
ABSTRACT
Macroautophagy/autophagy, which is one of the main degradation systems in the cell, is mediated by a specialized organelle, the autophagosome. Purification of autophagosomes before fusion with lysosomes is important for both mechanistic and physiological studies of the autophagosome. Here, we report a simple method to accumulate undigested autophagosomes. Overexpression of the autophagosomal Qa-SNARE STX17 (syntaxin 17) lacking the N-terminal domain (NTD) or N-terminally tagged GFP-STX17 causes accumulation of autophagosomes. A HeLa cell line, which expresses GFP-STX17ΔNTD or full-length GFP-STX17 under the control of the tetracycline-responsive promoter, accumulates a large number of undigested autophagosomes devoid of lysosomal markers or early autophagy factors upon treatment with doxycycline. Using this inducible cell line, nascent autophagosomes can be easily purified by OptiPrep density-gradient centrifugation and immunoprecipitation. This novel method should be useful for further characterization of nascent autophagosomes.
Introduction
Macroautophagy (hereafter referred to as autophagy) is one of the main degradation systems in eukaryotic cells. At an initial step of autophagy, a flat membrane sac termed the phagophore appears on or close to the endoplasmic reticulum (ER). As the phagophore develops it engulfs a small part of the cytoplasm and matures to become a double-membraned structure termed the autophagosome. In more complex eukaryotes, at least some of the autophagosomes fuse with endocytic vesicles to generate hybrid organelle called amphisomes.Citation1 Finally, the autophagosomes or amphisomes fuse with lysosomes to become autolysosomes, and engulfed cytoplasmic materials are degraded by lysosomal enzymes.Citation2-Citation4
Intracellular membrane fusion generally depends on RAB GTPases, tethering factors, and SNARE proteins.Citation5,Citation6 In the process of membrane fusion, tethering factors interact with RABs on each membrane and then SNARE proteins form a 4-helix bundle to drive specific membrane fusion. SNARE proteins are classified into 4 groups: Qa-, Qb-, Qc-, and R-SNAREs. Four SNARE proteins from each group are assembled to mediate membrane fusion in general. Fusion between autophagosomes and lysosomes uses these SNARE and tethering complex systems. We recently found that STX17 (syntaxin 17; Qa) on autophagosomes interacts with SNAP29 (synaptosome associated protein 29; Qbc) and lysosomal VAMP8 (vesicle associated membrane protein 8; R) to mediate autophagosome-lysosome fusion in mammalian cells.Citation7 In addition, STX17 interacts with the tethering homotypic fusion and protein sorting (HOPS) complex, which is also required for autophagosome-lysosome fusion as well as endosome-lysosome fusion.Citation8 This mechanism is conserved in Drosophila,Citation9,Citation10 and Caenorhabditis elegans.Citation11 Autophagy-related (ATG) 14, an essential autophagy-specific regulator of the class III phosphatidylinositol 3-kinase complex, and EPG5, a RAB7- and LC3-binding protein, also facilitate the fusion between autophagosomes and endosomes or lysosomes through interaction with STX17.Citation12,Citation13
Purification of undegraded nascent autophagosomes, which have not yet fused with lysosomes, is critically important to understand the mechanisms and physiological functions of autophagy. However, methods to accumulate these structures for purification have been limited. Several chemicals have been used to attempt to block the fusion between autophagosomes and lysosomes. For example, microtubule-depolymerizing reagents such as nocodazole are somewhat effective but their effects are controversial and indirect.Citation14-Citation16 The vacuolar-type H+-ATPase inhibitor bafilomycin A1 was initially reported to block the fusion step, but the fusion-inhibitory effect appears to be dependent on treatment timeCitation17 and could be a secondary effect of inhibition of lysosomal acidificationCitation17 or a calcium pump.Citation18 In many cases, partially degraded autolysosomes accumulate in bafilomycin A1-treated cells. Other possible accumulation techniques are knockout of STX17 or VPS33A (VPS33A, CORVET/HOPS core subunit), but as these factors are also important for ER-mitochondria contact sitesCitation19,Citation20 or the endocytic pathway,Citation8,Citation10 respectively, it would be more ideal to induce acute inhibition, rather than constitutive blockade, of these pathways; however, siRNA-mediated knockdown is not suitable for obtaining a large amount of cellular sample for biochemical studies.
The cytosolic portion of SNARE proteins lacking their transmembrane domain (TMD) act in a dominant-negative manner when overexpressed.Citation21-Citation27 Thus, we sought to generate a dominant-negative mutant of STX17. We found that, in contrast to previously reported SNAREs, STX17 lacking its TMDs did not exert a dominant-negative effect. Instead, overexpression of STX17 lacking the N-terminal domain (STX17ΔNTD) or STX17 tagged with GFP at the N terminus effectively blocked autophagosome-lysosome fusion and autophagic flux. We established a doxycycline (DOX)-inducible GFP-STX17ΔNTD- or full-length GFP-STX17-expressing cell line. Undegraded nascent autophagosomes can accumulate upon simple DOX treatment, and can then be subjected to purification and biochemical studies of autophagosomes.
Results
LC3-positive puncta accumulate by overexpression of STX17ΔNTD
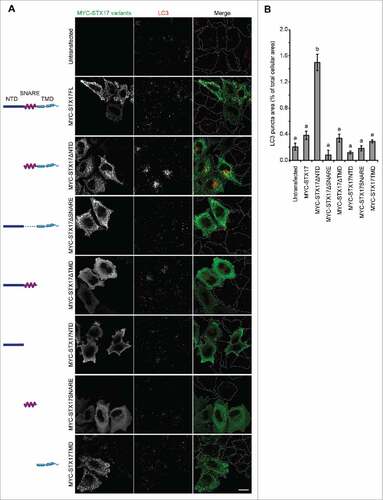
To block autophagosome-lysosome fusion and accumulate nascent autophagosomes, we sought to create a dominant-negative mutant of STX17 (). We created several deletion mutants of MYC-tagged STX17 lacking the NTD, SNARE domain, and/or TMDs. When expressed in HeLa cells, mutants containing the TMDs formed punctate structures, which could represent mitochondria and autophagosomes,Citation7 whereas those without the TMDs showed a cytosolic pattern (). In contrast to previous reports showing dominant-negative effects of SNAREs lacking TMDs, MYC-STX17ΔTMD did not act as a dominant-negative mutant. Instead, we observed clear accumulation of puncta positive for MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β), an established marker for autophagic membranes, only in cells overexpressing MYC-STX17ΔNTD ( and ).
Figure 1. LC3-positive puncta accumulate upon overexpression of STX17ΔNTD. (A) Wild-type (WT) HeLa cells were transiently transfected with plasmids expressing the indicated STX17 fragments. Two d later, cells were fixed under growing conditions and analyzed by immuofluorescence microscopy using anti-LC3 and anti-MYC antibodies. Scale bars: 20 μm. (B) Quantification of the ratio of the total area of LC3-positive puncta to the total cellular area expressing the indicated STX17 variants. Cells showing significant MYC signal intensities (10% or more of the highest signal among all of the variants) were selected from 5 or 6 randomly selected regions and 18–46 cells were analyzed. Data represent mean ± SEM. Different letters indicate significant differences between MYC-tagged STX17 variants at p < 0.01 by one-way ANOVA followed by Tukey's test.
Overexpression of GFP-STX17FL causes accumulation of LC3-positive puncta
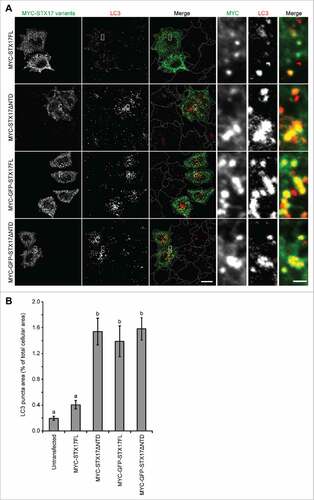
We also tested whether addition of a large tag such as GFP at the N terminus of STX17 produced a dominant-negative effect. To make direct comparison possible, we inserted GFP between the MYC tag and STX17. In contrast to full-length MYC-STX17 (MYC-STX17FL), MYC-GFP-STX17FL showed an accumulation of LC3 puncta in a level comparable to that caused by MYC-STX17ΔNTD ( and ). MYC-GFP-STX17ΔNTD showed a similar effect ( and ). All of MYC-GFP-STX17FL, MYC-STX17ΔNTD, and MYC-GFP-STX17ΔNTD mostly colocalized with LC3, suggesting that recruitment to the autophagosome was not affected. These results suggest that either deletion of the NTD or addition of a large molecule such as GFP at the N terminus causes a dominant-negative effect with STX17.
Figure 2. Overexpression of GFP-STX17FL causes accumulation of LC3-positive puncta. (A) WT HeLa cells were transiently transfected with the indicated plasmids. Two d later, cells were fixed under growing conditions and analyzed by immuofluorescence microscopy using anti-LC3 and anti-MYC antibodies. Scale bars: 20 μm; 2 μm in the magnified images (right). (B) Quantification was performed as in and 26–39 cells were analyzed. Different letters indicate significant differences between MYC- or MYC-GFP-tagged STX17 variants at p < 0.01 by one-way ANOVA followed by Tukey's test. The experiments were successfully performed twice.
Tetracycline-inducible GFP-STX17ΔNTD and GFP-STX17FL demonstrate a dominant-negative effect
To stably achieve overexpression of GFP-STX17ΔNTD or GFP-STX17FL to induce the dominant-negative effect, we established HeLa cell lines that can overexpress GFP-STX17ΔNTD or GFP-STX17FL under the control of the tetracycline-responsive promoter (TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cell lines). Although these cells expressed a low level of GFP-STX17ΔNTD and GFP-STX17FL even in the absence of DOX, they did not show the accumulation of LC3 puncta ( and ). However, when these cells were treated with 1.5 μg/ml DOX for 2 d, many LC3 puncta accumulated under both growing and starvation conditions (). These puncta were mostly positive for GFP-STX17ΔNTD or GFP-STX17FL.
Figure 3. The dominant-negative effect of GFP-STX17ΔNTD and GFP-STX17FL can be controlled using the tetracyclin-inducible system in HeLa cells. (A) WT and TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells were cultured with or without DOX (1.5 μg/ml). Two d later, cells were cultured in regular or starvation medium for 1 h and analyzed by immunofluorescence microscopy using anti-LC3 antibody. Scale bars: 20 μm; 2 μm in insets. The experiments were successfully performed twice. (B) WT and TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells were cultured with or without DOX (1.5 μg/ml) for 3 or 6 d. Subsequently, cells were cultured in regular or starvation medium with or without 100 nM bafilomycin A1 for 2 h and analyzed by immunoblotting using the indicated antibodies.
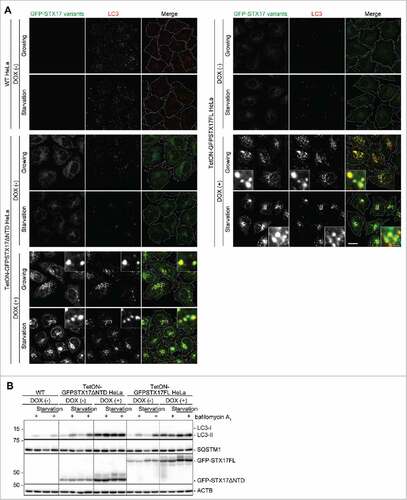
As accumulation of LC3 can represent either the activation of autophagy induction or suppression of autophagosome turnover at a late step, we measured the autophagic flux in TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells. When lysosomal degradation and/or fusion were blocked by treatment with bafilomycin A1, the level of LC3-II (the membrane form that is degraded by autophagy) was increased in wild-type HeLa cells and DOX-untreated TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells ().Citation28 When treated with DOX, LC3-II accumulated in both TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells under both growing and starvation conditions, but the amount of LC3-II was not increased further by bafilomycin A1 treatment (). In addition, SQSTM1/p62, another autophagy-specific substrate, also accumulated under growing conditions, but the amount of SQSTM1 was not reduced by starvation and was not increased further by bafilomycin A1 treatment. Thus, the autophagic flux was blocked almost completely following DOX treatment in TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells (). DOX treatment itself did not inhibit autophagic flux in wild-type cells (data not shown). These results suggest that simple addition of DOX can induce a dominant-negative effect against autophagy in both TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells.
Accumulated GFP-STX17ΔNTD and GFP-STX17FL puncta represent completed autophagosomes but not phagophores or autolysosomes
As LC3 can be present on phagophores (i.e., the elongating unclosed structure), completed autophagosomes (the closed form), and autolysosomes, we next determined whether the accumulated LC3-positive GFP-STX17ΔNTD or GFP-STX17FL structures indeed represented completed autophagosomes in DOX-treated TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells. GFP-STX17ΔNTD and GFP-STX17FL puncta rarely colocalized with phagophore markers such as RB1CC1/FIP200 and WIPI2 ( and ). The distribution patterns of RB1CC1 and WIPI2 were not affected by treatment with DOX (data not shown), suggesting that overexpression of GFP-STX17ΔNTD did not affect the formation of autophagosomes. These results suggest that phagophores do not accumulate upon DOX treatment.
Figure 4. Accumulated LC3-positive structures did not colocalize with phagophore and lysosome markers in DOX-treated TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells. (A-C) TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells were cultured with DOX (1.5 μg/ml) for 2 d. Cells were cultured in regular (Gr) or starvation (St) medium for 1 h and analyzed by immunofluorescence microscopy using antibodies against RB1CC1 (A), WIPI2 (B), and LAMP1 (C). Colocalization analysis was performed as described in Materials and Methods. Each correlation plot is derived from 30–61 cells in 3 different fields of view. The mean Pearson's correlation coefficient values ± SEM are shown on the plots and the graphs. The intensities of GFP-STX17 variants are represented on the x-axis. Asterisks indicate significant differences between MYC-tagged STX17 variants at p < 0.01 by t-test. Scale bars: 10 μm; 2 μm in insets.
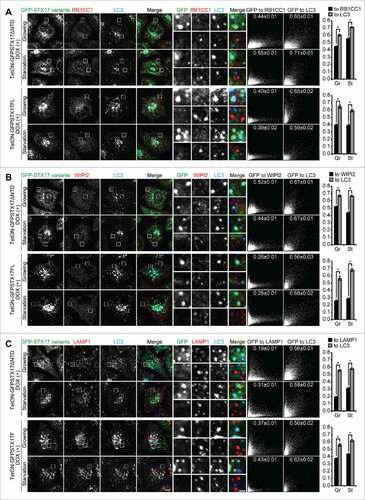
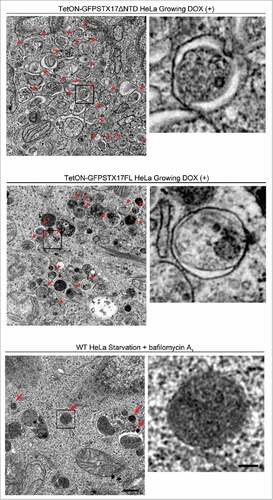
GFP-STX17ΔNTD and GFP-STX17FL puncta did not colocalize with the lysosomal marker LAMP1 (), even under chloroquine-treated conditions (Fig. S1). These structures of STX17 variants also did not colocalize with the early endosome marker EEA1, recycling endosome marker TFRC (transferrin receptor), and fluorescent dextran that localizes inside endocytic compartments (Fig. S2). Electron microscopy of DOX-treated cells confirmed accumulation of completed autophagosomes, which contained intact cytoplasmic materials without any features of degradation (). This appearance was different from autophagic structures generated in wild-type HeLa cells after treatment with bafilomycin A1, which were more electron dense (). These structures likely represented autolysosomes with impaired degradation of the internal contents. Furthermore, the number of accumulated autophagosomes in TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells was much larger than that of accumulated autolysosomes in wild-type HeLa cells treated with bafilomycin A1, indicating that autophagosome-lysosome fusion was much more efficiently blocked in DOX-treated TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells than in bafilomycin A1-treated wild-type HeLa cells. These results suggest that TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells effectively accumulate completed autophagosomes, not phagophores or autolysosomes, upon DOX treatment.
Figure 5. Undegraded autophagosomes but not phagophores or autolysosomes accumulate in TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells. TetON-GFPSTX17ΔNTD (top) and TetON-GFPSTX17FL (middle) HeLa cells were cultured with DOX (1.5 μg/ml) for 2 d. Wild-type (WT) HeLa cells were cultured in starvation medium with 100 nM bafilomycin A1 for 2 h (bottom). Cells were subjected to conventional electron microscopy analysis. Autophagosomes and autolysosomes are indicated by red arrowheads and arrows, respectively. Scale bars: 500 nm; 100 nm in insets. Representative images are shown.
Isolation of autophagosomes from TetON-GFPSTX17ΔNTD HeLa cells
To separate autophagosomes from other organelles, cell homogenates derived from TetON-GFPSTX17ΔNTD HeLa cells stably expressing FLAG-tagged LC3 were subjected to flotation analysis using OptiPrep density gradients. In the absence of DOX, LC3-II and a part of GFP-STX17ΔNTD and endogenous STX17 were distributed into medium-density fractions (fractions 7–11) together with other organelle markers, such as SEC61B (ER), LMAN1/ERGIC53 (ER-Golgi intermediate compartment), STX5 (cis-Golgi), TOMM20 (mitochondria), and LAMP1 (lysosome) (). Endogenous STX17, LC3-I, the autophagic substrate SQSTM1, and cytosolic markers HSP90AA1/HSP90 and LDH (lactate dehydrogenase) were mainly distributed in unfloated bottom fractions (fractions 12–14) (). By contrast, after DOX treatment, part of the population of LC3-II, GFP-STX17ΔNTD, endogenous STX17, and SQSTM1 were shifted to a lighter-density fraction (fraction 3), which did not contain the lysosome marker LAMP1 (). Distributions of the other organelle markers were mostly unchanged, though they were contained to a small extent in the lighter fractions; they can be considered as cytosol and organelles engulfed by autophagosomes (see below). These results suggest that autophagosomes can be separated from other organelles including autolysosomes.
Figure 6. Purification of autophagosomes. (A-D) TetON-GFPSTX17ΔNTD HeLa cells stably expressing FLAG-LC3 were cultured in the absence (A, C) or presence (B, D) of 1.5 μg/ml DOX for 2 d. After separation by the OptiPrep membrane flotation method, each fraction was analyzed by immunoblotting (A, B). The LDH activity of each fraction was measured under nontreated (closed circle) and Triton X-100-treated (open circle) conditions. Insets show magnified images of fractions 2–5 (C, D). The experiments were successfully performed twice. (E) FLAG-LC3-enriched membranes were precipitated from light-density fractions (fractions [Fr.] 3 and 4) in the absence of detergent using anti-FLAG antibody-coated magnetic beads and analyzed by immunoblotting.
![Figure 6. Purification of autophagosomes. (A-D) TetON-GFPSTX17ΔNTD HeLa cells stably expressing FLAG-LC3 were cultured in the absence (A, C) or presence (B, D) of 1.5 μg/ml DOX for 2 d. After separation by the OptiPrep membrane flotation method, each fraction was analyzed by immunoblotting (A, B). The LDH activity of each fraction was measured under nontreated (closed circle) and Triton X-100-treated (open circle) conditions. Insets show magnified images of fractions 2–5 (C, D). The experiments were successfully performed twice. (E) FLAG-LC3-enriched membranes were precipitated from light-density fractions (fractions [Fr.] 3 and 4) in the absence of detergent using anti-FLAG antibody-coated magnetic beads and analyzed by immunoblotting.](/cms/asset/5de73a81-8d9c-4d52-875b-00c367ec13c2/kaup_a_1327940_f0006_b.gif)
We next determined whether the autophagosomes enriched in the light-density fractions were completely closed. Because LDH, a cytosolic enzyme, can be nonselectively engulfed by phagophores, the activity of floated LDH reflects the existence of completely closed autophagosomes.Citation29 After separation by the OptiPrep density flotation method, the LDH activity of each fraction was measured under both nontreated and detergent-treated conditions. In the absence of DOX, LDH activity was mainly detected in unfloated cytosolic fractions (fractions 12–14) but not in light-density fractions (). Conversely, in the presence of DOX, weak but significant LDH activity was found in a light-density fraction (fraction 3) only after detergent treatment (). These LDH activities corresponded well to the distribution of the LDH protein in flotation analysis ( and ), suggesting that closed autophagosomes that accumulate in fraction 3 sequestered LDH.
To further confirm that closed autophagosomes accumulated in fraction 3, we performed a protease protection assay after membrane flotation. When each fraction was treated with proteinase K, all markers distributed into medium to bottom fractions (fractions 7–14) were completely or mostly degraded, suggesting that these proteins were not enclosed within membrane structures (Fig. S3). Conversely, the accumulated markers in fraction 3 were protected from the proteinase K degradation, suggesting that these markers were indeed contained within closed autophagosomes (Fig. S3).
Next, we sought to exclude the possibility of contamination by autolysosomes in fraction 3 by observing the degradation of mCherry-LC3 (Fig. S3). Since mCherry but not LC3 is resistant to lysosomal degradation, mCherry-LC3 should be degraded to mCherry inside autolysosomes. In fact, the mCherry fragment was detected in fractions 7 and 8, where LAMP1 was primarily distributed, but not in fraction 3, even though the amount of mCherry-LC3 in fraction 3 was larger than that in fractions 7 and 8 (Fig. S3). These results exclude the possibility that autolysosomes are contaminating the separated autophagosomal fraction.
Finally, we isolated autophagosomes from these light-density fractions. FLAG-LC3-enriched membranes were precipitated with anti-FLAG magnetic beads from fractions 3 and 4. GFP-STX17ΔNTD, endogenous LC3-II, SQSTM1, and a small amount of potentially engulfed TOMM20 were coprecipitated with FLAG-LC3-II-enriched membranes, whereas LAMP1 was not included in the immunoprecipitants (). Together these data suggest that completely sealed autophagosomes can be efficiently purified using DOX-treated TetON-GFPSTX17ΔNTD HeLa cells.
Potential mechanisms of the dominant-negative effect
Although the primary aim of this study was to generate dominant-negative STX17 mutants, we sought to reveal potential mechanisms underlying the dominant-negative effect of GFP-STX17ΔNTD. We first tested whether the autophagosomal localization is necessary to exert the dominant-negative effect. STX17 has glycine-zipper motifs in the 2 TMDs, which are required for its autophagosomal localization.Citation7 Thus, we introduced mutations in the glycine-zipper motif of the first TMD (G244,248L) in GFP-STX17ΔNTD. Unlike nonmutated GFP-STX17ΔNTD, the overexpressed form of this mutant failed to localize on autophagosomes and to induce the accumulation of LC3 puncta under both growing and starvation conditions (). Autophagic flux (bafilomycin A1-induced LC3-II increase) was also not affected by overexpression of GFP-STX17G244,248LΔNTD, although it was affected by overexpression of GFP-STX17FL or GFP-STX17ΔNTD (). These results suggest that the dominant-negative effect of GFP-STX17ΔNTD takes place not in the cytosol, but on the autophagosomal membrane.
Figure 7. Localization of GFP-STX17ΔNTD on autophagosomes is necessary for the dominant-negative effect. (A) WT HeLa cells were transiently transfected with the indicated plasmids. Two d later, cells were cultured in regular or starvation medium for 1 h and analyzed by immunofluorescence microscopy using anti-LC3 antibody. Representative images of 5 randomly selected regions are shown. Scale bars: 20 μm; 2 μm in insets. (B) WT HeLa cells were transiently transfected with the indicated plasmids. Two d later, cells were cultured in regular or starvation medium with or without 100 nM bafilomycin A1 for 2 h and analyzed by immunoblotting using the indicated antibodies. (C) TetON-GFPSTX17ΔNTD HeLa cells were transiently transfected with a plasmid expressing MYC-SNAP29 or MYC-VAMP8, or stably transfected with a plasmid encoding MYC-VPS33A. Cells were fixed and stained using anti-MYC antibody and analyzed by immunofluorescence microscopy. Representative images of 5 randomly selected regions are shown. Scale bars: 10 μm; 2 μm in insets. (D) HEK293T cells were stably transfected with the indicated plasmids. Cells were lysed and separated into 2 samples, which were immunoprecipitated with (indicated with “+”) and without anti-MYC antibody in combination with protein G-Sepharose, followed by immunoblotting using anti-VPS33A, anti-MYC, and anti-ACTB antibodies.
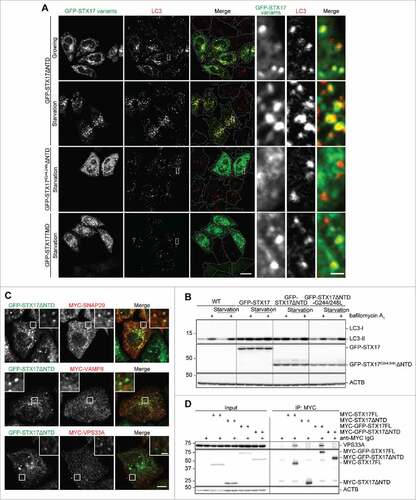
We next tested whether GFP-STX17ΔNTD could bind with its cognate SNAREs and HOPS. In DOX-treated TetON-GFPSTX17ΔNTD HeLa cells, SNAP29 formed many punctate structures, most of which colocalized with GFP-STX17ΔNTD, suggesting that recruitment of SNAP29 was not inhibited (). This interaction might be important because STX17TMD lacking the SNARE domain failed to show the dominant-negative effect (, and ). VAMP8 and VPS33A, a HOPS subunit, did not colocalize with GFP-STX17ΔNTD, confirming that autophagosome-lysosome fusion was blocked (). As previously reported,Citation8 STX17FL tagged with either MYC or GFP interacted with VPS33A based on co-immunoprecipitation (). Conversely, GFP-STX17ΔNTD failed to interact with VPS33A (). This impaired binding with the HOPS complex may contribute to the dominant-negative effect of STX17ΔNTD, whereas N-terminal GFP-tagging may have another effect.
Discussion
In the present study we found that STX17ΔNTD, GFP-STX17FL, and GFP-STX17ΔNTD showed dominant-negative effects on the fusion between the autophagosome and lysosome when they were overexpressed. In previous reports regarding other SNAREs,Citation21,Citation22,Citation25-Citation27,Citation30 overexpression of cytoplasmic portions of SNAREs causes a dominant-negative effect. However, deletion of the TMDs of STX17 did not produce a dominant-negative effect ( and ). Instead, translocation to autophagosomes was necessary for this dominant-negative effect ( and ), suggesting the presence of a mechanism that is different from the previous idea that the overexpressed cytoplasmic domain competes with the endogenous one for its partner SNAREs.Citation21
It has been reported that expression of an NTD-deletion mutant of STX1 causes a dominant-negative effect by an unknown mechanism.Citation31 The NTD of many Qa-SNAREs is composed of a Habc domain. The Habc domain of the yeast Qa-SNARE Vam3 was shown to be important for interaction with the HOPS complex, which promotes SNARE assembly.Citation32,Citation33 We also observed that the NTD of STX17 is important for the binding with HOPS in mammalian cells (). Conversely, the Habc domain interacts intramolecularly with the SNARE motif and negatively regulates binding with its partner SNAREs.Citation34,Citation35 Thus, STX17ΔNTD, which is defective in HOPS binding, may retrieve SNAP29 from endogenous STX17, leading to accumulation of a dysfunctional STX17ΔNTD-SNAP29 Qa-Qbc-SNARE complex. In contrast, the mechanism behind the dominant negative effect of GFP-STX17FL may be different because the latter can interact with VPS33A. We suspect that a large molecular tag such as GFP causes a steric hindrance at the N terminus of STX17 and could inhibit the fusogenic function of the SNARE protein.
The use of the dominant-negative STX17 mutant, especially TetON-GFPSTX17ΔNTD HeLa cells, allows us to purify nascent autophagosomes. Both morphological and biochemical studies showed that accumulated autophagosomes are sealed but not fused with lysosomes. Compared to previous methods to block autophagosome-lysosome fusion (i.e., using lysosome inhibitors or microtubule depolarizing drugs), our method is not only much easier and more efficient but also more direct in blocking the fusion of autophagosomes at a molecular level. This point is important to obtain pure autophagosomes and avoid contamination such as ER-associating phagophores and autolysosomes.
The present study also provides a caution in using GFP-STX17 as a marker. As STX17 is mostly absent on the phagophore, it serves as a good marker for the completed autophagosome.Citation7 However, our data show that overexpression of GFP-STX17 could inhibit autophagosome-lysosome fusion, which may affect experimental results. Therefore, it would be safer to use GFP-STX17TMD that lacks both NTD and SNARE domains for live imaging analysis, because it can still localize to autophagosomesCitation7 but does not show a dominant-negative effect (). For immunostaining, STX17FL with small tags such as MYC could be used.
Application of this method would provide new insight into the nature of autophagosomes. Furthermore, if this method can be applied to in vivo analysis for example by using viral vectors, it will provide a good strategy to block autophagosome-lysosome fusion in vivo.
Materials and methods
Plasmids
cDNAs encoding full-length STX17 and its fragments were amplified by PCR and subcloned into the pMRXIPCitation36 vector together with enhanced GFP and/or a 3xMYC tag to construct the following plasmids: pMRXIP-3xMyc-STX17ΔNTD, pMRXIP-3xMyc-STX17ΔTMD, pMRXIP-3xMyc-STX17NTD, pMRXIP-3xMyc-STX17SNARE, pMRXIP-3xMyc-STX17TMD, pMRXIP-3xMyc-GFP-STX17FL, pMRXIP-3xMyc-GFP-STX17ΔNTD, and pMRXIP-GFP-STX17ΔNTD. Point mutations were induced in STX17ΔNTD by inverse PCR to construct pMRXIP-GFP-STX17G244,248LΔNTD as described previously.Citation7 SNAP29, VAMP8, and VPS33A were amplified by PCR from described previously plasmids,78 and subcloned into pMRXIP together with a 3xMYC tag to construct pMRXIP-3xMyc-SNAP29, pMRXIP-3xMyc-VAMP8, and pMRXIP-3xMyc-VPS33A. pMRXIP-3xMyc-STX17FL, pMRXIP-3xMyc-STX17ΔSNARE and pMRXIP-GFP-STX17TMD have been described previously.Citation7 pTRE-d2EGFP was purchased from Clontech (632346). The original CMV promoter and neomycin-resistant gene in pTet-ON (Clontech, 631018) were replaced with the CAG promoter and puromycin-resistant gene, respectively, to generate pTet-ON-CAG-puro. GFP-STX17ΔNTD and GFP-STX17FL were amplified by PCR from pMRXIP-GFP-STX17ΔNTD and pMRXIP-GFP-STX17FL and subcloned into pTRE2hyg (Clontech, 631014) to construct pTRE2hyg-GFP-STX17ΔNTD and pTRE2hyg-GFP-STX17FL, respectively. LC3 was subcloned into the N-TAP (Flag-Strep-Strep)-pMXs-puro vectorCitation37 to construct pMXs-puro-TAP-LC3. The puromycin-resistant gene in pMXs-puroCitation38 was replaced with the neomycin-resistant gene to generate the pMXs-neo vector. LC3 and mCherry were subcloned into the pMXs-neo vector to construct pMXs-neo-mCherry-LC3.
Antibodies
Mouse monoclonal anti-LC3 (Cosmo Bio Co., clone 1703), anti-MYC (Covance Research Products, 9E10), anti-FLAG (Sigma-Aldrich, clone M2), anti-ACTB/β-actin (Sigma-Aldrich, clone AC74), anti-HSP90AA1 (BD Biosciences, 610419), anti-EEA1 (BD Biosciences, 610456), and anti-TFRC/TfR (Invitrogen, 13–6800) antibodies, rabbit polyclonal anti-LC3,Citation39 anti-MYC (Protein Tech, 16286–1-AP), anti-SQSTM1 (MBL, PM045), anti-GFP (Thermo Fisher Scientific, A-6455), anti-RB1CC1/FIP200 (Protein Tech, 10043–2-AP), anti-WIPI2 (Sigma-Aldrich, SAB4200400), anti-LAMP1 (abcam, ab24170), anti-STX17 (Sigma-Aldrich, HPA001204), anti-SEC61B (Millipore, 07–205), anti-LMAN1/ERGIC53 (Sigma-Aldrich, E1031), anti-STX5 (Synaptic Systems, 110 053), anti-TOMM20 (Santa Cruz, sc-11415), anti-mCherry (abcam, ab125096), and anti-VPS33A (GeneTex, GTX119416) antibodies, and goat polyclonal anti-LDH (abcam, ab2101) were used.
Cell culture
HeLa cells and human embryonic kidney (HEK) 293T cells were cultured in Dulbecco's modified Eagle's medium (Sigma-Aldrich, D6546) supplemented with 10% fetal bovine serum (Sigma-Aldrich, 172012), 50 units/ml penicillin-streptomycin (Thermo Fisher Scientific, 15070063), and 2 mM L-glutamine (Thermo Fisher Scientific, 25030081) in a 5% CO2 incubator. For starvation treatment, cells were washed twice with phosphate-buffered saline (Thermo Fisher Scientific, 14200075) and incubated in amino acid-free Dulbecco's modified Eagle's medium without serum (starvation medium).
Transient transfection
Cells were cultured on coverslips in 24-well plates (40–50% confluency) with 0.5-ml medium per well. For each transfection, 0.8 μg of plasmids and 2.5 μl of FuGENE HD reagent (Promega, E2311) were diluted into 50 μl of Gibco Opti-MEMI Reduced Serum Media (Thermo Fisher Scientific, 31985070) and incubated for 15 min at room temperature to prepare transfection complexes. The transfection complexes were directly added to each well. After 4 h of incubation, the medium containing transfection complexes was removed and changed with fresh medium.
Immunocytochemistry
Cells grown on coverslips were washed with phosphate-buffered saline and fixed in 4% paraformaldehyde for 10 min at room temperature. Fixed cells were permeabilized with 50 μg/ml digitonin (Wako, 043–21376) for 5 min, blocked with 3% bovine serum albumin (BSA; Sigma-Aldrich, 10735094001), and incubated with primary antibodies for 1 h. After washing thoroughly, cells were incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen, A11034), Alexa Fluor 568-conjugated goat anti-rabbit (Invitrogen, A11036) or anti-mouse (Invitrogen, A11031) IgG, and/or Alexa Fluor 660-conjugated goat anti-rabbit (Invitrogen, A21074) or anti-mouse (Invitrogen, A21055) IgG secondary antibodies for 1 h. The coverslips were observed using a confocal laser microscope (Olympus, FV1000D IX81) with a 60x oil-immersion objective lens with a numerical aperture of 1.42, and captured with FluoView software (Olympus). For final output, the images were processed using ImageJ software.
Quantification of correlation efficiency
Colocalization analysis was performed by calculating Pearson's correlation coefficient as described previously using ImageJ software.Citation40 A correlation plot was depicted by plotting the intensities of one channel against the intensities of a second channel. Values near 1 represent almost perfect correlation, whereas values near 0 reflect noncorrelation between 2 channels.
Development of TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cell lines and DOX treatment
HeLa cells were transfected with the pTet-ON-CAG-puro plasmid using Lipofectamine 2000 (Invitrogen, 11668019). Untransfected and transiently transfected cells were removed by puromycin selection. A clone that can highly induce Tet-ON-CAG-puro genes was selected by transient transfection with pTRE-d2EGFP. Then, the cells were transfected with pTRE2hyg-GFP-STX17ΔNTD or pTRE2hyg-GFP-STX17FL and treated with hygromycin (Invitrogen, 10687010). Clones showing high expression of GFP-STX17ΔNTD or GFP-STX17FL in response to DOX (Sigma-Aldrich, D3447) treatment were chosen, and termed TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cell lines, respectively. These cell lines were cultured with or without 1.5 μg/ml DOX for 2 d, followed by confocal microscopy or biochemical analysis.
Immunoblotting
Cells were lysed with lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100 [Nacalai Tesque, 3550115], 1 mM phenylmethanesulfonyl fluoride [Sigma-Aldrich, P7626], and complete EDTA-free protease inhibitor [Roche, 05056489001]). Cell lysates were resolved by SDS-PAGE, transferred to an Immobilon-P PVDF membrane (Millipore, IPVH00010) and blotted with primary and secondary antibodies. The protein bands were visualized with Super-Signal West Pico Chemiluminescent substrate (Thermo Fisher Scientific, 34080) or Immobilon Western Chemiluminescent HRP substrate (Millipore, WBKLS0500). Signal intensities were captured using a LAS-4000 mini imaging analyzer and ImageQuant LAS 4000 software version 1.2 (Fujifilm), or Fusion SOLO S (Vilber Lourmat). The images were processed using Photoshop CS6 (Adobe).
Electron microscopy
WT HeLa or TetON-GFPSTX17ΔNTD and TetON-GFPSTX17FL HeLa cells were cultured on collagen-coated plastic coverslips (Sumitomo Bakelite, MS-0113K) and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 2 h. The cells were washed with the same buffer 3 times, postfixed in 1% osmium tetroxide in 0.1 M phosphate buffer (pH 7.4) for 2 h, dehydrated, and embedded in Epon 812 (TAAB, T022) according to a standard procedure.Citation41 Ultrathin sections were stained with uranyl acetate and lead citrate and observed using a Hitachi H-7100 electron microscope.
OptiPrep flotation assay, LDH assay, and proteinase K protection assay
Cells from 4 10-cm dishes were collected, resuspended in 2 ml ice-cold homogenization buffer (250 mM sucrose [Wako, 195–07925], 20 mM HEPES-KOH, pH 7.4, 1 mM EDTA, 1 mM phenylmethanesulfonyl fluoride, and complete EDTA-free protease inhibitor), and disrupted by N2 cavitation (Parr Instrument, 4639; 800 psi, 10 min, 4°C). The homogenized cells were centrifuged twice at 3,000 × g for 10 min to remove cell debris and undisrupted cells. The supernatant was diluted with an equal volume of OptiPrep (Sigma-Aldrich, D1556). A discontinuous OptiPrep gradient was generated in a SW41 tube for ultracentrifuge rotors (Beckman Coulter, 344059) by overlaying the following OptiPrep solutions all in homogenization buffer: 2.4 ml of the diluted supernatant in 30% OptiPrep, 1.8 ml 20% OptiPrep, 2 ml 15% OptiPrep, 2 ml 10% OptiPrep, 2 ml 5% OptiPrep, and 2 ml 0% OptiPrep. The gradient was centrifuged at 150,200 × g in a SW41Ti rotor (Beckman Coulter) using an Optima XE-90 ultracentrifuge (Beckman Coulter) for 3 h and subsequently 14 fractions (0.85 ml each) were collected from the top. For the protease protection assay, each fraction was incubated with and without 100-μg/ml proteinase K (Sigma-Aldrich, P6556) for 30 min on ice before proceeding to the next step. Then, each fraction was mixed with 50 μl of TCA and incubated for 20 min on ice, followed by centrifugation at 20,000 × g for 20 min to precipitate proteins. The precipitates were washed with mixtures of acetone and ethanol in a 1:1 ratio. After that, they were dried, solubilized with SDS-PAGE sample buffer and heated (55°C) for 10 min. For the LDH assay, 100 μl of each fraction was treated with or without 0.18% Triton X-100 for 15 min, mixed with 100 μl CytoTox-ONE reagent (Promega, G7891), and incubated for 30 min. The LDH activity was determined by measuring the fluorescence (560EX:590EM).
Immunoisolation of FLAG-LC3-II-enriched membranes
Anti-FLAG magnetic beads were prepared according to the manufacturer's instructions. In brief, NHS-FG magnetic beads (Tamagawa Seiki, TAS8848N1141) were pre-treated with methanol and incubated with anti-FLAG M2 antibody. The magnetic beads were treated with 1 M ethanolamine (Sigma-Aldrich, E9508) to block unreacted NHS-ester groups. Anti-FLAG magnetic beads and 1.5-ml tubes (Sumitomo Bakelite, MS-4215M) were blocked with homogenization buffer containing 250 mM NaCl and 1% BSA. Cells were subjected to OptiPrep flotation analysis as described above. Aliquots (400 µl) of the indicated fractions were mixed with 20 µl 5 M NaCl and 20 µl anti-FLAG magnetic beads (50% slurry), and rotated for 2 h at 4°C. The magnetic beads were washed 4 times with homogenization buffer containing 250 mM NaCl and 1% BSA. The magnetic beads were transferred to a new 1.5-ml tube and washed with homogenization buffer containing 250 mM NaCl. The immunoprecipitated membranes were solubilized with 1 x SDS-PAGE sample buffer and heated (55°C) for 10 min.
Retroviral preparation and development of stable cell line
To prepare retroviruses, HEK293T cells were transiently transfected using Lipofectamine 2000 reagent with retroviral plasmids together with the pCG-VSV-G and pCG-gag-pol plasmids. HEK293T cells or HeLa cells were incubated with the culture medium containing retroviruses with 8 μg/ml polybrene (Sigma-Aldrich, H9268). Uninfected cells were removed by puromycin (Sigma-Aldrich, P8833) selection or a clone successfully infected was selected.
Immunoprecipitation
Cells were lysed with lysis buffer and centrifuged at 20,000 × g for 20 min. The supernatants were collected and the protein concentrations were adjusted to equivalency. The supernatants were subjected to immunoprecipitation using anti-FLAG M2 antibody-coated affinity gel (50% slurry; Sigma-Aldrich, A2220) or anti-MYC antibody in combination with protein G-Sepharose (50% slurry; GE Healthcare, 17061801). The immunoprecipitated complexes were washed 5 times with washing buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100) and boiled in sample buffer (46.7 mM Tris HCl, pH 6.8, 5% glycerol, 1.67% SDS [Nacalai Tesque, 02873–75], 1.55% dithiothreitol, 0.003% bromophenol blue).
Treatment with dextran-Alexa 594
Cells were cultured with 50 μg/ml dextran-Alexa Fluor 594 (Thermo Fisher Scientific, D22913) for 24 h and treated with or without starvation medium containing the same concentration of dextran-Alexa Fluor 594 for 1 h, followed by confocal microscopy imaging.
Statistical analysis
All results were expressed as mean ± SEM. Comparisons between 2 groups were done with an unpaired t test. A one-way ANOVA followed by Tukey's test was performed for multiple-comparison testing. Data were considered statistically significant when p < 0.01. Statistical analysis was processed using the R software.
Disclosure of potential conflict of interests
The authors declare that they have no conflicts of interest related to the contents of this article.
Supplemental_Material.docx
Download MS Word (3.2 MB)Acknowledgments
We thank Shoji Yamaoka for the retroviral vectors, Teruhito Yasui for the pCG-VSV-G and pCG-gag-pol plasmids, Noritaka Yamaguchi for the N-TAP-pMXs-puro plasmid, Takahide Matsui for preparing the pMRXIP-3xMyc-VAMP8 and pMRXIP-3xMyc-SNAP29 plasmids, and Norito Tamura for sorting TetON-GFPSTX17FL HeLa cells. We also thank Peidu Jiang and Yoh Wada for helpful discussion and Lo Yu Chia for technical assistance.
Funding
This work was supported by the Japan Society for the Promotion of Science KAKENHI Grant-in-Aid for Young Scientists (A) 26711011 (to TN) and the Grant-in-Aid for Scientific Research on Innovative Areas 25111005 (to NM) from MEXT, a grant from the Japan Foundation for Applied Enzymology (to TN), and Government Subsidies for Management Expense of the University of Tokyo Medical Scientist Training Program (to MU).
References
- Berg TO, Fengsrud M, Strømhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem 1998; 273:21883-92; PMID:9705327
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; https://doi.org/10.1016/j.cell.2011.10.026
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol 2010; 12:831-35; PMID:20811355; https://doi.org/10.1038/ncb0910-831
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102-9; PMID:17909521; https://doi.org/10.1038/ncb1007-1102
- Hong W, Lev S. Tethering the assembly of SNARE complexes. Trends Cell Biol 2014; 24:35-43; PMID:24119662; https://doi.org/10.1016/j.tcb.2013.09.006
- Jahn R, Scheller RH. SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol 2006; 7:631-43; PMID:16912714; https://doi.org/10.1038/nrm2002
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012; 151:1256-69; PMID:23217709; https://doi.org/10.1016/j.cell.2012.11.001
- Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, Mizushima N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell 2014; 25:1327-37; PMID:24554770; https://doi.org/10.1091/mbc.E13-08-0447
- Takáts S, Nagy P, Varga Á, Pircs K, Kárpáti M, Varga K, Kovács AL, Hegedűs K, Juhász G. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol 2013; 201:531-39; PMID:23671310; https://doi.org/10.1083/jcb.201211160
- Takáts S, Pircs K, Nagy P, Varga Á, Kárpáti M, Hegedűs K, Kramer H, Kovács AL, Sass M, Juhász G. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell 2014; 25:1338-54; PMID:24554766; https://doi.org/10.1091/mbc.E13-08-0449
- Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovács AL, Zhang Z, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol 2014; 16:1215-26; PMID:25419848; https://doi.org/10.1038/ncb3066
- Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015; 520:563-66; PMID:25686604; https://doi.org/10.1038/nature14147
- Wang Z, Miao G, Xue X, Guo X, Yuan C, Wang Z, Zhang G, Chen Y, Feng D, Hu J, et al. The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol Cell 2016; 63:781-95; PMID:27588602; https://doi.org/10.1016/j.molcel.2016.08.021
- Köchl R, Hu XW, Chan EYW, Tooze S a. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006; 7:129-45; PMID:16420522; https://doi.org/10.1111/j.1600-0854.2005.00368.x
- Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem 2006; 281:36303-16; PMID:16963441; https://doi.org/10.1074/jbc.M607031200
- Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct 2008; 33:109-22; PMID:18388399; https://doi.org/10.1247/csf.08005
- Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008; 4:849-50; PMID:18758232; doi.org/6845 [pii]
- Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015; 11:1437-38; PMID:26156798; https://doi.org/10.1080/15548627.2015.1066957
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013; 495:389-93; PMID:23455425; https://doi.org/10.1038/nature11910
- Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, Kudo Y, Baba M, Baba N, Cheng J, et al. A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell 2015; 32:304-17; PMID:25619926; https://doi.org/10.1016/j.devcel.2014.12.011
- Fujita T, Chen MJ, Li B, Smith NA, Peng W, Sun W, Toner MJ, Kress BT, Wang L, Benraiss A, et al. Neuronal transgene expression in dominant-negative SNARE mice. J Neurosci 2014; 34:16594-604; PMID:25505312; https://doi.org/10.1523/JNEUROSCI.2585-14.2014
- Kean MJ, Williams KC, Skalski M, Myers D, Burtnik A, Foster D, Coppolino MG. VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J Cell Sci 2009; 122:4089-98; PMID:19910495; https://doi.org/10.1242/jcs.052761
- Pattu V, Qu B, Marshall M, Becherer U, Junker C, Matti U, Schwarz EC, Krause E, Hoth M, Rettig J. Syntaxin7 is required for lytic granule release from cytotoxic T lymphocytes. Traffic 2011; 12:890-901; PMID:21438968; https://doi.org/10.1111/j.1600-0854.2011.01193.x
- Rossi G, Salminen A, Rice LM, Brünger a T, Brennwald P. Analysis of a yeast SNARE complex reveals remarkable similarity to the neuronal SNARE complex and a novel function for the C terminus of the SNAP-25 homolog, Sec 9. J Biol Chem 1997; 272:16610-17; PMID:9195974; https://doi.org/10.1074/jbc.272.26.16610
- Skalski M, Yi Q, Kean MJ, Myers DW, Williams KC, Burtnik A, Coppolino MG. Lamellipodium extension and membrane ruffling require different SNARE-mediated trafficking pathways. BMC Cell Biol 2010; 11:62; PMID:20698987; https://doi.org/10.1186/1471-2121-11-62
- Nakamura N, Yamamoto A, Wada Y, Futai M. Syntaxin 7 mediates endocytic trafficking to late endosomes. J Biol Chem 2000; 275:6523-29; PMID:10692457; https://doi.org/10.1074/jbc.275.9.6523
- Geelen D, Leyman B, Batoko H, Di Sansebastiano GP, Moore I, Blatt MR. The abscisic acid-related SNARE homolog NtSyr1 contributes to secretion and growth: evidence from competition with its cytosolic domain. Plant Cell 2002; 14:387-406; PMID:11884682; https://doi.org/10.1105/tpc.010328
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; https://doi.org/10.1016/j.cell.2010.01.028
- Kopitz J, Kisen GO, Gordon PB, Bohley P, Seglen PO. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J Cell Biol 1990; 111:941-53; PMID:2391370; https://doi.org/10.1083/jcb.111.3.941
- Murray RZ, Wylie FG, Khromykh T, Hume DA, Stow JL. Syntaxin 6 and Vti1b form a novel SNARE complex, which is up-regulated in activated macrophages to facilitate exocytosis of tumor necrosis Factor-α. J Biol Chem 2005; 280:10478-83; PMID:15640147; https://doi.org/10.1074/jbc.M414420200
- Cotrufo T, Pérez-Brangulí F, Muhaisen A, Ros O, Andrés R, Baeriswyl T, Fuschini G, Tarrago T, Pascual M, Ureña J, et al. A signaling mechanism coupling netrin-1/deleted in colorectal cancer chemoattraction to SNARE-mediated exocytosis in axonal growth cones. J Neurosci 2011; 31:14463-80; PMID:21994363; https://doi.org/10.1523/JNEUROSCI.3018-11.2011
- Lürick A, Kuhlee A, Bröcker C, Kümmel D, Raunser S, Ungermann C. The Habc domain of the SNARE Vam3 interacts with the HOPS tethering complex to facilitate vacuole fusion. J Biol Chem 2015; 290:5405-13; PMID:25564619; https://doi.org/10.1074/jbc.M114.631465
- Laage R, Ungermann C. The N-terminal domain of the t-SNARE Vam3p coordinates priming and docking in yeast vacuole fusion. Mol Biol Cell 2001; 12:3375-85; PMID:11694574; https://doi.org/10.1091/mbc.12.11.3375
- Dietrich LEP, Boeddinghaus C, LaGrassa TJ, Ungermann C. Control of eukaryotic membrane fusion by N-terminal domains of SNARE proteins. Biochim Biophys Acta 2003; 1641:111-19; PMID:12914952; https://doi.org/10.1016/S0167-4889(03)00094-6
- Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec 1-syntaxin 1a complex. Nature 2000; 404:355-62; PMID:10746715; https://doi.org/10.1038/35006120
- Saitoh T, Nakano H, Yamamoto N, Yamaoka S. Lymphotoxin-β receptor mediates NEMO-independent NF-кB activation. FEBS Lett 2002; 532:45-51; PMID:12459460; https://doi.org/10.1016/S0014-5793(02)03622-0
- Yamaguchi N, Oyama M, Kozuka-Hata H, Inoue J. Involvement of A20 in the molecular switch that activates the non-canonical NF-кB pathway. Sci Rep 2013; 3:2568; PMID:24008839; https://doi.org/10.1038/srep02568
- Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6:764-76; PMID:20639694; https://doi.org/10.4161/auto.6.6.12709
- Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett 2006; 580:2623-29; PMID:16647067; https://doi.org/10.1016/j.febslet.2006.04.008
- Bampton ETW, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy 2005; 1:23-36; PMID:16874023; https://doi.org/10.4161/auto.1.1.1495
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497-510; PMID:18443221; https://doi.org/10.1083/jcb.200712064