
ABSTRACT
Colorectal cancer (CRC), despite numerous therapeutic and screening attempts, still remains a major life-threatening malignancy. CRC etiology entails both genetic and environmental factors. Macroautophagy/autophagy and the unfolded protein response (UPR) are fundamental mechanisms involved in the regulation of cellular responses to environmental and genetic stresses. Both pathways are interconnected and regulate cellular responses to apoptotic stimuli. In this review, we address the epidemiology and risk factors of CRC, including genetic mutations leading to the occurrence of the disease. Next, we discuss mutations of genes related to autophagy and the UPR in CRC. Then, we discuss how autophagy and the UPR are involved in the regulation of CRC and how they associate with obesity and inflammatory responses in CRC. Finally, we provide perspectives for the modulation of autophagy and the UPR as new therapeutic options for CRC treatment.
Introduction and epidemiology
Colorectal cancer (CRC) is the second and third most common type of cancer in females and males, respectively, with 1.24 million new cases diagnosed in 2008 alone.Citation1 According to the Canadian Cancer Society, CRC has the third highest cancer incidence in both men and women.Citation2 Countries with the highest incidence include those in Europe, North America, and Oceania, while the lowest incidence is found in some South and Central Asian countries and in Africa.Citation3 In Saudi Arabia, CRC ranks first and third among males and females, respectively, of all cancers diagnosed in 2011.Citation4 According to the latest data by the Iran National Cancer Registry (INCR), the age-standardized incidence rate of Iranian CRC patients is 11.6 and 10.5 for men and women, respectively. The overall 5-year survival rate is 41%, and the proportion of CRC among the younger age group is higher than that of Western countries.Citation5 In developed countries, CRC occurrence is higher in nonsmokers of both males and females combined.Citation6 In Europe, CRC is the second leading cause of death among all cancer types in both men and women.Citation7 In the United States of America, CRC is the third leading cause of death and the 5-year overall survival (OS) of this disease is nearly 65%.
Seventy percent of CRC cases are sporadic with the presence of somatic mutations,Citation8 while about 20–30% of CRC are associated with a family history,Citation9,Citation10 and 5–15% show hereditary diseases, including polyposis and nonpolyposis CRC (). The common somatic mutations of CRC patients have been summarized in . There are several types of inherited CRC including hereditary nonpolyposis colorectal cancer (HNPCC), familial adenomatous polyposis (FAP), attenuated FAP, MUTYH-associated polyposis (MAP), hamartomatous polyps as the primary lesions in Peutz-Jeghers syndrome (PJS) and juvenile polyposis syndrome (JPS), hyperplastic polyposis (HPP) and familial CRC (FCC) syndrome X.Citation11 The etiologies of the remaining familial CRCs, which are more common compared with the well-characterized inherited syndromes, are not completely understood. However, common single nucleotide polymorphism (SNP) in genes that regulate metabolic pathways or affecting genes regulated by environmental or other genetic factors influence the incidence of this type of CRC.Citation11
Figure 1. CRC distribution—relation to the genetic background. The graph shows percentages of sporadic, familial, and hereditary (familial adenomatous polyposis, FAP; Lynch syndrome) subtypes of CRC.
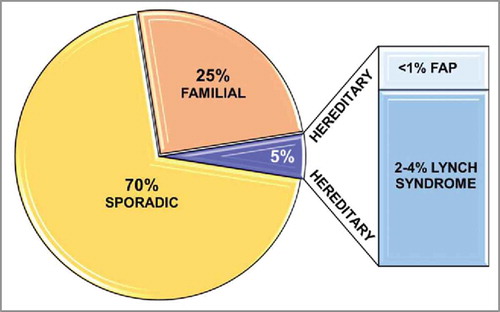
Table 1. Common somatic mutations in colorectal cancer (data extracted from: https://civic.genome.wustl.edu/).
Familial CRC is classified to familial adenomatous polyposis (FAP) and Lynch syndrome. FAP is an autosomal dominant hereditary disease that occurs in <1% of all CRC and is associated with a germline mutation in the tumor suppressor gene APC (APC, WNT signaling pathway regulator).Citation12 FAP is characterized by the presence of numerous adenomatous polyps (< 100) in the colon and rectum,Citation8 and is usually diagnosed between 20 and 30 y of age.Citation13 Lynch syndrome makes up approximately 2–4% of all CRC,Citation12 and is associated with autosomal dominant alterations in one of the DNA mismatch repair genes: MLH1, PMS2, MSH2, or MSH6.Citation14 This disease is characterized by early-onset CRC and an increased risk of other cancers, including skin, endometrium, stomach, ovary, upper urinary tract, pancreas, hepatobiliary tract, small bowel, and to a lesser extent, brain tumors.Citation15
Development of sporadic CRC involves different molecular pathways that lead to the transformation of normal epithelium to adenoma and carcinoma with diverse phenotypes. The 3 major genetic pathways distinguished in CRC are the chromosomal instability (CIN) pathway, CPG island methylator phenotype (CIMP; the “serrated” pathway), and microsatellite instability (MSI) pathway.Citation15-Citation17 In cases of sporadic CRC, epigenetic changes, including DNA methylation in gene promoters, leads to MSI because of inactivation of mismatch repair genes. Mutations in MMR genes can evoke similar genomic instability results.Citation14,Citation17 Based on these alterations, sporadic CRC is classified into 4 groups including, hypermutated, non-hypermutated, CpG island methylator phenotype and elevated microsatellite alterations at tetranucleotide repeats with metastatic behavior.Citation17,Citation18
Etiology and risk factors
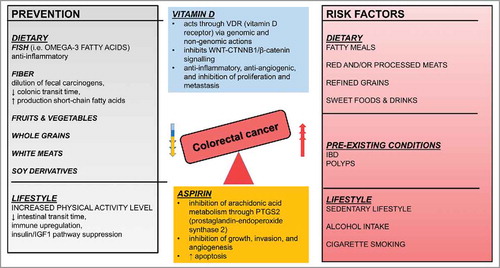
Presently, lifestyle and dietary patterns around the world are shifting toward the Western (high-fat) diet pattern.Citation19 Apart from age and sex, dietary patterns that include the high intake of red meat and/or processed meat, fatty meals, refined grains, and sweet foods increase the risk of CRC.Citation19,Citation20 Diets with high intake of fiber, fruits, vegetables, whole grain cereals, fish, white meats, soy derivatives, vitamin D, calcium, and omega-3 fatty acids have the ability to favorably modulate the development of CRC.Citation19 A number of observational studies showed that regular physical exercise decreases the risk of CRC by 40%.Citation20 In addition, regular moderate exercise of 150 min per wk increases CRC survival rates by 28%.Citation21 Aside from lifestyle modifications, chemoprevention with aspirin is effective in reducing the incidence and mortality, without significant adverse systemic effects.Citation22
Alcohol consumption has a positive dose-response correlation with the incidence of CRC; the higher the intake of alcohol, the higher the risk of CRC.Citation23 Cigarette smoking is associated with a wide variety of malignancies, and recently, has also been linked to CRC. Male smokers, especially those who smoke more than 20 cigarettes per d, are at the highest risk.Citation24 Another important risk factor is the presence of inflammatory bowel disease (IBD), a chronic inflammatory disorder of the gastrointestinal (GI) tract that includes Crohn disease and ulcerative colitis.Citation25 Patients with IBD are 6 times more susceptible to contracting CRC than the general population.Citation26 Regular colonoscopy is recommended after diagnosis.Citation27 Depending on their size, histology, and degree of dysplasia, the presence of GI tract polyps, including hyperplastic polyps, tubular adenomas, tubule-villous and villous adenomas, adenoma with high-grade dysplasia, and malignant adenomas, increases the risk of CRC.Citation28 summarizes preventive and promoting factors affecting the risk of CRC.
Figure 2. Schematic representation of factors that increase risk or prevent CRC. Both lifestyle and diet affect incidence of CRC as they can be both preventive and risk factors.
Screening and treatment
CRC screening is an effective strategy, leading to early diagnosis and prevention of disease-associated death. Screening procedures include colonoscopy, stool occult blood testing, barium enema, and digital rectal examination. Colonoscopy is the most accurate method of screening,Citation29 and often recommended as a first-line screening approach.Citation30 Stool occult blood testing reduces CRC mortality,Citation31,Citation32 and is currently the most widely used method of noninvasive screening for CRC. Advances in genomics, epigenetics, and proteomics will likely lead to the discovery of novel noninvasive biomarkers for the identification of CRC in the stool and/or blood.Citation33,Citation34
Early detection of CRC may result in CRC treatment with surgery alone, whereas late-stage advanced and/or metastasized CRC requires additional chemotherapy and radiotherapy. To ensure high quality treatment, a multidisciplinary team approach with radiologists, oncologists, surgeons, and pathologists is imperative. CRC treatment can be in the form of adjuvant therapy (AT) administered following primary tumor resection with the aim of reducing the risk of recurrence or as neo-adjuvant therapy (NAT) before tumor resection.Citation35 AT is highly recommended for CRC patients with stage III and ‘high risk’ stage II patients.Citation35 A study by Sauer et al. concluded that regardless of radiotherapy timing, NAT is favored over AT in terms of rates of local recurrences and toxic effects.Citation36 There are 2 possible NAT strategies. One approach is short-course radiotherapy without chemotherapy, followed by surgery within 1 wk.Citation37 Another approach is long-course pre-surgical chemotherapy and radiotherapy, while concurrently administrating 5-fluorouracil (5-FU)-based chemotherapy followed by surgery 8 to 12 wk later.Citation38 For the past 2 decades, standard chemotherapy for patients undergoing AT has been 5-FU in combination with levamisole and leucovorin.Citation39 Several treatment regimens have been developed since the millennium and include 5-FU in combination with leucovorin and oxaliplatin (FOLFOX), capecitabine in combination with oxaliplatin (CAPOX), and intravenous application of 5-FU and leucovorin in combination with irinotecan (FOLFIRI).Citation40 Administration of FOLFOX in 3-weekly cycles over 24 wk shows a 5–25% improvement in overall survival (OS).Citation41,Citation42 Despite the ongoing development of new therapeutic regimens and the inclusion of novel antitumor agents, the primary treatment of patients with CRC continues to be systemic chemotherapy involving infusions of 5-FU and leucovorin.Citation40
In stage IV CRC patients with unresectable metastatic lesions, a median OS of 6 mo was reported. After treatment with 5-FU and leucovorin, OS increased to 12 mo.Citation43 The GOLF regimen (gemcitabine, oxaliplatin, leucovorin, 5-FU) is another combination highly synergistic in inducing both growth inhibition and apoptosis of colon cancer cells.Citation44 Introduction of infusion regimens, such as FOLFIRI, has raised OS to a median of about 20 mo.Citation45 Regorafenib is a drug that targets and inhibits several tyrosine kinases involved in angiogenesis (FLT1/VEGFR1, KDR/VEGFR2, and FLT4/VEGFR3), oncogenesis (KIT, RET, RAF, BRAF), and the tumor microenvironment (PDGFR, FGFR), and it has been approved for use in patients that have relapsed or are refractory to all other systemic therapies.Citation46
Personalized medicine involves the tailoring of medical treatment to an individual patient depending on the specific genetic makeup of their cancer, taking into account different stages of care, which includes prevention, diagnosis, treatment, and followup. An example for this is temozolomide, which is used in pretreated patients with advanced CRC and MGMT promoter methylation. Patients with KRAS, BRAF, and NRAS wild-type (WT) CRC show significantly higher response when compared with CRC containing KRAS or BRAF mutations (44% versus 0%; P = 0.004).Citation17 Lists of chemotherapeutic drugs and regimens are presented in and , respectively.
Table 2. Summary of the chemotherapeutic drugs and their mechanism of action in CRC.
Table 3. Chemotherapeutic regimens (combination therapy) and their effect in CRC.
General aspects of autophagy
New therapeutic strategies are being designed to target autophagy to improve treatment options of different diseases, including cancer. In the context of cancer, autophagy may prevent cellular transformation in normal tissue by decreasing reactive oxygen species (ROS) content of the cells. Conversely, it can also promote cancer progression depending on the stage of cancer.Citation47,Citation48 Recent investigations revealed that autophagy has diverse functions in the development, maintenance, and progression of tumors.Citation48 While genetic evidence indicates that autophagy functions as a tumor suppressor mechanism, it is also apparent that autophagy can promote the survival of established tumors under stress conditions and in response to chemotherapyCitation49-Citation51 ( and ). Recent findings show that modulation of autophagy affects the immune response and the biology of cancer in general.Citation52-Citation56 Genetic alterations in autophagy may predispose individuals to autoimmune, auto-inflammatory, or infectious diseases. For instance, ATG5 mutations are associated with systemic lupus erythematosus and Crohn disease.Citation57,Citation58 Furthermore, stimulation or suppression of genes important for autophagy can regulate immune responses via antigen donor cells, antigen presenting cells, or downstream effectors of the immune system.Citation59 From an immunological point of view, cancer can progress when malignant cells escape the control of the immune system by altering their antigenic properties or by reducing or suppressing antitumor immune responses.Citation59 They accumulate genetic and epigenetic alterations, including, among others, loss of heterozygosity of BECN1, constitutive signaling via MTOR, activating phosphoinisotide 3-kinase (PI3K) mutations, loss of PTEN, accumulation of mutant TP53, or the overexpression of anti-apoptotic BCL2-family proteins. Such changes facilitate (directly or indirectly) genomic instability in cancerous cells, leading to malignant cells escaping immunosurveillance.Citation59
Figure 3. Dual role of autophagy in cancer chemotherapy. Autophagy may induce stress adaptation (A) in cancer cells allowing them to obtain a resistance phenotype against cancer chemotherapy agents, or it may induce cytotoxicity (B) resulting in autophagic cell death of cancer cells (adapted from ref. Citation49).
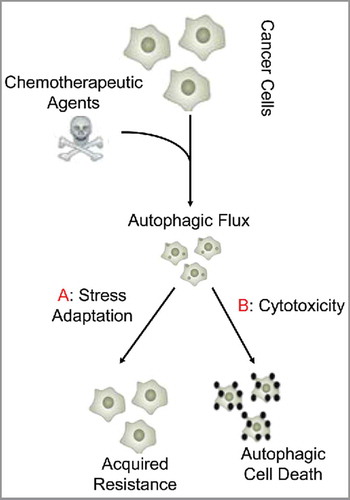
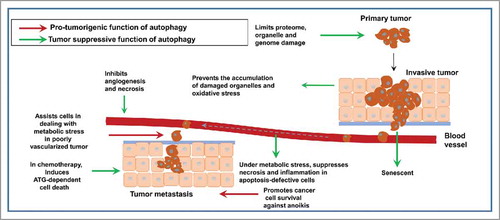
Figure 4. Dual role of autophagy during tumorigenesis: Autophagy may suppress tumorigenesis by eliminating damaged organelles in transformed cells and protect them against oxidative stress, resulting in subsequent genome stabilization and prevention of malignant transformation. Autophagy may also initiate an oncogene-induced senescence, thus preventing malignant transformation. It may prevent necrosis in apoptosis-deficient cells in tumors in response to metabolic stress. This reduces pro-tumorigenic inflammation and release of tumorigenic compounds from necrotic tumor cells. Tumor-supportive functions of autophagy are fulfilled mainly by stimulating tumor cell survival and protection against detachment-induced apoptosis (anoikis), which can facilitate chemoresistance and EMT induced-metastasis (adapted from. refs. Citation50,Citation51).
Manipulation of key elements of the autophagy pathway can be exploited as a novel therapeutic approach for CRC.Citation60 In eukaryotic cells, autophagy is an important protein degradation system and mainly responsible for the degradation of long-lived proteins and damaged organelles.Citation61 Autophagy refers to a collection of tightly regulated catabolic processes, all of which deliver cytoplasmic components to the lysosome for degradation. These are broadly classified into 3 types: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA).Citation62 Macroautophagy involves the formation of phagophores that engulf cytoplasmic proteins and organelles, maturing into double-membrane-bound vesicles called autophagosomes. These autophagosomes are trafficked to lysosomes and the sequestered cargo is degraded.Citation62 Microautophagy refers to the invagination of the lysosomal or endosomal membrane, resulting in the direct engulfment of substrates that are subsequently degraded by lysosomal proteases.Citation62-Citation64 CMA is distinct from macroautophagy and microautophagy because the cargo is not sequestered within a membrane vesicle. Instead, proteins targeted by CMA contain a KFERQ-like pentapetide motif that is recognized by HSPA8/HSC70 (heat shock protein family A [Hsp70] member 8). HSPA8 promotes the translocation of these targets across lysosomal membranes into the lysosomal lumen via LAMP2A (lysosomal-associated membrane protein 2A).Citation65
Usually the term “autophagy” refers to “macroautophagy” in the literature.Citation65 Autophagy dysregulation leads to various human diseases, including neurodegenerative disorders and cancer.Citation63,Citation66 In both normal and malignant cells, autophagy may be induced in response to cellular stress,Citation62 including nutrient deprivation, hypoxia, and toxin accumulation.Citation62 The outcome of autophagy induction however, affects the cell in various ways, being protective, and promoting survival, or causing growth arrest and triggering programmed cell death.Citation67 The molecular components of this pathway were first discovered in yeasts and include more than 40 autophagy-related (ATG) proteins. Most autophagy stimuli converge at MTOR (mechanistic target of rapamycin) and the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, which serve as autophagy-related key regulators. Several core autophagy machineries are required for autophagosome formation.Citation62,Citation65 The core machinery of the initiation stage during induction of autophagy is the ULK (unc51-like autophagy activating kinase) complex consisting of ULK1, ATG13, ATG101 and RB1CC1/FIP200. Upon initiation of autophagy, a complex nucleation arises when the PtdIns3K complex binds to its core units, such as BECN1/Beclin-1 (the human ortholog of yeast Vps30/Atg6) and PIK3R4/p150.Citation65,Citation68 This complex resides on the phagophore membrane and facilitates recruitment of other ATGs to the unit ().Citation62,Citation69 During phagophore elongation and maturation, the Atg8/LC3 protein, a ubiquitin-like protein, is conjugated to the membrane lipid phosphatidylethanolamine (PE) or possibly to phosphatidylserine.Citation70 In yeast, and several other organisms, the conjugated form is referred to as Atg8–PE. The mammalian homologs of Atg8 constitute a family of proteins subdivided into 2 major subfamilies: MAP1LC3/LC3 and GABARAP. The former consists of LC3A, B, B2 and C, whereas the latter family includes GABARAP, GABARAPL1 and GABARAPL2/GATE-16.Citation71 After cleavage of the precursor protein, mostly by the cysteine protease ATG4B,Citation72 the nonlipidated and lipidated forms are usually referred to as LC3-I and LC3-II, and GABARAP-I and GABARAP-II (), respectively. The increased level of LC3-II in the presence of lysosomal proteases inhibitors (bafilomycin A1 or chloroquine) typically serve as an analytical marker of autophagic flux because it confirms the autophagy flow from autophagosome formation to recycling in lysosomes (reviewed in refs. Citation65,Citation69). In the final stage, cargo is degraded by lysosomal hydrolases in the autolysosomes () and the resulting products are transported back to the cytosol by lysosomal permeases.
Figure 5. Autophagy and the UPR signaling pathways. (A) Depiction of autophagy pathways. Autophagy is a catabolic process that sequesters specific intracellular cargo by engulfing them within a cytosolic double-membraned vesicle, called an autophagosome. Extracellular stimuli or recognition of a cargo material induces the formation of the phagophore. ULK1 is an important upstream initiator that induces activation of nucleation complex, including PtdIns3K and BECN1, to engage phagophores for autophagy. LC3 is conjugated to the phagophores and controls their maturation and elongation. Upon vesicle completion, the autophagosome fuses with a lysosome, releasing its contents to be degraded by hydrolases. (B) Initiation of the UPR: The domain structures of ERN1, EIF2AK3, and ATF6 and their associations with HSPA5 are illustrated. ERN1, EIF2AK3, and ATF6 are docked and inactive in non-ER stress condition by binding to HSPA5. Upon ER stress, HSPA5 is released from the lumenal domain of ERN1, EIF2AK3 and ATF6 and this initiates the UPR.Citation64,Citation416
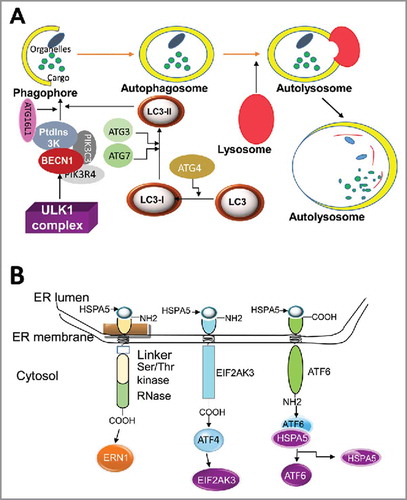
General aspects of the unfolded protein response
The endoplasmic reticulum serves as a subcellular compartment involved in maturation and folding of proteins, and plays important roles in maintaining normal cellular functions.Citation73,Citation74 An imbalance between cellular demand for ER function and ER capacity can lead to ER stress.Citation75 To cope with ER stress, mammalian cells are able to activate the unfolded protein response (UPR) which aims to maintain the homeostasis of proteins within the ER.Citation76 The UPR is initially associated with a stress-inducible chaperone, a glucose-regulated protein, which mainly resides in the ER and is encoded by the HSPA5/GRP78/BIP (heat shock protein family A [Hsp70] member 5) gene ().Citation77 The ER contains 3 transmembrane receptors () including EIF2AK3/PERK (eukaryotic translation initiation factor 2 α kinase 3), ATF6 (activating transcription factor 6) and ERN1/IRE1α (endoplasmic reticulum to nucleus signaling 1).Citation77 These 3 arms of the UPR sense the protein-folding status in the ER and transmit the information to the cytosol to regulate UPR-related gene expression.Citation78
Activation of ERN1 starts from the dissociation from HSPA5 and results in the splicing of XBP1 to form its active form (XBP1s). This modulates prosurvival signals by regulating genes involved in protein folding, maturation and ER-associated degradation.Citation79 Activation of ERN1 also targets MAP3K5/ASK1 and MAPK/JNK proteins, followed by triggering of TRAF2, which subsequently can promote apoptosis.Citation80 ERN1 is much more activated at the beginning of stress and its activity fades over time.Citation79
ATF6 is a basic leucine zipper (bZIP)-containing transcription factor in the ER which include ATF6/ATF6α, ATF6B/ATF6β, CREB3L1/OASIS, CREB3/LUMAN, CREB3L2/BBF2H7, CREB3L3/CREBH and CREB3L4.Citation81 ER stress causes dissociation of HSPA5 from ATF6 () and the translocation of ATF6 from the ER to the Golgi apparatus where it is processed by serine protease MBTPS1/S1P and the metalloprotease MBTPS2/S2P to produce an active cytosolic fragment.Citation82 This active product translocates to the nucleus and activates the expression of several genes that are involved in protein folding, including the ER chaperone proteins DDIT3/CHOP/GADD153, PDIA4/ERp72, PDI, EDEM1 and XBP1.Citation83
The third transducer of the UPR is EIF2AK3, which is the most immediate sensor to respond to ER stress.Citation84 Under ER stress condition, EIF2AK3 is released from HSPA5 (). Upon activation, EIF2AK3 phosphorylates EIF2A (eukaryotic translation initiation factor 2A) and subsequently inhibits protein synthesis by reducing activity of the EIF2A complex.Citation85 Despite global inhibition of protein synthesis, ATF4 is translationally upregulated by EIF2AK3 to increase the expression of stress-related genes and downstream ER chaperones.Citation86 Moreover, EIF2AK3 triggers antioxidant activity via phosphorylation of NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2).Citation87 NFE2L2 is a pro-survival factor and cells without NFE2L2 display increased cell death during ER stress.Citation87
CMA and its relevance to CRC
Chaperone-mediated autophagy (CMA) is a selective mechanism for the degradation of proteins through a lysosomal-dependent machinery.Citation88 Basal CMA activity is evident in most cells but is highly stimulated in response to cellular stress.Citation88,Citation89 CMA contributes to the degradation of proteins that are no longer needed under stress conditions, leading to recycling and promoting of cell survival.Citation90,Citation91 The cellular pathways and physiological importance of CMA in cancer still needs to be delineated.Citation91 It has been reported that high basal CMA activity is a common feature among different types of human tumors.Citation92 In contrast to normal cells, this upregulation of CMA occurs independent of the macroautophagy status of cancerous cells. For example, inhibition of CMA reduces cell proliferation and induces cell death in human lung cancer cell lines. In contrast to nontumor cells, cancer cells with blocked CMA upregulate their ubiquitin-proteasome system to ensure protein quality control. Blockade of CMA delays tumor growth and induces regression of already formed human lung cancer xenografts in mice. The fact that similar manipulations of CMA reduce tumor growth of other human cancer cell lines, such as melanoma, highlights that targeting this autophagic pathway may have broad antitumor activity.Citation93
Recently, an increased level of CMA activity was detected by immunostaining for LAMP2A in primary tumors of different human tissues (e.g., liver, lung, skin, stomach, colon, uterus, ovary). Although the intensity of LAMP2A staining varied depending on the type of tumor, the overall LAMP2A signal was significantly higher in all tumor samples when compared with respective normal tissues. In some cases, a gradual increase in LAMP2A staining was observed in parallel with the transition from a normal region to peri-neoplastic and neoplastic regions and with the stage of malignancy. The observed increase in LAMP2A reflects an expansion of the lysosomal compartment in tumor cells because control and tumor samples revealed no difference in staining for LAMP2B, a lysosomal protein splice variant of the same LAMP2 geneCitation94 with 95% sequence homology to LAMP2A. Importantly, LAMP2B is not involved in CMA.Citation93 In another study on HCT116 human colorectal cancer cells, inhibition of autophagy by the compound spautin-1 or genetic knockdown of autophagy-related genes promotes degradation of accumulated missense mutant TP53 proteins through the CMA pathway. These findings suggest that degradation of mutant TP53 is specifically mediated by the CMA-lysosomal pathway during stress conditions and reveals involvement of CMA in a unique pathway that regulates mutant TP53 expression-dependent cell death.Citation91
Organellophagy and its importance in CRC
Macroautophagy can also selectively eliminate organelles, a process termed organellophagy.Citation95 Organellophagy is common for organelles such as mitochondrion (mitophagy), ER (reticulophagy/ER-phagy), peroxisomes (pexophagy), lysosomes (lysophagy), nucleus (nucleophagy), and even ribosomes (ribophagy).Citation95 Mitophagy is a specific form of macroautophagy by which damaged mitochondria are selectively degraded.Citation96,Citation97 Previous investigations demonstrated that mitophagy prevents the accumulation of damaged organelles that are sources of ROS.Citation98 To maintain proliferative capacity and constantly generate progeny, cancer cells must continuously supply sufficient energy and building blocks such as amino acids, lipids and sugars.Citation99 Many solid tumors depend on activated glycolysis to cope with the energy requirement for faster proliferation (the Warburg effect). This process requires efficient glucose uptake even in a stressful environment, such as hypoxia frequently experienced by tumor cells.Citation100 If glycolysis can meet the cellular energy requirement for cancer cells, then the maintenance of a high level of mitochondrial mass is not essential for ATP production. Therefore, autophagy-dependent degradation of unnecessary mitochondria may serve as a useful mechanism to resupply nutrients and expedite glycolysis. This hypothesis is well supported by recent reports that autophagy facilitates glycolysis;Citation101 hence, transformed cells maintain small numbers of mitochondria during periods of rapid proliferation.Citation102 Further support in favor of this hypothesis comes from electron microscopy images which show a decreased number of intracellular organelles within the cytosol of proliferating cancer cells. This could mean that cancer cells may activate autophagy-mediated organelle degradation to maintain cellular ATP levels and resupply nutrients when glucose levels are insufficient.Citation102 Selective autophagy is also a backup mechanism for the failed proteasomal degradation of ubiquitinated aggregation-prone and misfolded proteins. Because ubiquitination has also been implicated in mitophagy,Citation103 modulation of the expression levels of the ubiquitin-binding autophagic receptors SQSTM1/p62 and NBR1 (cargo receptors for selective autophagy) by selective autophagy might also play a role in mitophagy. Thus, failures in selective autophagy may cause accumulation of protein aggregates and damaged organelles that mediate neoplastic transformation. In contrast, established tumors depend on autophagy to fuel their increased metabolic demands. Selective autophagy may ensure tumor survival via degradation of misfolded proteins and damaged organelles that accumulate in genetically unstable tumor cells.Citation104
Defective autophagy is linked to colonic tumor formation through a mechanism involving the aberrant activation of WNT-signaling from impaired degradation of DVL (disheveled segment polarity protein) by autophagy.Citation105 Therefore, pharmacological activators of autophagy may be of potential benefit for cancer chemoprevention.Citation106,Citation107 However, there is only indirect evidence for a role of organellophagy in CRC. In a recent report, TP53 inactivation in HCT116 colon cancer cells induced both reticulophagy and mitophagy.Citation108 When TP53 was inhibited in an acute fashion by addition of pifithrin-α,Citation107 reticulophagy was induced more rapidly than mitophagy, suggesting an intimate relationship between TP53 inhibition and ER stress-induced reticulophagy.Citation108 Interestingly, new findings show that some tumor suppressor proteins play a role in organellophagy, especially mitophagy, in various types of cancers, including CRC.Citation109
Autophagy and the microbiome in CRC
A growing body of evidence suggests that alterations in the population of gut microorganisms, the microbiome, contribute to the development of CRC.Citation110,Citation111 CRC patients show a distinct microbial signature in their gut which may predispose them to a tumor-promoting inflammation.Citation112,Citation113 Analysis of next-generation sequencing dataCitation112,Citation114 indicate that the colonic mucosa is initially colonized by pathogenic bacteria driving CRC via inducing persistent inflammation. This fuels increased cell proliferation and/or production of genotoxic substances involved in development of premalignant lesions and the accumulation of gene mutations such as in TP53.Citation115 As a consequence of alterations in colonic barrier permeability and cellular metabolism, pathogenic “driver” bacteria are replaced by “passenger” bacteria such as tumor-feeding opportunistic and commensal bacteria. Collectively, a “driver-passenger model” has been proposed to explain the role of the gut microbiome in CRC. The intestinal bacteria are more likely to play a “driver” role in the course of tumorigenesis rather than being passive “passengers.”Citation116
Basic studies in a mouse model of CRC have revealed that perturbations to the gut microbiota can lead to colon tumorigenesis in which transfer of the tumor-associated microbiome to the germ-free mice exacerbates tumor formation compared with the control germ-free mice that received microbiota from healthy mice.Citation117 Modulation of the gut microbiome has been proposed as a therapeutic or preventative approach for CRC. However, several mechanistic issues need to be precisely addressed before translational modification of the gut microbiome in CRC.
From an immunological point of view, IL23A produced by tumor-associated myeloid cells is a master initiator of the inflammatory response to tumor-infiltrating microbes and this induces expression of IL17 as a pro-tumorigenic mediator. Interestingly, prolonged administration of antibiotics suppresses tumor growth induced by IL23A.Citation118 Furthermore, colonic innate lymphoid cells (ILCs) play a pivotal role in microbiome-influenced CRC via regulation of IL23A-dependent on IL22 secretion, which is mediated by inducing phosphorylation of STAT3 in a mouse model.Citation119
Autophagy is activated in the intestinal epithelium of CRC patients and a mouse model of CRC.Citation120 Specific genetic ablation of Atg7 in murine intestinal epithelial cells leads to a significant suppression of pre-cancerous lesion development. The role of ATG7 in CRC is mediated by intestinal dysbiosis in which the gut microbiome is an essential component for an effective antitumor immune response. Inhibition of autophagy by epithelial deletion of Atg7 leads to bacterial invasion of the crypts which dramatically changes microbiome composition in the gut. This effect is mediated by controlling a stress response associated with activation of AMP-activated protein kinase (AMPK) signaling and TP53-mediated cell-cycle arrest specifically in tumor cells.Citation120 The lack of a protective response against colonic tumor upon antibiotic treatment further confirms the crucial role of the gut microbiome in ATG7-deficient mice.Citation120 Decreased levels of antimicrobial defenses mediated by Paneth cells (secretory cells of the intestinal crypts) and goblet cells may explain how the inhibition of autophagy in intestinal epithelial cells can downregulate host immunity.Citation121
The role of the microbiome in energy homeostasis is significantly more pronounced in colon than other tissues, which is mainly because of consuming bacterial butyrate as the primary energy source in colon cells. Interestingly, enhanced levels of autophagy are observed in colon cells of mice lacking a microbiome and this increase is rescued upon addition of butyrate to germ-free colon cells.Citation122 It has been suggested that butyrate, as a short chain fatty acid, has a protective role in colon tumorigenesis via induction of apoptosis and inhibition of proliferation and regulation of cell differentiation.Citation123
While investigating the mechanisms underlying the regulation of the gut microbiome by VDR (vitamin D [1,25 dihydroxyvitamin D3] receptor), Jin et al. demonstrated that VDR status influences the intestinal bacteria at both the taxonomic and functional level, and correlates with the VDR-associated bacterial changes in clinical diseases. Since VDR is a nuclear receptor that regulates the expression of antimicrobial peptides and the autophagy regulator ATG16L1, future studies need to address a crucial unknown link between the gut microbiome and autophagy mediated by VDR in the context of CRC.Citation124 Understanding the precise impact of the microbiome on autophagy in CRC will open new avenues, leading to the development of novel therapeutic strategies for CRC.
Mutations in autophagy-related genes in CRC
UVRAG
Recently, the role of UVRAG (UV radiation resistance associated) as a tumor suppressor gene has been described and the first reports of cancer-specific mutations in UVRAG have been published.Citation125 A 10-polyadenine repeat in exon 8 in MSI colorectal tumors was identified with mono-allelic frame-shift mutations. UVRAG positively regulates BECN1, suggesting that the interaction with BECN1 is necessary for the tumor suppressor function of UVRAG. In a colon cancer cell line carrying a deletion in UVRAG (c.709delA), a reduction in endogenous UVRAG levels and impaired autophagy induction were observed.Citation50,Citation125
ATG16L1
ATG16L1 is an autophagy gene that also controls host immune responses against bacteria and viruses. The nonsynonymous SNP in ATG16L1 (Thr300Ala) is associated with improved OS in human CRC and increased basal production of type I IFN, providing a mechanism to influence clinical outcome.Citation126 SNP may also explain why some patients have a higher risk of CRC or are prone to more mucosal inflammation than others. Autophagy gene polymorphisms correlate with the development of human CRC. The ATG16L1 (+898A>G [Thr300Ala] SNP) GG genotype is found at higher frequencies in moderately and poorly differentiated CRC cases. Whereas the AA genotype is correlated with a lower risk for CRC, the SNP switch to the GG genotype is correlated with a higher risk for CRC.Citation127
Autophagy and the UPR pathways provide a link between inflammation and cancer in CRC
Malignancies are the second most common cause of death after cardiovascular disease in both genders in patients with IBD.Citation128 IBD has etiological links to CRC at multiple levels and autophagy plays a crucial protective role.Citation129 The intestinal tract is the interface between the organism and its outer environment and a potential site of infection/inflammation and cancer formation. Clearance of invading microbes and intracellular waste components seems to be a protective function of autophagy in inflammatory disease.Citation130
Entero-pathogenic Escherichia coli (EPEC) are equipped with a well-developed infectious machinery by which they evade the host defenses and deplete host DNA mismatch repair (MMR) proteins in colonic cell lines. Alterations in the MutS or MutL complexes of mammalian cells may be associated with EPEC pathogenesis and the development of CRC. The MMR proteins of E. coli have been considered as potential therapeutic targets and early detection biomarkers for CRC.Citation131 The role of gut microbiota in the development of human CRC is influenced by diet and inflammation.Citation132 Importantly, autophagy deregulation in IBD and CRC development is associated with alterations in immune responses, defects in bacterial clearance, and malfunction of goblet and Paneth cells.Citation60
There is tight crosstalk between inflammation and the ER stress pathway, which can influence the pathology and progression of several diseases. ER stress-induced inflammation may aid the progression of type 2 diabetes, obesity, and cause IBD progression in Crohn disease and ulcerative colitis.Citation133 In addition, pro-inflammatory diets (i.e., high carbohydrates along with low antioxidants) are associated with increased risk of CRC.Citation134
Chronic inflammation in IBD can lead to prostaglandin release, production of ROS, and secretion of tumor-promoting cytokines. These cytokines promote the survival, growth, and metastasis of tumor cells through NFKB/NFκB (nuclear factor kappa B; mediators downstream of the UPR), STAT3 (signal transducer and activator of transcription 3) and AP-1 (AP-1 transcription factor) signaling pathways as well as cytokines such as IL1B/IL1β, IL6, IL11, and IL23A.Citation131 Prostaglandin E2 (PGE2) induces cancer stem cell (CSC) expansion by activating NFKB via E-type PTGER4/EP4-PtdIns3K and PTGER4-MAPK (mitogen activated protein kinase) signaling and promotes the formation of CRC liver metastases in mice. The PGE2 signaling pathway may serve as a therapeutic target to counteract CRC metastasis.Citation135 Dysregulation of PTGS/COX (prostaglandin-endoperoxide synthase) pathway may lead to the accumulation of pro-inflammatory mediators such as PGE2. In particular, PTGS, PTGER3 (prostaglandin E receptor 3), PTGFR (prostaglandin F receptor), and AKR1B1 (aldo-keto reductase family 1 member B) were found hyper-methylated in more than 40% of colorectal tumors.Citation136 A recent study on 618 participants diagnosed with CRC showed that CRC-specific mortality was higher in patients with PTGS2-positive tumors, when GDF15/MIC1 (growth differentiation factor 15) plasma levels were high preceding diagnosis.Citation137 When visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT) compartments were analyzed for metabolic and transcriptomic differences to elucidate a link between obesity and colorectal carcinogenesis, results showed that VAT compartments displayed elevated markers of inflammatory lipid metabolism, prostaglandin synthesis-related enzymes (PTGDS/PGS2S), PLA2G10 (phospholipase A2 group X), and free arachidonic acid. The presence of these inflammation markers in VAT supports a role of visceral adiposity in promoting cancer.Citation138,Citation139
CRC incidence is higher in wild type than in ppp1r15a/gadd34 (protein phosphatase 1, regulatory [inhibitor] subunit 15A) knockout mice.Citation140 PPP1R15A/GADD34 is part of a family of DNA damage-inducible proteins and it is a target of ATF4 during ER stress that regulates inflammation and host defense systems. For example, dextran sodium sulfate-induced inflammatory responses are curbed as a result of PPP1R15A deficiency. In addition, both expression of pro-inflammatory mediators and epithelial cell proliferation are lower in ppp1r15a KO mice.Citation140
Various ER-stress-associated proteins, such as HSPA5, ATF6, HSP90B1/GRP94, and XBP1s, are upregulated in cancer.Citation141 Therefore, a model of association between ER stress-induced tumor and pro-inflammatory gene pathways has been proposed.Citation133 In this model, ER stress induces NFKB and AP-1 activation in tumor cells followed by cytokine secretion in cancer cells.Citation142,Citation143 Continuous infiltration of immune cells into the tumor microenvironment,Citation142,Citation144 and induction of ER stress in tumor cells can affect both immune cells and cancer cells by triggering cytokine secretion from tumor cells.Citation145,Citation146 Finally, ER stress is also induced in tumor-infiltrating immune cells because of their high cytokine production in the tumor microenvironment.Citation147 Hence, it has been shown that inflammation can induce the UPR via pathways that are activated by ER stress. All 3 sensors of the UPR, EIF2AK3, ERN1, and also ATF6, are involved in activation of inflammatory processes. ER stress-induced inflammation contributes in the pathogenesis and progression of several diseases, including obesity, type 2 diabetes, and cancer. However, based on the type of stress, the UPR arms might either promote or prevent cancer progression, depending on activated inflammatory pathways, cell type and stage of disease.Citation133
Neutrophils have critical roles in tumorigenesis through their production of cytokines and chemokines, which influence inflammatory cell recruitment and regulate tumor cell proliferation, angiogenesis, and metastasis. Thus, neutrophils have been recognized as new targets for cancer therapy.Citation148 Infiltration of natural killer cells and CD8+ T lymphocytes into the CRC micro-environment is a good prognostic sign in CRC patients and suggests potential antitumor effects of natural killer cells and CD8+ T cells.Citation149 In addition to elevated levels of cytokines, chemokines, and ROS production, inflammasomes are strongly linked to increased rates of epithelial proliferation and angiogenesis, and play critical roles in colitis-associated CRC progression.Citation150 Furthermore, the role of proteases and their receptors on intestinal inflammation and cancer provide a rationale to explore the potential role of protease-activated receptor-induced PTGS2/COX2 in colitis-associated cancer.Citation151 American ginseng may have potential value in CRC chemoprevention via reduction of gene expression of inflammatory cytokines, including IL1A/IL1α (interleukin 1 α), IL1B, IL6, TNF, CSF3/G-CSF, and CSF2/GM-CSF in both the small intestine and the colon.Citation152
Changes in molecular mediators of the UPR in CRC
Alteration of ER stress-associated molecules has been extensively studies by various genetic and pharmacological approaches (both inhibitors and inducers) in different cells.Citation153 In the context of CRC, Lu et al., showed that dihydroartemisinin can trigger ER stress in human colorectal carcinoma HCT116 cells through inducing the expression of HSPA5 and DDIT3 at both mRNA and protein levels.Citation153 Paclitaxel induces all 3 arms of the endoplasmic reticulum stress response in CRC cells by upregulating HSPA5 and phosphorylation of EIF2A.Citation154 Other studies revealed that HSPA5 expression is elevated in CRC.Citation155,Citation156 Knocking down EIF2AK3, ERN1, or ATF6 in CRC HCT116 cells shows that EIF2AK3 has an important role in hypoxia-dependent induction of MAP1LC3B and ATG5.Citation157 SELENOS (selenoprotein S), which is involved in the metabolism of unfolded or misfolded proteins, is also associated with increased CRC risk.Citation158 In addition, high expression levels of XBP1 have been detected in CRC cells, emphasizing that upregulation of the corresponding gene may be one of key players in colon carcinogenesis.Citation159 Moreover, STC2 (stanniocalcin 2), as a main survival component of the UPR, is overexpressed in CRC to provide tolerance to ER stress.Citation160
Upon treatment of HCT116 cells with celecoxib, ER chaperones and particularly HSPA5 are upregulated, followed by an increased level of VEGF production, and finally, apoptosis.Citation161 Similarly, dihydroartemisinin chemotherapy induces mitochondria-dependent apoptosis via ER stress pathways in CRC HCT116 cells.Citation162 The ERN1-XBP1 pathway is also important for promotion and progression of CRC.Citation163 Recent report shows a pivotal role of XBP1 in CRC invasion.Citation164 Inhibiting ATF4 sensitizes CRC cells to chemotherapy and counteracts drug-induced apoptosis, showing that the HSPA5-EIF2AK3-ATF4 pathway is active in CRC.Citation165 The above-mentioned ER stress-associated molecules seem to have therapeutic potential in CRC and more research is required to elucidate their importance.
Inhibitors and activators of autophagy and the UPR in CRC
Autophagy modulators
HMGB1 (high mobility group box 1) may have different functions including proliferation, invasion, and metastasis by ligating its multi-ligand AGER/RAGE (advanced glycosylation end product specific receptor) in different cancer models.Citation166,Citation167 Necrotic cells release HMGB1 and are involved in inflammation induction.Citation168,Citation169 HMGB1 plays a fundamental role in the carcinogenesis and progression of CRC.Citation170,Citation171 HMGB1 colonic mucosa concentration is continuously increased in a rat azoxymethane model of CRC.Citation171 Several reports have shown that HMGB1 activates autophagy.Citation172,Citation173 HMGB1 can also bind to TLR4 (toll like receptor 4), which subsequently activates innate immunity and immunological autophagy by triggering the dissociation of BECN1 from BCL2.Citation174,Citation175 Cytosolic HMGB1 can directly bind to BECN1 to assist in the dissociation from BCL2 and consequently initiate an autophagic response.Citation176
Luo et al. reported that proteolysis associated with autophagy is induced in the presence of HMGB1.Citation177 Autophagy selectively degrades cellular components, including aged proteins, protein debris, and damaged organelles, which contributes to energy production, and to supply amino acids.Citation178,Citation179 HMGB1 is involved in the dephosphorylation of MTOR, which subsequently induces the proteins involved in autophagy, including BECN1 and LC3-II via AGER-mediated MAPK p38 phosphorylation.Citation177 An autophagy-induced glutamine supply might be important for maintaining mitochondrial energy production in muscles.Citation180 In contrast, cancer cells use glucose and glutamine as a source of energy for the lactate fermentation pathway.Citation181 Cancer cells use plasma glutamine released from the muscle, and HMGB1 treatment increases lactate fermentation in colorectal cancer cells in culture conditions.Citation177,Citation182 This underlines a cancer-host interaction in energy acquisition for cancer progression.
Initiation of the autophagy pathway involves BECN1, and the interaction with several cofactors, including AMBRA1, SH3GLB1/BIF (SH3 domain containing GRB2 like endophilin B1), and UVRAG, to activate the lipid kinase PIK3C3/VPS34.Citation62,Citation68 Immortalized kidney and mammary epithelial cells that harbor a mono-allelic deletion of BECN1 show increased growth rates when compared with their wild-type counterparts. Conversely, the tumor-suppressing property of BECN1 is associated with interactions of BECN1 and autophagy-related proteins downstream of BECN1, such as UVRAG and ATG4.Citation183 UVRAG overexpression suppresses the tumorigenicity of human colon cancer cells and, not surprisingly, one copy of the UVRAG gene is often deleted in human CRC. Furthermore, bi-allelic deletion of Atg4 in mice favors the development of chemically induced fibrosarcomas as a result of tissue-specific defects in the autophagy pathway.Citation183 BECN1 expression is low in human breast tumors, glioblastoma multiforme and other high-grade brain tumors.Citation184 In contrast, high expression of BECN1 is observed in the majority of colorectal (95%) and gastric (83%) carcinomas when compared with normal stomach and colon mucosa.Citation185 In another study, 363 colorectal tissues from CRC patients were evaluated by tissue microarray and immunohistochemistry to investigate the expression and prognostic role of BECN1 in CRC. The findings link high expression of BECN1 with better OS and disease-free survival, suggesting that BECN1 may serve as an independent prognostic marker in CRC.Citation184
Ectopic expression of the essential autophagy protein BECN1 reduces proliferation of cancer cells, suggesting its tumor suppressor properties. Indeed, BECN1 has been identified as a tumor suppressor complex. BECN1 serves as a scaffold for the formation of autophagosomes. MicroRNA (miRNA)-dependent decrease of BECN1 expression is an indication of poor prognosis and presumably promotes anti-apoptotic pathways.Citation184 Conversely, overexpression of BECN1 is associated with tumor hypoxia and these subgroups of tumors exhibit aggressive clinical behavior. In CRC tissues, BECN1 can be either up- or downregulated.Citation184 The activation of autophagy by overexpressing BECN1 may be an effective treatment of CRC with defects in BECN1.Citation186 In one study, the expression and significance of 3 autophagy-related proteins, namely BECN1, LC3, and MTOR, were investigated in the tumorigenesis and development of CRC. Immuno-histochemical studies revealed that the expression of these 3 proteins was significantly higher in CRC than in adjacent normal tissues.Citation186 In CRC tissues, the expression of LC3 was positively correlated with BECN1 and cell differentiation, but negatively correlated with MTOR, whereas the expression of MTOR was positively associated with cell differentiation and lymph node metastasis.Citation187 BECN1 and LC3 can predict the efficacy of cetuximab therapy, as low levels of autophagy are associated with a high antitumor efficacy of cetuximab.Citation188
Besides BECN1, alterations in other autophagy genes, such as deletion of the ATG5 gene or mutations in the key autophagic tumor suppressor UVRAG gene have been detected in colon cancer. We conclude that different components of the autophagic pathway mutually contribute to the regulation of cancer cell fate. The cancer-associated frame-shift mutation of UVRAG leads to the expression of its truncated form in CRC with MSI, and promotes tumorigenesis. The expression of truncated UVRAG can cause CRC metastatic spread through activation of the mall GTPase RAC1 and the epithelial-to-mesenchymal transition (EMT).Citation189
Increased expression of ATG10 in CRC is associated with lympho-vascular invasion and lymph node metastasis. ATG10 may serve as a potential prognostic maker in CRC.Citation190 ATG5 expression is lost in 23% of CRC patients and plays important roles in intestinal tumor growth. Heterozygous deletion of Atg5 in ApcMin mice increased the number and size of adenomas when compared with ApcMin Atg5+/+ mice.Citation189 Early treatment of ApcMin Atg5+/- mice with IFNG lowered the tumor incidence to 16.7% and reduced the number of adenomas by 95.5%.Citation189 IFNG treatment also led to tumor regression.Citation189 Heterozygous deletion of Atg5 activates EGFR-MAPK1/ERK2-MAPK3/ERK1 and WNT-CTNNB1/β-catenin pathways in adenomas of ApcMin mice and enhances the effects of IFNG-dependent inhibition of tumor growth. A combination of IFNG and ATG5 deficiency or ATG5-targeted inhibition may offer promising strategies for the prevention and treatment of CRC.Citation189 Besides the paradoxical role of autophagy in tumorigenesis and cancer progression, the lack of expression of 3 autophagy-related proteins (ATG5, BECN1, and MAP1LC3B/LC3B [microtubule associated protein 1 light chain 3 β) is associated with poor prognosis in CRC, suggesting that these proteins have a potential to serve as new prognostic markers in CRC.Citation191
Identified as a key autophagy-related protein and prosurvival factor in CRC cell lines, VMP1 (vacuole membrane protein 1) promotes autophagy via binding to BECN1 and triggering the BECN1-autophagy pathway. Upon specific VMP1 knockdown, CRC cells become more susceptible to apoptosis suggesting that VMP1 is an important negative regulator of the apoptotic pathways.Citation192
Ectopic expression of MIR140–5p in colorectal CSC inhibits CSC growth and sphere formation in vitro by disrupting autophagy. There is progressive loss of MIR140–5p expression from normal colorectal mucosa to CRC tissues and a further reduction in liver metastatic tissues. The functional and clinical significance of MIR140–5p suggests that it is an important regulator of CRC progression and metastatic potential, and may serve as a lead for the development of novel therapeutic molecules to treat CRC.Citation193
UPR modulators
Elevated HSPA5, a marker of the UPR, correlates with higher pathological grade, tumor recurrence, and poor survival in patients with breast, gastric, liver, colon, and prostate cancer.Citation194 The activation of several members of the UPR pathway, including HSPA5, has been reported in colon cancer.Citation195 In a cancer xenograft animal model, reduction of HSPA5 inhibited tumor formation and growth. In early tumor stages, increased expression of HSPA5 may be responsible for controlling local tumor growth, whereas in advanced stages high expression of HSPA5 and HSP90B1 is dependent on other cellular stress reactions such as glucose deprivation and hypoxia.Citation194
SEL1L is a member of the ER-associated protein degradation (ERAD) and UPR pathways. When associated with the E3-ligase SYVN1/HRD1, SEL1L assists in clearing unfolded proteins in the ER.Citation196 SEL1L expression is low in the normal gut mucosa but significantly correlates with the progression from adenoma to carcinoma, suggesting that it may become a potential target for CRC therapy.Citation196
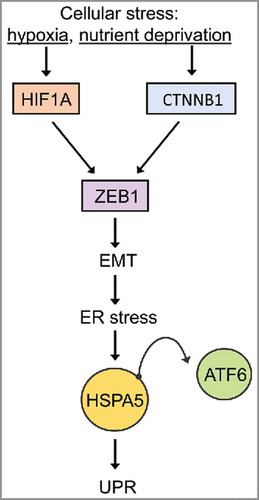
Hypoxia-like conditions (oxygen deprivation and nutrient stress) lead to EMT and ER stress in CRC cells and alter the localization of CTNNB1/β-catenin and CDH1/E-cadherin in SW480 and HCT116 colon cancer cells. Nuclear CTNNB1/β-catenin is an inducer of EMT and serves as an indicator for CRC stem cells (CSC), which promote tumor progression and a chemoresistance phenotype.Citation197 When cultured under hypoxia conditions, CRC upregulate the mesenchymal marker VIM (vimentin) as well as HSPA5, HIF1A/HIF1α, ZEB1, and the 50-kD ATF6 fragment. It can be inferred that cellular stress activates HIF1A and/or CTNNB1/β-catenin signaling pathways, resulting in the induction of the EMT and ER stress. Several methods based on differential live staining of cells are being developed that should allow for the identification of CSC.Citation198 depicts an interconnected network of cellular stress-EMT-ER stress.Citation199
Figure 6. Hypoxia- and nutrient deprivation-induced stress cause EMT-mediated UPR. The relationship between EMT and ER stress. At the invasive front of CRCs, cellular stress conditions (hypoxia or changes in the microenvironment) induce EMT via activation of HIF1A or CTNNB1/β-catenin, which consequently leads to ZEB1 activation and EMT induction. EMT activates the UPR which induces the activation of UPR-related transcription factors (ATF6) (adapted from ref. Citation199).
Oncoproteins and tumor suppressor proteins involved in the UPR and autophagy in CRC
MYB/cMyb
The expression of ER-located HSPA5 is mandatory for protein folding in most cells.Citation200 MYB is a conserved transcription factor involved in normal colon development and hematopoiesis.Citation201 Overexpression of MYB induces HSPA5 gene expression. The promoters of human and murine HSPA5 and HSP90B1 contain functional MYB binding sites as demonstrated by chromatin immunoprecipitation assays using recombinant MYB and nuclear extracts of colon cell lines. Amplification of MYB in tumor cells may lead to HSPA5 gene induction, and in turn, this promotes cell survival during oxygen deprivation and nutrient stress conditions.Citation202 This reinforces the view that UPR modulation may be a new attractive therapeutic target for the eradication of glucose-deprived solid tumors. shows a list of oncogenes and tumor suppressors involved in CRC.
Table 4. Oncogenes and tumor suppressors and their link to autophagy and tumorigenesis.
TAGLN/SM22
TAGLN (transgelin) is considered a tumor suppressor and its expression changes under many pathological conditions, including CRC.Citation203 TAGLN binds to the actin protein network and is also considered a marker for smooth muscle differentiation.Citation204 The expression of the tumor suppressor TAGLN is significantly decreased in CRC tissues and a link exists between low TAGLN expression and inhibition of autophagy in human CRC tissues and CRC cell lines.Citation203
SQSTM1/p62 (sequestosome 1) is a multifunctional receptor protein implicated in the delivery of cargo to phagophores.Citation63 SQSTM1 is a ubiquitin-binding scaffold protein that colocalizes with and guides ubiquitinated proteins to the autophagic machinery via binding to LC3.Citation205 SQSTM1 is a marker of autophagy flux and its upregulation could be interpreted as inhibition of lysosomal digestion of autophagosomes.Citation206 SQSTM1 is also involved in autophagy-related cell signaling pathways and tumorigenesis. SQSTM1 and LC3 are upregulated in a subset of CRC.Citation207 The increase in SQSTM1 is thought to be a crucial contributing factor in CRC tumorigenesis. Knockdown of Sqstm1 expression significantly inhibits autophagy activation and tumor growth, both in vitro and in xenograft tumor models.Citation207 SQSTM1 and autophagy have been suggested as therapeutic targets for the treatment of CRC.Citation207
SH3GLB1/BIF1 is a tumor suppressor gene of the endophilin protein family. SH3GLB1 colocalizes with ATG5 and LC3, which suggests its involvement in early autophagosome formation. Loss of SH3GLB1 reduces PIK3CVPS34 kinase activity and suppresses autophagy induction in response to nutrient starvation.Citation50 SH3GLB1 regulates autophagy by forming a multiprotein complex with the PtdIns3K and BECN1 through UVRAG.Citation208 The transition from normal epithelium to CRC coincides with a downregulation of SH3GLB1.Citation209 Furthermore, SH3GLB1 interacts with BAX to regulate apoptosis. Loss of SH3GLB1 suppresses apoptotic cell death by inhibiting BAX-BAK1 conformational change and caspase activation, hence promoting tumorigenesis.Citation209-Citation211
PCDH17
PCDH17 (protocadherin 17) exerts its tumor-suppressing activity through apoptosis and autophagy induction. PCDH17 is frequently silenced by promoter methylation in most gastric and colorectal tumor cell lines, as well as in 95% of primary CRC tumors. PCDH17 deletion was detected in only 18% of gastric and 12% of CRC tissues, suggesting that both epigenetic and genetic inactivation of PCDH17 are involved in gastric and colorectal tumorigenesis. PCDH17 methylation status has been suggested as an epigenetic biomarker for these tumors.Citation212 In gastric and CRC patients, high PCDH17 expression was significantly correlated with low tumor stage and lower frequency of lymph node metastasis, indicating a promising role for PCDH17 as a prognostic marker.Citation212 Restoring PCDH17 expression promotes apoptosis and blocks tumor cell growth both in vitro and in vivo.Citation212 Furthermore, PCDH17 induces autophagy through the upregulation of autophagic proteins (such as ATG5, ATG12 and LC3B-II) and formation of autophagic vacuoles.
PARK2/parkin
Frequent loss of heterozygosity and deletions in the PARK2 gene are found in several cancers. Consistent with PARK2s property as a tumor suppressor, several studies showed that ectopic expression of PARK2 reduces cell growth and increases apoptosis in hepatocellular and lung cancer. In a colon cancer model, alternatively spliced variants of PARK2 failed to degrade CCNE (cyclin E). This finding suggests that loss of CCNE regulation by PARK2 contributes to colon cancer. Tumor cells often suppress their mitochondria in response to hypoxia and ER stress to reduce oxidative stress, a process that also utilizes autophagy. PARK2's role in mitophagy might also in part account for its tumor suppressor functions. PARK2 induces autophagy via BNIP3L/NIX, which causes mitochondrial depolarization and MTOR inhibition. PARK2 ubiquitinates effector proteins and can select mitochondria for autophagy. The HSP90/HSP70-based chaperone machinery plays a key role in the degradation of aberrant proteins via the ubiquitin-proteasome pathway.Citation213 The ability of PARK2 to ubiquitinate HSP70 proteins suggests that PARK2 may play a role in the degradation of substrates normally stabilized by HSP90 and important for tumor cell survival and proliferation.Citation214
Class I PI3K
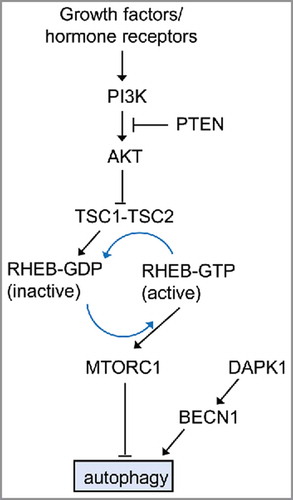
Class I PI3Ks phosphorylate PtdIns4P and PtdIns(4,5)P2. The deregulation of class I PI3Ks has been described in the course of tumorigenesis and resistance to therapy in cancer. Class I PI3K activation and its products PtdIns(3,4)P2 and PtdIns(3,4,5)P3 inhibit autophagy in HT-29 cells.Citation142,Citation185 The activation of MTOR and the resulting inhibition of autophagy in response to cellular stress can occur through the activation of the class I PI3K and its downstream effector AKT/PKB. The inhibition of AKT potently induces autophagy through inactivation of MTORC1. Likewise, overexpression of PTEN, a dual lipid/protein phosphatase, tumor suppressor, and negative regulator of the PI3K-AKT pathway, induces autophagy.Citation50 The PI3K-AKT pathway is a potent activator of cell proliferation and cell survival, and it is regulated at multiple levels.Citation215 A dominant negative AKT mutant enhances autophagy whereas expression of active AKT decreases autophagy. depicts the role of MTOR in cancer and its association with autophagy.
Figure 7. The role of MTOR in cancer-associated signaling pathways that regulate autophagy in mammalian cells. The best known regulator of autophagy is MTOR (mechanistic target of rapamycin), a serine/threonine kinase conserved throughout eukaryotes. The activity of MTORC1 is inversely correlated with autophagy induction. The µTORC1 inhibitor rapamycin potently induces autophagy, even in the presence of abundant nutrients (adapted from ref. Citation50). The PI3K-AKT regulates autophagy. This regulation is mediated via the small RHO-GTPase RHEB.
Class III PtdIns3K
In contrast to class I PI3Ks, class III PtdIns3Ks are autophagy stimulators. Inhibition of class III PtdIns3K (by e.g. 3-methyladenine; 3-MA) decreases the rate of autophagy, whereas the class III PtdIns3K adaptor (PIK3R4/p150) overexpression or the addition of PtdIns3P induces autophagy.Citation185,Citation216 BECN1 plays an integral role in the class III PtdIns3K pathway.Citation185 Knockdown of BECN1 inhibits autophagy and promotes cell death through nutrient starvation. BECN1 and PTEN are important for autophagy induction and are therefore considered potential targets in the treatment of cancer.Citation185 Justicidin A (JA), a novel and pure arylnaphthalide lignan isolated from Justicia procumbens, induces class III PtdIns3K-dependent autophagy in a colorectal cancer cell line (HT29). This enhances JA-mediated apoptotic activity and antitumor effects in these cells.Citation217
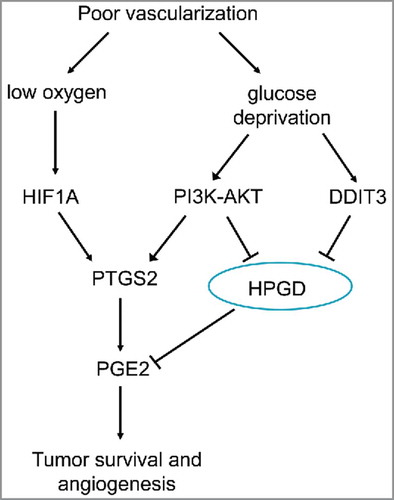
HPGD/15-PGDH
Recently, HPGD (hydroxyprostaglandin dehydrogenase 15-[NAD]), a key enzyme in PGE2 degradation, has been indicated as a tumor suppressor in several cancers, including colon cancer.Citation218 Glucose deprivation in colon tumors elevates PTGS2/COX2 expression and simultaneously reduces the expression of HPGD.Citation218 Depriving colon tumor cells of glucose, results in upregulation of PGE2 with both an increase in PTGS2 expression and a decrease in HPGD expression, which is mediated via enhanced PI3K-AKT signaling. Glucose deprivation leads to activation of the UPR, which, through increased levels of DDIT3, can lead to the suppression of the key tumor suppressor gene HPGD. This inverse regulation between DDIT3 and HPGD suggests that tumor cells could manage to survive in the presence of therapeutic agents that activate the UPR. In this way, regulation of PTGS2 and HPGD might be critical via effective PGE2 target-based chemotherapy approaches to suppress tumor development. shows how glucose deprivation increases PGE2 expression during tumorigenesis.Citation218
Figure 8. Regulation of tumor survival and angiogenesis via glucose and oxygen supply. Glucose deprivation increases PGE2 by upregulating PTGS2 and downregulating HPGD expression via PI3K-AKT and DDIT3-dependent mechanisms. Hypoxia increases PGE2 levels by upregulating PTGS2 expression via HIF1A). Elevated PGE2 increases survival of colon cancer cells exposed to both glucose deprivation and hypoxic conditions (adapted from ref. Citation218).
Association between obesity and CRC through autophagy and the UPR
Obesity is a significant risk factor for various types of cancers.Citation138,Citation219 Diets high in fat and genetic predisposition to obesity in Apc1638N mice, a mouse model for familial adenomatous polyposis, differentially alter the composition of microorganisms and metabolites in the intestine. A reduction in P. distasonis and adenosine is anti-inflammatory in the colon and could promote tumorigenesis.Citation220 A study has been recently conducted on 451 Hispanic participants, of whom 218 had CRC, 77 had colorectal adenomas, and 156 were colonoscopy-negative controls. The study found an increased risk of adenoma, especially in proximal locations, among Hispanic women with type 2 diabetes providing a rationale for increased screening in this population.Citation221
The MTOR pathway integrates signals from growth factors, nutrients, mutagens, and hormones, to induce cell proliferation and resistance to apoptosis, and autophagy.Citation222 Glucose deprivation is a form of nutritional stress in tumor cells. ADIPOQ (adiponectin, C1Q and collagen domain containing) negatively influences cancer progression during glucose deprivation.Citation130 Under normal conditions, ADIPOQ inhibits IGF1 (insulin like growth factor 1) signaling in tumor cells and activates both PRKAA/AMPKα and PPARA/PPARα (peroxisome proliferator activated receptor α) to inhibit the PI3K-AKT-MTOR pathway and enhance autophagy.Citation130 Hence, ADIPOQ provides an important molecular link between cancer and obesity. Epidemiological and clinical data show a relationship between obesity-related inflammation via pro-inflammatory cytokines, such as TNF secreted by macrophages. Low level of LEP (leptin) and ADIPOQ are additional factors that play an important role in physiological responses to inflammation and can promote the development of CRC in obese individuals. The role of LEP and ADIPOQ in carcinogenesis is attributed to several signaling pathways, including the activation of JAK-STAT, MAPK, PI3K, MTOR, and AMPK, and downregulation of PTGS2,Citation223 and upregulation of CDH13/T-cadherin (cadherin 13), a unique member of the cadherin superfamily lacking the transmembrane and cytoplasmic domains that anchors to the cell membrane of HCT116 cells.Citation224
Hypoglycemic agents and autophagy and the UPR in CRC
Metformin, an oral hypoglycemic agent, has recently being receiving increased attention due to its antitumorigenic effects in breast and colon malignancies that have an association with obesity and hyper-insulinemia. Chemotherapy with metformin is associated with decreased incidence of colon and pancreatic cancer but does not affect the outcomes in breast or prostate cancer. A randomized pilot study involving nondiabetic patients showed that low-dose metformin (250 mg/d) given for 1 mo suppresses the formation of aberrant crypt foci (ACF), an early indicator of colon cancer.Citation225
Mechanisms of action of metformin include activation of the STK11/LKB1 (serine/threonine kinase 11)-AMPK pathway, induction of cell cycle arrest and/or apoptosis, inhibition of protein synthesis, reduction in circulating insulin, inhibition of the UPR, activation of the immune system, and eradication of cancer stem cells. Using a tumor xenograft model, Buzzai et al. showed that metformin was able to selectively inhibit cell growth and induce autophagy in TP53-deficient colon cancer cells. In glucose-starved cultures of human colon, fibrosarcoma, renal, and stomach cancer, gene expression profiling techniques revealed that metformin was able to inhibit UPR activators and lead to cell death.Citation225
Aspirin, in combination with metformin, enhances AMPK activation and this leads to MTOR suppression and autophagy induction, which could contribute to the tumor supressor role of AMPK in the development of CRC. The PI3K-MTOR signaling pathway controls cell survival and regulates cell metabolism, and deregulated PI3K-MTOR signaling is associated with CRC development. Pharmacological AMPK activators, such as 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR) and metformin, inhibit growth and delay tumor initiation.Citation226 These findings suggest a possible mechanism by which metformin and aspirin may inhibit cancer growth through MTOR signaling/autophagy and the UPR.
The UPR and autophagy pathways as potential treatment strategies for IBD and CRC
Autophagic adaptive responses in CRC can increase the sensitivity against autophagy inhibition and improve the efficacy of chemotherapy. Autophagosomes are actively produced in CRC cells under conditions of nutrient starvation. Autolysosome inhibitors suppress autophagosome formation and enhance apoptosis under amino acid- and glucose-deprived conditions.Citation227 We will also discuss the critical role of combinatorial therapies with particular attention to genetic and molecular markers associated with the autophagy and UPR pathways.
Therapeutic targeting of the autophagy pathway
Silibinin, a flavonolignan isolated from the milk thistle plant (Silybum marianum), inhibits autophagy and enhances apoptotic pathways in the SW480 and SW620 CRC cell lines.Citation228 Also, curcumin and a curcumin analog G0-Y030 inhibit tumor sphere formation in ALDHA+ CD133+ colon CSCs.Citation229 Multiple signaling pathways are inhibited by curcumin in epithelial cancers and this contributes to apoptotic cell death. Besides apoptosis, curcumin induces autophagy in cancer cells.Citation229 Chemo-protective properties of plant-derived phytochemicals (e.g., curcumin) may be induced through several effects, including cell restorative processes, stimulation of antimetastatic and anti-angiogenic responses and/or increased antioxidant and anti-inflammatory activity.Citation230,Citation231 In the future, it may be possible to avoid toxic effects of radio/chemotherapy by using combinatorial strategies with nontoxic agents such as curcumin, which can target CSC.Citation232 The oncogenic microRNA MIR22 is thought to be a switch between apoptosis and autophagy. MIR22 also inhibits autophagy and promotes apoptosis both in vitro and in vivo and increases CRC cell sensitivity to 5-FU treatment.Citation149 The oncogenic MIR22 may be considered a predictor of 5-FU sensitivity and a target for CRC therapy.Citation149
Mammalian MAPK14/p38α activity is required for CRC cell growth in vitro and in mouse models of human colon cancer. Inhibition of MAPK14 in CRC cells reduces tumor growth and induces autophagic cell death. Combination therapy using inhibitors of MAPK14 (SB202190) and MAP2K1/MEK1 (PD98059) significantly reduces cell survival and induces apoptosis through TNFSF10/TRAIL signaling in both HT-29 and HCT-116 cells. Several MAPK14 and MAP2K1 inhibitors are in phase II clinical trials for the treatment of inflammation and cancer.Citation233,Citation234 Also, MAPK14 is required to sustain the expression of HIF1A target genes. Inhibition of MAPK14 causes a rapid drop in ATP levels in CRC cells. The AMPK-FOXO3 (forkhead box O3) axis is a metabolic switch that senses variations in the AMP:ATP ratio. Manipulation of this pathway in combination with drugs targeting the ‘Warburg effect’ and/or autophagy may be an effective strategy for selective targeting of cancer cells.Citation235-Citation237 In addition, AMPK is a major regulator of energy metabolism with key roles in the inhibition of biosynthetic pathways and enhancement of ATP-generating pathways. Compound C, a small molecule inhibitor of AMPK, causes an increase in the sub-G1 cell population (apoptotic cells), in HCT116 and KM12C cells. Compound C also triggers acidic vesicular formation, conversion of LC3-I to autophagosome-associated LC3-II, and finally autophagic cell death in DLD1 and SW480 cells.Citation238
Ectopic expression of MIR124–2HG, a modulator of energy metabolism and tumor suppressor, enhances oxidative stress. The MIR124–2HG-PTBP1/PTB1-PKLR/PKM1-PKM/PKM2 axis induces apoptosis and autophagy in colon cancer cells.Citation239 MAPKs are activated by 5-FU. SB203580 compound-mediated inhibition, or the shRNA-specific knockdown of MAPK p38, are associated with resistance to 5-FU-induced apoptosis in HCT116 cells.Citation240 This resistance is correlated with an autophagic response mediated by a decrease in TP53-induced apoptosis but does not affect TP53-dependent autophagy. The critical role of the MAPK p38 signaling pathway in modulating the rates of autophagy and apoptosis in response to 5-FU has been outlined in .Citation240 We have summarized agents targeting autophagy pathways in vivo and in vitro in CRC in .
Figure 9. The role for the MAPK p38 signaling pathway in cellular response to 5-FU. There is a critical role for the MAPK p38-signaling pathway in the cellular response to 5-FU by controlling the balance between apoptosis and autophagy (adapted from ref. Citation240). This pathway is tightly controlled by the TP53-mediated regulation of autophagy.
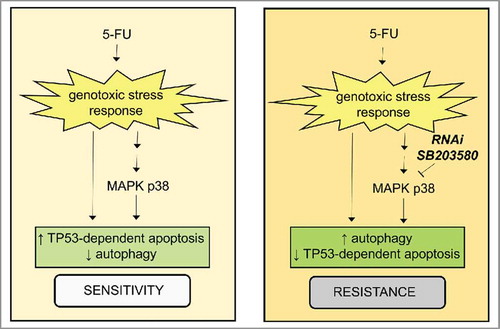
Figure 10. Autophagy targeting strategies in in vitro and in vivo models of CRC. All chemical compounds, drugs, and inhibitors have been introduced in the section “Therapeutic targeting of the autophagy pathway.”

Autophagy and Immunotherapy in CRC
Immunotherapy has emerged as a powerful weapon to combat different types of cancer, as it targets tumor-specific antigens.Citation241 In the context of CRC, considering drawbacks of current treatment options such as chemo- and radiotherapy, development of novel alternative specific strategies with more efficacies and less side effects is an unmet clinical need. In general, immunotherapeutic approaches to treat CRC include peptide vaccines, dendritic cell (DC)-based vaccines, whole tumor cell vaccines, viral vector-based vaccines,Citation242 adoptive cell transfer therapy, antibody-based cancer therapy,Citation243 cytokine therapy,Citation241 checkpoint inhibitors, and combined therapy.Citation241,Citation244
CRC peptide vaccines are well-characterized epitopes able to elicit a specific immune response against colorectal tumor-associated antigens (TAAs). For example, CRC cells often express CEACAM5/CEA (carcinoembryonic antigen related cell adhesion molecule 5),Citation245 EGFR (epidermal growth factor receptor),Citation246 TP53,Citation247 or KRAS,Citation248 which are potential targets for CRC immunotherapy. DCs can provide necessary signals to induce an efficient antitumor immune response.Citation249 Therefore, several DC-based immunotherapeutic approaches have been developed by using TAA-pulsed DCs. These approaches include the known TAAs,Citation250 tumor cell lysates,Citation251 apoptotic tumor cells,Citation252 and tumor RNA.Citation253
Adoptive cell transfer therapy is a passive immunotherapy in which specific effector cells, e.g. cytotoxic T lymphocytes, are directly infused within the CRC patient. Autologous T cells are removed from CRC patients, activated, expanded to large numbers in vitro and transferred back into the patients.Citation254,Citation255 Immune checkpoint blockade by targeting the inhibitory immune receptors CTLA4 (cytotoxic T-lymphocyte associated protein 4), PDCD1/PD1 (programmed cell death 1), and CD274/PDL1 is a novel immunotherapeutic approach to treat CRC patients.Citation256,Citation257 A combined approach by using both chemo/radiotherapy and immunotherapy seems to be more effective for CRC.Citation258 For instance, it has been shown that chemotherapy increases the antitumor effects of cancer immunotherapy by depleting regulatory T cells (Treg).Citation259,Citation260
Several lines of evidence suggest that autophagy is involved in development and progression of CRC and it could be considered as a potential target for treatment of CRC.Citation261 However, there are several issues, which remain to be addressed. It is not clear whether targeting autophagy machinery in CRC patients or experimental models will affect the antitumor immune response. Furthermore, current CRC immunotherapeutic modalities might function at least in part through modulation of autophagy. In addition to its direct antitumorigenic roles, autophagy can inhibit CRC by attenuating the inflammatory response in the tumor microenvironment. Autophagy could increase the processing and presentation of TAAs that result in antitumor immunity. Tumor cells have the ability to escape immunosurveillance by tuning down autophagy, though some chemotherapies have been revealed to exert immunogenic antitumor properties via inducing autophagic cell death.Citation262
It has been demonstrated that the cell wall of Mycobacterium bovis, and Bacillus Calmette-Guerin (BCG) induces a radiosensitizing effect on colorectal cell lines via induction of autophagic cell death through TLR2 and TLR4 signaling. In vivo evidence further supports the idea that BCG-mediated radiosensitization is an autophagy-dependent phenomenon. These data suggest that the BCG cell wall in combination with ionizing radiation provides a promising strategy for enhancing radiation therapy in CRC through the induction of autophagy.Citation263
Wei et al. have recently reported that autophagy is active in Treg cells, and involved in their lineage stability and survival fitness. Genetic abrogation of autophagy in these cells has led to loss of Treg cells, greater tumor resistance and development of inflammatory disorders in a mouse model of colon cancer.Citation264 Specific deletion of the Atg7 gene in Treg cells has been associated with increased apoptosis and downregulation of transcription factor FOXP3. Loss of autophagy leads to upregulation of metabolic mediators such as MTORC1 and MYC as the mechanism underlying the defective Treg phenotype in this CRC model.Citation264 These findings indicate that targeting autophagy in Treg cells along with tumor cells could heighten the efficacy of treatment in CRC.
Considering the inhibitory effect of chloroquine as an autophagy inhibitor on colon cancer cell growth,Citation265 and also development of systemic autophagic syndrome upon recombinant IL2 immunotherapy, Liang et al. demonstrated that co-administration of chloroquine increases IL2 immunotherapeutic efficacy and also limits toxicity in an advanced murine metastatic liver tumor model. This beneficial dose-dependent combinatorial therapy is associated with increased long-term survival, vascular leakage and enhanced immune cell proliferation and infiltration.Citation266 From a mechanistic point of view, IL2 treatment alone induces autophagy and overexpression of HMGB1, IFNG, IL6, and IL18 within the liver and translocation of HMGB1 from the nucleus to the cytosol in hepatocytes, which is significantly inhibited upon addition of chloroquine.Citation266 Furthermore, the chloroquine effect could be directly mediated on tumor cells by several mechanisms such as increased autophagic vacuoles and LC3-II levels, cell death, CASP3/caspase-3 cleavage and CYCS (cytochrome c, somatic) release from mitochondria as well as decreased oxidative phosphorylation and ATP production.Citation266
Collectively, in combination with chemotherapeutics and immune checkpoint inhibitors, autophagy regulators might strengthen the efficacy of CRC immunotherapy in a more targeted manner. One has to keep in mind that the interaction between cell proliferation and immunity is a complex one.Citation267 Future investigations aiming to understand the effect of these combinatorial approaches on antitumor immunity could address several unknown issues regarding this complex problem.
Therapeutic targeting of ER stress and the UPR
Falcarindiol (FAD) is a natural polyacetylene in carrots that induces intracellular buildup of ubiquitinated proteins in cancer cells, including CRC. This leads to the accumulation of unfolded/misfolded proteins in the ER and causes an increase in ER stress-induced cell death via FAD.Citation268 Treatments leading to increased ER stress enhance FAD-induced cell death.Citation269
Cancer cells, in poorly vascularized solid tumors, are frequently exposed to nutrient starvation, which activates the UPR pathway. In a glucose-deprived environment, anticancer agents, such as arctigenin (ARC-G), which targets the UPR, could preferentially cause tumor cell death.Citation34 Another target for cancer therapy is HSPA5 because it is considered a main target of UPR signaling for survival and often upregulated in most cancers. Verrucosidin can disrupt HSPA5 expression during glucose deprivation in HT-29 human colon cancer cells.Citation270 In addition to being an inhibitor of HSPA5 expression, versipelostatin, blocks the UPR and induces cytotoxicity in glucose-deprived colon tumor cells both in vitro and in vivo.Citation269 This cytotoxic effect of versipelostatin is mediated through suppression of HSP90B1 expression and the UPR transcriptional activators XBP1 and ATF4.Citation271,Citation272
Brefeldin A (BFA), an inhibitor of protein transfer from the ER to the Golgi, leads to an accumulation of proteins in the ER and this results in the activation of apoptotic UPR signals. BFA triggers apoptosis in HT-29 cells.Citation195 These results suggest 2 approaches to target the UPR: (i) designing inhibitors of the UPR pathway to block the adaptive response needed for survival of tumor cells, and (ii) using inducers of the UPR to overload stress and induce UPR-mediated cell death pathways in cancerous cells.Citation195 Targeting autophagy leads to increased cell death in HCT116 colon cancer cells.Citation273 lists the drugs targeting ER stress and autophagy for anticancer therapy. High doses of selenium induce ER stress and cause subsequent EIF2AK3-dependent ElF2 phosphorylation and apoptotic cell death. Selenium can also contribute to an increase in ER stress upon irradiation and may promote radio-sensitization.Citation273
Table 5. Drugs that target ER stress and autophagy for anticancer therapy.
The expression of FASN (fatty acid synthase) and the autophagy marker SQSTM1 is increased specifically in primary CRCs and liver metastases of CRC.Citation274 The activation of fatty acid oxidation and the downregulation of stress-response signaling pathways may be key adaptation mechanisms to facilitate the effects of FASN on cancer cell survival and metastasis. This provides a strong rationale for targeting this pathway in advanced CRC.Citation274 Importantly, ER stress can be induced in tumor cells by FASN inhibitors, such as cerulenin or C75, but not in normal cells. FASN inhibitors exhibit selective apoptosis-inducing cytotoxicity particularly in therapy-resistant, TP53-deficient cells. The selective activation of the FASN pathway could be responsible directly for CRC destruction by apoptosis and/or target them for immunological attack. FASN inhibitors were toxic to HT-29 cells expressing a dominant negative EIF2AK3. This was also observed in cells expressing normal levels of EIF2AK3 and is mediated by FASN inhibitor-induced persistent phosphorylation of EIF2A by EIF2AK3.Citation275 In this context, FASN causes inhibition of protein synthesis and the activation of ERN1 to increase the expression of the ER stress regulated genes ATF4, DDIT3, and HSPA5. This suggests that an approach combining irradiation-induced ER stress with FASN inhibitors could potentially improve the outcome of cancer treatment.Citation275
To prevent protein misfolding and degradation, cells upregulate protein chaperone members of the heat shock protein (HSP) family. HSPs form interactions with key proteins in the UPR pathway. HSP90 inhibitors, such as 17-allylamino-17-demethoxygeldanamycin (17AAG), act as UPR activators and can activate all 3 major UPR arms (). All HSP90 inhibitors tested repress cell proliferation and increase the expression of the chaperones HSP90B1 and HSPA5. In 17AAG-treated myeloma cells, exposure to HSP90 inhibitors changed the LC3 expression levels consistent with autophagosome formation. HSP90 inhibitors may be interesting targets for the treatment of myeloma, breast cancer, and CSC.Citation194 Analogous to the ER responses initiated by defective protein folding, a mitochondrial UPR may play a role in HSP90- and proteasome inhibitor-mediated apoptotic pathways. Application of HSP inhibitors blocks chaperone function and increases mitochondrial protein expression. This is associated with increased cytochrome oxidase activity, leading to mitochondrial dysfunction.
ER stress inducers, such as thapsigargin or bortezomib, exhibit significantly higher cytotoxicity along with enhanced UPR activation when used under hypoxic conditions. Despite being generally more resistant to genotoxic agents, these drugs may induce hypersensitivity to proteasome inhibitors via increased UPR signals in hypoxic tumor cells.Citation276
N-3 poly-unsaturated fatty acids, such as docosahexaenoic acid (DHA), induce apoptosis by altering the expression and localization of HSPA5 in colon cancer cell lines (i.e., HT-29, HCT116 and SW480). Transfection of SW480 cells with HSPA5-GFP induced increased cell growth and inhibited the DHA induced apoptosis.Citation277 As shown in , DHA induces key mediators of ER stress and the UPR and it may also coordinate many downstream pathways, including the regulators of cholesterol metabolism, calcium homeostasis, ubiquitination, and proteasomal degradation. DHA may cause cholesterol depletion in the ER due to reduced de novo cholesterol synthesis and inhibition of cholesterol transport to the ER through redistribution of cholesterol from the ER to DHA-cholesteryl ester-enriched lipid droplets.Citation278 Accompanied by increased cholesterol esterification, this is an important factor for the initiation of calcium mobilization. Subsequent ER stress may lead to growth inhibition and cell death. Therefore, compounds that decrease cellular cholesterol stores activate a network of stress responses leading to cell death.Citation278
Figure 11. Endoplasmic reticulum (ER) stress induction by docosahexaenoic acid (DHA). DHA induces ER stress in colorectal cells. Diagram showing transcripts affected by DHA treatment in SW620 colon cancer cells by gene expression analysis in the main pathways of ER stress signaling. Three transmembrane proteins mediate the unfolded protein response (UPR) across the ER membrane after dissociation from HSPA5-ATF6, EIF2AK3 and ERN1 (adapted from ref. Citation278).
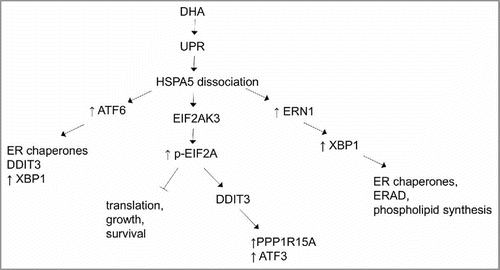
Synthetic 3-thia fatty acid, tetradecylthioacetic acid, can inhibit proliferation in SW620 cells. Like DHA, tetradecylthioacetic acid induces growth inhibition, which is mediated via ER stress and the UPR and involves EIF2A phosphorylation and downstream regulation of ATF4.Citation279 Riproximin (RPX) is a component of a plant extract and cytotoxic to breast and colorectal cancer cells by targeting type II ribosome inactivating proteins with high selectivity in certain tumor cell lines. This increases the expression of the UPR genes ATF6 and ERN1. A higher concentration of RPX induces the UPR pathway via the EIF2AK3 branch, which results in numerous complex effects, such as translational arrest, growth inhibition, and apoptosis by enhanced EIF2A phosphorylation that includes elevated expression of transcription factors such as ATF3 and DDIT3.Citation280 The antioxidant 2-(3, 4-dihydroxyphenyl) ethanol (DPE) derived from olive oil induces growth arrest and apoptosis in HT-29 cells. Like DHA, DPE acts via ER stress induction and leads to the activation of 2 routes of the UPR, the ERN1-XBP1-HSPA5 and EIF2AK3-EIF2A pathway, resulting in enhanced expression of the pro-apoptotic factor DDIT3.Citation281
Treatment of colon cancer cells with the RRM (ribonucleotide reductase) inhibitor Triapine (3-AP; 3-aminopyridine-2-carboxaldehyde thiosemicarbazone) activated all 3 ER stress pathways (EIF2AK3, ERN1, ATF6). In particular, 3-AP-Me led to 16-fold upregulation of an mRNA variant of XBP1. 3-AP and 3-AP-Me activated the cellular stress kinases, MAPK/JNK and MAPK p38 with subsequent UPR activation and apoptosis. These data suggest that 3-AP and 3-AP-Me could induce apoptosis via ER stress in colon cancer cells.Citation282
The nonsteroid anti-inflammatory drug tolfenamic acid (TFA) suppresses cancer cell growth and tumorigenesis in various cancer models. TFA markedly reduced the number of polyps and tumor load in an experimental rodent model of CRC. TFA promotes ER stress and UPR activation, which leads to CCND1 (cyclin D1) translation inhibition. The EIF2AK3-EIF2A-ATF4 autophagy branch plays a role in TFA-induced apoptosis in CRC cells since the silencing of ATF4 attenuated TFA-induced apoptosis. This implicates ER stress being involved in TFA-induced inhibition of CRC cell growth in mice.Citation283
The EIF2AK3-EIF2A-ATF4 pathway is critical for the adaptation to hypoxic stress in tumor cells. HT29 cells expressing dominant negative EIF2AK3 are more sensitive to hypoxic conditions and die by apoptosis. Autophagy acts in a prosurvival manner by removing aggregated proteins accumulating in the cytosol, thereby preventing manifestation of the UPR. The accumulation of polyglutamine proteins causes the activation of autophagy by a EIF2AK3-EIF2A-ATF4 mediated upregulation of ATG12 after ER stress. Although this may qualify the EIF2AK3 pathway as a bona fide target to impede the survival of tumor cells under hypoxia, EIF2AK3 targeting does not always produce desirable effects. For example, activation of EIF2AK3 and EIF2A signaling in highly malignant squamous THEP3 or SW620 CRC cells induces both survival and suppression of tumor growth both in vitro and in vivo. Moreover, despite its prosurvival properties, EIF2AK3 may also suppress advanced tumor growth. This dual function of EIF2AK3 should be considered when developing EIF2AK3-targeted anticancer strategies, as EIF2AK3 inhibition might stimulate the proliferation of quiescent state tumor cells.Citation142 We have summarized agents targeting ER stress and the UPR pathway in vivo and in vitro in CRC in .
Figure 12. ER stress and UPR targeting strategies in in vitro and in vivo models for CRC therapy. All chemical compounds, drugs, and inhibitors have been introduced in the section “Therapeutic targeting of ER stress and the UPR.”

Cancer therapy using lysosomal targeting
There is profound interest in using demethylating agents, such as 5-aza-dC or 5-azacytidine, in the treatment of several tumors, including CRC and other malignancies harboring KRAS mutations. 5-aza-dC also serves as a chemosensitizer via interconnection between BNIP3 protein and hypoxia. BNIP3 expression is initially silenced via methylation but becomes activated in a KRAS-dependent manner in colon cancer cells. Reactivated BNIP3 contributes to 5-FU resistance.Citation284 Furthermore, mutants of KRAS act via an inflammatory pathway, involving the kinase IKK, which activates NFKB. In contrast to mutant KRAS, the BRAF (V600E) mutant triggered the phosphorylation of a proteolytic fragment of CHUK/IKKα (CHUK p45) in CRC cells, which is necessary for transformation of NIH-3T3 cells and BRAF-dependent transcription. CHUK p45 is further phosphorylated by MAP3K7/TAK1, which is associated with the endosomal compartment. Bafilomycin A1 or chloroquine-induced inhibition of the endosomal vacuolar-type H+-translocating ATPase (V-ATPase), blocked CHUK p45 phosphorylation and induced apoptosis in BRAF-mutant CRC cells independent of autophagy.Citation16 CRC cells harboring mutant KRAS, but not mutant BRAF, show resistance to chemotherapy and EGFR-targeted therapy. However, these CRC cells are sensitive to the treatment with recombinant LGALS9/Galectin-9 (rLGALS9), a lysosomal inhibitor. Treatment with rLGALS9 leads to elevated autophagic flux, excessive lysosomal swelling and death in KRAS mutant CRC cells.Citation285
In addition to lysosomal membrane permeabilization, acetate, a short-chain fatty acid secreted by Propionibacteria in the human intestine, induces mitochondrial apoptotic death in CRC cells. Lysosomal membrane permeabilization results in the release of the anti-apoptotic protease CTSD (cathepsin D). Moreover, pepstatin A (CTSD inhibitor) can increase acetate-induced apoptosis. Hence, CTSD inhibitors could serve as novel strategy for the prevention and/or treatment of CRC by enhancing acetate-mediated cancer cell death.Citation286 We have summarized agents targeting the lysosomal pathway in vivo and in vitro in CRC in .
Figure 13. Lysosomal targeting strategies in in vitro and in vivo models for CRC therapy. All chemical compounds, drugs, and inhibitors have been introduced in the section “Cancer therapy using lysosomal targeting.”
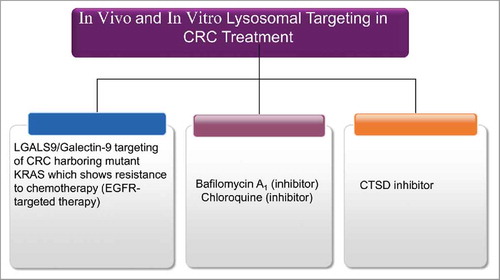
Anticancer potential of MTOR inhibitors
Inhibitors of TORC1 (rapamycin and rapalogs) have been effective in IBD and in many CRC models. Second generation MTOR inhibitors are more effective, particularly when combined with proteasome inhibitors or histone deacetylase inhibitors (HDACi).Citation287 Studies also showed an inverse association between TYMP (thymidine phosphorylase), deoxyribose, and rapamycin. TYMP has an important role in the MTOR-RPS6KB/p70S6K pathway and activation of RPS6KB and subsequent inhibition of autophagy was observed in the human Colo320 cells and transfected variant Colo320 TYMP1/TP1+/- cells when treated with deoxyribose and rapamycin. Thus, deoxyribose protects from rapamycin-induced cytotoxicity in CRC cells.Citation130 Conversely, the MTOR inhibitor Torin-1 negatively affected the growth, motility, invasion, and survival of CSCs in vitro, and suppressed tumor growth in vivo. Thus, Torin-1 may serve as a potential lead compound for the treatment of metastatic CRC therapy.Citation288
The second-generation MTOR inhibitor AZD-2014 blocks MTORC1 and MTORC2 and induces autophagic death in CRC cells.Citation289 AZD-2014 can inhibit the growth of tumors in SCID mice bearing HT-29 xenografts.Citation289 Oral AZD-2014 administration inhibits MTORC1/2 activation and HT-29 cell growth, while inducing autophagy in vivo by upregulation of LC3B-II and BECN1.Citation289 In apoptosis-resistant CRC cells, autophagic cell death is known as a major contributor of growth inhibition.Citation289 Aspirin also induces autophagic cell death in CRC cells through inhibition of MTOR signaling.Citation226,Citation289 Similar to AZD-2014 and aspirin, bufalin induced autophagic cell death in HT-29 and Caco-2 colon cells and this involves both ROS and the MAPK/JNK pathway. MAPK/JNK activation is required for the upregulation of ATG5 and BECN1, subsequent ROS generation, and autophagy-mediated cell death.Citation203 We have summarized agents targeting the MTOR pathway in CRC in vivo and in vitro in .
Figure 14. MTOR targeting strategies in in vitro and in vivo models for CRC therapy. All chemical compounds, drugs, and inhibitors have been introduced in the section “Anticancer potential of MTOR inhibitors.”
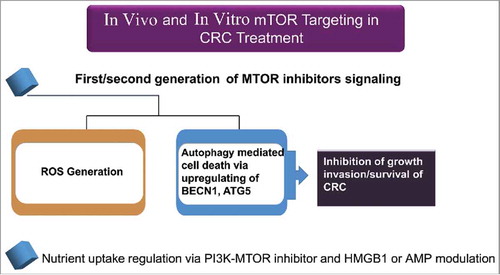
Therapeutic targeting of epigenetic regulators
Recent findings show that the anticancer effects of HDACi compound LBH589 are augmented by the activity of the tumor suppressor DAPK (death associated protein kinase). In autophagy-deficient cells, DAPK is necessary for HDACi-induced apoptosis. In in vitro and in vivo CRC studies, LBH589 upregulated and activated DAPK, inhibited cell proliferation, and reduced cell survival.Citation290 LBH589 induced an accumulation of LC3-II, promoted acidic vesicular organelle formation, and enhanced the degradation of SQSTM1, thus, causing DAPK-dependent autophagy. Conversely, autophagy inhibition sensitized tumor cells to LBH589-induced apoptosis, which involves DAPK. Hence, upon autophagy inhibition, DAPK acts as a switch between autophagy and apoptosis.Citation290 We have summarized agents targeting the epigenetic pathway in CRC in vivo and in vitro in .
Figure 15. Therapeutic targeting of epigenetic regulators in in vitro and in vivo models for CRC therapy. All chemical compounds, drugs, and inhibitors have been introduced in the section “Therapeutic targeting of epigenetic regulators.”

Other pharmacological autophagy inducers
Other drugs contributing to autophagy induction include the chimeric anti-EGFR antibody, cetuximab, which exerts its anticancer effect, at least in part, via autophagy-induced cell death. The rapamycin derivative everolimus that has recently been proposed for the treatment of neuroendocrine and colorectal tumors may function as autophagy inducer. Blockade of VEGF can inhibit vascularization of tumor cells and subsequent tumor growth. Co-administration of anti-angiogenic therapy (i.e., bevazucimab or avastin) with irinotecan may produce a favorable treatment response and prolong the progression-free survival.Citation291 A combination of the VEGFR tyrosine kinase inhibitor tivozanib and everolimus resulted in stabilization of the disease in 50% of all patients with metastatic colon cancer.Citation292
Due to a broad overlap of apoptosis and autophagy signaling networks, BH3-mimetics induce both apoptosis and autophagy. For instance, in CRC cells, ABT-737 along with the PTGS2 inhibitor celecoxib synergistically induce cell death by modulating autophagy and apoptosis.Citation292 Cellular senescence acts as a physiological barrier to tumor development and many studies have proposed its exploitation as a potential therapeutic strategy in cancer. In HCT116 cells, addition of melatonin might inhibit overgrowth through regulation of cell death and senescence in a time-dependent manner. Within 18 h of melatonin treatment, HCT116 cells upregulate both pro-apoptotic BAX and anti-apoptotic BCL2L1/BCL−XL, thus, activating both the autophagic and apoptotic machinery.Citation293 Pathways affecting cell death in senescent cells include: (i) methylation of HIST2H2/histone H2 lysine9 by the enzyme SUV39H1, (ii) telomerase-based therapies involve the use of gene promoters of the various telomerases for gene-therapy “suicide” strategies, and telomerase-derived peptides, proteins, or RNA as vaccines for immunotherapy, (iii) telomerase inhibitor, GRN163L, a lipidated 13-mer oligonucleotide complementary to the RNA template region of human telomerase RNA.Citation294 We have summarized this section in .
Figure 16. Miscellaneous drugs targeting autophagy in in vitro and in vivo models for CRC therapy. All chemical compounds, drugs, and inhibitors have been introduced in the section “Other pharmacological autophagy inducers.”

TP53 status-dependent therapeutic strategies
Zebularine (ZEB), a cytidine deaminase inhibitor, inhibits CRC tumorigenesis via TP53-dependent ER stress. ZEB is very stable and preferentially targets cancer cells in human and mouse models. Microarray analysis revealed that ZEB causes the upregulation of ER stress-related genes as well as UPR genes and stabilizes TP53 through RPS7 (ribosomal protein S7-MDM2. ZEB also causes DNA damage and induces TP53-dependent apoptosis and autophagy. Colonospheres enriched in cancer stem cells derived from HCT116 ‘side populations’ expressed higher levels of HSPA5 and SQSTM1. Treatment with ZEB induced TP53 stabilization, EIF2A phosphorylation, and blocked SQSTM1 expression. Hence, ZEB downregulates pro-survival markers of ER stress and the UPR and these result in autophagy induction in tumor tissues of CRC patients, mice with azoxymethane- or dextran sodium sulfate-induced CRC, and HCT116-derived colonospheres.Citation284,Citation295 ZEB-mediated modulation of epigenetic signals converts ER stress-mediated prosurvival into a pro-apoptotic response.
TP53 has been linked to regulation of autophagy by affecting nutrient availability. The loss of TP53 drives LC3 overproduction and forces cells to maintain high autophagy rates. A study involving HCT116 cells showed that TP53 promotes selective downregulation of LC3 mRNA and protein under conditions of prolonged nutrient deprivation. Initially, autophagy occurs to protect the cells. However, after complete nutrient depletion, this process becomes stalled, causing the accumulation of aberrant metabolic intermediates which eventually leads to apoptotic cell death.Citation296 TP53 dysfunction commonly occurs in human cancers and contributes to disease progression and chemotherapy resistance. Using HCT116 (TP53−/−) and HT-29 (TP53WT) colon cancer cells, it was shown that 5-FU treatment causes aberrant autophagosome accumulation and augmented autophagy in HCT116 cells. This counteracted 5-FU toxicity but 5-FU resistance can be overcome by specific inhibition of autophagy by 3-MA, chloroquine, or siRNA-targeted knockdown of ATG5 and BECN1. MAPK/JNK activation and BCL2 phosphorylation are key events in 5-FU-induced autophagy. MAPK/JNK inhibition by siRNA or SP600125 suppressed autophagy by blocking the phosphorylation of JUN; it also blocked phosphorylation of BCL2, leading to increased 5-FU-induced apoptosis. Thus, targeting key proteins within the autophagy pathway in CRC patients harboring TP53-mutation may be a promising strategy to improve 5-FU efficacy.Citation232
Recent studies have demonstrated that a combination of nutlin (an MMD2 antagonist and inducer of E2F1 transcription) with conventional antitumor agents or irradiation may afford therapeutic benefit in cancers harboring mutant TP53. A large proportion of human cancers have defective or hyper-ubiquitinated TP53 and are partially resistant to nutlin due to MDM2 overexpression. However, even in cancer cells overexpressing MDM2, nutlin and CDK inhibitors (roscovitine and 5,6-dichloro-1-ribofuranosylbenzimidazole) still exert anti-proliferative activity by inhibition of TP53-MDM2 interaction.Citation297 This has been observed in a variety of cancers, including melanoma, colon carcinoma, breast adenocarcinoma, and hepatocellular-carcinoma.Citation297 Another approach will be the delivery of adenoviral vectors containing wild-type TP53 directly to tumors.Citation294
CRC with mutated TP53 is resistant to 5-FU. Ursolic acid (UA), a triterpenoid in fruits and herbs, has anticarcinogenic potential through inhibitory effects on the PI3K pathway in HCT15, an MSI mutant TP53 CRC cell line. UA also induces caspase-independent apoptosis in HCT15 cells, enhances 5-FU toxicity related to MAPK/JNK activation, promotes the induction of BECN1 expression, and enhances TP53 phosphorylation. UA induces autophagy in a MAPK/JNK-dependent manner. Experiments in xenografted nude mice showed that UA simultaneously decreased tumor growth while increasing expression of autophagy markers SQSTM1 and MAPK/JNK.Citation298 Saffron is a natural compound and toxic toward a TP53-nonmutated CRC. In TP53−/− tumors, saffron induces a prosurvival autophagic response.Citation127 Interestingly, curcumin (diferuloylmethane), the yellow pigment in Indian saffron, can sensitize tumors to different chemotherapeutic agents including doxorubicin, 5-FU, paclitaxel, vincristine, melphalan, butyrate, cisplatin, celecoxib, vinorelbine, gemcitabine, oxaliplatin, etoposide, sulfinosine, thalidomide, and bortezomib.Citation299 Overall, 5-FU may induce prosurvival autophagy that partly reverses its apoptosis-inducing effect. This may explain why the inhibition of autophagy by 3-MA or ATG7 siRNA significantly augments the induction of apoptosis by 5-FU.Citation300 We have summarized agents targeting TP53 in CRC in vivo and in vitro in .
Figure 17. TP53 status of CRC and the respective targeting strategies in in vitro and in vivo models for CRC therapy. All chemical compounds, drugs, and inhibitors have been introduced in the section “TP53 status-dependent therapeutic strategies.”
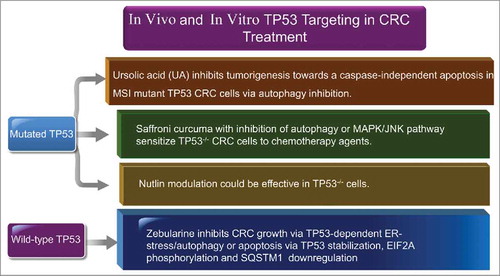
Proteasome inhibitor-based antitumor strategies
Epoxomicin, one of the earliest documented proteasome inhibitors, also activates the transcription of both BBC3/PUMA (a TP53 upregulated modulator of apoptosis) and BCL2L11/BIM in human colon cancer cells. Upregulation of BBC3 and BCL2L11 are typical features of ER stress-induced apoptosis. The combination of bortezomib and the death receptor ligand TNFS10, provoked a synergistic apoptotic response in prostate and colon cancer cell lines. In tumor-bearing mice, the mechanistic synergism between bortezomib and TNF involves CASP3 and CASP12 proteolytic activation, TP53 accumulation, increased MAPK9/SAPK-MAPK/JNK phosphorylation, and upregulation of HSPA5, and PDI. These events collectively contribute to the suppression of tumor growth.Citation301 We have schematically depicted proteasome inhibitors affecting CRC in vivo and in vitro in .
Figure 18. Proteasome activity modulators in in vitro and in vivo models for CRC therapy. All chemical compounds, drugs, and inhibitors have been introduced in the section “Proteasome inhibitor-based antitumor strategies.”

Targeting CRC via combinatorial therapy strategies focusing on autophagy and the UPR/ER stress pathway
The inhibition of autophagy is an attractive new therapeutic target in colon cancer. Targeting autophagy leads to increased cell death in HCT116 cancer cells.Citation272,Citation273 Inhibition of autophagy in combination with modern anticancer therapies are being tested in colon cancer.Citation50 A list of autophagy inhibitors in combination with chemotherapeutic agents for the treatment of CRC is shown in . Autophagy inhibition is an effective way to promote the anticancer activity of agents such as sulforaphane (SUL) and fluorouracil (5-FU).Citation300,Citation302 Trifluorothymidine (TFT) is a more potent inducer of cell death than 5-FU because it induces higher levels of cell death without autophagic survival responses in colon cancer. Thus, TFT is an attractive candidate for new treatment strategy in CRC.Citation303 Many studies support the combinatorial use of chloroquine as a novel therapeutic agent to improve the efficacy of 5-FU to inhibit autophagy-dependent resistance to chemotherapy. HT-29 cells activate autophagy as a defense mechanism against 5-FU. Hence, chloroquine-induced inhibition of autophagy may potentiate the anticancer effect of 5-FU.Citation300,Citation304
Table 6. Autophagy inhibition during cancer chemotherapy.
Autophagy inhibition by chloroquine, which prevents the fusion of autophagosomes and lysosomes sensitizes HT-29 CRC cells to chemotherapy and irradiation. Presurgical treatment with hydroxychloroquine improves the treatment response to 5-FU and irradiation in patients with advanced CRC.Citation305 Resistance to oxaliplatin has been shown in Caco-2 cells. Agents such as 3-MA, bafilomycin A1, or RNAi knockdown of essential autophagy genes, such as ATG5 or BECN1, enhanced cell death and ROS production in Caco-2 treated with oxaliplatin. Hence, increasing ROS production via inhibiting autophagy may be a therapeutic strategy for the sensitization of cells to oxaliplatin in the management of CRC.Citation300,Citation306 Furthermore, bufalin enhances colon cancer sensitivity through ROS-mediated autophagy.Citation307 Common therapeutic agents may promote autophagy in cancer cells, while disruption of autophagy alone does not necessarily enhance cell death. Blocking ROS production by scavengers such as NAC or Tiron decreased autophagy in tumor cells. However, blocking ER stress by RNAi targeting NUPR1/COM1/p8 (nuclear protein 1, transcriptional regulator) and DDIT3 decreased autophagy and ROS production. Hence, ER stress is upstream of autophagy and ROS generation. A combination therapy that causes a subsequent increase in ROS production is more efficient.Citation304
Other treatments exploit the antineoplastic activation of apoptosis and autophagy using iron core-gold shell nanoparticles,Citation308 and anti-VEGF therapy.Citation291,Citation309,Citation310 When used in combination with drugs like cisplatin, doxorubicin, docetaxel, or 5-FU, Solanum nigrum leaves, a common component in traditional Chinese medicine for the treatment of cancer; treatment efficacy in colorectal DLD-1 and HT-29 cells improves by inducing autophagy and enhancing cytotoxicity. Since DLD-1 and HT-29 cell lines have TP53 mutations and are resistant to the TP53-mediated apoptosis, cell death might be induced in a CASP3-independent manner with the activation of autophagy. The inclusion of Solanum nigrum leaves in chemotherapeutic therapies has been suggested to improve the efficacy of CRC treatment.Citation311 Hence, rather than autophagy inhibition, activation of the autophagy pathway that leads to cell death is able to improve CRC treatment. In HCT116 colon cells, combined use of oxaliplatin and bortezomib resulted in increased caspase activation and subsequent induction of apoptosis. This treatment modulated the synergistic effect through the mitochondria-dependent apoptotic pathway by promoting the MAPK/JNK-BCL2L1-BAX pathway. BCL2L1 affects autophagy through alteration of its interaction with BECN1. BECN1 dissociates from BCL2L1 and initiates autophagy during combined oxaliplatin- bortezomib treatment, which was shown to significantly inhibit tumor growth in CRC xenografts.Citation312 A better understanding of the crosstalk between apoptosis and autophagy may lead to new and improved treatment options for CRC.Citation312 Furthermore, tumor recurrence was significantly delayed with chloroquine cotreatment, and chloroquine enhanced TP53-mediated apoptosis via inhibition of autophagy. Chloroquine and its derivatives have been used in clinical trials to evaluate its use as a sensitizing agent for tumor, which would otherwise be unresponsive to standard chemotherapy.Citation50
The MTOR inhibitor temsirolimus and chloroquine, with an autophagy-inhibitor function, exhibit a potent cooperative antitumor effect against CRC cells. Temsirolimus was effective in inhibiting tumor growth in CaR-1 and HT-29 cells, possibly through the induction of G1 cell cycle arrest and a reduction in HIF1A and VEGF levels. Chloroquine significantly potentiated this antitumor activity. The combined therapy with temsirolimus and chloroquine enhanced the level of apoptosis and increased the BAX:BCL2 ratio.Citation313 Temsirolimus and chloroquine are already in clinical use as anticancer and antimalarial drugs, respectively, and represent a new option in the treatment of CRC.
Photodynamic therapy (PDT) combined with bortezomib could be a potential therapeutic strategy in CRC. Bortezomib and other proteasome inhibitors effectively sensitize cells to other therapeutics and enhance their cytotoxicity. PDT can lead to the accumulation of carbonylated proteins, normally degraded by proteasomes in the ER. This leads to ER stress due to oxidative damage of cellular macromolecules, resulting in cytotoxicity toward tumor cells.Citation314 It has been recently reported that Protoporphyrin IX-mediated PDT induced autophagy in colorectal CSC. The inhibition of PDT-induced autophagy by genetic and pharmacological means induced apoptosis in colorectal CSC, decreased their clonogenic potential and tumorigenicity in vitro and in vivo, respectively.Citation315 These findings suggest that targeting autophagy increases the PDT sensitivity of CSC, and thus can aid in designing new therapeutic approaches for targeting this population of cancerous cells that show high resistance to current therapies.
UPR induction can cause tumor cell sensitization to cisplatin-induced death. The mechanism of action of cisplatin is thought to involve DNA binding and interference with DNA-repair processes. In addition, a correlation between the UPR and sensitization to other DNA-crosslinking agents, such as carboplatin, melphalan, and BCNU, has been observed.Citation269 It seems that manipulation of other signaling pathways rather than autophagy such as MTOR or using proteasome inhibitors, can increase cytotoxicity in tumor cells through inducing apoptosis or ER stress.
Conclusions and future direction
When searching for new CRC therapeutic strategies aimed at manipulating autophagy, particular attention should be paid to the specific type, stage, and metabolic characteristics of CRC. During the course of CRC, autophagy could either promote tumor survival or cause cancer cell death, depending on the tumor type, CRC stage and the metabolic context. In human CRC cells, autophagy is activated in response to high-energy demands in the initial stage of cell transformation or as an adaptive tumor cell response at later stages. Combinatorial therapeutic approaches have great potential in the treatment of colorectal tumors. Hence a better understanding of the molecular mechanisms of crosstalk between apoptosis and autophagy will be the key in identifying novel applications of combinatorial treatment to CRC. Beside important diet modifications, chemopreventive measures, such as, for example, administration of low-dose aspirin, especially in combination with metformin, and/or intake of curcumin and statins lowers cancer incidence.Citation230,Citation316 It is unlikely that any type of cancer, including CRC could easily be defeated by applying a singular approach, therefore combined effort, using the plethora of available interventions, is the most likely path to successful CRC-prevention and treatment. Novel experimental therapies that utilize natural and modified biologics inspired by derivates from the animal kingdom, and from plants and viruses, are a rich source of potential anticancer therapeutics.Citation317-Citation319
Abbreviations
| 3-AP-Me | = | NCitation4,N4-dimethyl-triapine |
| 5-Aza-Cd | = | 5-aza-2′-deoxycytidine |
| 3-MA | = | 3-methyladenine |
| 5-FU | = | 5-fluorouracil |
| 6TG | = | 6-thioguanine |
| AGER/RAGE | = | advanced glycosylation end product specific receptor |
| AICAR | = | 5-aminoimidazole-4-carboxyamide ribonucleoside |
| AKR1B1 | = | aldo-keto reductase family 1 member B1 |
| AMBRA1 | = | autophagy and Beclin 1 regulator 1 |
| AMPK | = | 5′-adenosine monophosphate-activated protein kinase |
| AP-1 | = | AP-1 transcription factor |
| AKR | = | aldo-keto reductase |
| ATF6 | = | activating transcription factor 6 |
| BBC3/PUMA | = | BCL2 binding component 3 |
| BCL2, BCL2 | = | apoptosis regulator |
| BCL2L1 | = | BCL2 like 1 |
| BECN1 | = | Beclin 1 |
| BFA | = | brefeldin A |
| BNIP3 | = | BCL2 interacting protein 3 |
| CQ | = | chloroquine |
| CRC | = | colorectal cancer |
| CSC | = | cancer stem cell |
| DC | = | dendritic cell |
| DDIT3/ CHOP | = | DNA damage inducible transcript 3 |
| DHA | = | docosahexaenoic acid |
| DPE | = | 2–3,4-dihydroxyphenylethanol |
| EIF2AK3/PERK | = | eukaryotic translation initiation factor 2 α kinase 3 |
| EMT | = | epithelial-to-mesenchymal transition |
| ERAD | = | ER-associated degradation |
| ERN1/IRE1a | = | endoplasmic reticulum to nucleus signaling 1 |
| FASN | = | fatty acid synthase |
| GDF15/MIC1 | = | growth/differentiation factor 15 |
| HDACi | = | histone deacetylase inhibitor |
| HMGB1 | = | high mobility group box 1 |
| HPGD/15PGDH | = | hydroxyprostaglandin dehydrogenase 15-(NAD) |
| MLH1 | = | mutL homolog 1 |
| HPD | = | high-protein diet |
| HSP | = | heat shock protein |
| HSPA5 | = | heat shock protein family A (Hsp70) member 5 |
| IBD | = | inflammatory bowel disease |
| IDO1 | = | indoleamine 2,3-dioxygenase 1 |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MetS | = | metabolic syndrome |
| MMR | = | mismatch repair protein |
| MSI | = | microsatellite instability |
| MTOR | = | mechanistic target of rapamycin |
| NFKB/NF□B | = | nuclear factor kappa B |
| NOS2/iNOS | = | nitric oxide synthase 2 |
| OS | = | overall survival |
| OR | = | odds ratios |
| PCD | = | programmed cell death |
| PCDH17 | = | protocadherin 17 |
| PDI | = | protein disulfide isomerase |
| PDT | = | photodynamic therapy |
| PE | = | phosphatidylethanolamine |
| PG | = | prostaglandin |
| PGE2 | = | prostaglandin E2 |
| PLA2G10 | = | phospholipase A2 group X |
| PLAUR/uPAR | = | plasminogen activator, urokinase receptor |
| PPP1R15A/GADD34 | = | protein phosphatase 1 regulatory subunit 15A |
| PTGER3 | = | prostaglandin E receptor 3 |
| PTGER4/EP4 | = | prostaglandin E receptor 4 |
| PTGFR | = | prostaglandin F receptor |
| PTGS2/COX2 | = | prostaglandin-endoperoxide synthase 2 |
| RIP | = | ribosome-inactivating protein |
| ROS | = | reactive oxygen species |
| SAT | = | subcutaneous adipose tissue |
| SEL1L | = | SEL1L ERAD E3 ligase adaptor subunit |
| SH3GLB1/BIF-1 | = | SH3 domain containing GRB2 like, endophilin B1 |
| SIRT1 | = | sirtuin 1 |
| SNP | = | single nucleotide polymorphism |
| STAT3 | = | signal transducer and activator of transcription 3 |
| STK11/LKB1 | = | serine/threonine kinase 11 |
| SUL | = | sulforaphane |
| TAA | = | tumor-associated antigen |
| TFA | = | tolfenamic acid |
| TNF/TNFα | = | tumor necrosis factor |
| TNFSF10/TRAIL | = | tumor necrosis factor superfamily member 10 |
| TP53 | = | tumor protein p53 |
| Treg | = | regulatory T cells |
| UPR | = | unfolded protein response |
| UVRAG | = | UV radiation resistance associated |
| VAT | = | visceral adipose tissue |
| VEGF | = | vascular endothelial growth factor |
| VMP1 | = | vacuole membrane protein 1 |
| XBP1 | = | X-box binding protein 1. |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
SG acknowledges Manitoba Medical Service Foundation, and University of Manitoba Research Grant Program. AM acknowledges National Institute for Genetic Engineering and Biotechnology Research Grants. TK extends his gratitude to the Natural Sciences and Engineering Council of Canada (NSERC) for funding. MA was supported by MITACS Globalink award.
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90; PMID:21296855; http://dx.doi.org/10.3322/caac.20107
- Canada CCSNCIo. Canadian Cancer Statistics 2008. Toronto: Canadian Cancer Society, 2008
- Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin 2009; 59:366-78; PMID:19897840; http://dx.doi.org/10.3322/caac.20038
- Saudi Moh. Health awareness center: “Cancer incidence in the Kingdom, Compared to the global incidence, Is still low. Saudi Arabia: Ministry of health, 2013
- Dolatkhah R, Somi MH, Bonyadi MJ, Asvadi Kermani I, Farassati F, Dastgiri S. Colorectal cancer in iran: Molecular epidemiology and screening strategies. J Cancer Epidemiol 2015; 2015:643020; PMID:25685149; http://dx.doi.org/10.1155/2015/643020
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55:74-108; PMID:15761078; http://dx.doi.org/10.3322/canjclin.55.2.74
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur J Cancer 2013; 49:1374-403; PMID:23485231; http://dx.doi.org/10.1016/j.ejca.2012.12.027
- Vasen HF, Moslein G, Alonso A, Aretz S, Bernstein I, Bertario L, Blanco I, Bülow S, Burn J, Capella G, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57:704-13; PMID:18194984; http://dx.doi.org/10.1136/gut.2007.136127
- Burt RW. Colon cancer screening. Gastroenterology 2000; 119:837-53; PMID:10982778; http://dx.doi.org/10.1053/gast.2000.16508
- Rustgi AK. The genetics of hereditary colon cancer. Genes Dev 2007; 21:2525-38; PMID:17938238; http://dx.doi.org/10.1101/gad.1593107
- Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology 2010; 138:2044-58; PMID:20420945; http://dx.doi.org/10.1053/j.gastro.2010.01.054
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med 2003; 348:919-32; PMID:12621137; http://dx.doi.org/10.1056/NEJMra012242
- Knudsen AL, Bisgaard ML, Bulow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam Cancer 2003; 2:43-55; PMID:14574166; http://dx.doi.org/10.1023/A:1023286520725
- Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomäki P, Mecklin JP, Järvinen HJ. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999; 81:214-8; PMID:10188721; http://dx.doi.org/10.1002/(SICI)1097-0215(19990412)81:2%3c214::AID-IJC8%3e3.0.CO;2-L
- Broaddus RR, Lynch PM, Lu KH, Luthra R, Michelson SJ. Unusual tumors associated with the hereditary nonpolyposis colorectal cancer syndrome. Mod Pathol 2004; 17:981-9; PMID:15143336; http://dx.doi.org/10.1038/modpathol.3800150
- Mokarram P, Asghari Estiar M, Ashktorab H. Epigenetics Territory and Cancer (methylation in colorectal cancer). New York, NY: Springer; 2015; 373–455
- Cherry LM. The genetic etiology of familial and nonfamilial colorectal cancer. Proc (Bayl Univ Med Cent) 2011; 24:139-41; PMID:21566761
- Carethers JM, Jung BH. Genetics and genetic biomarkers in sporadic colorectal cancer. Gastroenterology 2015; 149:1177-90 e3; PMID:26216840; http://dx.doi.org/10.1053/j.gastro.2015.06.047
- Yusof AS, Isa ZM, Shah SA. Dietary patterns and risk of colorectal cancer: A systematic review of cohort studies (2000–2011). Asian Pac J Cancer Prev 2012; 13:4713-7; PMID:23167408; http://dx.doi.org/10.7314/APJCP.2012.13.9.4713
- Vanio H, Bianchini F. IARC handbooks of cancer prevention. Volume 6: weight control and physical activity. Lyon, France: International Agency for Research on Cancer 2002
- Schmid D, Leitzmann MF. Cardiorespiratory fitness as predictor of cancer mortality: A systematic review and meta-analysis. Ann Oncol 2015; 26:272-8; PMID:25009011; http://dx.doi.org/10.1093/annonc/mdu250
- Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010; 376:1741-50; PMID:20970847; http://dx.doi.org/10.1016/S0140-6736(10)61543-7
- Tarraga Lopez PJ, Albero JS, Rodriguez-Montes JA. Primary and secondary prevention of colorectal cancer. Clin Med Insights Gastroenterol 2014; 7:33-46; PMID:25093007; http://dx.doi.org/10.4137/CGast.S14039
- Tsoi KK, Pau CY, Wu WK, Chan FK, Griffiths S, Sung JJ. Cigarette smoking and the risk of colorectal cancer: A meta-analysis of prospective cohort studies. Clin Gastroenterol Hepatol 2009; 7:682-8. e1–5; PMID:19245853; http://dx.doi.org/10.1016/j.cgh.2009.02.016
- Rhodes JM, Campbell BJ. Inflammation and colorectal cancer: IBD-associated and sporadic cancer compared. Trend Mol Med 2002; 8:10-6; PMID:11796261; http://dx.doi.org/10.1016/S1471-4914(01)02194-3
- Mattar MC, Lough D, Pishvaian MJ, Charabaty A. Current management of inflammatory bowel disease and colorectal cancer. Gastrointest Cancer Res 2011; 4:53-61; PMID:21673876
- Farraye FA, Odze RD, Eaden J, Itzkowitz SH. AGA technical review on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology 2010; 138:746-74, 74.e1-4; quiz e12-3; PMID:20141809; http://dx.doi.org/10.1053/j.gastro.2009.12.035
- Anderson JC. Risk factors and diagnosis of flat adenomas of the colon. Expert Rev Gastroenterol Hepatol 2011; 5:25-32; PMID:21309669; http://dx.doi.org/10.1586/egh.10.86
- Whitlock E, Lin J, Liles E, Beil T, Fu R, O'Connor E, Thompson RN, Cardenas T. Screening for colorectal cancer: An updated systematic review. Evidence Syntheses, N0.65.1. Agency for Healthcare Research and Quality (US); 2008 Oct. Report No.: 08-05-05124-EF-1
- Brenner H, Chang-Claude J, Seiler CM, Rickert A, Hoffmeister M. Protection from colorectal cancer after colonoscopy: A population-based, case-control study. Ann Intern Med 2011; 154:22-30; PMID:21200035; http://dx.doi.org/10.7326/0003-4819-154-1-201101040-00004
- Hardcastle JD, Chamberlain JO, Robinson MH, Moss SM, Amar SS, Balfour TW, James PD, Mangham CM. Randomised controlled trial of faecal-occult-blood screening for colorectal cancer. Lancet 1996; 348:1472-7; PMID:8942775; http://dx.doi.org/10.1016/S0140-6736(96)03386-7
- Mandel JS, Church TR, Bond JH, Ederer F, Geisser MS, Mongin SJ, Snover DC, Schuman LM. The effect of fecal occult-blood screening on the incidence of colorectal cancer. N Eng J Med 2000; 343:1603-7;PMID:11096167;http://dx.doi.org/10.1056/NEJM200011303432203
- Wu CW, Sung JJ. Colorectal cancer screening: Are stool and blood based tests good enough?. Chin Clin Oncol 2013; 2:2304-3865
- Kim JY, Hwang JH, Cha MR, Yoon MY, Son ES, Tomida A, Ko B, Song SW, Shin-ya K, Hwang YI, et al. Arctigenin blocks the unfolded protein response and shows therapeutic antitumor activity. J Cell Physiol 2010; 224:33-40; PMID:20232300
- Labianca R, Nordlinger B, Beretta GD, Brouquet A, Cervantes A. Primary colon cancer: ESMO Clinical practice guidelines for diagnosis, adjuvant treatment and follow-up. Annal Oncol 2010; 21 Suppl 5:70-7; http://dx.doi.org/10.1093/annonc/mdq168
- Sauer R, Liersch T, Merkel S, Fietkau R, Hohenberger W, Hess C, Becker H, Raab HR, Villanueva MT, Witzigmann H, et al. Preoperative versus postoperative chemoradiotherapy for locally advanced rectal cancer: Results of the German CAO/ARO/AIO-94 randomized phase III trial after a median follow-up of 11 years. J Clin Oncol 2012; 30:1926-33; PMID:22529255; http://dx.doi.org/10.1200/JCO.2011.40.1836
- Sebag-Montefiore D, Stephens RJ, Steele R, Monson J, Grieve R, Khanna S, Quirke P, Couture J, de Metz C, Myint AS, et al. Preoperative radiotherapy versus selective postoperative chemoradiotherapy in patients with rectal cancer (MRC CR07 and NCIC-CTG C016): A multicentre, randomised trial. Lancet 2009; 373:811-20; PMID:19269519; http://dx.doi.org/10.1016/S0140-6736(09)60484-0
- Bosset JF, Collette L, Calais G, Mineur L, Maingon P, Radosevic-Jelic L, Daban A, Bardet E, Beny A, Ollier JC, et al. Chemotherapy with preoperative radiotherapy in rectal cancer. N Engl J Med 2006; 355:1114-23;PMID:16971718;http://dx.doi.org/10.1056/NEJMoa060829
- Haller DG, Catalano PJ, Macdonald JS, O'Rourke MA, Frontiera MS, Jackson DV, Mayer RJ. Phase III study of fluorouracil, leucovorin, and levamisole in high-risk stage II and III colon cancer: Final report of Intergroup 0089. J Clin Oncol 2005; 23:8671-8; PMID:16314627; http://dx.doi.org/10.1200/JCO.2004.00.5686
- Gustavsson B, Carlsson G, Machover D, Petrelli N, Roth A, Schmoll HJ, Tveit KM, Gibson F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin Colorectal Cancer 2015; 14:1-10; PMID:25579803; http://dx.doi.org/10.1016/j.clcc.2014.11.002
- Andre T, Iveson T, Labianca R, Meyerhardt JA, Souglakos I, Yoshino T, Paul J, Sobrero A, Taieb J, Shields AF, et al. The IDEA (International duration evaluation of adjuvant chemotherapy) collaboration: Prospective combined analysis of phase III trials investigating duration of adjuvant therapy with the FOLFOX (FOLFOX4 or Modified FOLFOX6) or XELOX (3 versus 6 months) regimen for patients with stage III colon cancer: Trial design and current status. Curr Colorectal Cancer Rep 2013; 9:261-9; PMID:24032000; http://dx.doi.org/10.1007/s11888-013-0181-6
- Sargent D, Sobrero A, Grothey A, O'Connell MJ, Buyse M, Andre T, Zheng Y, Green E, Labianca R, O'Callaghan C, et al. Evidence for cure by adjuvant therapy in colon cancer: Observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol 2009; 27:872-7; PMID:19124803; http://dx.doi.org/10.1200/JCO.2008.19.5362
- Petrelli N, Douglass HO, Jr., Herrera L, Russell D, Stablein DM, Bruckner HW, Mayer RJ, Schinella R, Green MD, Muggia FM, et al. The modulation of fluorouracil with leucovorin in metastatic colorectal carcinoma: A prospective randomized phase III trial. Gastrointestinal tumor study group. J Clin Oncol 1989; 7:1419-26; PMID:2674331
- Caraglia M, Marra M, Budillon A, Meo G, Ricciardiello F, Bismuto E, Brachelente G, Francini G, Giordano A, Correale P, et al. Chemotherapy regimen GOLF induces apoptosis in colon cancer cells through multi-chaperone complex inactivation and increased Raf-1 ubiquitin-dependent degradation. Cancer Biol Ther 2005; 4:1159-67; PMID:16294035; http://dx.doi.org/10.4161/cbt.4.10.2206
- Tournigand C, Andre T, Achille E, Lledo G, Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: A randomized GERCOR study. J Clin Oncol 2004; 22:229-37; PMID:14657227; http://dx.doi.org/10.1200/JCO.2004.05.113
- Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013; 381:303-12; PMID:23177514; http://dx.doi.org/10.1016/S0140-6736(12)61900-X
- Liu EY, Ryan KM. Autophagy and cancer–issues we need to digest. J Cell Sci 2012; 125:2349-58; PMID:22641689; http://dx.doi.org/10.1242/jcs.093708
- Mah LY, Ryan KM. Autophagy and cancer. Cold Spring Harb Perspect Biol 2012; 4:a008821; PMID:22166310; http://dx.doi.org/10.1101/cshperspect.a008821
- Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Zhang Q, et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis 2013; 4:e838; PMID:24113172; http://dx.doi.org/10.1038/cddis.2013.350
- Chen N, Debnath J. Autophagy and tumorigenesis. FEBS Lett 2010; 584:1427-35; PMID:20035753; http://dx.doi.org/10.1016/j.febslet.2009.12.034
- Roy S, Debnath J. Autophagy and tumorigenesis. Semin Immunopathol 2010; 32:383-96; PMID:20589500; http://dx.doi.org/10.1007/s00281-010-0213-0
- Tsai WT, Lo YC, Wu MS, Li CY, Kuo YP, Lai YH, Tsai Y, Chen KC, Chuang TH, Yao CH, et al. Mycotoxin patulin suppresses innate immune responses by mitochondrial dysfunction and p62/sequestosome-1-dependent mitophagy. J Biol Chem 2016; 291(37):19299-311; PMID:27458013; http://dx.doi.org/10.1074/jbc.M115.686683
- Stranks AJ, Hansen AL, Panse I, Mortensen M, Ferguson DJ, Puleston DJ, Shenderov K, Watson AS, Veldhoen M, Phadwal K, et al. Autophagy controls acquisition of aging features in macrophages. J Innate Immun 2015; 7:375-91; PMID:25764971; http://dx.doi.org/10.1159/000370112
- Kim YH, Baek SH, Kim EK, Ha JM, Jin SY, Lee HS, Ha HK, Song SH, Kim SJ, Shin HK, et al. Uncoordinated 51-like kinase 2 signaling pathway regulates epithelial-mesenchymal transition in A549 lung cancer cells. FEBS Lett 2016; 590:1365-74; PMID:27062295; http://dx.doi.org/10.1002/1873-3468.12172
- Zhou Y, Rucker EB, 3rd, Zhou BP. Autophagy regulation in the development and treatment of breast cancer. Acta Biochim Biophys Sin (Shanghai) 2016; 48:60-74; PMID:26637829; http://dx.doi.org/10.1093/abbs/gmw063
- Zarzynska JM. The importance of autophagy regulation in breast cancer development and treatment. Biomed Res Int 2014; 2014:710345; PMID:25317422; http://dx.doi.org/10.1155/2014/710345
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal paneth cells. Nature 2008; 456:259-63; PMID:18849966; http://dx.doi.org/10.1038/nature07416
- Lopez P, Alonso-Perez E, Rodriguez-Carrio J, Suarez A. Influence of Atg5 mutation in SLE depends on functional IL-10 genotype. PloS one 2013; 8:e78756; PMID:24205307; http://dx.doi.org/10.1371/journal.pone.0078756
- Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity 2013; 39:211-27; PMID:23973220; http://dx.doi.org/10.1016/j.immuni.2013.07.017
- Madia F, Grossi V, Peserico A, Simone C. Updates from the intestinal front line: Autophagic weapons against inflammation and cancer. Cells 2012; 1:535-57; PMID:24710489; http://dx.doi.org/10.3390/cells1030535
- Liton PB. The autophagic lysosomal system in outflow pathway physiology and pathophysiology. Exp Eye Res 2016; 144:29-37; http://dx.doi.org/10.1016/j.exer.2015.07.013
- Wen X, Klionsky DJ. Autophagy is a key factor in maintaining the regenerative capacity of muscle stem cells by promoting quiescence and preventing senescence. Autophagy 2016; 12:617-8; PMID:27050452; http://dx.doi.org/10.1080/15548627.2016.1158373
- Wasik AM, Grabarek J, Pantovic A, Cieslar-Pobuda A, Asgari HR, Bundgaard-Nielsen C, Rafat M, Dixon IM, Ghavami S, Łos MJ. Reprogramming and carcinogenesis–parallels and distinctions. Int Rev Cell Mol Biol 2014; 308:167-203; PMID:24411172
- Iranpour M, Moghadam AR, Yazdi M, Ande SR, Alizadeh J, Wiechec E, Lindsay R, Drebot M, Coombs KM, Ghavami S. Apoptosis, autophagy and unfolded protein response pathways in Arbovirus replication and pathogenesis. Expert Rev Mol Med 2016; 18:e1; PMID:26781343; http://dx.doi.org/10.1017/erm.2015.19
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 2014; 112:24-49; PMID:24211851; http://dx.doi.org/10.1016/j.pneurobio.2013.10.004
- Yeganeh B, Ghavami S, Kroeker AL, Mahood TH, Stelmack GL, Klonisch T, Coombs KM, Halayko AJ. Suppression of influenza A virus replication in human lung epithelial cells by noncytotoxic concentrations bafilomycin A1. Am J Physiol Lung Cell Mol Physiol 2015; 308:L270-86; PMID:25361566; http://dx.doi.org/10.1152/ajplung.00011.2014
- Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene 2013; 32:2261-72, 72e 1–11; PMID:22733132; http://dx.doi.org/10.1038/onc.2012.252
- Zeki AA, Yeganeh B, Kenyon NJ, Post M, Ghavami S. Autophagy in airway diseases: a new frontier in human asthma?. Allergy 2016; 71:5-14; PMID:26335713; http://dx.doi.org/10.1111/all.12761
- Sou YS, Tanida I, Komatsu M, Ueno T, Kominami E. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J Biol Chem 2006; 281:3017-24; PMID:16303767; http://dx.doi.org/10.1074/jbc.M505888200
- Le Grand JN, Chakrama FZ, Seguin-Py S, Fraichard A, Delage-Mourroux R, Jouvenot M, Boyer-Guittaut M. GABARAPL1 (GEC1): Original or copycat?. Autophagy 2011; 7:1098-107; PMID:21597319; http://dx.doi.org/10.4161/auto.7.10.15904
- Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem 2004; 279:36268-76; PMID:15187094; http://dx.doi.org/10.1074/jbc.M401461200
- Yeganeh B, Moghadam AR, Tran AT, Rahim MN, Ande SR, Hashemi M, Coombs KM, Ghavami S. Asthma and influenza virus infection: Focusing on cell death and stress pathways in influenza virus replication. Iran J Allergy Asthma Immunol 2013; 12:1; PMID:23454774
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519-29; PMID:17565364; http://dx.doi.org/10.1038/nrm2199
- Iranpour M, Moghadam AR, Yazdi M, Ande SR, Alizadeh J, Wiechec E, Lindsay R, Drebot M, Coombs KM, Ghavami S. Apoptosis, autophagy and unfolded protein response pathways in Arbovirus replication and pathogenesis. Expert Rev Mol Med 2016; 18:e1; PMID:26781343; http://dx.doi.org/10.1017/erm.2015.19
- Cao SS, Kaufman RJ. Unfolded protein response. Curr Biol 2012; 22:R622-R6;PMID:22917505;http://dx.doi.org/10.1016/j.cub.2012.07.004
- Chevet E, Hetz C, Samali A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov 2015; 5:586-97; PMID:25977222; http://dx.doi.org/10.1158/2159-8290.CD-14-1490
- Walter P, Ron D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011; 334:1081-6; PMID:22116877; http://dx.doi.org/10.1126/science.1209038
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318:944-9; PMID:17991856; http://dx.doi.org/10.1126/science.1146361
- Forman MS, Lee VM, Trojanowski JQ. Unfolding'pathways in neurodegenerative disease. Trends Neurosci 2003; 26:407-10; PMID:12900170; http://dx.doi.org/10.1016/S0166-2236(03)00197-8
- Hetz C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nature Rev Mol Cell Biol 2012; 13:89-102
- Namba T, Ishihara T, Tanaka KI, Hoshino T, Mizushima T. Transcriptional activation of ATF6 by endoplasmic reticulum stressors. Biochem Biophys Res Commun 2007; 355:543-8; PMID:17307147; http://dx.doi.org/10.1016/j.bbrc.2007.02.004
- Sovolyova N, Healy S, Samali A, Logue SE. Stressed to death–mechanisms of ER stress-induced cell death. Biol Chem 2014; 395:1-13; PMID:24002662; http://dx.doi.org/10.1515/hsz-2013-0174
- Kaufman RJ. Regulation of mRNA translation by protein folding in the endoplasmic reticulum. Trends Biochem Sci 2004; 29:152-8; PMID:15003273; http://dx.doi.org/10.1016/j.tibs.2004.01.004
- Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nature Rev Drug Discov 2013; 12:703-19; http://dx.doi.org/10.1038/nrd3976
- Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci 2013; 70:2425-41; PMID:23052213; http://dx.doi.org/10.1007/s00018-012-1173-4
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 2003; 23:7198-209; PMID:14517290; http://dx.doi.org/10.1128/MCB.23.20.7198-7209.2003
- Sato M, Seki T, Konno A, Hirai H, Kurauchi Y, Hisatsune A, Katsuki H. Fluorescent-based evaluation of chaperone-mediated autophagy and microautophagy activities in cultured cells. Genes Cells 2016; 21:861-73; PMID:27377049; http://dx.doi.org/10.1111/gtc.12390
- Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, et al. Corrigendum: Chaperone-mediated autophagy degrades mutant p53. Genes Dev 2016; 30:870; PMID:27036968; http://dx.doi.org/10.1101/gad.280453.116
- Kaushik S, Massey AC, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 2008; 19:2179-92; PMID:18337468; http://dx.doi.org/10.1091/mbc.E07-11-1155
- Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev 2013; 27:1718-30; PMID:23913924; http://dx.doi.org/10.1101/gad.220897.113
- Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong J, Zhang S, Shang Y, Jiang X, Guo H, et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 2016; 12:515-28;PMID:26761524;http://dx.doi.org/10.1080/15548627.2015.1136770
- Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med 2011; 3:109ra17-ra17; http://dx.doi.org/10.1126/scitranslmed.3003182
- Eskelinen EL, Cuervo AM, Taylor MR, Nishino I, Blum JS, Dice JF, Sandoval IV, Lippincott-Schwartz J, August JT, Saftig P. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic 2005; 6:1058-61; PMID:16190986; http://dx.doi.org/10.1111/j.1600-0854.2005.00337.x
- Okamoto K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J Cell Biol 2014; 205:435-45; PMID:24862571; http://dx.doi.org/10.1083/jcb.201402054
- Jangamreddy JR, Los MJ. Mitoptosis, a novel mitochondrial death mechanism leading predominantly to activation of autophagy. Hepat Mon 2012; 12:e6159; PMID:23087751; http://dx.doi.org/10.5812/hepatmon.6159
- Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Tan J, Tan Y, Han H, Tian R, et al. BNIP3 Suppresses PINK1 Proteolytic cleavage to promote mitophagy. J Biol Chem 2016; 291(41):21616-629; PMID:27528605; http://dx.doi.org/10.1074/jbc.M116.733410
- McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 2016; 214:333-45; PMID:27458135; http://dx.doi.org/10.1083/jcb.201603039
- Bondanese VP, Lamboux A, Simon M, Lafont JE, Albalat E, Pichat S, Vanacker JM, Telouk P, Balter V, Oger P, et al. Hypoxia induces copper stable isotope fractionation in hepatocellular carcinoma, in a HIF-independent manner. Metallomics 2016; 8(11):1177-84; PMID:27500357; http://dx.doi.org/10.1039/C6MT00102E
- Ko YS, Cho SJ, Park J, Choi Y, Lee JS, Youn HD, Kim WH, Kim MA, Park JW, Lee BL. Hypoxic inactivation of glycogen synthase kinase-3beta promotes gastric tumor growth and angiogenesis by facilitating hypoxia-inducible factor-1 signaling. APMIS 2016; 124(9):748-56; PMID:27365055; http://dx.doi.org/10.1111/apm.12569
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011; 22:165-78; PMID:21119005; http://dx.doi.org/10.1091/mbc.E10-06-0500
- Kim JH, Kim HY, Lee YK, Yoon YS, Xu WG, Yoon JK, Choi SE, Ko YG, Kim MJ, Lee SJ, et al. Involvement of mitophagy in oncogenic K-Ras-induced transformation: Overcoming a cellular energy deficit from glucose deficiency. Autophagy 2011; 7:1187-98; PMID:21738012; http://dx.doi.org/10.4161/auto.7.10.16643
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259-69; PMID:19450525; http://dx.doi.org/10.1016/j.molcel.2009.04.026
- Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res 2010; 70:3431-4; PMID:20424122; http://dx.doi.org/10.1158/0008-5472.CAN-09-4027
- Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, Fu W, Zhang J, Wu W, Zhang X, et al. Autophagy negatively regulates Wnt signalling by promoting dishevelled degradation. Nature cell biology 2010; 12:781-90; PMID:20639871; http://dx.doi.org/10.1038/ncb2082
- Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: Therapeutic implications. Mol Cancer Ther 2011; 10:1533-41; PMID:21878654; http://dx.doi.org/10.1158/1535-7163.MCT-11-0047
- Ghavami S, Mutawe MM, Hauff K, Stelmack GL, Schaafsma D, Sharma P, McNeill KD, Hynes TS, Kung SK, Unruh H, et al. Statin-triggered cell death in primary human lung mesenchymal cells involves p53-PUMA and release of Smac and Omi but not cytochrome c. Biochim Biophys Acta 2010; 1803:452-67; PMID:20045437; http://dx.doi.org/10.1016/j.bbamcr.2009.12.005
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10:676-87; PMID:18454141; http://dx.doi.org/10.1038/ncb1730
- Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, et al. Autophagy in malignant transformation and cancer progression. EMBO J 2015; 34:856-80; PMID:25712477; http://dx.doi.org/10.15252/embj.201490784
- Bultman SJ. The microbiome and its potential as a cancer preventive intervention. Semin Oncol 2016; 43:97-106; PMID:26970128; http://dx.doi.org/10.1053/j.seminoncol.2015.09.001
- Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, O'Riordain M, Shanahan F, O'Toole PW. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2016; PMID:26992426
- Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 2012; 22:292-8; PMID:22009990; http://dx.doi.org/10.1101/gr.126573.111
- Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WK, Ng SC, Tsoi H, Dong Y, Zhang N, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nature Commun 2015; 6:8727; http://dx.doi.org/10.1038/ncomms9727
- Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 2012; 22:299-306; PMID:22009989; http://dx.doi.org/10.1101/gr.126516.111
- Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol 2011; 6:479-507; PMID:21090969; http://dx.doi.org/10.1146/annurev-pathol-011110-130235
- Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat Rev Microbiol 2012; 10:575-82; PMID:22728587; http://dx.doi.org/10.1038/nrmicro2819
- Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, Schloss PD. The gut microbiome modulates colon tumorigenesis. mBio 2013; 4:e00692-13; PMID:24194538; http://dx.doi.org/10.1128/mBio.00692-13
- Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, Zhang W, Zhong Z, Sanchez-Lopez E, Wu LW, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 2014; 41:1052-63; PMID:25526314; http://dx.doi.org/10.1016/j.immuni.2014.11.009
- Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med 2013; 210:917-31; PMID:23589566; http://dx.doi.org/10.1084/jem.20122308
- Levy J, Cacheux W, Bara MA, L'Hermitte A, Lepage P, Fraudeau M, Trentesaux C, Lemarchand J, Durand A, Crain AM, et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nature Cell Biol 2015; 17:1062-73; PMID:26214133; http://dx.doi.org/10.1038/ncb3206
- Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell 2014; 54:224-33; PMID:24766886; http://dx.doi.org/10.1016/j.molcel.2014.03.009
- Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, Bultman SJ. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 2011; 13:517-26; PMID:21531334; http://dx.doi.org/10.1016/j.cmet.2011.02.018
- Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr 2002; 132:1012-7; PMID:11983830
- Jin D, Wu S, Zhang YG, Lu R, Xia Y, Dong H, Sun J. Lack of Vitamin D receptor causes dysbiosis and changes the functions of the murine intestinal microbiome. Clin Ther 2015; 37:996-1009 e7; PMID:26046242; http://dx.doi.org/10.1016/j.clinthera.2015.04.004
- Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol 2009; 3:366-75; PMID:19559660; http://dx.doi.org/10.1016/j.molonc.2009.05.007
- Grimm WA, Messer JS, Murphy SF, Nero T, Lodolce JP, Weber CR, Logsdon MF, Bartulis S, Sylvester BE, Springer A, et al. The Thr300Ala variant in ATG16L1 is associated with improved survival in human colorectal cancer and enhanced production of type I interferon. Gut 2015; 2:2014-308735
- Nicoli ER, Dumitrescu T, Uscatu CD, Popescu FD, Streata I, Serban Sosoi S, Ivanov P, Dumitrescu A, Bărbălan A, Lungulescu D, et al. Determination of autophagy gene ATG16L1 polymorphism in human colorectal cancer. Rom J Morphol Embryol 2014; 55:57-62; PMID:24715166
- Lorenzo G, Lopez-Gil E, Warimwe GM, Brun A. Understanding Rift Valley fever: Contributions of animal models to disease characterization and control. Mol Immunol 2015; 66:78-88; PMID:25725948; http://dx.doi.org/10.1016/j.molimm.2015.02.001
- Boelens J, Lust S, Offner F, Bracke ME, Vanhoecke BW. Review. The endoplasmic reticulum: A target for new anticancer drugs. In Vivo 2007; 21:215-26; PMID:17436569
- Habeeb BS, Kitayama J, Nagawa H. Adiponectin supports cell survival in glucose deprivation through enhancement of autophagic response in colorectal cancer cells. Cancer Sci 2011; 102:999-1006; PMID:21299716; http://dx.doi.org/10.1111/j.1349-7006.2011.01902.x
- Khan S. Potential role of Escherichia coli DNA mismatch repair proteins in colon cancer. Crit Rev Oncol Hematol 2015; 15:00091-8
- Keku TO, Dulal S, Deveaux A, Jovov B, Han X. The gastrointestinal microbiota and colorectal cancer. Am J Physiol Gastrointest Liver Physiol 2015; 308:24; http://dx.doi.org/10.1152/ajpgi.00360.2012
- Garg AD, Kaczmarek A, Krysko O, Vandenabeele P, Krysko DV, Agostinis P. ER stress-induced inflammation: does it aid or impede disease progression?. Trend Mol Med 2012; 18:589-98; PMID:22883813; http://dx.doi.org/10.1016/j.molmed.2012.06.010
- Wirth MD, Shivappa N, Steck SE, Hurley TG, Hebert JR. The dietary inflammatory index is associated with colorectal cancer in the National Institutes of Health-American Association of Retired Persons Diet and Health Study. Br J Nutr 2015; 113:1819-27; PMID:25871645; http://dx.doi.org/10.1017/S000711451500104X
- Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology 2015; 7:01097-5
- Cebola I, Custodio J, Munoz M, Diez-Villanueva A, Pare L, Prieto P, Aussó S, Coll-Mulet L, Boscá L, Moreno V, et al. Epigenetics override pro-inflammatory PTGS transcriptomic signature towards selective hyperactivation of PGE2 in colorectal cancer. Clin Epigenetics 2015; 7:015-0110; http://dx.doi.org/10.1186/s13148-015-0110-4
- Mehta RS, Chong DQ, Song M, Meyerhardt JA, Ng K, Nishihara R, Qian Z, Morikawa T, Wu K, Giovannucci EL, et al. Association between plasma levels of macrophage inhibitory cytokine-1 before diagnosis of colorectal cancer and mortality. Gastroenterology 2015; 149:614-22; PMID:26026393; http://dx.doi.org/10.1053/j.gastro.2015.05.038
- Gurevich-Panigrahi T, Panigrahi S, Wiechec E, Los M. Obesity: Pathophysiology and clinical management. Curr Med Chem 2009; 16:506-21; PMID:19199918; http://dx.doi.org/10.2174/092986709787315568
- Liesenfeld DB, Grapov D, Fahrmann JF, Salou M, Scherer D, Toth R, Habermann N, Böhm J, Schrotz-King P, Gigic B, et al. Metabolomics and transcriptomics identify pathway differences between visceral and subcutaneous adipose tissue in colorectal cancer patients: The colo care study. Am J Clin Nutr 2015; 102:433-43; PMID:26156741; http://dx.doi.org/10.3945/ajcn.114.103804
- Tanaka Y, Ito S, Oshino R, Chen N, Nishio N, Isobe K. Effects of growth arrest and DNA damage-inducible protein 34 (GADD34) on inflammation-induced colon cancer in mice. Br J Cancer 2015; 113:669-79; PMID:26196182;http://dx.doi.org/10.1038/bjc.2015.263
- Yeganeh B, Jager R, Gorman AM, Samali A, Ghavami S. Induction of autophagy: Role of endoplasmic reticulum stress and unfolded protein response. In: Hayat E, ed. Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging. V.7: Role of Autophagy in Therapeutic Applications. San Diego, CA: Elsevier; 2015; 91-101
- Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett 2013; 332:249-64; PMID:20732741; http://dx.doi.org/10.1016/j.canlet.2010.07.016
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140:883-99; PMID:20303878; http://dx.doi.org/10.1016/j.cell.2010.01.025
- Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochimica Et Biophysica Acta 2010; 1805:53-71; PMID:; PMID:19720113
- Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol 2008; 8:923-34; PMID:19029988; http://dx.doi.org/10.1038/nri2449
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010; 140:900-17; PMID:20303879; http://dx.doi.org/10.1016/j.cell.2010.02.034
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008; 454:455-62; PMID:18650916; http://dx.doi.org/10.1038/nature07203
- Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Research 2011; 71:2411-6; PMID:21427354; http://dx.doi.org/10.1158/0008-5472.CAN-10-2583
- Coppola A, Arriga R, Lauro D, Del Principe M, Buccisano F, Maurillo L, Palomba P, Venditti A, Sconocchia G. NK Cell Inflammation in the Clinical Outcome of Colorectal Carcinoma. Front Med 2015; 2; http://dx.doi.org/10.3389/fmed.2015.00033
- Elinav E, Henao-Mejia JF, Flavell RA. Integrative inflammasome activity in the regulation of intestinal mucosal immune responses. Mucosal Immunol 2013; 6:4-13; PMID:23212196; http://dx.doi.org/10.1038/mi.2012.115
- Trusevych EF, MacNaughton WK. Proteases and their receptors as mediators of inflammation-associated colon cancer. Curr Pharm Des 2015; 21:2983-92; PMID:26004412; http://dx.doi.org/10.2174/1381612821666150514104800
- Yu C, Wen XD, Zhang Z, Zhang CF, Wu X, He X, Liao Y, Wu N, Wang CZ, Du W, et al. American ginseng significantly reduced the progression of high-fat-diet-enhanced colon carcinogenesis in Apc (Min/+) mice. J Ginseng Res 2015; 39:230-7; PMID:26199554; http://dx.doi.org/10.1016/j.jgr.2014.12.004
- Lu J-J, Chen S-M, Zhang X-W, Ding J, Meng LH. The anti-cancer activity of dihydroartemisinin is associated with induction of iron-dependent endoplasmic reticulum stress in colorectal carcinoma HCT116 cells. Invest New Drugs 2011; 29:1276-83; PMID:20607588; http://dx.doi.org/10.1007/s10637-010-9481-8
- Mhaidat NM, Alali FQ, Matalqah SM, Matalka II, Jaradat SA, Al-sawalha NA, Thorne RF. Inhibition of MEK sensitizes paclitaxel-induced apoptosis of human colorectal cancer cells by downregulation of GRP78. Anticancer Drugs 2009; 20:601-6; PMID:19521235; http://dx.doi.org/10.1097/CAD.0b013e32832e3120
- Thornton M, Aslam MA, Tweedle EM, Ang C, Campbell F, Jackson R, Costello E, Rooney PS, Vlatković N, Boyd MT. The unfolded protein response regulator GRP78 is a novel predictive biomarker in colorectal cancer. Int J Cancer 2013; 133:1408-18; PMID:23456958; http://dx.doi.org/10.1002/ijc.28137
- Mhaidat NM, Alzoubi KH, Khabour OF, Banihani MN, Al-Balas QA, Swaidan S. GRP78 regulates sensitivity of human colorectal cancer cells to DNA targeting agents. Cytotechnology 2016; 68:459-67; PMID:25399254; http://dx.doi.org/10.1007/s10616-014-9799-8
- Rouschop KM, Van Den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010; 120:127-41; PMID:20038797; http://dx.doi.org/10.1172/JCI40027
- Sutherland A, Kim D-H, Relton C, Ahn Y-O, Hesketh J. Polymorphisms in the selenoprotein S and 15-kDa selenoprotein genes are associated with altered susceptibility to colorectal cancer. Genes Nutr 2010; 5:215-23; PMID:21052528; http://dx.doi.org/10.1007/s12263-010-0176-8
- Fujimoto T, Yoshimatsu K, Watanabe K, Yokomizo H, Otani T, Matsumoto A, Osawa G, Onda M, Ogawa K. Overexpression of human X-box binding protein 1 (XBP-1) in colorectal adenomas and adenocarcinomas. Anticancer Res 2007; 27:127-31; PMID:17352224
- Ieta K, Tanaka F, Yokobori T, Kita Y, Haraguchi N, Mimori K, Kato H, Asao T, Inoue H, Kuwano H, Mori M. Clinicopathological significance of stanniocalcin 2 gene expression in colorectal cancer. Int J Cancer 2009; 125:926-31; PMID:19415750; http://dx.doi.org/10.1002/ijc.24453
- Du H, Li W, Wang Y, Chen S, Zhang Y. Celecoxib induces cell apoptosis coupled with up-regulation of the expression of VEGF by a mechanism involving ER stress in human colorectal cancer cells. Oncol Rep 2011; 26:495-502; PMID:21567098
- Lu M, Sun L, Zhou J, Zhao Y, Deng X. Dihydroartemisinin-induced apoptosis is associated with inhibition of sarco/endoplasmic reticulum calcium ATPase activity in colorectal cancer. Cell Biochem Biophys 2015; 73:137-45; PMID:25701954; http://dx.doi.org/10.1007/s12013-015-0643-3
- Jin C, Jin Z, Chen N-z, Lu M, Liu CB, Hu W-L, Zheng CG. Activation of IRE1α-XBP1 pathway induces cell proliferation and invasion in colorectal carcinoma. Biochem Biophys Res Commun 2016; 470:75-81; PMID:26742428; http://dx.doi.org/10.1016/j.bbrc.2015.12.119
- Mhaidat NM, Alzoubi KH, Abushbak A. X-box binding protein 1 (XBP-1) enhances colorectal cancer cell invasion. J Chemother 2015; 27:167-73; PMID:25692573; http://dx.doi.org/10.1179/1973947815Y.0000000006
- Bao W, Gu Y, Ta L, Wang K, Xu Z. Induction of autophagy by the MG-132 proteasome inhibitor is associated with endoplasmic reticulum stress in MCF-7 cells. Mol Med Rep 2016; 13:796-804; PMID:26648402
- Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000; 405:354-60; PMID:10830965; http://dx.doi.org/10.1038/35012626
- Ohmori H, Luo Y, Kuniyasu H. Non-histone nuclear factor HMGB1 as a therapeutic target in colorectal cancer. Expert Opin Ther Targets 2011; 15:183-93; PMID:21204727; http://dx.doi.org/10.1517/14728222.2011.546785
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418:191-5; PMID:12110890; http://dx.doi.org/10.1038/nature00858
- Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 2002; 3:995-1001; PMID:12231511; http://dx.doi.org/10.1093/embo-reports/kvf198
- Kuniyasu H, Chihara Y, Takahashi T. Co-expression of receptor for advanced glycation end products and the ligand amphoterin associates closely with metastasis of colorectal cancer. Oncol Rep 2003; 10:445-8; PMID:12579287
- Ohmori H, Luo Y, Fujii K, Sasahira T, Shimomoto T, Denda A, Kuniyasu H. Dietary linoleic acid and glucose enhances azoxymethane-induced colon cancer and metastases via the expression of high-mobility group box 1. Pathobiology 2010; 77:210-7; PMID:20616616; http://dx.doi.org/10.1159/000296305
- Ouyang F, Huang H, Zhang M, Chen M, Huang H, Huang F, Zhou S. HMGB1 induces apoptosis and EMT in association with increased autophagy following H/R injury in cardiomyocytes. Int J Mol Med 2016; 37:679-89; PMID:26847839
- Zhu L, Huang G, Sheng J, Fu Q, Chen A. High-mobility group box 1 induces neuron autophagy in a rat spinal root avulsion model. Neuroscience 2016; 315:286-95; PMID:26705737; http://dx.doi.org/10.1016/j.neuroscience.2015.12.020
- Ioannou S, Voulgarelis M. Toll-like receptors, tissue injury, and tumourigenesis. Mediators Inflamm; Epub 2010 Sep. 14; Article ID:581837; PMID:20871832; http://dx.doi.org/10.1155/2010/581837
- Delgado MA, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ 2009; 16:976-83; PMID:19444282; http://dx.doi.org/10.1038/cdd.2009.40
- Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ, 3rd, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol 2010; 190:881-92; PMID:20819940; http://dx.doi.org/10.1083/jcb.200911078
- Luo Y, Yoneda J, Ohmori H, Sasaki T, Shimbo K, Eto S, Kato Y, Miyano H, Kobayashi T, Sasahira T, et al. Cancer usurps skeletal muscle as an energy repository. Cancer Research 2014; 74:330-40; PMID:24197136; http://dx.doi.org/10.1158/0008-5472.CAN-13-1052
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287-95; PMID:20083114; http://dx.doi.org/10.1016/j.febslet.2010.01.017
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463-77; PMID:15068787; http://dx.doi.org/10.1016/S1534-5807(04)00099-1
- Bluemlein K, Gluckmann M, Gruning NM, Feichtinger R, Kruger A, Wamelink M, Lehrach H, Tate S, Neureiter D, Kofler B, et al. Pyruvate kinase is a dosage-dependent regulator of cellular amino acid homeostasis. Oncotarget 2012; 3:1356-69; PMID:23154538; http://dx.doi.org/10.18632/oncotarget.730
- Dang CV. PKM2 tyrosine phosphorylation and glutamine metabolism signal a different view of the Warburg effect. Sci Signal 2009; 2:pe75; PMID:19920249; http://dx.doi.org/10.1126/scisignal.297pe75
- Luo Y, Chihara Y, Fujimoto K, Sasahira T, Kuwada M, Fujiwara R, Fujii K, Ohmori H, Kuniyasu H. High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur J Cancer 2013; 49:741-51; PMID:23040637; http://dx.doi.org/10.1016/j.ejca.2012.09.016
- Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC, Kroemer G. Anti- and pro-tumor functions of autophagy. Biochimica Et Biophysica Acta 2009; 1793:1524-32; PMID:19371598; http://dx.doi.org/10.1016/j.bbamcr.2009.01.006
- Yang Z, Ghoorun RA, Fan X, Wu PC, Bai Y, Li J, Yang Z, Ghoorun RA, Fan X, Wu P, Bai Y, Li J. High expression of Beclin-1 predicts favorable prognosis for patients with colorectal cancer. Clin Res Hepatol Gastroenterol 2015; 39:98-106; PMID:25130795; http://dx.doi.org/10.1016/j.clinre.2014.06.014
- Chen N, Karantza-Wadsworth V. Role and regulation of autophagy in cancer. Biochimica Et Biophysica Acta 2009; 1793:1516-23; PMID:19167434; http://dx.doi.org/10.1016/j.bbamcr.2008.12.013
- Chen Z, Li Y, Zhang C, Yi H, Wu C, Wang J, Liu Y, Tan J, Wen J. Downregulation of Beclin 1 and impairment of autophagy in a small population of colorectal cancer. Dig Dis Sci 2013; 58:2887-94; PMID:23812859; http://dx.doi.org/10.1007/s10620-013-2732-8
- Wu S, Sun C, Tian D, Li Y, Gao X, He S, Li T. Expression and clinical significances of Beclin1, LC3 and mTOR in colorectal cancer. Int J Clin Exp Pathol 2015; 8:3882-91; PMID:26097572
- Guo GF, Jiang WQ, Zhang B, Cai YC, Xu RH, Chen XX, Wang F, Xia LP. Autophagy-related proteins Beclin-1 and LC3 predict cetuximab efficacy in advanced colorectal cancer. World J Gastroenterol 2011; 17:4779-86; PMID:22147978; http://dx.doi.org/10.3748/wjg.v17.i43.4779
- Reyes-Gibby CC, Wang J, Yeung S-CJ, Shete S. Informative gene network for chemotherapy-induced peripheral neuropathy. BioData mining 2015; 8:1; PMID:25621011; http://dx.doi.org/10.1186/s13040-015-0058-0
- Jo YK, Kim SC, Park IJ, Park SJ, Jin DH, Hong SW, Cho DH, Kim JC. Increased expression of ATG10 in colorectal cancer is associated with lymphovascular invasion and lymph node metastasis. PloS One 2012; 7:e52705; PMID:23285162; http://dx.doi.org/10.1371/journal.pone.0052705
- Choi JH, Cho Y-S, Ko YH, Hong SU, Park JH, Lee MA. Absence of autophagy-related proteins expression is associated with poor prognosis in patients with colorectal adenocarcinoma. Gastroenterol Res Pract 2014; 2014:10; http://dx.doi.org/10.1155/2014/179586
- Qian Q, Zhou H, Chen Y, Shen C, He S, Zhao H, Wang L, Wan D, Gu W. VMP1 related autophagy and apoptosis in colorectal cancer cells: VMP1 regulates cell death. Biochem Biophys Res Commun 2014; 443:1041-7; PMID:24365149; http://dx.doi.org/10.1016/j.bbrc.2013.12.090
- Zhai H, Fesler A, Ba Y, Wu S, Ju J. Inhibition of colorectal cancer stem cell survival and invasive potential by hsa-miR-140-5p mediated suppression of Smad2 and autophagy. Oncotarget 2015; 6:19735-46; PMID:25980495; http://dx.doi.org/10.18632/oncotarget.3771
- Li X, Zhang K, Li Z. Unfolded protein response in cancer: the physician's perspective. J Hematol Oncol 2011; 4:8; PMID:21345215; http://dx.doi.org/10.1186/1756-8722-4-8
- Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol 2009; 625:234-46; PMID:19835867; http://dx.doi.org/10.1016/j.ejphar.2009.06.064
- Ashktorab H, Green W, Finzi G, Sessa F, Nouraie M, Lee EL, Morgano A, Moschetta A, Cattaneo M, Mariani-Costantini R, et al. SEL1L, an UPR response protein, a potential marker of colonic cell transformation. Dig Dis Sci 2012; 57:905-12; PMID:22350780; http://dx.doi.org/10.1007/s10620-011-2026-y
- Akbari-Birgani S, Paranjothy T, Zuse A, Janikowski T, Cieslar-Pobuda A, Likus W, Urasińska E, Schweizer F, Ghavami S, Klonisch T, et al. Cancer stem cells, cancer-initiating cells and methods for their detection. Drug Discov today 2016; 21:836-42; PMID:26976692; http://dx.doi.org/10.1016/j.drudis.2016.03.004
- Cieslar-Pobuda A, Back M, Magnusson K, Jain MV, Rafat M, Ghavami S, Nilsson KP, Łos MJ. Cell type related differences in staining with pentameric thiophene derivatives. Cytometry A 2014; 85:628-35; PMID:24500794; http://dx.doi.org/10.1002/cyto.a.22437
- Zeindl-Eberhart E, Brandl L, Liebmann S, Ormanns S, Scheel SK, Brabletz T, Kirchner T, Jung A. Epithelial-mesenchymal transition induces endoplasmic-reticulum-stress response in human colorectal tumor cells. PloS One 2014; 9:e87386; PMID:24498091; http://dx.doi.org/10.1371/journal.pone.0087386
- Munro S, Pelham HR. An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 1986; 46:291-300; PMID:3087629; http://dx.doi.org/10.1016/0092-8674(86)90746-4
- Zorbas M, Sicurella C, Bertoncello I, Venter D, Ellis S, Mucenski ML, Ramsay RG. c-Myb is critical for murine colon development. Oncogene 1999; 18:5821-30; PMID:10523863; http://dx.doi.org/10.1038/sj.onc.1202971
- Ramsay RG, Ciznadija D, Mantamadiotis T, Anderson R, Pearson R. Expression of stress response protein glucose regulated protein-78 mediated by c-Myb. Int J Biochem Cell Biol 2005; 37:1254-68; PMID:15778089; http://dx.doi.org/10.1016/j.biocel.2004.12.011
- Xie XL, Liu YB, Liu YP, Du BL, Li Y, Han M, Li BH. Reduced expression of SM22 is correlated with low autophagy activity in human colorectal cancer. Pathol Res Pract 2013; 209:237-43; PMID:23538046; http://dx.doi.org/10.1016/j.prp.2013.02.007
- Zhang JC, Kim S, Helmke BP, Yu WW, Du KL, Lu MM, et al. Analysis of SM22alpha-deficient mice reveals unanticipated insights into smooth muscle cell differentiation and function. Mol Cell Biol 2001; 21:1336-44; PMID:11158319; http://dx.doi.org/10.1128/MCB.2001.21.4.1336-1344.2001
- Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181-97; PMID:19200883
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/10.1080/15548627.2015.1100356
- Ren F, Shu G, Liu G, Liu D, Zhou J, Yuan L, Zhou J. Knockdown of p62/sequestosome 1 attenuates autophagy and inhibits colorectal cancer cell growth. Mol Cell Biochem 2014; 385:95-102; PMID:24065390; http://dx.doi.org/10.1007/s11010-013-1818-0
- McKnight NC, Zhenyu Y. Beclin 1, an essential component and master regulator of PI3K-III in health and disease. Curr Pathobiol Rep 2013; 1:231-8; PMID:24729948; http://dx.doi.org/10.1007/s40139-013-0028-5
- Ko YH, Cho YS, Won HS, An HJ, Sun DS, Hong SU, Park JH, Lee MA. Stage-stratified analysis of prognostic significance of bax-interacting Factor-1 expression in resected colorectal cancer. Biomed Res Int 2013; 2013:8
- Coppola D, Khalil F, Eschrich SA, Boulware D, Yeatman T, Wang HG. Down-regulation of Bax-interacting factor-1 in colorectal adenocarcinoma. Cancer 2008; 113:2665-70; PMID:18833585; http://dx.doi.org/10.1002/cncr.23892
- Shakeri R, Hosseinkhani S, Los MJ, Davoodi J, Jain MV, Cieslar-Pobuda A, Rafat M, Ardestani SK. Role of the salt bridge between glutamate 546 and arginine 907 in preservation of autoinhibited form of Apaf-1. Inter J Biol Macromol 2015; 81:370-4; PMID:26277751; http://dx.doi.org/10.1016/j.ijbiomac.2015.08.027
- Wu X-Y, Chen J, Cao Q-H, Dong M, Lin Q, Fan XJ, Xia Q, Chen ZH, Liu Q, Wan X. Beclin 1 activation enhances chemosensitivity and predicts a favorable outcome for primary duodenal adenocarcinoma. Tumour Biol 2013; 34:713-22; PMID:23225331; http://dx.doi.org/10.1007/s13277-012-0599-5
- Pratt WB, Morishima Y, Peng HM, Osawa Y. Proposal for a role of the Hsp90/Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp Biol Med (Maywood) 2010; 235:278-89; PMID:20404045; http://dx.doi.org/10.1258/ebm.2009.009250
- Tsai YC, Weissman AM. The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer 2010; 1:764-78; PMID:21331300; http://dx.doi.org/10.1177/1947601910383011
- Jain MV, Shareef A, Likus W, Cieslar-Pobuda A, Ghavami S, Los MJ. Inhibition of miR301 enhances Akt-mediated cell proliferation by accumulation of PTEN in nucleus and its effects on cell-cycle regulatory proteins. Oncotarget 2016; 7(15):20953-65; PMID:26967567; http://dx.doi.org/10.18632/oncotarget.7996
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 2000; 275:992-8; PMID:10625637; http://dx.doi.org/10.1074/jbc.275.2.992
- Won SJ, Yen CH, Liu HS, Wu SY, Lan SH, Jiang-Shieh YF, Lin CN, Su CL. Justicidin A-induced autophagy flux enhances apoptosis of human colorectal cancer cells via class III PI3K and Atg5 pathway. J Cell Physiol 2015; 230:930-46; PMID:25216025; http://dx.doi.org/10.1002/jcp.24825
- Roberts HR, Smartt HJM, Greenhough A, Moore AE, Williams AC, Paraskeva C. Colon tumour cells increase PGE2 by regulating COX-2 and 15-PGDH to promote survival during the microenvironmental stress of glucose deprivation. Carcinogenesis 2011; 32:1741-7; PMID:21926111; http://dx.doi.org/10.1093/carcin/bgr210
- De Pergola G, Silvestris F. Obesity as a major risk factor for cancer. J Obesity 2013; 2013:291546; PMID:24073332; http://dx.doi.org/10.1155/2013/291546
- Pfalzer AC, Nesbeth PD, Parnell LD, Iyer LK, Liu Z, Kane AV, Chen CY, Tai AK, Bowman TA, Obin MS, et al. - Diet- and Genetically-induced obesity differentially affect the fecal microbiome and metabolome in Apc1638N Mice. PLoS One 2015; 10; PMID:26284788; http://dx.doi.org/10.1371/journal.pone.0135758
- Diaz-Algorri Y, Lozada ME, Lopez SM, Bertran-Rodriguez CE, Gonzalez-Hernandez CM, Gonzalez D, Pérez-Cardona CM, Hernández J, Pedrosa C, Toro DH, et al. Type 2 diabetes mellitus and colorectal neoplasia risk in Hispanics: a case-control study. J Diabetes Complications 2015; 29:502-7; PMID:25784088; http://dx.doi.org/10.1016/j.jdiacomp.2015.01.010
- Weijenberg MP, Hughes LA, Bours MJ, Simons CC, van Engeland M, van den Brandt PA. The mTOR Pathway and the Role of Energy Balance Throughout Life in Colorectal Cancer Etiology and Prognosis: Unravelling Mechanisms Through a Multidimensional Molecular Epidemiologic Approach. Curr Nutr Rep 2013; 2:19-26; PMID:23396869; http://dx.doi.org/10.1007/s13668-012-0038-7
- Pietrzyk L, Torres A, Maciejewski R, Torres K. Obesity and Obese-related Chronic Low-grade inflammation in promotion of colorectal cancer development. Asian Pac J Cancer Prev 2015; 16:4161-8; PMID:26028066; http://dx.doi.org/10.7314/APJCP.2015.16.10.4161
- Tae CH, Kim SE, Jung SA, Joo YH, Shim KN, Jung HK, Kim TH, Cho MS, Kim KH, Kim JS. Involvement of adiponectin in early stage of colorectal carcinogenesis. BMC Cancer 2014; 14:811; PMID:25370174; http://dx.doi.org/10.1186/1471-2407-14-811
- Kourelis TV, Siegel RD. Metformin and cancer: new applications for an old drug. Med Oncol 2012; 29:1314-27; PMID:21301998; http://dx.doi.org/10.1007/s12032-011-9846-7
- Din FV, Valanciute A, Houde VP, Zibrova D, Green KA, Sakamoto K, Alessi DR, Dunlop MG. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012; 142:1504-15 e3; PMID:22406476; http://dx.doi.org/10.1053/j.gastro.2012.02.050
- Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, Ueno T, Ochiai A, Esumi H. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res 2007; 67:9677-84; PMID:17942897; http://dx.doi.org/10.1158/0008-5472.CAN-07-1462
- Kauntz H, Bousserouel S, Gosse F, Raul F. Silibinin triggers apoptotic signaling pathways and autophagic survival response in human colon adenocarcinoma cells and their derived metastatic cells. Apoptosis 2011; 16:1042-53; PMID:21779837; http://dx.doi.org/10.1007/s10495-011-0631-z
- Kantara C, O'Connell M, Sarkar S, Moya S, Ullrich R, Singh P. Curcumin promotes autophagic survival of a subset of colon cancer stem cells, which are ablated by DCLK1-siRNA. Cancer Res 2014; 74:2487-98; PMID:24626093; http://dx.doi.org/10.1158/0008-5472.CAN-13-3536
- Moghadam AR, Tutunchi S, Namvaran-Abbas-Abad A, Yazdi M, Bonyadi F, Mohajeri D, Mazani M, Marzban H, Łos MJ, Ghavami S. Pre-administration of turmeric prevents methotrexate-induced liver toxicity and oxidative stress. BMC Complementary Altern Med 2015; 15:246; PMID:26199067; http://dx.doi.org/10.1186/s12906-015-0773-6
- Talero E, Avila-Roman J, Motilva V. Chemoprevention with phytonutrients and microalgae products in chronic inflammation and colon cancer. Curr Pharm Des 2012; 18:3939-65; PMID:22632755; http://dx.doi.org/10.2174/138161212802083725
- Sui X, Kong N, Wang X, Fang Y, Hu X, Xu Y, Chen W, Wang K, Li D, Jin W, et al. JNK confers 5-fluorouracil resistance in p53-deficient and mutant p53-expressing colon cancer cells by inducing survival autophagy. Sci Rep 2014; 4:4694; PMID:24733045; http://dx.doi.org/10.1038/srep04694
- Chiacchiera F, Simone C. Signal-dependent regulation of gene expression as a target for cancer treatment: inhibiting p38alpha in colorectal tumors. Cancer Lett 2008; 265:16-26; PMID:18395970; http://dx.doi.org/10.1016/j.canlet.2008.02.061
- Comes F, Matrone A, Lastella P, Nico B, Susca FC, Bagnulo R, Ingravallo G, Modica S, Lo Sasso G, Moschetta A, et al. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ 2007; 14:693-702; PMID:17159917; http://dx.doi.org/10.1038/sj.cdd.4402076
- Chiacchiera F, Matrone A, Ferrari E, Ingravallo G, Lo Sasso G, Murzilli S, Petruzzelli M, Salvatore L, Moschetta A, Simone C. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ 2009; 16:1203-14; PMID:19343039; http://dx.doi.org/10.1038/cdd.2009.36
- Chiacchiera F, Simone C. Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy 2009; 5:1030-3; PMID:19587525; http://dx.doi.org/10.4161/auto.5.7.9252
- Simone C. Signal-dependent control of autophagy and cell death in colorectal cancer cell: the role of the p38 pathway. Autophagy 2007; 3:468-71; PMID:17495519; http://dx.doi.org/10.4161/auto.4319
- Yang WL, Perillo W, Liou D, Marambaud P, Wang P. AMPK inhibitor compound C suppresses cell proliferation by induction of apoptosis and autophagy in human colorectal cancer cells. J Surg Oncol 2012; 106:680-8; PMID:22674626; http://dx.doi.org/10.1002/jso.23184
- Taniguchi K, Sugito N, Kumazaki M, Shinohara H, Yamada N, Nakagawa Y, Ito Y, Otsuki Y, Uno B, Uchiyama K, et al. MicroRNA-124 inhibits cancer cell growth through PTB1/PKM1/PKM2 feedback cascade in colorectal cancer. Cancer Lett 2015; 363:17-27; PMID:25818238; http://dx.doi.org/10.1016/j.canlet.2015.03.026
- de la Cruz-Morcillo MA, Valero ML, Callejas-Valera JL, Arias-Gonzalez L, Melgar-Rojas P, Galan-Moya EM, García-Gil E, García-Cano J, Sánchez-Prieto R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: implication in resistance. Oncogene 2012; 31:1073-85; PMID:21841826; http://dx.doi.org/10.1038/onc.2011.321
- Koido S, Ohkusa T, Homma S, Namiki Y, Takakura K, Saito K, Ito Z, Kobayashi H, Kajihara M, Uchiyama K, et al. Immunotherapy for colorectal cancer. World J Gastroenterol 2013; 19:8531-42; PMID:24379570; http://dx.doi.org/10.3748/wjg.v19.i46.8531
- Marshall JL, Gulley JL, Arlen PM, Beetham PK, Tsang KY, Slack R, Hodge JW, Doren S, Grosenbach DW, Hwang J, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol 2005; 23:720-31; PMID:15613691; http://dx.doi.org/10.1200/JCO.2005.10.206
- Noguchi T, Ritter G, Nishikawa H. Antibody-based therapy in colorectal cancer. Immunotherapy 2013; 5:533-45; PMID:23638747; http://dx.doi.org/10.2217/imt.13.35
- Sanchez-Castanon M, Er TK, Bujanda L, Herreros-Villanueva M. Immunotherapy in colorectal cancer: What have we learned so far? Clin Chim Acta 2016; 460:78-87; PMID:27350293; http://dx.doi.org/10.1016/j.cca.2016.06.027
- Koido S, Hara E, Homma S, Torii A, Toyama Y, Kawahara H, Watanabe M, Yanaga K, Fujise K, Tajiri H, et al. Dendritic cells fused with allogeneic colorectal cancer cell line present multiple colorectal cancer-specific antigens and induce antitumor immunity against autologous tumor cells. Clin Cancer Res 2005; 11:7891-900; PMID:16278414; http://dx.doi.org/10.1158/1078-0432.CCR-05-1330
- Gonzalez G, Crombet T, Catala M, Mirabal V, Hernandez JC, Gonzalez Y, et al. A novel cancer vaccine composed of human-recombinant epidermal growth factor linked to a carrier protein: report of a pilot clinical trial. Annal Oncol 1998; 9:431-5; http://dx.doi.org/10.1023/A:1008261031034
- Speetjens FM, Kuppen PJ, Welters MJ, Essahsah F, Voet van den Brink AM, Lantrua MG, Valentijn AR, Oostendorp J, Fathers LM, Nijman HW, et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin Cancer Res 2009; 15:1086-95; PMID:19188184; http://dx.doi.org/10.1158/1078-0432.CCR-08-2227
- Paulsen JE, Bjorheim J, Roe J, Eide TJ, Alexander J, Gaudernack G. Effect of vaccination with mutant KRAS peptides on rat colon carcinogenesis induced by azoxymethane. Anticancer Res 2002; 22:171-5; PMID:12017282
- Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol 2005; 5:296-306; PMID:15803149; http://dx.doi.org/10.1038/nri1592
- Celluzzi CM, Mayordomo JI, Storkus WJ, Lotze MT, Falo LD, Jr. Peptide-pulsed dendritic cells induce antigen-specific CTL-mediated protective tumor immunity. J Exp Med 1996; 183:283-7; PMID:8551233; http://dx.doi.org/10.1084/jem.183.1.283
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nature Med 1998; 4:328-32
- Berard F, Blanco P, Davoust J, Neidhart-Berard EM, Nouri-Shirazi M, Taquet N, Rimoldi D, Cerottini JC, Banchereau J, Palucka AK. Cross-priming of naive CD8 T cells against melanoma antigens using dendritic cells loaded with killed allogeneic melanoma cells. J Exp Med 2000; 192:1535-44
- Koido S, Kashiwaba M, Chen D, Gendler S, Kufe D, Gong J. Induction of antitumor immunity by vaccination of dendritic cells transfected with MUC1 RNA. J Immunol 2000; 165:5713-9
- Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011; 19:620-6
- Soda H, Koda K, Yasutomi J, Oda K, Takiguchi N, Saito N, Nakajima N. Adoptive immunotherapy for advanced cancer patients using in vitro activated cytotoxic T lymphocytes. J Surg Oncol 1999; 72:211-7; PMID:10589036
- Moehler M, Delic M, Goepfert K, Aust D, Grabsch HI, Halama N, Heinrich B, Julie C, Lordick F, Lutz MP, et al. Immunotherapy in gastrointestinal cancer: Recent results, current studies and future perspectives. Eur J Cancer 2016; 59:160-70; PMID:27039171
- Paul B, O'Neil BH, McRee AJ. Checkpoint inhibition for colorectal cancer: progress and possibilities. Immunotherapy 2016; 8:693-704; PMID:27197538
- Koido S, Homma S, Takahara A, Namiki Y, Komita H, Uchiyama K, Ito M, Gong J, Ohkusa T, Tajiri H. Immunotherapy synergizes with chemotherapy targeting pancreatic cancer. Immunotherapy 2012; 4:5-7; PMID:22149993
- Kan S, Hazama S, Maeda K, Inoue Y, Homma S, Koido S, Okamoto M, Oka M. Suppressive effects of cyclophosphamide and gemcitabine on regulatory T-cell induction in vitro. Anticancer Res 2012; 32:5363-9; PMID:23225438
- Nowak AK, Robinson BW, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res 2003; 63:4490-6; PMID:12907622
- Zhou H, Yuan M, Yu Q, Zhou X, Min W, Gao D. Autophagy regulation and its role in gastric cancer and colorectal cancer. Cancer Biomark 2016; 17:1-10; PMID:27314289
- Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell 2016; 166:288-98; PMID:27419869
- Yuk JM, Shin DM, Song KS, Lim K, Kim KH, Lee SH, Paik TH, Kim JS, Jo EK. Bacillus calmette-guerin cell wall cytoskeleton enhances colon cancer radiosensitivity through autophagy. Autophagy 2010; 6:46-60; PMID:19901560
- Akbari-Birgani S, Paranjothy T, Zuse A, Janikowski T, Cieslar-Pobuda A, Likus W, Urasińska E, Schweizer F, Ghavami S, Klonisch T, et al. Cancer stem cells, cancer-initiating cells and methods for their detection. Drug Discovery Today 2016; 21(5):836-42; PMID:26976692
- Zheng Y, Zhao YL, Deng X, Yang S, Mao Y, Li Z, Jiang P, Zhao X, Wei Y. Chloroquine inhibits colon cancer cell growth in vitro and tumor growth in vivo via induction of apoptosis. Cancer Investgation 2009; 27:286-92; PMID:19194831
- Liang X, De Vera ME, Buchser WJ, Romo de Vivar Chavez A, Loughran P, Beer Stolz D, Basse P, Wang T, Van Houten B, Zeh HJ, 3rd, et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res 2012; 72:2791-801; PMID:22472122; http://dx.doi.org/10.1158/0008-5472.CAN-12-0320
- Likus W, Siemianowicz K, Markowski J, Wiaderkiewicz J, Kostrzab-Zdebel A, Jura-Szoltys E, Dziubdziela W, Wiaderkiewicz R, Łos MJ, et al. Bacterial infections and osteoclastogenesis regulators in men and women with cholesteatoma. Archivum Immunol Ther Exp 2016, 64:241-7; PMID:26584851; http://dx.doi.org/10.1007/s00005-015-0373-7
- Jin HR, Zhao J, Zhang Z, Liao Y, Wang CZ, Huang WH, Li SP, He TC, Yuan CS, Du W. The antitumor natural compound falcarindiol promotes cancer cell death by inducing endoplasmic reticulum stress. Cell Death Dis 2012; 3:e376; PMID:22914324; http://dx.doi.org/10.1038/cddis.2012.122
- Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol 2013; 301:215-90; PMID:23317820
- Park HR, Ryoo IJ, Choo SJ, Hwang JH, Kim JY, Cha MR, Shin-Ya K, Yoo ID. Glucose-deprived HT-29 human colon carcinoma cells are sensitive to verrucosidin as a GRP78 down-regulator. Toxicology 2007; 229:253-61; PMID:17161515; http://dx.doi.org/10.1016/j.tox.2006.11.049
- Park HR, Tomida A, Sato S, Tsukumo Y, Yun J, Yamori T, Hayakawa Y, Tsuruo T, Shin-ya K. Effect on tumor cells of blocking survival response to glucose deprivation. J Natl Cancer Inst 2004; 96:1300-10; PMID:15339968; http://dx.doi.org/10.1093/jnci/djh243
- Suh DH, Kim MK, Kim HS, Chung HH, Song YS. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann N Y Acad Sci 2012; 1271:20-32; PMID:23050960; http://dx.doi.org/10.1111/j.1749-6632.2012.06739.x
- Schleicher SM, Moretti L, Varki V, Lu B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: implications for future therapeutic approaches. Drug Resist Updat 2010; 13:79-86; PMID:20471904; http://dx.doi.org/10.1016/j.drup.2010.04.002
- Zaytseva YY, Harris JW, Mitov MI, Kim JT, Butterfield DA, Lee EY, Weiss HL, Gao T, Evers BM. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget 2015; 6:18891-904; PMID:25970773; http://dx.doi.org/10.18632/oncotarget.3783
- Moretti L, Yang ES, Kim KW, Lu B. Autophagy signaling in cancer and its potential as novel target to improve anticancer therapy. Drug Resist Updat 2007; 10:135-43; PMID:17627865; http://dx.doi.org/10.1016/j.drup.2007.05.001
- Fels DR, Ye J, Segan AT, Kridel SJ, Spiotto M, Olson M, Koong AC, Koumenis C. Preferential cytotoxicity of bortezomib toward hypoxic tumor cells via overactivation of endoplasmic reticulum stress pathways. Cancer Res 2008; 68:9323-30; PMID:19010906; http://dx.doi.org/10.1158/0008-5472.CAN-08-2873
- Fasano E, Serini S, Piccioni E, Toesca A, Monego G, Cittadini AR, Ranelletti FO, Calviello G. DHA induces apoptosis by altering the expression and cellular location of GRP78 in colon cancer cell lines. Biochimica Et Biophysica Acta 2012; 1822:1762-72; PMID:22898250; http://dx.doi.org/10.1016/j.bbadis.2012.08.003
- Jakobsen CH, Storvold GL, Bremseth H, Follestad T, Sand K, Mack M, Olsen KS, Lundemo AG, Iversen JG, Krokan HE, et al. DHA induces ER stress and growth arrest in human colon cancer cells: associations with cholesterol and calcium homeostasis. J Lipid Res 2008; 49:2089-100; PMID:18566476; http://dx.doi.org/10.1194/jlr.M700389-JLR200
- Lundemo AG, Pettersen CH, Berge K, Berge RK, Schonberg SA. Tetradecylthioacetic acid inhibits proliferation of human SW620 colon cancer cells–gene expression profiling implies endoplasmic reticulum stress. Lipids Health Dis 2011; 10:190; PMID:22027281; http://dx.doi.org/10.1186/1476-511X-10-190
- Adwan H, Bayer H, Pervaiz A, Sagini M, Berger MR. Riproximin is a recently discovered type II ribosome inactivating protein with potential for treating cancer. Biotechnol Adv 2014; 32:1077-90; PMID:24699434; http://dx.doi.org/10.1016/j.biotechadv.2014.03.008
- Guichard C, Pedruzzi E, Fay M, Marie JC, Braut-Boucher F, Daniel F, Grodet A, Gougerot-Pocidalo MA, Chastre E, Kotelevets L, et al. Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis 2006; 27:1812-27; PMID:16524888; http://dx.doi.org/10.1093/carcin/bgl009
- Trondl R, Flocke LS, Kowol CR, Heffeter P, Jungwirth U, Mair GE, Steinborn R, Enyedy ÉA, Jakupec MA, Berger W, et al. Triapine and a more potent dimethyl derivative induce endoplasmic reticulum stress in cancer cells. Mol Pharmacol 2014; 85:451-9; PMID:24378333; http://dx.doi.org/10.1124/mol.113.090605
- Zhang X, Lee SH, Min KW, McEntee MF, Jeong JB, Li Q, Baek SJ. The involvement of endoplasmic reticulum stress in the suppression of colorectal tumorigenesis by tolfenamic acid. Cancer Prev Res (Phila) 2013; 6:1337-47; PMID:24104354; http://dx.doi.org/10.1158/1940-6207.CAPR-13-0220
- Symonds EL, Konczak I, Fenech M. The Australian fruit Illawarra plum (Podocarpus elatus Endl., Podocarpaceae) inhibits telomerase, increases histone deacetylase activity and decreases proliferation of colon cancer cells. Br J Nutr 2013; 109:2117-25; PMID:23069328; http://dx.doi.org/10.1017/S0007114512004333
- Wiersma VR, de Bruyn M, Wei Y, van Ginkel RJ, Hirashima M, Niki T, Nishi N, Zhou J, Pouwels SD, Samplonius DF, et al. The epithelial polarity regulator LGALS9/galectin-9 induces fatal frustrated autophagy in KRAS mutant colon carcinoma that depends on elevated basal autophagic flux. Autophagy 2015; 11:1373-88; PMID:26086204; http://dx.doi.org/10.1080/15548627.2015.1063767
- Oliveira CS, Pereira H, Alves S, Castro L, Baltazar F, Chaves SR, Preto A, Côrte-Real M. Cathepsin D protects colorectal cancer cells from acetate-induced apoptosis through autophagy-independent degradation of damaged mitochondria. Cell Death Dis 2015; 18:157
- Garcia-Maurino S, Alcaide A, Dominguez C. Pharmacological control of autophagy: therapeutic perspectives in inflammatory bowel disease and colorectal cancer. Curr Pharm Des 2012; 18:3853-73; PMID:22632751; http://dx.doi.org/10.2174/138161212802083653
- Francipane MG, Lagasse E. Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget 2013; 4:1948-62; PMID:24185040; http://dx.doi.org/10.18632/oncotarget.1310
- Huo HZ, Zhou ZY, Wang B, Qin J, Liu WY, Gu Y. Dramatic suppression of colorectal cancer cell growth by the dual mTORC1 and mTORC2 inhibitor AZD-2014. Biochem Biophys Res Commun 2014; 443:406-12; PMID:24309100; http://dx.doi.org/10.1016/j.bbrc.2013.11.099
- Gandesiri M, Chakilam S, Ivanovska J, Benderska N, Ocker M, Di Fazio P, Feoktistova M, Gali-Muhtasib H, Rave-Fränk M, Prante O, et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis 2012; 17:1300-15; PMID:23011180; http://dx.doi.org/10.1007/s10495-012-0757-7
- Chekhonin VP, Shein SA, Korchagina AA, Gurina OI. VEGF in tumor progression and targeted therapy. Curr Cancer Drug Targets 2013; 13:423-43; PMID:23167597; http://dx.doi.org/10.2174/15680096113139990074
- Koehler BC, Jager D, Schulze-Bergkamen H. Targeting cell death signaling in colorectal cancer: current strategies and future perspectives. World J Gastroenterol 2014; 20:1923-34; PMID:24587670; http://dx.doi.org/10.3748/wjg.v20.i8.1923
- Hong Y, Won J, Lee Y, Lee S, Park K, Chang K-T, Hong Y. Melatonin treatment induces interplay of apoptosis, autophagy, and senescence in human colorectal cancer cells. J Pineal Res 2014; 56:264-74; PMID:24484372; http://dx.doi.org/10.1111/jpi.12119
- Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist 2006; 11:342-57; PMID:16614230; http://dx.doi.org/10.1634/theoncologist.11-4-342
- Yang PM, Lin YT, Shun CT, Lin SH, Wei TT, Chuang SH, Wu MS, Chen CC. Zebularine inhibits tumorigenesis and stemness of colorectal cancer via p53-dependent endoplasmic reticulum stress. Sci Rep 2013; 3:3219; PMID:24225777
- Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A 2010; 107(43):18511-6; PMID:20937856; http://dx.doi.org/10.1073/pnas.1006124107
- Hiss DC, Gabriels GA. Implications of endoplasmic reticulum stress, the unfolded protein response and apoptosis for molecular cancer therapy. Part I: targeting p53, Mdm2, GADD153/CHOP, GRP78/BiP and heat shock proteins. Expert Opin Drug Discov 2009; 4:799-821; PMID:23496268; http://dx.doi.org/10.1517/17460440903052559
- Koukourakis MI, Giatromanolaki A, Sivridis E, Pitiakoudis M, Gatter KC, Harris AL. Beclin 1 over- and underexpression in colorectal cancer: distinct patterns relate to prognosis and tumour hypoxia. Br J Cancer 2010; 103:1209-14; PMID:20842118; http://dx.doi.org/10.1038/sj.bjc.6605904
- Goel A, Aggarwal BB. Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr Cancer 2010; 62:919-30; PMID:20924967; http://dx.doi.org/10.1080/01635581.2010.509835
- Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010; 10:1-11; PMID:20047689; http://dx.doi.org/10.1186/1471-2407-10-370
- Hiss DC, Gabriels GA. Implications of endoplasmic reticulum stress, the unfolded protein response and apoptosis for molecular cancer therapy. Part II: targeting cell cycle events, caspases, NF-κB and the proteasome. Exp Opin Drug Dis 2009; 4:907-21; PMID:23480539; http://dx.doi.org/10.1517/17460440903055032
- Nishikawa T, Tsuno NH, Okaji Y, Shuno Y, Sasaki K, Hongo K, Sunami E, Kitayama J, Takahashi K, Nagawa H. Inhibition of autophagy potentiates sulforaphane-induced apoptosis in human colon cancer cells. Ann Surg Oncol 2010; 17:592-602; PMID:19830499; http://dx.doi.org/10.1245/s10434-009-0696-x
- Bijnsdorp IV, Peters GJ, Temmink OH, Fukushima M, Kruyt FA. Differential activation of cell death and autophagy results in an increased cytotoxic potential for trifluorothymidine compared to 5-fluorouracil in colon cancer cells. Int J Cancer 2010; 126:2457-68; PMID:19816940
- Sasaki K, Tsuno NH, Sunami E, Kawai K, Hongo K, Hiyoshi M, Kaneko M, Murono K, Tada N, Nirei T, et al. Resistance of colon cancer to 5-fluorouracil may be overcome by combination with chloroquine, an in vivo study. Anticancer Drugs 2012; 23:675-82; PMID:22561420; http://dx.doi.org/10.1097/CAD.0b013e328353f8c7
- Schonewolf CA, Mehta M, Schiff D, Wu H, Haffty BG, Karantza V, Jabbour SK. Autophagy inhibition by chloroquine sensitizes HT-29 colorectal cancer cells to concurrent chemoradiation. World J Gastrointest Oncol 2014; 6:74-82; PMID:24653797
- Shi Y, Tang B, Yu PW, Hao YX, Lei X, Luo HX, Zeng DZ. Autophagy protects against oxaliplatin-induced cell death via ER stress and ROS in Caco-2 cells. PloS one 2012; 7:e51076; PMID:23226467; http://dx.doi.org/10.1371/journal.pone.0051076
- Xie CM, Chan WY, Yu S, Zhao J, Cheng CH. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Radic Biol Med 2011; 51:1365-75; PMID:21763418; http://dx.doi.org/10.1016/j.freeradbiomed.2011.06.016
- Wu YN, Wu PC, Yang LX, Ratinac KR, Thordarson P, Jahn KA, Chen DH, Shieh DB, Braet F. The anticancer properties of iron core-gold shell nanoparticles in colorectal cancer cells. Int J Nanomedicine 2013; 8:3321-31; PMID:24039416; http://dx.doi.org/10.2147/IJN.S47742
- Fels DR, Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther 2006; 5:723-8; PMID:16861899; http://dx.doi.org/10.4161/cbt.5.7.2967
- Paridaens A, Laukens D, Vandewynckel YP, Coulon S, Van Vlierberghe H, Geerts A, Colle I. Endoplasmic reticulum stress and angiogenesis: is there an interaction between them? Liver Int 2014; 34:e10-8; PMID:24393274; http://dx.doi.org/10.1111/liv.12457
- Tai CJ, Wang CK, Tai CJ, Lin YF, Lin CS, Jian JY, Chang YJ, Chang CC. Aqueous extract of solanum nigrum leaves induces autophagy and enhances cytotoxicity of cisplatin, doxorubicin, docetaxel, and 5-Fluorouracil in human colorectal carcinoma cells. Evid Based Complement Alternat Med 2013; 2013:12
- Kim SY, Song X, Zhang L, Bartlett DL, Lee YJ. Role of Bcl-xL/Beclin-1 in interplay between apoptosis and autophagy in oxaliplatin and bortezomib-induced cell death. Biochem Pharmacol 2014; 88:178-88; PMID:24486574; http://dx.doi.org/10.1016/j.bcp.2014.01.027
- Kaneko M, Nozawa H, Hiyoshi M, Tada N, Murono K, Nirei T, Emoto S, Kishikawa J, Iida Y, Sunami E, et al. Temsirolimus and chloroquine cooperatively exhibit a potent antitumor effect against colorectal cancer cells. J Cancer Res Clin Oncol 2014; 140:769-81; PMID:24619662; http://dx.doi.org/10.1007/s00432-014-1628-0
- Szokalska A, Makowski M, Nowis D, Wilczynski GM, Kujawa M, Wojcik C, Mlynarczuk-Bialy I, Salwa P, Bil J, Janowska S, et al. Proteasome inhibition potentiates antitumor effects of photodynamic therapy in mice through induction of endoplasmic reticulum stress and unfolded protein response. Cancer Res 2009; 69:4235-43; PMID:19435917; http://dx.doi.org/10.1158/0008-5472.CAN-08-3439
- Wei M-F, Chen M-W, Chen K-C, Lou P-J, Lin SY-F, Hung S-C, Hsiao M, Yao CJ, Shieh MJ. Autophagy promotes resistance to photodynamic therapy-induced apoptosis selectively in colorectal cancer stem-like cells. Autophagy 2014; 10:1179-92; PMID:24905352; http://dx.doi.org/10.4161/auto.28679
- Likus W, Siemianowicz K, Bieńk K, Pakuła M, Pathak H, Dutta C, Wang Q, Shojaei S, Assaraf YG, Ghavami S, et al. Mevalonate signaling cascade and (cancer) cell stemness. Drug Resist Updat 2016; 25:13-25; PMID:27155373; http://dx.doi.org/10.1016/j.drup.2016.02.001
- Chaabane W, Cieslar-Pobuda A, El-Gazzah M, Jain MV, Rzeszowska-Wolny J, Rafat M, et al. Human-gyrovirus-Apoptin triggers mitochondrial death pathway–Nur77 is required for apoptosis triggering. Neoplasia (New York, NY) 2014; 16:679-93; http://dx.doi.org/10.1016/j.neo.2014.08.001
- Jain MV, Jangamreddy JR, Grabarek J, Schweizer F, Klonisch T, Cieslar-Pobuda A, Łos MJ. Nuclear localized Akt enhances breast cancer stem-like cells through counter-regulation of p21(Waf1/Cip1) and p27(kip1). Cell Cycle (Georgetown, Tex) 2015; 14:2109-20; PMID:26030190; http://dx.doi.org/10.1080/15384101.2015.1041692
- Savelyeva A, Ghavami S, Davoodpour P, Asoodeh A, Los MJ. An overview of Brevinin superfamily: structure, function and clinical perspectives. Adv Exp Med Biol 2014; 818:197-212; PMID:25001538
- Chen D, Huang JF, Liu K, Zhang LQ, Yang Z, Chuai ZR, Wang YX, Shi DC, Huang Q, Fu WL. BRAFV600E mutation and its association with clinicopathological features of colorectal cancer: a systematic review and meta-analysis. PloS One 2014; 9:e90607; PMID:24594804; http://dx.doi.org/10.1371/journal.pone.0090607
- Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, Hamid O, Messersmith WA, Daud A, Kurzrock R, Pierobon M, et al. Combined BRAF and MEK inhibition with Dabrafenib and Trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol 2015; 33:4023-31; PMID:26392102; http://dx.doi.org/10.1200/JCO.2015.63.2471
- Kopetz S, Desai J, Chan E, Hecht JR, O'Dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Chung W, et al. Phase II pilot study of Vemurafenib in patients with Metastatic BRAF-mutated colorectal cancer. J Clin Oncol 2015; 33:4032-8; PMID:26460303; http://dx.doi.org/10.1200/JCO.2015.63.2497
- Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 2012; 379:1893-901; PMID:22608338; http://dx.doi.org/10.1016/S0140-6736(12)60398-5
- Yaeger R, Cercek A, O'Reilly EM, Reidy DL, Kemeny N, Wolinsky T, Capanu M, Gollub MJ, Rosen N, Berger MF, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res 2015; 21:1313-20; PMID:25589621; http://dx.doi.org/10.1158/1078-0432.CCR-14-2779
- Rad R, Cadinanos J, Rad L, Varela I, Strong A, Kriegl L, Constantino-Casas F, Eser S, Hieber M, Seidler B, et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell 2013; 24:15-29; PMID:23845441; http://dx.doi.org/10.1016/j.ccr.2013.05.014
- Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L, Wang WV, Vecchione L, de Vriendt V, Weinstein BJ, et al. Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF(V600E) colorectal cancer. Clin Cancer Res 2013; 19:2688-98; PMID:23549875; http://dx.doi.org/10.1158/1078-0432.CCR-12-2556
- Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757-65; PMID:18946061; http://dx.doi.org/10.1056/NEJMoa0804385
- De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010; 11:753-62; PMID:20619739; http://dx.doi.org/10.1016/S1470-2045(10)70130-3
- De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010; 304:1812-20; PMID:20978259; http://dx.doi.org/10.1001/jama.2010.1535
- Modest DP, Brodowicz T, Stintzing S, Jung A, Neumann J, Laubender RP, Ocvirk J, Kurteva G, Papai Z, Knittelfelder R, et al. Impact of the specific mutation in KRAS codon 12 mutated tumors on treatment efficacy in patients with metastatic colorectal cancer receiving cetuximab-based first-line therapy: a pooled analysis of three trials. Oncology 2012; 83:241-7; PMID:22948721; http://dx.doi.org/10.1159/000339534
- Fiala O, Pesek M, Finek J, Benesova L, Belsanova B, Minarik M. The dominant role of G12C over other KRAS mutation types in the negative prediction of efficacy of epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Cancer Genet 2013; 206:26-31; PMID:23313110; http://dx.doi.org/10.1016/j.cancergen.2012.12.003
- Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 2009; 69:1851-7; PMID:19223544; http://dx.doi.org/10.1158/0008-5472.CAN-08-2466
- Cushman SM, Jiang C, Hatch AJ, Shterev I, Sibley AB, Niedzwiecki D, Venook AP, Owzar K, Hurwitz HI, Nixon AB. Gene expression markers of efficacy and resistance to cetuximab treatment in metastatic colorectal cancer: results from CALGB 80203 (Alliance). Clini Cancer Res 2015; 21:1078-86; PMID:25520391; http://dx.doi.org/10.1158/1078-0432.CCR-14-2313
- Dalerba P, Sahoo D, Paik S, Guo X, Yothers G, Song N, Wilcox-Fogel N, Forgó E, Rajendran PS, Miranda SP, et al. CDX2 as a prognostic biomarker in Stage II and Stage III colon cancer. N Engl J Med 2016; 374:211-22; PMID:26789870; http://dx.doi.org/10.1056/NEJMoa1506597
- Sanchez-Martin FJ, Bellosillo B, Gelabert-Baldrich M, Dalmases A, Canadas I, Vidal J, Martinez A, Argilés G, Siravegna G, Arena S, et al. The First-in-class Anti-EGFR antibody mixture Sym004 overcomes cetuximab resistance mediated by EGFR extracellular domain mutations in colorectal cancer. Clin Cancer Res 2016; 22:3260-7; PMID:26888827; http://dx.doi.org/10.1158/1078-0432.CCR-15-2400
- Cho J, Bass AJ, Lawrence MS, Cibulskis K, Cho A, Lee SN, Yamauchi M, Wagle N, Pochanard P, Kim N, et al. Colon cancer-derived oncogenic EGFR G724S mutant identified by whole genome sequence analysis is dependent on asymmetric dimerization and sensitive to cetuximab. Mol Cancer 2014; 13:141; PMID:24894453; http://dx.doi.org/10.1186/1476-4598-13-141
- Shen WD, Chen HL, Liu PF. EGFR gene copy number as a predictive biomarker for resistance to anti-EGFR monoclonal antibodies in metastatic colorectal cancer treatment: a meta-analysis. Chin J Cancer Res 2014; 26:59-71; PMID:24653627
- Lau T, Chan E, Callow M, Waaler J, Boggs J, Blake RA, Magnuson S, Sambrone A, Schutten M, Firestein R, et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res 2013; 73:3132-44; PMID:23539443; http://dx.doi.org/10.1158/0008-5472.CAN-12-4562
- Boidot R, Chevrier S, Julie V, Ladoire S, Ghiringhelli F. HRAS G13D, a new mutation implicated in the resistance to anti-EGFR therapies in colorectal cancer, a case report. Int J Colorectal Dis 2016; 31:1245-6; PMID:26561417; http://dx.doi.org/10.1007/s00384-015-2448-7
- Al-Aidaroos AQ, Yuen HF, Guo K, Zhang SD, Chung TH, Chng WJ, Chng WJ, Zeng Q. Metastasis-associated PRL-3 induces EGFR activation and addiction in cancer cells. J Clin Invest 2013; 123:3459-71; PMID:23867504; http://dx.doi.org/10.1172/JCI66824
- Lupini L, Bassi C, Mlcochova J, Musa G, Russo M, Vychytilova-Faltejskova P, Svoboda M, Sabbioni S, Nemecek R, Slaby O, et al. Prediction of response to anti-EGFR antibody-based therapies by multigene sequencing in colorectal cancer patients. BMC Cancer 2015; 15:808; PMID:26508446; http://dx.doi.org/10.1186/s12885-015-1752-5
- Collura A, Lagrange A, Svrcek M, Marisa L, Buhard O, Guilloux A, Wanherdrick K, Dorard C, Taieb A, Saget A, et al. Patients with colorectal tumors with microsatellite instability and large deletions in HSP110 T17 have improved response to 5-fluorouracil-based chemotherapy. Gastroenterology 2014; 146:401-11 e1; PMID:24512910; http://dx.doi.org/10.1053/j.gastro.2013.10.054
- Lee SJ, Li GG, Kim ST, Hong ME, Jang J, Yoon N, Ahn SM, Murphy D, Christiansen J, Wei G, et al. NTRK1 rearrangement in colorectal cancer patients: evidence for actionable target using patient-derived tumor cell line. Oncotarget 2015; 6:39028-35; PMID:26472021
- Arcaroli JJ, Powell RW, Varella-Garcia M, McManus M, Tan AC, Quackenbush KS, Pitts TM, Gao D, Spreafico A, Dasari A, et al. ALDH+ tumor-initiating cells exhibiting gain in NOTCH1 gene copy number have enhanced regrowth sensitivity to a gamma-secretase inhibitor and irinotecan in colorectal cancer. Mol Oncol 2012; 6:370-81; PMID:22521243; http://dx.doi.org/10.1016/j.molonc.2012.03.004
- Tian L, Song S, Liu X, Wang Y, Xu X, Hu Y, Xu J. Schlafen-11 sensitizes colorectal carcinoma cells to irinotecan. Anticancer Drugs 2014; 25:1175-81; PMID:25089570; http://dx.doi.org/10.1097/CAD.0000000000000151
- Nygard SB, Christensen IJ, Nielsen SL, Nielsen HJ, Brunner N, Spindler KL. Assessment of the topoisomerase I gene copy number as a predictive biomarker of objective response to irinotecan in metastatic colorectal cancer. Scand J Gastroenterol 2014; 49:84-91; PMID:24256029; http://dx.doi.org/10.3109/00365521.2013.856464
- Santi DV, McHenry CS, Sommer H. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry 1974; 13:471-81; PMID:4203910; http://dx.doi.org/10.1021/bi00700a012
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nature Rev Cancer 2003; 3:330-8; http://dx.doi.org/10.1038/nrc1074
- Abdallah EA, Fanelli MF, Buim MEC, Machado Netto MC, Gasparini Junior JL, Souza e Silva V, Dettino AL, Mingues NB, Romero JV, Ocea LM, et al. Thymidylate synthase expression in circulating tumor cells: A new tool to predict 5‐fluorouracil resistance in metastatic colorectal cancer patients. Inter J Cancer 2015; 137:1397-405; PMID:25721610; http://dx.doi.org/10.1002/ijc.29495
- Wyatt MD, Wilson DM, III. Participation of DNA repair in the response to 5-fluorouracil. Cell Mol Life Sci 2009; 66:788-99; PMID:18979208; http://dx.doi.org/10.1007/s00018-008-8557-5
- Ghoshal K, Jacob ST. An alternative molecular mechanism of action of 5-fluorouracil, a potent anticancer drug. Biochem Pharmacol 1997; 53:1569-75; PMID:9264308; http://dx.doi.org/10.1016/S0006-2952(97)00040-3
- Zhou X, Wang W, Li P, Zheng Z, Tu Y, Zhang Y, You T. Curcumin Enhances the effects of 5-Fluorouracil and oxaliplatin in inducing gastric cancer cell apoptosis both in vitro and in vivo. Oncol Res 2016; 23:29-34; PMID:26802648; http://dx.doi.org/10.3727/096504015X14452563486011
- Amankwatia E, Chakravarty P, Carey F, Weidlich S, Steele R, Munro A, Wolf CR, Smith G. MicroRNA-224 is associated with colorectal cancer progression and response to 5-fluorouracil-based chemotherapy by KRAS-dependent and-independent mechanisms. Br J Cancer 2015; 112:1480-90; PMID:25919696; http://dx.doi.org/10.1038/bjc.2015.125
- Sun Z, Zhou N, Han Q, Zhao L, Bai C, Chen Y, Zhou J, Zhao RC. MicroRNA-197 influences 5-fluorouracil resistance via thymidylate synthase in colorectal cancer. Clin Transl Oncol 2015; 17:876-83; PMID:26055341; http://dx.doi.org/10.1007/s12094-015-1318-7
- Rosmarin D, Palles C, Pagnamenta A, Kaur K, Pita G, Martin M, Domingo E, Jones A, Howarth K, Freeman-Mills L, et al. A candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMS. Gut 2015; 64:111-20; PMID:24647007; http://dx.doi.org/10.1136/gutjnl-2013-306571
- Buhrmann C, Shayan P, Kraehe P, Popper B, Goel A, Shakibaei M. Resveratrol induces chemosensitization to 5-fluorouracil through up-regulation of intercellular junctions, Epithelial-to-mesenchymal transition and apoptosis in colorectal cancer. Biochem Pharmacol 2015; 98:51-68; PMID:26310874; http://dx.doi.org/10.1016/j.bcp.2015.08.105
- Li S, Gao J, Gu J, Yuan J, Hua D, Shen L. MicroRNA-215 inhibits relapse of colorectal cancer patients following radical surgery. Med Oncol 2013; 30:1-5
- Nishida N, Yamashita S, Mimori K, Sudo T, Tanaka F, Shibata K, Yamamoto H, Ishii H, Doki Y, Mori M. MicroRNA-10b is a prognostic indicator in colorectal cancer and confers resistance to the chemotherapeutic agent 5-fluorouracil in colorectal cancer cells. Annal Surg Oncol 2012; 19:3065-71; PMID:22322955; http://dx.doi.org/10.1245/s10434-012-2246-1
- Allen WL, Stevenson L, Coyle VM, Jithesh PV, Proutski I, Carson G, Gordon MA, Lenz HJ, Van Schaeybroeck S, Longley DB, et al. A systems biology approach identifies SART1 as a novel determinant of both 5-fluorouracil and SN38 drug resistance in colorectal cancer. Mol Cancer Ther 2012; 11:119-31; PMID:22027693; http://dx.doi.org/10.1158/1535-7163.MCT-11-0510
- Yang C, Cui X, Dai X, Liao W. Downregulation of Foxc2 enhances apoptosis induced by 5-fluorouracil through activation of MAPK and AKT pathways in colorectal cancer. Oncol Lett 2016; 11:1549-54; PMID:26893778
- Berindan-Neagoe I, Braicu C, Pileczki V, Petric RC, Miron N, Balacescu O, Iancu D, Ciuleanu T. 5-fluorouracil potentiates the anti-cancer effect of oxaliplatin on Colo320 colorectal adenocarcinoma cells. J Gastrointestin Liver Dis 2013; 22:37-43; PMID:23539389
- Paschall A, Yang D, Li X, Choi J-H, Liu F, Figueroa M, Oberlies NH, Pearce C, Bollag WB, Nayak-Kapoor A, et al. H3K9 trimethylation silences Fas expression to confer colon carcinoma immune escape and chemoresistance (IRM6P. 654). J Immunol 2015; 194:1868-82; http://dx.doi.org/10.4049/jimmunol.1402243
- Lourencao BC, Medeiros RA, Thomasi SS, Ferreira AG, Rocha-Filho RC, Fatibello-Filho O. Amperometric flow-injection determination of the anthelmintic drugs ivermectin and levamisole using electrochemically pretreated boron-doped diamond electrodes. Sensors and Actuators B: Chemical 2016; 222:181-9; http://dx.doi.org/10.1016/j.snb.2015.08.036
- Maude RJ, Silamut K, Plewes K, Charunwatthana P, Ho M, Faiz MA, Hassan MU, Bin Yunus E, Hoque G, Islam F, et al. Randomized controlled trial of levamisole hydrochloride as adjunctive therapy in severe falciparum malaria with high parasitemia. J Infect Dis 2014; 209:120-9; PMID:23943850; http://dx.doi.org/10.1093/infdis/jit410
- Patil U, Jaydeokar A, Bandawane D. Immunomodulators: A pharmacological review. Int J Pharm Pharm Sci 2012; 4:30-6
- Buchanan JA, Lavonas EJ. Agranulocytosis and other consequences due to use of illicit cocaine contaminated with levamisole. Curr Opin Hematol 2012; 19:27-31; PMID:22143075; http://dx.doi.org/10.1097/MOH.0b013e32834da9ef
- Sakurai H, Kubota K, Inaba S-i, Takanaka K, Shinagawa A. Identification of a metabolizing enzyme in human kidney by proteomic correlation profiling. Mol Cell Proteomics 2013; 12:2313-23; PMID:23674616; http://dx.doi.org/10.1074/mcp.M112.023853
- Allegra CJ, Parr AL, Wold LE, Mahoney MR, Sargent DJ, Johnston P, Klein P, Behan K, O'Connell MJ, Levitt R, et al. Investigation of the prognostic and predictive value of thymidylate synthase, p53, and Ki-67 in patients with locally advanced colon cancer. J Clin Oncol 2002; 20:1735-43; PMID:11919229; http://dx.doi.org/10.1200/JCO.2002.07.080
- McLeod H, Murray G. Tumour markers of prognosis in colorectal cancer. Br J Cancer 1999; 79:191; PMID:9888457; http://dx.doi.org/10.1038/sj.bjc.6690033
- Liao VH-C, Liu J-T, Li W-H, Yu C-W, Hsieh Y-C. Caenorhabditis elegans bicarbonate transporter ABTS-1 is involved in arsenite toxicity and cholinergic signaling. Chem Res Toxicol 2010; 23:926-32; PMID:20423156; http://dx.doi.org/10.1021/tx100016e
- Kozlenkov A, Le Du MH, Cuniasse P, Ny T, Hoylaerts MF, Millán JL. Residues determining the binding specificity of uncompetitive inhibitors to tissue‐nonspecific alkaline phosphatase. J Bone Miner Res 2004; 19:1862-72; PMID:15476587; http://dx.doi.org/10.1359/JBMR.040608
- Sakai H, Kokura S, Ishikawa T, Tsuchiya R, Okajima M, Matsuyama T, Adachi S, Katada K, Kamada K, Uchiyama K, et al. Effects of anticancer agents on cell viability, proliferative activity and cytokine production of peripheral blood mononuclear cells. J Clin Biochem Nutr 2013; 52:64; PMID:23341700; http://dx.doi.org/10.3164/jcbn.12-60
- Chia WK, Ali R, Toh HC. Aspirin as adjuvant therapy for colorectal cancer—reinterpreting paradigms. Nat Rev Clin Oncol 2012; 9:561-70; PMID:22910681; http://dx.doi.org/10.1038/nrclinonc.2012.137
- Kandioler D, Mittlböck M, Kappel S, Puhalla H, Herbst F, Langner C, Wolf B, Tschmelitsch J, Schippinger W, Steger G, et al. TP53 mutational status and prediction of benefit from adjuvant 5-fluorouracil in stage III colon cancer patients. EBioMedicine 2015; 2:823-8; http://dx.doi.org/10.1016/j.ebiom.2015.06.003
- Siegelin MD, Altieri DC. Combination therapies with mitochondrial-targeted anti-tumor agents. us20110268722A1; 2011 Nov. 3; patent application publication; 65 pages.
- Marin JJ, Sanchez de Medina F, Castaño B, Bujanda L, Romero MR, Martinez-Augustin O, Moral-Avila RD, Briz O. Chemoprevention, chemotherapy, and chemoresistance in colorectal cancer. Drug Metab Rev 2012; 44:148-72; PMID:22497631; http://dx.doi.org/10.3109/03602532.2011.638303
- Ge R, Rajeev V, Ray P, Lattime E, Rittling S, Medicherla S, Protter A, Murphy A, Chakravarty J, Dugar S, et al. Inhibition of growth and metastasis of mouse mammary carcinoma by selective inhibitor of transforming growth factor-β type I receptor kinase in vivo. Clin Cancer Res 2006; 12:4315-30; PMID:16857807; http://dx.doi.org/10.1158/1078-0432.CCR-06-0162
- Caruso M-E, Jenna S, Bouchecareilh M, Baillie DL, Boismenu D, Halawani D, Latterich M, Chevet E. GTPase-mediated regulation of the unfolded protein response in Caenorhabditis elegans is dependent on the AAA+ ATPase CDC-48. Mol Cell Biol 2008; 28:4261-74; PMID:18458060; http://dx.doi.org/10.1128/MCB.02252-07
- Li W, Zou W, Yang Y, Chai Y, Chen B, Cheng S, Tian D, Wang X, Vale RD, Ou G. Autophagy genes function sequentially to promote apoptotic cell corpse degradation in the engulfing cell. J Cell Biol 2012; 197:27-35; PMID:22451698; http://dx.doi.org/10.1083/jcb.201111053
- Latheef S, Devanabanda M, Sankati S, Madduri R. Differential expression of alkaline phosphatase gene in proliferating primary lymphocytes and malignant lymphoid cell lines. Immunol Lett 2016; 170:37-41; PMID:26730846; http://dx.doi.org/10.1016/j.imlet.2015.12.008
- Staab TA, Griffen TC, Corcoran C, Evgrafov O, Knowles JA, Sieburth D. The conserved SKN-1/Nrf2 stress response pathway regulates synaptic function in Caenorhabditis elegans. PLoS Genet 2013; 9:e1003354; PMID:23555279; http://dx.doi.org/10.1371/journal.pgen.1003354
- Prummel MF, Brokken LJ, Meduri G, Misrahi M, Bakker O, Wiersinga WM. Expression of the thyroid-stimulating hormone receptor in the folliculo-stellate cells of the human anterior pituitary. J Clin Endocrinol Metab 2000; 85:4347-53; PMID:11095478; http://dx.doi.org/10.1210/jcem.85.11.6991
- Bennett MK, Gross MI, Bromley SD, Li J, Chen L, Goyal B, et al. Treatment of cancer with heterocyclic inhibitors of glutaminase. us2015/0004134A1; 2015 1 Jan.; patent application publication; 326 pages
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nature Rev Cancer 2007; 7:573-84; http://dx.doi.org/10.1038/nrc2167
- Zhang S, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, Lapuk A, Chen Y, Komori T, Gray JW, Chen X, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Research 2006; 66:8847-57; PMID:16951202; http://dx.doi.org/10.1158/0008-5472.CAN-06-0769
- Maclean D, Khokhar A, Tyle P, Perez-Soler R. Intraliposomal chemical activation patterns of liposomal cis-bis-neodecanoato-trans-R, R-1, 2-diaminocyclohexane platinum (II)(L-NDDP)-a potential antitumour agent. J Microencapsul 2000; 17:307-22; PMID:10819419; http://dx.doi.org/10.1080/026520400288283
- Cui X, Clark DN, Liu K, Xu X-D, Guo J-T, Hu J. Viral DNA-dependent induction of innate immune response to hepatitis B virus in immortalized mouse hepatocytes. J Virol 2016; 90:486-96; http://dx.doi.org/10.1128/JVI.01263-15
- Oguri T, Mitsuma A, Inada-Inoue M, Morita S, Shibata T, Shimokata T, Sugishita M, Nakayama G, Uehara K, Hasegawa Y, et al. Genetic polymorphisms associated with oxaliplatin-induced peripheral neurotoxicity in Japanese patients with colorectal cancer. Int J Clin Pharmacol Ther 2013; 51:475-81; PMID:23547850; http://dx.doi.org/10.5414/CP201851
- Won HH, Lee J, Park JO, Park YS, Lim HY, Kang WK, Kim JW, Lee SY, Park SH. Polymorphic markers associated with severe oxaliplatin-induced, chronic peripheral neuropathy in colon cancer patients. Cancer 2012; 118:2828-36; PMID:22020760; http://dx.doi.org/10.1002/cncr.26614
- Geva R, Shamai S, Brazowsky E, Paoulas M, Ben-Haim M, Johnstone E, Alex B, Shacham-Shmueli E. The predictive role of ERCC1 status in Oxaliplatin based neoadjuvant therapy for metastatic Colorectal Cancer (mCRC) to the Liver. Cancer Invest 2015; 33:89-97;PMID:25723812;http://dx.doi.org/10.3109/07357907.2014.998834
- Perego P, Robert J. Oxaliplatin in the era of personalized medicine: from mechanistic studies to clinical efficacy. Cancer Chemother Pharmacol 2016; 77:5-18; PMID:26589793; http://dx.doi.org/10.1007/s00280-015-2901-x
- Tang D, Kang R, Cheh C-W, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010; 29:5299-310; PMID:20622903; http://dx.doi.org/10.1038/onc.2010.261
- Ishtikhar M, Khan MV, Khan S, Chaturvedi SK, Badr G, Mahmoud MH, Khan RH. Biophysical and molecular docking insight into interaction mechanism and thermal stability of human serum albumin isoforms with a semi-synthetic water-soluble camptothecin analog irinotecan hydrochloride. J Biomol Struct Dyn 2016; 34(7):1545-60; PMID:26309154; http://dx.doi.org/10.1080/07391102.2015.1082504
- Chaniyara R, Tala S, Chen C-W, Zang X, Kakadiya R, Lin LF, Chen CH, Chien SI, Chou TC, Tsai TH, et al. Novel antitumor indolizino [6, 7-b] indoles with multiple modes of action: DNA cross-linking and topoisomerase I and II inhibition. J Med Chem 2013; 56:1544-63; PMID:23360284; http://dx.doi.org/10.1021/jm301788a
- Kaczirek K, Ciuleanu TE, Vrbanec D, Marton E, Messinger D, Liegl-Atzwanger B, Wrba F, Knittelfelder R, Lindner E, Zielinski CC, et al. FOLFOX4 Plus Cetuximab for patients with previously untreated metastatic colorectal cancer according to Tumor RAS and BRAF mutation status: Updated analysis of the CECOG/CORE 1.2. 002 study. Clin Colorectal Cancer 2015; 14:91-8; PMID:25666295; http://dx.doi.org/10.1016/j.clcc.2014.12.003
- Liu X, Cheng D, Kuang Q, Liu G, Xu W. Association of UGT1A1* 28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: a meta-analysis in Caucasians. Pharmacogenomics J 2014; 14:120-9; PMID:23529007; http://dx.doi.org/10.1038/tpj.2013.10
- Zhang G, Wang Z, Qian F, Zhao C, Sun C. Silencing of the ABCC4 gene by RNA interference reverses multidrug resistance in human gastric cancer. Oncol Rep 2015; 33:1147-54; PMID:25572969
- Peters GJ, Giovannetti E. Research Highlights: Highlights from the latest articles in pharmacogenomics of irinotecan/cisplatin toxicity. Pharmacogenomics 2012; 13:1445; PMID:23057543; http://dx.doi.org/10.2217/pgs.12.134
- Van Cutsem E, Lenz H-J, Köhne C-H, Heinemann V, Tejpar S, Melezínek I, Beier F, Stroh C, Rougier P, van Krieken JH, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 2015; 33:692-700; PMID:25605843; http://dx.doi.org/10.1200/JCO.2014.59.4812
- Chaudhuri AR, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 2012; 19:417-23; PMID:22388737; http://dx.doi.org/10.1038/nsmb.2258
- Saif MW. Targeting cancers in the gastrointestinal tract: role of capecitabine. Onco Targets Ther 2009; 2:29; PMID:20616892; http://dx.doi.org/10.2147/OTT.S3469
- Abd-Elzaher MM, Labib AA, Mousa HA, Moustafa SA, Abdallah MM. Synthesis, characterization and cytotoxic activity of ferrocenyl hydrazone complexes containing a furan moiety. Res Chem Intermediat 2014; 40:1923-36; http://dx.doi.org/10.1007/s11164-013-1090-7
- Deenen MJ, Tol J, Burylo AM, Doodeman VD, de Boer A, Vincent A, Guchelaar HJ, Smits PH, Beijnen JH, Punt CJ, et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin Cancer Res 2011; 17:3455-68; PMID:21498394; http://dx.doi.org/10.1158/1078-0432.CCR-10-2209
- O'Donnell PH, Stark AL, Gamazon ER, Wheeler HE, McIlwee BE, Gorsic L, Im HK, Huang RS, Cox NJ, Dolan ME. Identification of novel germline polymorphisms governing capecitabine sensitivity. Cancer 2012; 118:4063-73; PMID:22864933; http://dx.doi.org/10.1002/cncr.26737
- Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013; 32:805-18; PMID:22508478; http://dx.doi.org/10.1038/onc.2012.130
- Marechal R, Mackey JR, Lai R, Demetter P, Peeters M, Polus M, Cass CE, Salmon I, Devière J, Van Laethem JL. Deoxycitidine kinase is associated with prolonged survival after adjuvant gemcitabine for resected pancreatic adenocarcinoma. Cancer 2010; 116:5200-6; PMID:20669326; http://dx.doi.org/10.1002/cncr.25303
- Rosell R, Lord RV, Taron M, Reguart N. DNA repair and cisplatin resistance in non-small-cell lung cancer. Lung Cancer 2002; 38:217-27; PMID:12445742; http://dx.doi.org/10.1016/S0169-5002(02)00224-6
- Honeywell RJ, van Haperen VWR, Veerman G, Smid K, Peters GJ. Inhibition of thymidylate synthase by 2′, 2′-difluoro-2′-deoxycytidine (Gemcitabine) and its metabolite 2′, 2′-difluoro-2′-deoxyuridine. Int J Biochem Cell Biol 2015; 60:73-81; PMID:25562513; http://dx.doi.org/10.1016/j.biocel.2014.12.010
- Giovannetti E, Mey V, Nannizzi S, Pasqualetti G, Marini L, Del Tacca M, Danesi R. Cellular and pharmacogenetics foundation of synergistic interaction of pemetrexed and gemcitabine in human non–small-cell lung cancer cells. Mol Pharmacol 2005; 68:110-8; PMID:15795320
- Sakharkar MK, Sakharkar KR. Targetability of human disease genes. Curr Drug Discov Technol 2007; 4:48-58; PMID:17630928; http://dx.doi.org/10.2174/157016307781115494
- Zhang X, Yashiro M, Ren J, Hirakawa K. Histone deacetylase inhibitor, trichostatin A, increases the chemosensitivity of anticancer drugs in gastric cancer cell lines. Oncol Rep 2006; 16:563-8; PMID:16865256
- Innocenti F, Owzar K, Cox NL, Evans P, Kubo M, Zembutsu H, et al. A genome-wide association study of overall survival in pancreatic cancer patients treated with gemcitabine in CALGB 80303. Clin Cancer Res 2012; 18:577-84; PMID:22142827; http://dx.doi.org/10.1158/1078-0432.CCR-11-1387
- Jin K, Teng L, Shen Y, He K, Xu Z, Li G. Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin Transl Oncol 2010; 12:473-80; PMID:20615824; http://dx.doi.org/10.1007/s12094-010-0540-6
- Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 2012; 90:85-94; PMID:22124371; http://dx.doi.org/10.1038/icb.2011.100
- Porcelli L, E Quatrale A, Mantuano P, Silvestris N, E Brunetti A, Calvert H, Paradiso A, Azzariti A. Synthetic lethality to overcome cancer drug resistance. Currnt Med Chem 2012; 19:3858-73; PMID:22788762; http://dx.doi.org/10.2174/092986712802002563
- Marzban H, Del Bigio MR, Alizadeh J, Ghavami S, Zachariah RM, Rastegar M. Cellular commitment in the developing cerebellum. Front Cell Neurosci 2014; 8:450