
ABSTRACT
The molecular basis of chronic morphine exposure remains unknown. In this study, we hypothesized that macroautophagy/autophagy of dopaminergic neurons would mediate the alterations of neuronal dendritic morphology and behavioral responses induced by morphine. Chronic morphine exposure caused Atg5 (autophagy-related 5)- and Atg7 (autophagy-related 7)-dependent and dopaminergic neuron-specific autophagy resulting in decreased neuron dendritic spines and the onset of addictive behaviors. In cultured primary midbrain neurons, morphine treatment significantly reduced total dendritic length and complexity, and this effect could be reversed by knockdown of Atg5 or Atg7. Mice deficient for Atg5 or Atg7 specifically in the dopaminergic neurons were less sensitive to developing a morphine reward response, behavioral sensitization, analgesic tolerance and physical dependence compared to wild-type mice. Taken together, our findings suggested that the Atg5- and Atg7-dependent autophagy of dopaminergic neurons contributed to cellular and behavioral responses to morphine and may have implications for the future treatment of drug addiction.
Introduction
Drug addiction is a chronic, relapsing brain disorderCitation1,2 characterized by compulsive drug use, sensitization and dependence.Citation2-4 Because of the impact on health, social cohesion, crime, comorbidity, and associated neuropsychiatric abnormalities, drug addiction has become one of the biggest health problems across the world and causes an enormous financial burden on society.Citation1 The exact mechanism underlying addiction has not been fully explained.
Repeated exposure to drugs can cause cellular adaptations in specific brain neurons, which might finally contribute to drug addiction.Citation5,6 Previous studies have demonstrated that morphological changes in neurons are important indicators of addictive behaviors.Citation7,8 Drug exposure alters the size of cell bodies,Citation9,10 and decreases the complexity of dendritic branching and the number of dendritic spinesCitation11,12 of neurons. The dopaminergic system projects from the ventral tegmental area (VTA) of the midbrain to the nucleus accumbens (NAC) and from VTA to the other forebrain regions including the dorsal striatum, and has a major role in reward and reinforcement for both natural reward and addictive drug effects.Citation4 The mesolimbic dopaminergic pathway and dopaminergic neurons act as the final common neural pathway for mediating the behavioral responses to drug abuse.Citation13-15 Chronic morphine exposure decreases the dendritic spine number and dendritic branching complexity of NAC medium spiny neurons,Citation11 hippocampus pyramidal neurons,Citation16 dentate gyrus granular cells,Citation16 and medial prefrontal cortex pyramidal neurons.Citation12,16 Similarly, chronic morphine treatment and withdrawal alter the morphology of dopaminergic neurons in the VTA region.Citation9,12
Autophagy is a lysosome-mediated degradation pathway actively involved in drug addiction.Citation17-20 It has been reported that autophagy regulates the axonal and dendritic degeneration of neurons.Citation21 Both ATG5 (autophagy-related 5) and ATG7 (autophagy-related 7) are required for the maintenance of axonal homeostasis.Citation22,23 It is possible that autophagy contributes to the neuronal dendritic morphological changes and the resulting behavioral consequences from morphine exposure. Indeed, chronic morphine treatment induces BECN1 (Beclin 1, autophagy related)- and ATG5-dependent autophagy.Citation17 Intracerebroventricular pretreatment with the autophagy inhibitor wortmannin or 3-methyladenine significantly attenuates the antinociception from morphineCitation18 and aggravates morphine-induced memory impairment.Citation20 Recently, we found that chronic morphine treatment can cause mitochondrial dysfunction and autophagy, and co-administration of melatonin with morphine ameliorates morphine-induced behavioral sensitization and analgesic tolerance.Citation19 But how autophagy contributes to the development of addictive behaviors has not been answered.
In this study, we hypothesized that autophagy of dopaminergic neurons would contribute to the alterations of neuronal dendritic morphology and behavioral responses induced by morphine. We provided direct evidence that chronic morphine treatment-induced autophagy was Atg5- and Atg7-dependent in dopaminergic neurons. Deficiency of Atg5 or Atg7 in dopaminergic neurons blunted morphine-induced behavioral responses such as reward, behavioral sensitization, analgesic tolerance and physical dependence. The results thereby established a crucial role for Atg5- and Atg7-dependent autophagy in dopaminergic neurons with regard to drug addiction.
Results
Morphine-induced autophagy was Atg5- and Atg7-dependent
ATG5 is an acceptor molecule for the ubiquitin-like molecule ATG12 (autophagy related 12),Citation24 whereas ATG7 is an E1-like activating enzymeCitation25 required for the conjugation of ATG12 to ATG5Citation26 and of MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta) to phosphatidylethanolamine (PE).Citation26 Both ATG5Citation27 and ATG7Citation28 are key components in the formation of autophagosomes, and are essential for macroautophagy.Citation29 Previously we showed that morphine induces autophagy in rat PC12 cells and mouse hippocampus.Citation19 To assess whether ATG5 and ATG7 played active roles in morphine-induced autophagy, we analyzed the expression of Atg5 and Atg7 in cultured mouse midbrain neurons. After morphine treatment, the mRNA expression levels of Atg5 (Fig. S1A) and Atg7 (Fig. S1B) were significantly upregulated and accompanied by increased levels of the LC3B-II (the PE-conjugated form), ATG5, ATG7, and ATG12–ATG5 complex proteins and a decreased level of the SQSTM1/p62 (sequestosome 1) protein in a dose-dependent manner ( and ). We confirmed these results in both rat PC12 cells (Fig. S2A and B) and mouse midbrain tissues (Fig. S2C and D).
Figure 1. Morphine treatment induces autophagy in mouse midbrain neurons and this effect is Atg5- and Atg7-dependent. Mouse primary midbrain neurons were cultured in Neurobasal® Medium containing 2% B27 and were treated with different concentrations of morphine (0, 50, 100, 200 μM) for 24 h. There was a dose-dependent increase of LC3B-II, ATG7, ATG5 and ATG12–ATG5 protein levels and a decrease of SQSTM1 protein level (A and B). Knockdown of the Atg5 gene reversed the altered protein levels of LC3B-II, ATG7, ATG5, ATG12–ATG5 and SQSTM1 in response to morphine treatment (C and D). A similar effect was observed for knockdown of the Atg7 gene (E and F). Pretreatment with naloxone for 1 h followed by morphine abolished the altered protein levels of LC3B-II, ATG7, ATG5, ATG12–ATG5 and SQSTM1 induced by morphine (G and H). Relative protein abundance was normalized to ACTB. Data are representative of 3 independent experiments with similar results. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA with the Tukey's post-hoc test. Bars represent mean ± SEM.
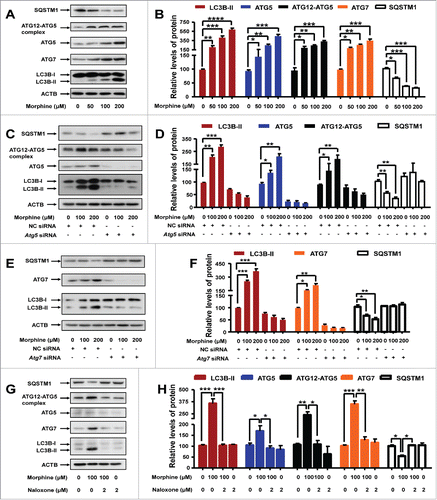
To characterize the function of ATG5 and ATG7 in morphine-induced autophagy, we performed experiments using siRNAs that targeted the Atg5 and Atg7 genes, respectively. We designed 3 siRNAs for each gene; Atg7-siRNA-3 (first reported in our previous studyCitation19) and Atg5-siRNA-2 (Fig. S3) had the best inhibitory effect, and we used these 2 siRNAs in the subsequent knockdown assays. As predicted, the midbrain neurons transfected with negative control (NC) siRNA showed a pronounced autophagy induction upon morphine treatment ( to ). In contrast, knockdown of Atg5 or Atg7 using the most efficient siRNA abolished the morphine-induced autophagy in the midbrain neurons, as demonstrated by the fact that the morphine-induced increase of ATG5 ( and ), ATG7 ( and ), ATG12–ATG5 complex ( and ) and LC3B-II ( to ) protein levels and the decrease of the SQSTM1 protein level ( to ) could be counteracted by Atg5 or Atg7 knockdown. These results could be confirmed in PC12 cells (Fig. S4).
Considering the fact that previous studies indicating morphine responsiveness in vitro used nanomolar concentrations,Citation30,31 whereas morphine concentrations used in our in vitro experiments were much higher and there was a possibility of off-target effects of morphine, we pretreated cultured midbrain neurons and PC12 cells with OPRM1 (opioid receptor mu 1) antagonist naloxone (2 µM) 1 h before morphine (100 µM) treatment. We found that autophagy induced by morphine could be abolished by pretreatment with naloxone in cultured midbrain neurons ( and ) and PC12 cells (Fig. S5), suggesting that the effects of morphine were mediated by OPRM1.
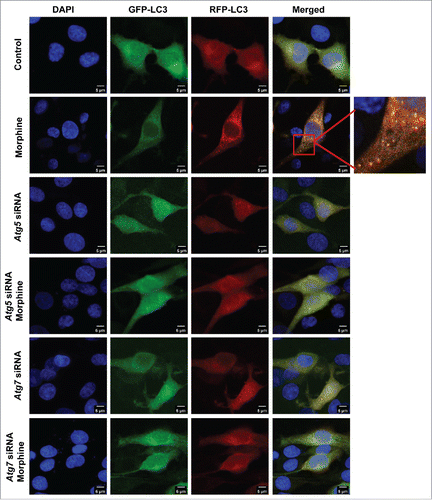
We further introduced the tandem monomeric RFP-GFP-tagged LC3 (mRFP-GFP-LC3) reporter into PC12 cells to determine the effect of morphine on autophagic flux.Citation32 RFP-GFP-LC3 distribution was evaluated in PC12 cells treated with rapamycin (an autophagy inducer) and/or morphine. When the RFP-GFP-LC3 was located in autolysosomes, it displayed red fluorescence as the GFP signal was sensitive to the acidic condition in the lysosome lumen, but the RFP signal was more stable.Citation32 Increased red puncta were observed in the merged section of the rapamycin-treated cells, indicating the maturation of autolysosomes in these cells (Fig. S6). Morphine treatment had a similar effect to rapamycin and increased the autophagic flux (). Knockdown of Atg5 or Atg7 resulted in a few red puncta () in PC12 cells with and without morphine treatment, suggesting that Atg5 or Atg7 knockdown blocked the autophagic flux induced by morphine. Collectively, these results demonstrated that autophagy induced by morphine is Atg5- and Atg7- dependent.
Figure 2. Knockdown of Atg5 or Atg7 reverses the increased autophagic flux in response to morphine treatment in PC12 cells. Cells were transfected with NC siRNA, Atg5 siRNA or Atg7 siRNA for 24 h, then were infected with mRFP-GFP-LC3 lentivirus for 24 h. Infected cells were treated with or without morphine (100 μM) for 24 h. Morphine treatment increased the maturation of autolysosomes as shown by the increased red puncta of mRFP-GFP-LC3 in cells, and this effect could be restored by knockdown of the Atg5 or Atg7 gene. Data shown are representative of 3 experiments.
The Atg5- and Atg7-dependent autophagy induced by morphine was specific for dopaminergic neurons in mouse midbrain
We examined whether morphine-induced autophagy has a neuron-specific pattern in mouse brain slices by using a double immunofluorescence assay. We used LC3 to label the autophagosomes, and TH (tyrosine hydroxylase) and GAD1/GAD67 (glutamate decarboxylase 1) to stain the dopaminergic neurons and GABAergic interneurons, respectively. There were no obvious LC3 puncta in the control brain slices ( and Fig. S7A). However, a large number of LC3 puncta were found in the midbrain slices of mice that had been given morphine by injection (10 mg/kg, once a d for 7 d) with many of the puncta colocalized with TH-positive neurons (approximately 40%) ( and ) and a few colocalized with GAD1-positive neurons (less than 5%) (Fig. S7). These results indicated that morphine-induced autophagy might be specific to dopaminergic neurons but not GABAergic interneurons in mouse midbrain. We performed the same tests with striatum and prefrontal cortex, but found no positive staining. Consistent with our previous observations that chronic morphine treatment increases the protein levels of autophagic markers in mouse hippocampus,Citation19 morphine increased the protein levels of LC3B-II, ATG7, ATG5 and the ATG12–ATG5 complex and decreased the protein level of SQSTM1 in the midbrain ( and ) and hippocampus (Fig. S8) of wild-type (WT) mice.
Figure 3. Morphine induces Atg5- and Atg7-dependent autophagy in dopaminergic neurons. Double-immunostaining of mouse brain slices with anti-MAP1LC3B/LC3B (red) and anti-TH (green) antibodies to indicate autophagy in dopaminergic neurons (A). The induced LC3 puncta by morphine were colocalized with TH-positive neurons (A and B) in WT mice. DAPI (blue) was used for nuclear staining. The protein levels of LC3B-II, ATG7, ATG5 and ATG12–ATG5 were increased and the protein level of SQSTM1 was decreased in the midbrain tissues (C and D) of WT mice with morphine treatment (10 mg/kg, once a d for 7 d). Knockout Atg5 or Atg7 in dopaminergic neurons restored the induced LC3 puncta (A and B) and the altered levels of LC3B-II, ATG7, ATG5, ATG12–ATG5 and SQSTM1 in response to morphine (C and D). Relative protein abundance was normalized to ACTB. Data are representative of 4 independent experiments with similar results. **, P < 0.01; ***, P < 0.001; one-way ANOVA with the Tukey's post-hoc test. Bars represent mean ± SEM.
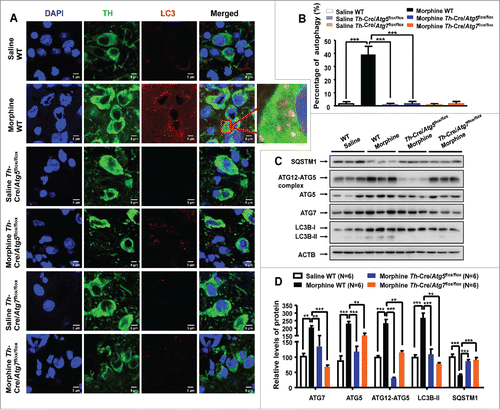
We further explored whether morphine selectively induced autophagy in dopaminergic neurons was Atg5- and Atg7-dependent by using conditional knockout mice. To overcome the potential obstacle of embryonic lethality and/or postnatal death of mice deficient for the autophagy-related genes, we crossed Atg5- or Atg7-conditional knockout mice (Atg5flox/flox, Atg7flox/flox) with transgenic mice expressing Cre recombinase under the control of the Th promoter (Th-Cre), to produce mice deficient for Atg5 and Atg7 specifically in dopaminergic neurons (hereafter called Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox), respectively (Fig. S9A and B). The Atg5 and Atg7 knockout efficiency in the midbrain tissues of Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice were quantified by using western blotting. We found that protein levels of ATG5 or ATG7 were dramatically reduced in the midbrain of Th-Cre/Atg5flox/flox or Th-Cre/Atg7flox/flox mice compared with Th-Cre/Atg5flox/+ or Th-Cre/Atg7flox/+ and WT littermates (Fig. S9C and D). Consistent with the suppression of autophagy due to deficiency of Atg5 and Atg7, no autophagosomes were observed in the dopaminergic neurons and GABAergic interneurons of Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice ( and ) with or without morphine treatment. Similarly, chronic morphine treatment for the Th-Cre/Atg5flox/flox or Th-Cre/Atg7flox/flox mice failed to induce autophagic markers, and there were defects in the degradation of SQSTM1 and the conjugation of ATG12 to ATG5 in the midbrain ( and ) and hippocampus (Fig. S8). Note that there were relatively weaker bands for ATG5 and ATG7 in western blots for Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice, which were likely caused by the expression of these proteins in Th-negative neuronal cells in tissue homogenates. These results suggested that morphine-induced Atg5- and Atg7-dependent autophagy was specific for dopaminergic neurons.
The Atg5- and Atg7-dependent autophagy regulated neuronal morphological changes
Dendritic arborization and spine formation are critical for neuron function. The degradation of axons and dendrites is a common and early pathological feature of many neurodegenerative diseases,Citation33,34 and dendritic changes have been implicated in drug addiction.Citation11,12,16 To confirm the impact of morphine treatment and autophagy on the branching of neurons, mouse primary midbrain neurons were treated with 100 μM morphine for 24 h with or without knockdown of Atg5 and Atg7, followed by staining of MAP2 (microtubule-associated protein 2) to evaluate the changes of total neuron dendritic length and complexity, and of TH to recognize the dopaminergic neurons, respectively. As expected, both total dendritic length ( and ) and dendritic complexity ( and ) were significantly reduced in morphine-treated dopaminergic neurons relative to untreated neurons. The decreased total dendritic length and dendritic complexity induced by morphine could be reversed by knockdown of Atg5 and Atg7 using siRNAs ( to ). It should be mentioned that knockdown of Atg5 and Atg7 had no significant impact on the morphological changes of cultured dopaminergic neurons ( to ).
Figure 4. Effect of Atg5- and Atg7-dependent autophagy on regulation of neuronal dendritic morphogenesis in cultured midbrain neurons and mouse brain tissues. Mouse primary midbrain neurons were cultured in Neurobasal® Medium containing 2% B27 and were treated with 100 μM morphine for 24 h. The dopaminergic neurons and dendrites were demonstrated by TH (green) and MAP2 (red) staining (A), respectively. Quantification of total MAP2-positive total dendritic length (B) and dendritic complexity (C) in TH-positive neurons (N = 20). Upon morphine treatment, total dendritic length (B) and dendritic complexity (C) of TH-positive neurons were reduced. DAPI (blue) was used for nuclear staining. These changes could be reversed by knockdown of Atg5 or Atg7 using siRNAs (A to C). Golgi-Cox staining showed that chronic morphine exposure decreased the number of neuronal dendritic spines in the midbrain (D and E) of WT mice, but there were no changes in the Th-Cre/Atg5flox/flox or Th-Cre/Atg7flox/flox mice with or without morphine administration (D and E). Data are representative of 3 independent experiments with similar results. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA with the Tukey's post-hoc test. Bars represent mean ± SEM.
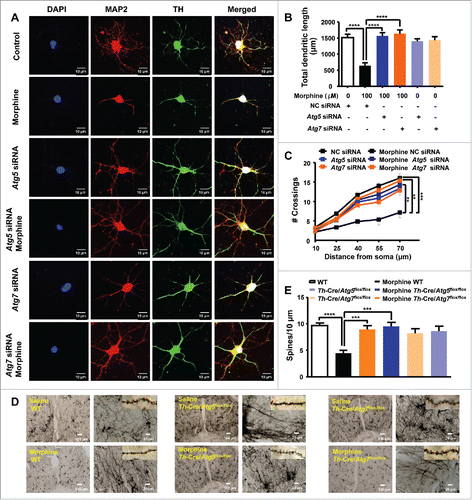
We next explored whether morphine regulates the morphological changes of neurons in vivo. Golgi-Cox impregnated neurons showed that morphine given by injection dramatically decreased the number of neuronal dendritic spines in the midbrain ( and ) and hippocampus (Fig. S10) compared to the untreated WT mice (control group). However, in mice with Atg5- and Atg7- deficiency specific for dopaminergic neurons, the presence or absence of morphine injections had no apparent effect on the alterations of dendritic spines in the midbrain ( and ) and hippocampus (Fig. S10). This result was consistent with a recent report showing no effect of Atg7 knockdown on the number of dendritic spines of cultured hippocampus neurons.Citation35 These findings indicated a critical role of the Atg5- and Atg7-dependent autophagy in regulating neuronal morphological changes triggered by morphine.
Specific deficiency of the Atg5 or Atg7 gene in the dopaminergic neurons had an impact on behaviors
We measured the behavioral changes in mice with deficiency of the Atg5 or Atg7 gene specifically in the dopaminergic neurons. There was no significant difference in locomotor activity (Fig. S11A) and the ability for exploratory activity in a new environment (Fig. S11B) between the WT and Th-Cre/Atg7flox/flox mice. However, the traveled ambulatory distance was significantly decreased in the Th-Cre/Atg5flox/flox mice compared to the WT or Th-Cre/Atg7flox/flox mice (Fig. S11A and B) during the habituation stage (at the 1st d that all animals received no treatment). The Th-Cre/Atg5flox/flox mice had a significantly higher central distance ratio than the WT or Th-Cre/Atg7 flox/flox mice (Fig. S11C); this pattern was negatively correlated with the level of anxiety.Citation36 Both the Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice had an increased number of jump counts compared to the WT littermates (Fig. S11D).
The conditioned place preference induced by morphine was impaired in mice with an Atg5 or Atg7 deficiency in dopaminergic neurons
We assessed the influence of the Atg5- and Atg7-dependent autophagy on the rewarding effects of morphine by using the conditioned place preference (CPP) paradigmCitation37,38 (), in which animals learn to prefer an environment paired with drug exposure. During the preconditioning phase, all WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice visited the 2 compartments of the place-preference apparatus and spent approximately the same amount of time in each compartment (Table S1), suggesting that the ATG5 or ATG7 deficiency had no effect on the preference. Morphine administration (10 mg/kg body weight, subcutaneously) induced a clear CPP in WT mice and Th-Cre/Atg7flox/flox mice, as indicated by a significant increase in the CPP score compared with control mice (with the same genotypes but without morphine administration) during the testing phase. Note that the Th-Cre/Atg7flox/flox mice generally had a poor performance compared to WT mice (). This conditioned behavior was almost completely absent in the Th-Cre/Atg5flox/flox mice (). This result implied that inhibition of the Atg5- and Atg7-dependent autophagy in the dopaminergic neurons reduced the preference elicited by morphine in mice.
Figure 5. Mice deficient in Atg5 and Atg7 in dopaminergic neurons display blunted morphine-induced rewarding properties and behavioral sensitization. A schematic illustrating the experimental design of the conditional place preference (CPP) paradigm (A). Injection with morphine in WT (N = 24) and Th-Cre/Atg7flox/flox mice (N = 19) produced a significant increase in CPP scores compared to the control mice (WT: N = 20, Th-Cre/Atg7flox/flox: N = 12) injected with saline (B). The Th-Cre/Atg5flox/flox mice injected with morphine (N = 17) did not show a rewarding effect compared to the Th-Cre/Atg5flox/flox mice injected with saline (N = 12) (B). Knockout of Atg5 or Atg7 in dopaminergic neurons had no effect on exploratory activity as measured by the number of chamber entries during the baseline and the testing phases with or without morphine injection (C). A schematic illustrating the experimental design of the behavioral sensitization paradigm (D). There was no genotype effect on ambulatory distance during the baseline phase of the open field test among WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice (E). A steady increase of locomotor activity was observed in the WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice upon repeated morphine injections compared to the control WT mice (F). Knockout of Atg5 or Atg7 in the dopaminergic neurons significantly reduced behavioral sensitization compared to the WT littermates with morphine injection (F). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ANOVA, followed by LSD for post-hoc comparisons. Bars represent mean ± SEM.
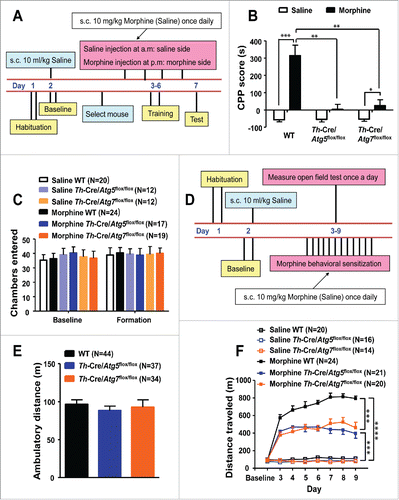
To test whether the inhibited rewarding properties elicited by morphine administration in the conditional knockout mice were affected by potentially impaired exploratory activity, we measured the number of chamber entries. There was no significant difference in the counts of chamber entries among the WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice with or without morphine administration (). Evidently, specific deficiency of the Atg5 or Atg7 genes in dopaminergic neurons reduced the sensitivity of animals to morphine rewards, and this effect was not related to the exploratory activity.
The Atg5 and Atg7 deficiency in mouse dopaminergic neurons prevented the development of morphine-induced behavioral sensitization
To understand the potential impact of the Atg5- and Atg7-dependent autophagy induced by morphine, we assessed the behavioral responses to morphine in the Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice based on behavioral sensitization, a well-documented effect of morphine that is characterized by a progressive increase of locomotor activity following repeated morphine exposure.Citation39 The WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice were given either saline (1 ml/kg) or morphine (10 mg/kg), and the total distance traveled in 1 h was measured by the open field test (). There was no significant difference in the baseline for the WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice () that received the saline before confinement to the test room. For the control group, we got similar locomotor activity among WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice with saline injection (). Behavioral sensitization was observed in all morphine-treated animals, with a significantly increased horizontal locomotor activity in mice given morphine (10 mg/kg; subcutaneously, once per d for 7 d) relative to the saline controls (). Specific deficiency of the Atg5 and Atg7 genes in the dopaminergic neurons resulted in reduced locomotor activity in response to the acute and repeated morphine administration (). This result suggested that inhibition of the Atg5- and Atg7-dependent autophagy in the dopaminergic neurons prevented the development of morphine-induced behavioral sensitization.
Mice deficient for the Atg5 or Atg7 gene specifically in the dopaminergic neurons had an impaired antinociceptive tolerance in response to morphine
Repeated opiate exposure causes tolerance to the analgesic effects of these drugs and this phenomenon limits their clinical use in the treatment of chronic pain. To evaluate whether the morphine-induced analgesic effects could be affected by inhibiting the Atg5- and Atg7-dependent autophagy in dopaminergic neurons, we examined the analgesic effects of morphine and the development of analgesic tolerance in Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice by using the standard hot-plate and tail-flick tests. The mice were tested 1 h after being given a morphine injection (). The Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice showed a higher nociceptive threshold than the WT littermates in the tail-flick (with a laser density of FOCUS 30; ) and the hot-plate tests (at 52.5°C; ). The duration of morphine action was not affected by the deficiency of Atg5 or Atg7 in dopaminergic neurons, because the analgesic effects persisted for similar amounts of time in Th-Cre/Atg5flox/flox, Th-Cre/Atg7flox/flox and WT mice ().
Figure 6. Deficiency of Atg5 or Atg7 in dopaminergic neurons in mice affects morphine-induced analgesia and tolerance. A schematic illustrating the experimental design of the analgesia tolerance paradigm (A). Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice have normal nociceptive thresholds compared with their WT littermates in the FOCUS 40 tail-flick test (B) and 47.5°C/50°C hot-plate assay (C). A higher nociceptive threshold was observed in the FOCUS 30 tail-flick test (B) and 52.5°C hot-plate assay (C) in the Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice compared to WT littermates. Analgesic responses to morphine (10 mg/kg, subcutaneously) in the 50°C hot-plate test were approximately same in the WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice, and the duration time of analgesia was also similar (D). The WT and Th-Cre/Atg5flox/flox mice with chronic morphine administration (10 mg/kg once daily) had an analgesic tolerance as mirrored by the gradually decreased maximal possible effect (MPE) % in the FOCUS 40 tail-flick test (E) and 50°C hot-plate assay (F), but Th-Cre/Atg7flox/flox mice showed a delayed tolerance to morphine (E and F). The antinociceptive response was calculated as the percentage of MPE, where MPE % = (test latency - baseline latency)/(cutoff latency- baseline latency) × 100. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ANOVA, followed by LSD for post-hoc comparisons. Bars represent mean ± SEM.
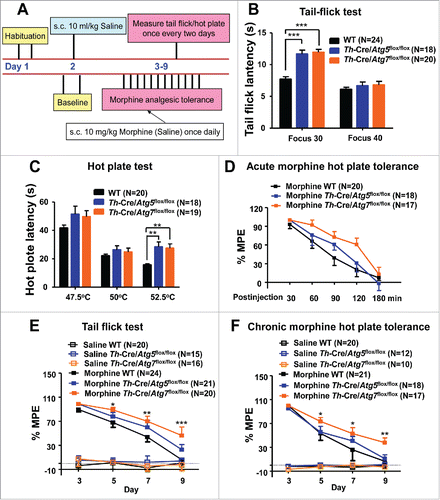
We next investigated the role of the Atg5- and Atg7-dependent autophagy induced by morphine in the development of analgesic tolerance. In order to avoid the potential genotype effect, we used the tail-flick test with a laser density of FOCUS 40 and the hot-plate test at 50°C. Tolerance to analgesic effects developed after repeated morphine administration over 7 d ( and ) in WT mice. Strikingly, the Th-Cre/Atg7flox/flox mice showed a delayed development of morphine tolerance in response to chronic morphine administration (10 mg/kg) compared to WT littermates in both the tail-flick () and hot-plate tests (). The Th-Cre/Atg5 flox/flox mice also had a delayed development of morphine antinocicepitive tolerance in the tail-flick test (), but not for the hot-plate test (). Together, these findings demonstrated that the Atg5- and Atg7-dependent autophagy in dopaminergic neurons was involved in the development of analgesic responses and tolerance to repeated morphine administration.
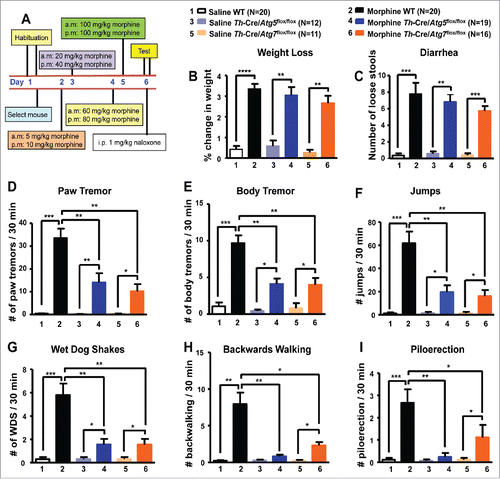
Deficiency of the Atg5 or Atg7 gene specifically in the dopaminergic neurons caused an attenuation of morphine withdrawal symptoms
Besides tolerance, chronic morphine exposure also causes a severe physical dependence.Citation4 The dopaminergic mesolimbic system has been reported to be actively involved in this process.Citation40,41 We investigated the effect of the Atg5- and Atg7-dependent autophagy in the opiate physical dependence and withdrawal by using a standard morphine withdrawal procedure (). Briefly, mice were treated with increasing doses of morphine, and signs of opiate withdrawal were monitored for 30 min after administration of naloxone (an OPRM1 antagonist; 1 mg/kg, given intraperitoneally). No significant withdrawal behaviors were observed in the WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice injected with saline (). Opiate withdrawal behaviors were significantly increased in the WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice with morphine withdrawal (). However, several but not all of these signs were attenuated in the Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice, and the overall withdrawal scores were significantly decreased as compared to the WT littermates (). There was a trend for a milder withdrawal effect on weight loss () and diarrhea () in the Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice. However, the withdrawal effects were significant for paw tremor (), body tremor (), jumps (), wet dog shakes (), backward walking () and piloerection () between the WT littermates and the Th-Cre/Atg5flox/flox or Th-Cre/Atg7flox/flox mice. These observations suggested that the Atg5- and Atg7-dependent autophagy might play an important role in modulating the development of morphine dependence.
Figure 7. Measurement of morphine physical dependence in mice deficient of Atg5 and Atg7 in dopaminergic neurons. A schematic illustrating the experimental design of morphine physical dependence (A). Morphine withdrawal behavior was significantly changed in the WT littermates after morphine withdrawal (B to I). There was no significant difference in weight loss (B) and diarrhea (C) between Th-Cre/Atg5flox/flox and WT littermates or between Th-Cre/Atg7flox/flox and WT littermates after morphine withdrawal. Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice show greater antagonist-precipitated morphine withdrawal effects than WT littermates, including paw tremor (D), body tremor (E), jumps (F), wet dog shakes (G), backward walking (H) and piloerection (I). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ANOVA, followed by LSD for post-hoc comparisons. Bars represent mean ± SEM.
Discussion
The results of the present study provided novel insights into the molecular basis of the morphine-induced changes in neuron morphology and animal behaviors. Our data established that upregulation of autophagy activity induced by repeated morphine usage is essential for the morphine-mediated decreases of the dendritic spine number, total dendritic length and complexity of dopaminergic neurons. Our findings further indicated that the upregulation of autophagy was Atg5- and Atg7-dependent. Moreover, the Atg5- and Atg7-dependent autophagy was dopaminergic neuron-specific in the midbrain, and directly related to an increased sensitivity to the addictive behaviors of morphine. Indeed, conditional knockout mice with a deficiency of the Atg5 or Atg7 genes in the dopaminergic neurons had a series of behavioral changes relative to the WT littermates when challenged by morphine.
A previous study found that chronic morphine administration induced autophagy in GABAergic interneurons from the spinal cord;Citation18 however, we did not observe autophagy in the midbrain GABAergic interneurons in this study. The apparent discrepancy might be caused by several factors that are crucial for drug-induced plasticity, such as the age of the mice (8-wk-old C57BL/6 male mice in our experiments versus 10-wk-old in ref. Citation18), the methodological differences in building morphine-addicted models (subcutaneous morphine over 7 d in our experiments versus intraperitoneally-injected morphine for 5 d in ref. Citation18), and brain region differences in measuring autophagy (midbrain in our study versus spinal cord in ref. Citation18). It would be rewarding to test the effect of morphine on autophagy in spinal interneurons in Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice. Nonetheless, both studies had consistently showed that morphine treatment increased autophagy activity, and inhibition of morphine-induced autophagy prevented the development of drug tolerance. Although our findings demonstrated a critical role of the Atg5- and Atg7-dependent autophagy in morphine addiction, the seemingly paradoxical observation that the autophagy only occurred in dopaminergic neurons, but not in GABAergic interneurons in the midbrain, and what is the biological implication for the dopamine imbalance caused by this neuron-specific change remain to be elucidated. A recent study showed that SLC35D3 (solute carrier family 35, member D3), as a regulator of tissue-specific autophagy, increases autophagic activity in midbrain dopaminergic neurons via enhancing BECN1-ATG14-PIK3C3 complex formation.Citation42 It would be rewarding to test whether this process was also involved in the morphine-induced dopaminergic neuron-specific autophagy in the midbrain.
Our findings are in good agreement with previous results demonstrating the importance of the autophagy pathway in regulating the axonal and dendritic degeneration of neurons.Citation21 Although previous reports have suggested that Atg5 and Atg7 are both required for the degeneration of axons,Citation22,23 the role of Atg5- and Atg7-dependent autophagy in the regulation of neuronal dendritic morphology in response to morphine has not yet been investigated. We demonstrated that inhibition of autophagy signaling in the dopaminergic neuron reversed the decrease of dendritic spine number, complexity and total dendritic length of neurons through the Atg5- and Atg7-mediated pathway (). It will be rewarding for future studies to determine whether the effect of the Atg5- and Atg7-dependent autophagy on morphine-induced neuronal morphological changes of the dopaminergic neurons has a selectivity for other (sub)types of neurons, which were reported to play distinct roles in the addiction process.Citation13 It should be noted that the changes in the Atg5- and Atg7-dependent autophagy observed here may not occur exclusively in the dendrites, but can occur in the soma and axon as well, where autophagy regulates the degradation of proteins and organelles.Citation43
Recently, morphine has been reported to affect HCRT/hypocretin,Citation44 and PLCG1 (phospholipase C, gamma 1)Citation45 and inhibit class I phosphoinositide 3-kinase-AKT,Citation45,46 MAPK/ERK (mitogen-activated protein kinase)Citation46 signaling and IRS2 (insulin receptor substrate 2)-AKT signaling,Citation10,47 as well as to cause mitochondrial dysfunction.Citation19 Disruption of the IRS2-AKT pathway alters the reduction of dopaminergic neuron size and morphine rewarding properties induced by morphine.Citation10 However, whether these findings are directly related to autophagy has not been fully understood. The activation of MAPK/ERK and phosphoinositide 3-kinase-AKT signaling pathways would inhibit autophagy activity,Citation48 but we showed here that upregulation, not inhibition, of autophagy by morphine caused dendritic changes and addictive behaviors. We speculated that autophagy was induced as a compensatory mechanism to the alterations/activations of these pathways induced by morphine to regulate the dendritic morphology of dopaminergic neurons.
The morphological changes in dopaminergic neurons induced by morphine () might play a central role in regulating addictive behaviors in mice with morphine administration, and this has been supported by evidence that neuron morphological changes contribute to morphine addictive behaviors.Citation7,8 Morphine-induced behavioral responses and synaptic plasticity are strongly associated with adaptations in dopaminergic neurons in the dopaminergic system.Citation13 Repeated administration of morphine induced dopaminergic morphological changes ().
Whereas recent studies have shown that autophagy might be involved in drug addiction,Citation17-19 very little is known about the effect of morphine on the activity of the Atg5- and Atg7-dependent autophagy pathway on dopaminergic neurons, and the role of autophagy in mediating morphine-induced addictive behaviors remains elusive. Several previous studies show that blockage of the morphine-induced autophagy by a variety of pharmacological and molecular approaches leads to blunted behavioral effects of morphine.Citation18,49 Concomitantly, we found that inhibition of autophagy by melatonin prevents the behavioral sensitization and antinociceptive tolerance.Citation19 In this study, we found that inhibition of autophagy by 3-methyladenine prevented the behavioral sensitization, whereas induction of autophagy by rapamycin promoted the behavioral sensitization induced by morphine (Fig. S12). These findings highlighted the need for further studies to delineate the Atg5- and Atg7-dependent autophagy of dopaminergic neurons in mediating distinct aspects of behavioral adaptations to morphine. It should be noted that most of the available studies that examined drug-induced autophagy did not use conditional knockout mice, which are best for demonstrating the potentially deleterious effect as we showed here. Deficiency of the Atg5 and Atg7 genes specifically in the dopaminergic neurons caused an increase of analgesic effect of acute morphine treatment, but a reduction of adaptation to repeated morphine exposure (including decreased morphine rewarding effects, locomotor sensitization, analgesic tolerance and physical dependence) (). The diverse effects of the Atg5- and Atg7-dependent autophagy might have resulted from its broad distribution and multiple actions in the brain. Further studies should be carried out to determine whether the molecular pathways revealed by the current investigator-administered drug experiments are essentially the same as those involved with self-administered drug usage.
An interesting finding in this study is that conditional knockout of Atg5 or Atg7 specifically in the dopaminergic neurons resulted in an inhibition of morphine-induced autophagy and associated dendritic reorganization of neurons in the hippocampus. As the Th-Cre line used to generate Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox mice did not express Cre in the hippocampus,Citation50 we thought that this effect might be indirect and be influenced by the dopaminergic projection in the midbrain. Further studies are essential to elucidate the exact mechanism.
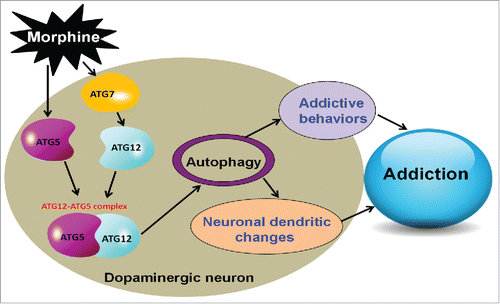
In summary, we found that repeated morphine administration led to upregulation of ATG5 and ATG7, which facilitated the formation of the ATG12–ATG5 conjugate and Atg5- and Atg7-dependent autophagy in the dopaminergic neurons. The detrimental effects of morphine-induced autophagy could be counteracted by a deficiency of ATG5 or ATG7 in dopaminergic neurons, and this protected neuron function and prevented the development of addictive behaviors (). These convergent lines of evidence describe a previously unknown dopaminergic neuron-specific autophagy pathway in the regulation of morphine-induced behavioral, cellular and morphological plasticity. Manipulation of Atg5- and Atg7-dependent autophagy might be explored as a potential therapeutic strategy in the future treatment of drug addiction.
Figure 8. A proposed mechanism of the Atg5- and Atg7-dependent autophagy for morphine addiction.
Materials and methods
Reagents, chemicals and cells
The following antibodies and chemicals were used in this study: rabbit monoclonal anti-MAP1LC3B/LC3B (Cell Signaling Technology, 3868), rabbit monoclonal anti-MAP1LC3B/LC3B (Abcam, ab64781), rabbit monoclonal anti-ATG5 (Cell Signaling Technology, 8540), rabbit monoclonal anti-ATG7 (Cell Signaling Technology, 8558), mouse monoclonal anti-SQSTM1/p62 (Elabscience, EAP3350), mouse polyclonal anti-TH (Merck Millipore, MAB318), mouse polyclonal anti-GAD1/GAD67 (Merck Millipore, AB152), rabbit polyclonal anti-MAP2 (Merck Millipore, AB5622), mouse monoclonal anti-ACTB/β-actin (Beijing Zhong Shan-Golden Bridge Biological Technology CO., LTD, TA-09), peroxidase-conjugated anti-rabbit antibody (KPL, 474–1516), peroxidase-conjugated anti-mouse antibody (KPL, 474–1086), FITC-conjugated anti-rabbit IgG (Life Technologies, A21207) and anti-mouse IgG (Invitrogen, A21202) secondary antibody. Poly-L-ornithine (Sigma, P4957), 3-methyladenine (Sigma, M9281), rapamycin (InvivoGen, trl-rap) and 4′, 6-diamidino-2-phenylindole (DAPI; Roche, 10236276001), pentobarbital (Sigma, P3761), procaine hydrochloride (Sigma, P9879), and naloxone hydrochloride (Sigma, N7758) were purchased as indicated. Morphine hydrochloride for injection was bought from Shenyang Pharmaceutical Co. Ltd. (State Drug Approval Document Number: H21022436; Shenyang, China).
Rat pheochromocytoma PC12 cells were obtained from the Kunming Cell Bank, Kunming Institute of Zoology. Cells were maintained in DMEM medium (Gibco-BRL, 11965-092) supplemented with 10% fetal bovine serum (Gibco-BRL, 10099–141) at 37°C in a humidified atmosphere incubator with 5% CO2 and 95% humidity. Mouse midbrain neurons were prepared and cultured as previously described.Citation51 Briefly, primary midbrain neurons were isolated from mouse embryonic day 15 (E15) embryos and were cultured in 12-well plates coated with poly-L-ornithine (Sigma, P4957) using Neurobasal® Medium supplement with 2% B27 (Gibco-BRL, 17504). Primary neurons were cultured for at least 2 wk to allow the normal development and elaboration of neurites. Drugs were applied directly to the culture medium for treatment. All experiments were repeated at least 3 times.
Animals
Th-Cre mice were generated and genotyped as described previously.Citation50 Atg5flox/flox miceCitation52 and Atg7flox/flox miceCitation53 were obtained from the RIKEN Bioresource Center, Japan. Th-Cre/Atg5flox/flox mice and Th-Cre/Atg7flox/flox mice were generated by mating Atg5flox/flox and Atg7flox/flox mice with Th-Cre mice, respectively. For all behavioral tests, WT, Th-Cre/Atg5flox/flox and Th-Cre/Atg7flox/flox male littermates at age of 8 wk were used. Mice were genotyped by PCR amplification (Table S2). Mice were housed in clear plastic cages with free access to water and food in an established animal house of 22 ± 2°C and 50% humidity, with a 12-h light/dark cycle.
Establishment of a chronic morphine-addicted model in mice
Experiments were performed on male C57BL/6 mice (weight 25 g to 30 g). All mice were acclimatized for at least 1 wk before experiments. Experiments were performed during 9:00 am to 17:00 pm each day. Animals were habituated in the test room for 2 h before starting the experiments. Mice were treated with morphine hydrochloride (10 mg/kg, subcutaneously) once a day for 7 consecutive d, following the previously described procedure.Citation19 Mice were then sacrificed for tissue collection (including midbrain, hippocampus and prefrontal cortex tissues).
Conditioned place preference
Place conditioning was performed as previously described,Citation37,38 with some modifications. Several visual and nonvisual (tactile) cues enable the animals to distinguish 2 side chambers of the apparatus. All conditioning and test sessions were performed under dim illumination. Mice did not show a preference for either side of the chamber at the baseline. The baseline preference was monitored at day 2 without treatment. On d 2, mice explored the 2 chambers freely for 15 min and they did not show a preference for any chamber before conditioning. During the next 4 d mice were conditioned to the saline-paired side for 30 min in the morning, where they received saline (10 ml/kg, subcutaneously). Five h later, mice were conditioned to the morphine-paired side, where they received morphine (10 mg/kg, subcutaneously). On d 7, mice were placed again in the central compartment at noon and allowed to move freely between the 2 chambers for 15 min. The CPP score was calculated by the time that a mouse spent in the morphine-paired chamber minus the time it spent in the saline-paired chamber.Citation10
Behavioral locomotor sensitization
Morphine-induced behavioral sensitization was measured in an open field test using the ENV-510 test environment and Activity Monitor software (Med Associates) on d 2 (baseline) and the following 7 d as described in our previous study.Citation19 Mice were injected subcutaneously with morphine (10 mg/kg) immediately before confinement to the test room. The control mice received 0.9% saline (10 ml/kg, subcutaneously) in all experiments.
Morphine analgesia and tolerance
Analgesia was assessed using the radiant heat tail-flick latency (LE 7106 Light beam analgesy-meter, Panlab HARVARD, MA, USA)Citation19 and a hot platform (BIO-CHP, Bioseb, France) as described previously.Citation37 For the tail-flick test, mice were placed in Plexiglas cages (9 × 6 × 3 cm) on a modified Hargreaves Device (Panlab, Harvard, MA, USA). Mice were habituated to the device for 2 min before each test session. A halogen lamp was focused on the tail and the withdrawal reflex time was determined by a photocell. We measured tail-flick latency at FOCUS 30 (decimal selector of heat intensity) and FOCUS 40 every 2 d from d 0 (baseline) to d 7. We used 20 sec as a cutoff time to avoid damaging the tail. Baseline responses were determined for each mouse before drug injection. For morphine hot-plate tolerance, repeated morphine injections (10 mg/kg subcutaneously) were given daily for 4 d. The hot-plate test was performed on a platform heated to 47.5°C, 50°C and 52.5°C with a cutoff of 60 sec, and the latency to paw lick or jump was recorded. The baseline response was determined for each animal before treatment. The analgesic response to morphine was assessed by the tail-flick test or hot-plate test at 1 h after morphine injection (10 mg/kg). The antinociceptive response was calculated as a percentage of maximal possible effect (MPE), where MPE % = (test latency - baseline latency)/ (cutoff latency- baseline latency) × 100.Citation37,38
Morphine withdrawal and dependence
Mice were injected with escalating doses of morphine (5, 10, 20, 40, 60, 80, 100, 100 mg/kg) or saline (10 ml/kg), twice daily for 4 d. To measure physical dependence, withdrawal was precipitated by naloxone (1 mg/kg) 12 h after the last morphine injection as described previously.Citation54 We waited 12 h to limit any influence of the last morphine injection. Mice were weighed before the final saline or naloxone injection, the behavior was then scored for 30 min, and mice were weighed again. Weight loss was measured as the percentage of the pre-injection weight. The number of loose stools, jumps, wet dog shakes, backward walking, piloerection, paw and body tremors were recorded for 30 min.Citation44,54
For all experiments in vivo, data acquisition and analysis were performed with a double-blind, controlled design. All experiments involving animals and the animal care and experimental protocol were approved by the institutional review board of Kunming Institute of Zoology, Chinese Academy of Sciences.
Western blot analysis
Western blot assays for the target proteins were performed using a common approach. In brief, cell lysates of different mouse brain tissues (hippocampus and midbrain), cultured mouse midbrain neurons and rat PC12 cells were prepared using protein lysis buffer (Beyotime Institute of Biotechnology, P0013). Protein concentration was determined using the BCA protein assay kit (Beyotime Institute of Biotechnology, P0012). A total of 25 μg protein was separated by 12% or 15% SDS-PAGE, and was transferred to a polyvinylidene difluoride membrane (Bio-Rad, L1620177 Rev D). The membrane was soaked with 5% (w:v) skim milk for 2 h at room temperature. The membrane was incubated with primary antibodies against LC3B (Cell Signaling Technology, 3868; 1:1000), ATG5 (Cell Signaling Technology, 8540, 1:1000), ATG7 (Cell Signaling Technology, 8558, 1:1000), SQSTM1/p62 (Elabscience, EAP3350; 1:1000) and ACTB (Beijing Zhong Shan-Golden Bridge Biological Technology CO., LTD, TA-09; 1:10000) overnight at 4°C. The membranes were washed 3 times with Tris-buffered saline (Cell Signaling Technology, 9997) with Tween 20 (0.1%; Sigma, P1379), each time for 5 min, followed by incubation with the peroxidase-conjugated anti-mouse (lot number 474–1806) or anti-rabbit (474-1516) IgG (1:5000; KPL) for 1 h at room temperature. The epitope was visualized using an ECL western blot detection kit (Millipore, WBKLS0500). ImageJ (National Institutes of Health, Bethesda, Maryland, USA) was used to evaluate the densitometry. Western blot for ACTB was used as a loading control to measure the densitometry of ATG7, ATG5, SQSTM1 and ATG12–ATG5. The LC3B-II densitometric signal is determined by the ratio of LC3B-II to LC3B-I before normalization to ACTB.
RNA interference and transfection
Three siRNAs targeting Atg5 or Atg7 and the negative control siRNA (NC siRNA) were obtained from RiboBio (Guangzhou, China). Cultured PC12 cells and mouse midbrain neurons were transfected according to the procedure in our previous study.Citation19 In brief, cells (1 × 105 per well) were seeded in 12-well plates to grow to 50% confluence. Before transfection, culture medium was removed and washed once with Opti-MEM medium (Gibco-BRL, 31985-070). The Atg5, Atg7 siRNA or NC siRNA was dissolved in Opti-MEM medium, and was then mixed with 3 μL Lipofectamine™ 3000 (Invitrogen, L3000008) to achieve a final volume of 100 μL. The siRNA-Lipofectamine mixture was incubated at room temperature for 20 min, then the mixture was added to each well together with an additional 400 μL Opti-MEM medium. After an incubation period of 6 h in the CO2 incubator (Thermo Fisher Scientific, MA, USA), the medium was removed and 1 mL fresh medium was added to each well for growth. We optimized the siRNA concentration for transfection and the time for cell harvest by testing 5 different concentrations of siRNA (6.25, 12.5, 25, 50, and 100 nM) and an optimized 48-h time frame. The efficiency of siRNA to reduce the expression of Atg5 or Atg7 mRNA was determined by real-time quantitative PCR analysis.
Tandem mRFP-GFP fluorescence microscopy
A tandem monomeric RFP-GFP-tagged LC3 (mRFP-GFP-LC3) lentivirus (GeneChem, GPL2001) was used to monitor autophagy flux as previously reported.Citation32 PC12 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum and were infected according to the manufacturer's specifications. For evaluating tandem fluorescent LC3 puncta, 48 h after mRFP-GFP-LC3 lentivirus infection, cell images were captured using a confocal microscope (Olympus, Melville, NY, USA).
Immunofluorescence, Golgi staining and quantification
For immunofluorescence assay, mice were anesthetized with pentobarbital immediately at the end of the cycle of drug treatment, and were intracardially perfused with saline, followed by 4% paraformaldehyde. The ventral midbrain was sectioned coronally at 10-μm thickness on a cryostat (Leica, CM1850UV-1-1, Amtzell, Germany). Sections were collected on slides and were incubated for 24 h with primary anti-rabbit LC3B (1:500; Abcam, ab64781), anti-mouse TH (1:500; Merck Millipore, MAB318) and anti-mouse GAD1/GAD67 (1:500; Merck Millipore, AB152). After 3 washes with PBST (phosphate- buffered saline [Beyotime Institute of Biotechnology, C0221A] containing Triton X-100 [0.1%; Sigma-Aldrich, 10789704001]), each for 5 min, sections were then incubated with a FITC-conjugated anti-Rabbit IgG (1:500; Life Technologies, A21207) or anti-mouse IgG (1:500; Invitrogen, A21202) secondary antibody, and nuclei were counterstained with DAPI. The slides were visualized under an Olympus FluoView™ 1000 confocal microscope (Olympus, America).
We detect the morphological changes of neurons as previously described.Citation55 In brief, primary midbrain neurons were fixed and stained with antibodies against MAP2 (Merck Millipore, AB5622; 1:500) and TH (Merck Millipore, MAB318, 1:1000). Immunoreactivity was detected with a FITC-conjugated anti-rabbit IgG (1:500; Life Technologies, A21207) or anti-mouse IgG (1:500; Invitrogen, A21202) secondary antibody, and nuclei were counterstained with DAPI. The slides were visualized under an Olympus FluoView™ 1000 confocal microscope (Olympus, America).
Golgi-Cox staining (FD NeuroTechnologies, PK401) was used to detect the dendritic spines of neurons. Mouse brain tissues were impregnated for 2 wk and processed according to the manufacturer's instructions. Approximately 20 neurons were randomly selected from each group and quantified with a double blind, controlled design. ImageJ (National Institutes of Health, Bethesda, MD, USA) and Image-Pro Plus (IPP; Media Cybernetics, Inc., Washington, MD, USA) were used to evaluate the number of spines, the total dendritic length and complexity of neurons.
Statistical analysis
Comparisons of the relative protein levels of LC3B-II, ATG5, ATG7, SQSTM1 and ATG12–ATG5 from animals of multiple groups and cells with different genotypes/treatments were conducted by one-way analysis of variance (ANOVA) with the Tukey's post-hoc test using PRISM software (GraphPad Software, Inc., La Jolla, CA, USA). Differences in animal rewarding properties, behavioral sensitization, analgesic tolerance and physical dependence were determined by ANOVA, followed by least significant difference (LSD) for post-hoc comparisons. Data were represented as mean ± SEM. A P value of < 0.05 was considered to be statistically significant.
Abbreviations
| ANOVA | = | one-way analysis of variance |
| ATG5 | = | autophagy-related 5 |
| ATG7 | = | autophagy-related 7 |
| ATG12 | = | autophagy-related 12 |
| CPP | = | conditioned place preference |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| GAD1/GAD67 | = | glutamate decarboxylase 1 |
| LSD | = | least significant difference |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 beta |
| MAP2 | = | microtubule-associated protein 2 |
| MPE | = | maximal possible effect |
| NAC | = | nucleus accumbens |
| NC | = | negative control |
| OPRM1 | = | opioid receptor mu 1 |
| TH | = | tyrosine hydroxylase |
| Th-Cre | = | Cre recombinase under the control of the endogenous Th promoter |
| VTA | = | ventral tegmental area |
| WT | = | wild type |
Disclosure of potential conflicts of interest
There were no potential conflicts of interest to be disclosed.
KAUP_A_1332549_supplemental.docx
Download MS Word (11.4 MB)Acknowledgments
We thank Dr. Noboru Mizushima and Dr. Masaaki Komatsu for sharing the floxed Atg5 and floxed Atg7 mice, respectively. We thank Dr. Yue-Mei Feng, Dr. Yun-Fang Jia, and Ms Li Lv for technical assistance. We are grateful to Ian Logan for language editing and the 3 anonymous reviewers for helpful comments on the early version of the manuscript.
Funding
The study was done at Kunming Institute of Zoology, Chinese Academy of Sciences (CAS), and was supported by the National Natural Science Foundation of China (31671050), the Strategic Priority Research Program (B) of CAS (XDB02020003) and the Bureau of Frontier Sciences and Education of CAS (QYZDJ-SSW-SMC005).
Related Research Data
References
- Leshner AI. Addiction is a brain disease, and it matters. Science 1997; 278:45-7; PMID:9311924; https://doi.org/10.1126/science.278.5335.45
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology 2010; 35:217-38; PMID:19710631; https://doi.org/10.1038/npp.2009.110
- Kreek MJ, LaForge KS, Butelman E. Pharmacotherapy of addictions. Nat Rev Drug Discov 2002; 1:710-26; PMID:12209151; https://doi.org/10.1038/nrd897
- Hyman SE, Malenka RC. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci 2001; 2:695-703; PMID:11584307; https://doi.org/10.1038/35094560
- Kenny PJ. Common cellular and molecular mechanisms in obesity and drug addiction. Nat Rev Neurosci 2011; 12:638-51; PMID:22011680; https://doi.org/10.1038/nrn3105
- Chao J, Nestler EJ. Molecular neurobiology of drug addiction. Annu Rev Med 2004; 55:113-32; PMID:14746512; https://doi.org/10.1146/annurev.med.55.091902.103730
- Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci 2010; 33:267-76; PMID:20207024; https://doi.org/10.1016/j.tins.2010.02.002
- Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology 2004; 47(Suppl 1):33-46; PMID:15464124; https://doi.org/S0028390804001959
- Sklair-Tavron L, Shi WX, Lane SB, Harris HW, Bunney BS, Nestler EJ. Chronic morphine induces visible changes in the morphology of mesolimbic dopamine neurons. Proc Natl Acad Sci U S A 1996; 93:11202-7; PMID:8855333; https://doi.org/10.1073/pnas.93.20.11202
- Russo SJ, Bolanos CA, Theobald DE, DeCarolis NA, Renthal W, Kumar A, Winstanley CA, Renthal NE, Wiley MD, Self DW, et al. IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat Neurosci 2007; 10:93-9; PMID:17143271; https://doi.org/nn1812
- Robinson TE, Kolb B. Morphine alters the structure of neurons in the nucleus accumbens and neocortex of rats. Synapse 1999; 33:160-2; PMID:10400894; https://doi.org/10.1002/(SICI)1098-2396(199908)33:2<160::AID-SYN6>3.0.CO;2-S
- Ricalde AA, Hammer RP, Jr. Perinatal opiate treatment delays growth of cortical dendrites. Neurosci Lett 1990; 115:137-43; PMID:2172870; https://doi.org/10.1016/0304-3940(90)90444-E
- Nutt DJ, Lingford-Hughes A, Erritzoe D, Stokes PR. The dopamine theory of addiction: 40 years of highs and lows. Nat Rev Neurosci 2015; 16:305-12; PMID:25873042; https://doi.org/10.1038/nrn3939
- Spanagel R, Weiss F. The dopamine hypothesis of reward: past and current status. Trends Neurosci 1999; 22:521-7; PMID:10529820; https://doi.org/S0166-2236(99)01447-2
- Volkow ND, Fowler JS, Wang GJ, Swanson JM, Telang F. Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Arch Neurol 2007; 64:1575-9; PMID:17998440; https://doi.org/10.1001/archneur.64.11.1575
- Robinson TE, Gorny G, Savage VR, Kolb B. Widespread but regionally specific effects of experimenter- versus self-administered morphine on dendritic spines in the nucleus accumbens, hippocampus, and neocortex of adult rats. Synapse 2002; 46:271-9; PMID:12373743; https://doi.org/10.1002/syn.1014617
- Zhao L, Zhu Y, Wang D, Chen M, Gao P, Xiao W, Rao G, Wang X, Jin H, Xu L, et al. Morphine induces Beclin 1- and ATG5-dependent autophagy in human neuroblastoma SH-SY5Y cells and in the rat hippocampus. Autophagy 2010; 6:386-94; PMID:20190558; https://doi.org/11289
- Hayashi Y, Koga Y, Zhang X, Peters C, Yanagawa Y, Wu Z, Yokoyama T, Nakanishi H. Autophagy in superficial spinal dorsal horn accelerates the cathepsin B-dependent morphine antinociceptive tolerance. Neuroscience 2014; 275:384-94; PMID:24973657; https://doi.org/10.1016/j.neuroscience.2014.06.037
- Feng YM, Jia YF, Su LY, Wang D, Lv L, Xu L, Yao YG. Decreased mitochondrial DNA copy number in the hippocampus and peripheral blood during opiate addiction is mediated by autophagy and can be salvaged by melatonin. Autophagy 2013; 9:1395-406; PMID:23800874; https://doi.org/10.4161/auto.25468
- Pan J, He L, Li X, Li M, Zhang X, Venesky J, et al. Activating autophagy in hippocampal cells alleviates the morphine-induced memory impairment. Mol Neurobiol 2017; 54(3):1710-1724; PMID:26873855; https://doi.org/10.1007/s12035-016-9735-3
- Yang Y, Coleman M, Zhang L, Zheng X, Yue Z. Autophagy in axonal and dendritic degeneration. Trends Neurosci 2013; 36:418-28; PMID:23639383; https://doi.org/10.1016/j.tins.2013.04.00122
- Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 2007; 3:591-6; PMID:17912025; https://doi.org/4964
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A 2007; 104:14489-94; PMID:17726112; https://doi.org/0701311104
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature 1998; 395:395-8; PMID:9759731; https://doi.org/10.1038/26506
- Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem 1998; 273:33889-92; PMID:9852036; https://doi.org/10.1074/jbc.273.51.33889
- Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol 2004; 15:231-6; PMID:15209383; https://doi.org/S1084-9521(03)00109-5
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657-68; PMID:11266458; https://doi.org/10.1083/jcb.152.4.657
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; https://doi.org/jcb.200412022
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena, A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; https://doi.org/10.1080/15548627.2015.1100356
- Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, Knapp PE. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at mu-opioid receptor-expressing glia. Brain 2011; 134:3616-31; PMID:22102648; https://doi.org/10.1093/brain/awr28131
- Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience 2001; 102:555-63; PMID:11226693; https://doi.org/S0306-4522(00)00461-9
- Feng L, Ma Y, Sun J, Shen Q, Liu L, Lu H, Wang F, Yue Y, Li J, Zhang S, et al. YY1-MIR372-SQSTM1 regulatory axis in autophagy. Autophagy 2014; 10:1442-53; PMID:24991827; https://doi.org/10.4161/auto.29486
- Coleman MP, Perry VH. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci 2002; 25:532-7; PMID:12220882; https://doi.org/S0166-2236(02)02255-5
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science 2002; 296:868-71; PMID:11988563; https://doi.org/10.1126/science.1068613
- Cai Y, Yang L, Hu G, Chen X, Niu F, Yuan L, Liu H, Xiong H, Arikkath J, Buch S. Regulation of morphine-induced synaptic alterations: Role of oxidative stress, ER stress, and autophagy. J Cell Biol 2016; 215:245-58; PMID:27810915; https://doi.org/jcb.201605065
- Seibenhener ML, Wooten MC. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J Vis Exp 2015; (96):e52434; PMID:25742564; https://doi.org/10.3791/52434
- Zachariou V, Georgescu D, Sanchez N, Rahman Z, DiLeone R, Berton O, Neve RL, Sim-Selley LJ, Selley DE, Gold SJ, et al. Essential role for RGS9 in opiate action. Proc Natl Acad Sci U S A 2003; 100:13656-61; PMID:14595021; https://doi.org/10.1073/pnas.2232594100
- Zachariou V, Bolanos CA, Selley DE, Theobald D, Cassidy MP, Kelz MB, Shaw-Lutchman T, Berton O, Sim-Selley LJ, Dileone RJ, et al. An essential role for DeltaFosB in the nucleus accumbens in morphine action. Nat Neurosci 2006; 9:205-11; PMID:16415864; https://doi.org/nn163639
- Steketee JD, Kalivas PW. Drug wanting: behavioral sensitization and relapse to drug-seeking behavior. Pharmacol Rev 2011; 63:348-65; PMID:21490129; https://doi.org/10.1124/pr.109.001933
- Harris GC, Aston-Jones G. Involvement of D2 dopamine receptors in the nucleus accumbens in the opiate withdrawal syndrome. Nature 1994; 371:155-7; PMID:7915401; https://doi.org/10.1038/371155a0
- Kaufling J, Aston-Jones G. Persistent adaptations in afferents to ventral tegmental dopamine neurons after opiate withdrawal. J Neurosci 2015; 35:10290-303; PMID:26180204; https://doi.org/10.1523/JNEUROSCI.0715-15.2015
- Wei ZB, Yuan YF, Jaouen F, Ma MS, Hao CJ, Zhang Z, Chen Q, Yuan Z, Yu L, Beurrier C, et al. SLC35D3 increases autophagic activity in midbrain dopaminergic neurons by enhancing BECN1-ATG14-PIK3C3 complex formation. Autophagy 2016; 12:1168-79; PMID:27171858; https://doi.org/10.1080/15548627.2016.117940243
- Wang D, Hiesinger PR. Autophagy, neuron-specific degradation and neurodegeneration. Autophagy 2012; 8:711-3; PMID:22498474; https://doi.org/10.4161/auto.19660
- Georgescu D, Zachariou V, Barrot M, Mieda M, Willie JT, Eisch AJ, Yanagisawa M, Nestler EJ, DiLeone RJ. Involvement of the lateral hypothalamic peptide orexin in morphine dependence and withdrawal. J Neurosci 2003; 23:3106-11; PMID:12716916; https://doi.org/ 23/8/3106
- Wolf DH, Numan S, Nestler EJ, Russell DS. Regulation of phospholipase Cγ in the mesolimbic dopamine system by chronic morphine administration. J Neurochem 1999; 73:1520-8; PMID:10501197; https://doi.org/10.1046/j.1471-4159.1999.0731520.x
- Muller DL, Unterwald EM. In vivo regulation of extracellular signal-regulated protein kinase (ERK) and protein kinase B (Akt) phosphorylation by acute and chronic morphine. J Pharmacol Exp Ther 2004; 310:774-82; PMID:15056728; https://doi.org/10.1124/jpet.104.066548
- Harvey BK, Hope BT, Shaham Y. Tolerance to opiate reward: role of midbrain IRS2-Akt pathway. Nat Neurosci 2007; 10:9-10; PMID:17189943; https://doi.org/nn0107-948
- Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy 2007; 3:635-7; PMID:17786026; https://doi.org/4916
- Cui Y, Zhang XQ, Xin WJ, Jing J, Liu XG. Activation of phosphatidylinositol 3-kinase/Akt-mammalian target of Rapamycin signaling pathway in the hippocampus is essential for the acquisition of morphine-induced place preference in rats. Neuroscience 2010; 171:134-43; PMID:20826199; https://doi.org/10.1016/j.neuroscience.2010.08.064
- Gelman DM, Noain D, Avale ME, Otero V, Low MJ, Rubinstein M. Transgenic mice engineered to target Cre/loxP-mediated DNA recombination into catecholaminergic neurons. Genesis 2003; 36:196-202; PMID:12929090; https://doi.org/10.1002/gene.10217
- Yang F, Feng L, Zheng F, Johnson SW, Du J, Shen L, Wu CP, Lu B. GDNF acutely modulates excitability and A-type K(+) channels in midbrain dopaminergic neurons. Nat Neurosci 2001; 4:1071-8; PMID:11593232; https://doi.org/10.1038/nn734
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; https://doi.org/nature04724
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; https://doi.org/nature0472354
- Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zachariou V, Impey S, Duman RS, Storm D, Nestler EJ. Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci 2002; 22:3663-72; PMID:11978842; https://doi.org/20026223
- Su LY, Li H, Lv L, Feng YM, Li GD, Luo R, Zhou HJ, Lei XG, Ma L, Li JL, et al. Melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA/α-synuclein aggregation. Autophagy 2015; 11:1745-59; PMID:26292069; https://doi.org/10.1080/15548627.2015.1082020