx.doi.org/10.4161/auto.28772
- Maycotte P, Gearheart CM, Barnard R, Aryal S, Mulcahy Levy JM, Fosmire SP, Hansen RJ, Morgan MJ, Porter CC, Gustafson DL, et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res 2014; 74:2579-90; PMID:24590058; http://dx.doi.org/10.1158/0008-5472.CAN-13-3470
- Yokoyama T, Kondo Y, Kondo S. Roles of mTOR and STAT3 in autophagy induced by telomere 3′ overhang-specific DNA oligonucleotides. Autophagy 2007; 3:496-8; PMID:17617738; http://dx.doi.org/10.4161/auto.4602
- Yoon S, Woo SU, Kang JH, Kim K, Kwon MH, Park S, Shin HJ, Gwak HS, Chwae YJ. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy 2010; 6:1125-38; PMID:20930550; http://dx.doi.org/10.4161/auto.6.8.13547
- Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib F, et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 2011; 71:5976-86; PMID:21810913; http://dx.doi.org/10.1158/0008-5472.CAN-11-1094
- Zhang T, Li Y, Park KA, Byun HS, Won M, Jeon J, Lee Y, Seok JH, Choi SW, Lee SH, et al. Cucurbitacin induces autophagy through mitochondrial ROS production which counteracts to limit caspase-dependent apoptosis. Autophagy 2012; 8:559-76; PMID:22441021; http://dx.doi.org/10.4161/auto.18867
- Yang D, Sun Y, Hu L, Zheng H, Ji P, Pecot CV, Zhao Y, Reynolds S, Cheng H, Rupaimoole R, et al. Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell 2013; 23:186-99; PMID:23410973; http://dx.doi.org/10.1016/j.ccr.2012.12.020
- Sun Y, Hu L, Zheng H, Bagnoli M, Guo Y, Rupaimoole R, Rodriguez-Aguayo C, Lopez-Berestein G, Ji P, Chen K, et al. MiR-506 inhibits multiple targets in the epithelial-to-mesenchymal transition network and is associated with good prognosis in epithelial ovarian cancer. J Pathol 2015; 235:25-36; PMID:25230372; http://dx.doi.org/10.1002/path.4443
- Liu G, Sun Y, Ji P, Li X, Cogdell D, Yang D, Parker Kerrigan BC, Shmulevich I, Chen K, Sood AK, et al. MiR-506 suppresses proliferation and induces senescence by directly targeting the CDK4/6-FOXM1 axis in ovarian cancer. J Pathol 2014; 233:308-18; PMID:24604117; http://dx.doi.org/10.1002/path.4348
- Wen SY, Lin Y, Yu YQ, Cao SJ, Zhang R, Yang XM, Li J, Zhang YL, Wang YH, Ma MZ, et al. miR-506 acts as a tumor suppressor by directly targeting the hedgehog pathway transcription factor Gli3 in human cervical cancer. Oncogene 2015; 34:717-25; PMID:24608427; http://dx.doi.org/10.1038/onc.2014.9
- Liu G, Yang D, Rupaimoole R, Pecot CV, Sun Y, Mangala LS, Li X, Ji P, Cogdell D, Hu L, et al. Augmentation of response to chemotherapy by microRNA-506 through regulation of RAD51 in serous ovarian cancers. J Natl Cancer Inst 2015; 107:djv108; PMID:25995442; http://dx.doi.org/10.1093/jnci/djv108
- Li J, Wu H, Li W, Yin L, Guo S, Xu X, Ouyang Y, Zhao Z, Liu S, Tian Y, et al. Downregulated miR-506 expression facilitates pancreatic cancer progression and chemoresistance via SPHK1/Akt/NF-κB signaling. Oncogene 2016; 35(42):5501-5514. [ Epub ahead of print]; PMID:27065335; http://dx.doi.org/10.1038/onc.2016.90
- Du J, Zheng X, Cai S, Zhu Z, Tan J, Hu B, Huang Z, Jiao H. MicroRNA-506 participates in pancreatic cancer pathogenesis by targeting PIM3. Mol Med Rep 2015; 12:5121-6; PMID:26238203
- Peng T, Zhou L, Zuo L, Luan Y. miR-506 functions as a tumor suppressor in glioma by targeting STAT3. Oncol Rep 2016; 35:1057-64; PMID:26554866
- Aits S, Kricker J, Liu B, Ellegaard AM, Hämälistö S, Tvingsholm S, Corcelle-Termeau E, Høgh S, Farkas T, Holm Jonassen A, et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015; 11:1408-24; PMID:26114578; http://dx.doi.org/10.1080/15548627.2015.1063871
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/10.1080/15548627.2015.1100356
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961-7; PMID:17972889; http://dx.doi.org/10.1038/nrc2254
- Høyer-Hansen M, Jäättelä M. Autophagy: an emerging target for cancer therapy. Autophagy 2008; 4:574-80; PMID:18362515; http://dx.doi.org/10.4161/auto.5921
- Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, et al. Stromal biology and therapy in pancreatic cancer. Gut 2011; 60:861-8; PMID:20966025; http://dx.doi.org/10.1136/gut.2010.226092
- Noman MZ, Janji B, Berchem G, Mami-Chouaib F, Chouaib S. Hypoxia-induced autophagy: a new player in cancer immunotherapy? Autophagy 2012; 8:704-6; PMID:22441015; http://dx.doi.org/10.4161/auto.19572
- Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, Gibson SB. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 2008; 4:195-204; PMID:18059169; http://dx.doi.org/10.4161/auto.5278
- Farrall AL, Whitelaw ML. The HIF1 alpha-inducible pro-cell death gene BNIP3 is a novel target of SIM2s repression through cross-talk on the hypoxia response element. Oncogene 2009; 28:3671-80; PMID:19668230; http://dx.doi.org/10.1038/onc.2009.228
- Morselli E, Galluzzi L, Kepp O, Mariño G, Michaud M, Vitale I, Maiuri MC, Kroemer G. Oncosuppressive functions of autophagy. Antioxid Redox Signal 2011; 14:2251-69; PMID:20712403; http://dx.doi.org/10.1089/ars.2010.3478
- Cuervo AM, Macian F. Autophagy, nutrition and immunology. Mol Aspects Med 2012; 33:2-13; PMID:21982744; http://dx.doi.org/10.1016/j.mam.2011.09.001
- Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, del Razo LM, Quintanilla-Vega B, Pappa A, Panayiotidis MI, Franco R. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal 2014; 21:66-85; PMID:24483238; http://dx.doi.org/10.1089/ars.2014.5837
- Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M, Kinoshita T, Ueno T, Esumi H, Ochiai A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci 2008; 99:1813-9; PMID:18616529; http://dx.doi.org/10.1111/j.1349-7006.2008.00743.x
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717-29; PMID:21406549; http://dx.doi.org/10.1101/gad.2016111
- Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, Curiel DT, Yacoub A, Graf M, Lee R, et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res 2008; 14:5385-99; PMID:18765530; http://dx.doi.org/10.1158/1078-0432.CCR-08-0469
- Pardo R, Lo Ré A, Archange C, Ropolo A, Papademetrio DL, Gonzalez CD, Alvarez EM, Iovanna JL, Vaccaro MI. Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology 2010; 10:19-26; PMID:20299819; http://dx.doi.org/10.1159/000264680
- Sheikh R, Walsh N, Clynes M, O'Connor R, McDermott R. Challenges of drug resistance in the management of pancreatic cancer. Expert Rev Anticancer Ther 2010; 10:1647-61; PMID:20942635; http://dx.doi.org/10.1586/era.10.148
- Donadelli M, Dando I, Zaniboni T, Costanzo C, DallaPozza E, Scupoli MT, Scarpa A, Zappavigna S, Marra M, Abbruzzese A, et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis 2011; 2:e152; PMID:21525939; http://dx.doi.org/10.1038/cddis.2011.36
- Udelnow A, Kreyes A, Ellinger S, Landfester K, Walther P, Klapperstueck T, Wohlrab J, Henne-Bruns D, Knippschild U, Würl P. Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells. PLoS One 2011; 6:e20143; PMID:21629657; http://dx.doi.org/10.1371/journal.pone.0020143
- Saeki K, Yuo A, Okuma E, Yazaki Y, Susin SA, Kroemer G, Takaku F. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ 2000; 7:1263-9; PMID:11175264; http://dx.doi.org/10.1038/sj.cdd.4400759
- Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS, Cheng AL, Chen PJ, Chen KF. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis 2013; 4:e485; PMID:23392173; http://dx.doi.org/10.1038/cddis.2013.18
- Kang Y, Zhang R, Suzuki R, Li SQ, Roife D, Truty MJ, Chatterjee D, Thomas RM, Cardwell J, Wang Y, et al. Two-dimensional culture of human pancreatic adenocarcinoma cells results in an irreversible transition from epithelial to mesenchymal phenotype. Lab Invest 2015; 95:207-22; PMID:25485535; http://dx.doi.org/10.1038/labinvest.2014.143