
ABSTRACT
The ingrained capacity of melanoma cells to rapidly evolve toward an aggressive phenotype is manifested by their increased ability to develop drug-resistance, evident in the case of vemurafenib, a therapeutic-agent targeting BRAFV600E. Previous studies indicated a tight correlation between heightened melanoma-associated macroautophagy/autophagy and acquired Vemurafenib resistance. However, how this vesicular trafficking pathway supports Vemurafenib resistance remains unclear. Here, using isogenic human and murine melanoma cell lines of Vemurafenib-resistant and patient-derived melanoma cells with primary resistance to the BRAFV600E inhibitor, we found that the enhanced migration and invasion of the resistant melanoma cells correlated with an enhanced autophagic capacity and autophagosome-mediated secretion of ATP. Extracellular ATP (eATP) was instrumental for the invasive phenotype and the expansion of a subset of Vemurafenib-resistant melanoma cells. Compromising the heightened autophagy in these BRAFV600E inhibitor-resistant melanoma cells through the knockdown of different autophagy genes (ATG5, ATG7, ULK1), reduced their invasive and eATP-secreting capacity. Furthermore, eATP promoted the aggressive nature of the BRAFV600E inhibitor-resistant melanoma cells by signaling through the purinergic receptor P2RX7. This autophagy-propelled eATP-dependent autocrine-paracrine pathway supported the maintenance and expansion of a drug-resistant melanoma phenotype. In conclusion, we have identified an autophagy-driven response that relies on the secretion of ATP to drive P2RX7-based migration and expansion of the Vemurafenib-resistant phenotype. This emphasizes the potential of targeting autophagy in the treatment and management of metastatic melanoma.
Introduction
Metastatic melanoma is the most aggressive form of skin cancer, as demonstrated by its extremely poor survival rates.Citation1 In recent years, one of the most significant advances in the field of melanoma-targeted therapy was the successful implementation of BRAFV600E-targeted therapy (Vemurafenib), which interrupts the prosurvival stimulation provided by continuous activation of the BRAF-MAP2K/MEK pathway in approximately 40–60% of cutaneous melanomas.Citation2-6 Unfortunately, most patients eventually develop resistance to Vemurafenib and relapse within a year, showing the propensity of melanoma cells to rapidly evolve a resistant phenotype with more aggressive features.Citation4 To date, a plethora of molecular signaling adaptations have been described to contribute to the development of a Vemurafenib-resistant phenotype.Citation4,7 These often rely on melanoma cells' ability to bypass BRAFV600E blockade and reactivate MAP2K-based signaling through alternative routes.Citation7,8 In addition to MAPK signaling-related alterations, melanoma cells can also acquire resistance against Vemurafenib through the potentiation of macroautophagy (hereafter referred to as autophagy).Citation8 Autophagy is an essential adaptive response that initiates the sequestration of damaged or aging intracellular content leading to their degradation.Citation9 This ensures important quality control of cytoplasmic materials along with the generation of building blocks to support growth and survival under conditions of nutrient deprivation or cellular stress.Citation10 This multistep catabolic process plays an especially important role in melanoma development and acquisition of resistance to anticancer therapies.Citation11 In fact, studies published by us as well as others, have established that autophagy can exert a cell-autonomous cytoprotective function against Vemurafenib-induced cell death in melanoma.Citation8,12
However, in recent times it has emerged that the prosurvival role of autophagy in cancer cells may not be limited to intracellular cell-autonomous functions but may extend to autocrine-paracrine signaling.Citation12 More specifically, autophagy may act as an unconventional secretory apparatus for extracellular exodus of certain biomolecules, which in turn can exert cancer-supportive functions, e.g. increased migration or invasion of cancer cells.Citation12 Notably, autophagy has emerged as a major pathway for unconventional secretion of extracellular ATP (eATP).Citation13 In the context of a tumor, eATP can exert pleiotropic effects depending on the target cells that interact with it; ranging from pro-chemotactic or pro-inflammatory effects (on immune cells) to the enablement of cell proliferation, migration and/or invasion (in cancer cells and some stromal cell types).Citation13-15 Moreover, eATP can also modulate the levels of a subpopulation of cancer cells known as the cancer stem-like side population (SP), which is often critical for the global fitness of the cancer cell-population,Citation16 retaining their clonogenicity under stress to underlie tumor cell survival.Citation17 Secreted ATP, which is observed in a variety of physiological and pathological conditions, exerts its functional effects predominantly through interaction with the purinergic receptors.Citation18-21 With respect to cancer cells, autophagy-mediated ATP secretion has been demonstrated to operate in both physiological stress conditions (i.e., during starvation) as well as following therapeutic stress exerted by various anticancer therapies.Citation12,22,23
Despite this ubiquitous tendency to get secreted and its pleiotropic functional activities, the effects of eATP on Vemurafenib-resistant melanoma cells remain completely unresolved. This knowledge is crucial considering that the BRAF inhibitor-induced tumor secretome has been recently shown to promote melanoma aggressiveness;Citation24 however, the molecular nature of the key secreted factors remained uncharacterized. To this end, we wondered whether heightened autophagy, activated in Vemurafenib-resistant melanoma cells, could be facilitating certain melanoma-associated aggressive features (e.g., migration, invasion and maintenance of a SP fraction) through the autocrine-paracrine interactions between eATP and purinergic receptors.
Here, we show that the increased autophagic capacity of Vemurafenib-resistant melanoma cells exacerbates the secretion of ATP that, via interaction with purinergic receptors, stimulates melanoma cell migration and invasion. Moreover, this autocrine response favors the presence and enrichment of a melanoma cell subpopulation hallmarked by a potentiated xenobiotic export, which may further solidify melanoma cell acquired resistance to Vemurafenib.
Results
Acquired BRAFV600E inhibitor resistance facilitates extracellular ATP-dependent enhancement of melanoma cell migration and invasion
Acquired resistance to Vemurafenib is a critical mechanism behind the aggressiveness of BRAFV600E melanoma cells. To investigate the mechanisms underlying the Vemurafenib or PLX4032-resistant phenotype (hereafter referred to as PLX-resistance), we compared 2 isogenic metastatic melanoma cell lines, i.e. 451-Lu and A375, to their PLX-resistant counterparts. Acquired resistance to PLX resulted in a noticeable alteration in cellular morphology toward a more mesenchymal-like phenotype (Fig. S1A). This was accompanied by an increase in the protein levels of typical mesenchymal markers (Fig. S1B), including a transition from CDH1/E-cadherin to CDH2/N-cadherin, and enhanced MITF, FN1/fibronectin and TJP1/ZO-1 expression, which are characteristics of highly invasive cancer cells.Citation25 Moreover, both PLX-resistant melanoma cell lines showed a significantly heightened cell migration, as measured by the scratch-wound ( and Fig. S1C) and transwell assays ( and Fig. S1D) and invasive capability (), as demonstrated by their ability to migrate through the extracellular matrix ( and Fig. S1E), as compared with their isogenic PLX-susceptible counterparts.
Figure 1. PLX-acquired resistance promotes migration, invasion and resistance in an eATP-dependent manner. The indicated isogenic cell models were assessed after a 72-h incubation by light microscopy-based scratch assays (A-B), transwell cell migration (C), invasion (D) and eATP (E). The capacity of 451-Lu/Res conditioned media (CM) to stimulate migration of the parental 451-Lu cells was assessed by transwell migration assays in the presence or absence of 2 U/ml apyrase (APY) (F). The ability of eATP to stimulate cell migration (G, I) or invasion (H, J) were monitored by transwell assays. Cells were exposed to either APY (G, H) or ATP (50 µM, I, J) and then monitored after 72 h. The SM1-based isogenic cell models were assessed for their eATP alone or following 72-h exposure to 10 µM PLX; RLU, relative luciferase units. (K). The effect of extracellular ATP modulation of migration (L) and invasion (M) of the SM1 isogenic cell lines was assessed 72 h post ATP or APY additions. Scale bars: 100 µm. All experiments are representative of 3 independent experiments and expressed as mean ± SD. */$ = P < 0.05, ** = P < 0.01, ***/$$$ = P < 0.001, **** P < 0.0001.
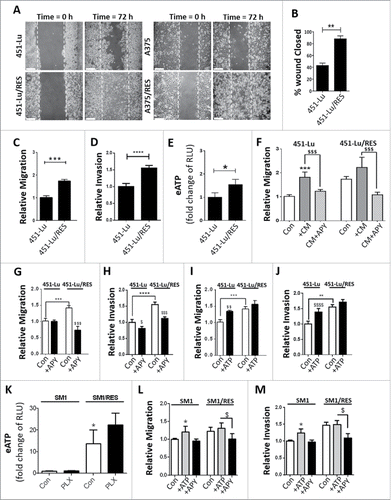
Recent studies have highlighted the importance of extracellular ATP (eATP) for chemotaxis and cellular migration.Citation26 Therefore, we wondered whether the above phenotype of PLX-resistant cells was regulated by eATP. Analysis of the basal eATP showed that both PLX-resistant melanoma cell lines displayed a higher secretion of ATP compared with their PLX-susceptible partners ( and Fig. S1F). Interestingly, the addition of PLX exacerbated ATP secretion (Fig. S1G) and stimulated migration of PLX-resistant cells (Fig. S1H). In contrast, the MAP2K inhibitor UO126 (MAP2Ki) suppressed eATP and reduced migration to a level observed for the PLX-susceptible cells (Fig. S1G, H), suggesting that heightened MAP2K activity, a characteristic of PLX-resistant melanoma cells,Citation8 fosters the active secretion of ATP.
Next, to confirm the role of eATP in favoring melanoma cell migration, PLX-susceptible and PLX-resistant melanoma cells were incubated with conditioned media (CM) derived from PLX-resistant cells either alone or in combination with apyrase (APY, ), an ATP-degrading enzymeCitation27 (Fig. S1I). Incubation of PLX-susceptible cells with the above CM increased their migratory capacity to the same level observed for PLX-resistant cells (). Moreover, the addition of PLX (Fig. S1J) phenocopied the effect of PLX-resistant cell-derived CM. These effects were dependent on eATP since the addition of the APY reduced the migration of PLX-resistant cells under basal ( and Fig. S1K), CM-supplemented () or PLX-stimulated (Fig. S1J) conditions, without affecting the PLX-susceptible cells. Furthermore, the PLX-driven heightened invasion capacity of the PLX-resistant cells, in comparison to their isogenic counterparts, was significantly reduced by the addition of APY ( and Fig. S1L). Conversely, the addition of ATP into the culture media of PLX-sensitive melanoma cells potentiated both their migration and invasion (, and Fig. S2A-C), without affecting these responses in their PLX-resistant counterparts. However, exogenously added ATP did not alter the proliferation rate of either PLX-sensitive or -resistant melanoma cells (Fig. S2D, E) nor did it induce cell death (Fig. S2F), thus formally excluding effects potentially due to changes in cell proliferation and viability.
Additionally, we used an isogenic mouse model (SM1 vs. SM1/RES) of PLX-acquired resistance (Fig. S2G) to further prove the species independence of this eATP-mediated autocrine-paracrine loop. Notably, a similar pattern of basal eATP (), as well as eATP-mediated enhancement of migration and invasion, but not proliferation, was confirmed in the isogenic murine melanoma cell lines, SM1 and its PLX-resistant counterpart SM1/RES (, and Fig. S2H).
Taken together these data show that PLX-resistant human or murine melanoma cells promote ATP secretion, which in turn stimulates their migratory and invasive phenotype.
Extracellular ATP-driven cell invasion is conserved in patient-derived melanoma cells displaying primary resistance to PLX
To further validate the role of eATP in supporting migration and invasion of PLX-resistant melanoma cells, we then expanded our observations in more relevant cellular models of melanoma PLX-resistance. Moreover, we wished to explore whether the eATP-fostered melanoma migratory/invasive phenotype was a specific hallmark of in vitro acquired PLX-resistance, or a more general phenomenon associated with primary PLX-resistance as well.Citation28,29 To this end, we used a pair of BRAFV600E patient-derived melanoma cell lines displaying PLX sensitivity or primary drug-resistance.Citation29,30
Initially we investigated the PLX-mediated cytotoxicity of patient-derived BRAFV600E PLX-sensitive melanoma cell lines, i.e. M229 and M249 cells, or with primary PLX-resistance, i.e. M233 and M263 cells.Citation29 Interestingly, the latter PLX-resistant melanoma cells, displayed the more elongated morphology characteristic of invasive, drug-resistant melanoma cells (Fig. S2I). In line with previous reports,Citation29 we found that while BRAFV600E M229 and M249 melanoma cells rapidly lost their viability upon exposure to increasing concentrations of PLX, the M233 and M263 melanoma cells demonstrated no significant inhibition of cell viability ().
Figure 2. Extracellular ATP phenotype extends in vitro to naturally resistant tumor cells. Patient-derived melanoma cells demonstrating natural PLX-sensitive (M229, M249) and -resistant (M233, M263) phenotypes were exposed to increasing doses of PLX4032 (PLX) for 72 h and resistance assessed by MUH assay-based cell viability assays (A) or extracellular ATP analyzed (B, 10 µM PLX for 72 h; RLU, relative luciferase units). The capacity of exogenous ATP to facilitate migration (C, E) or invasion (D, F) were assessed following either APY (C, D; 2 U/ml) or ATP (E, F; 50 µM) exposure for 72 h. Data are the mean ± SD of 3 independent experiments. * = P < 0.05, **/$$ = P < 0.01.
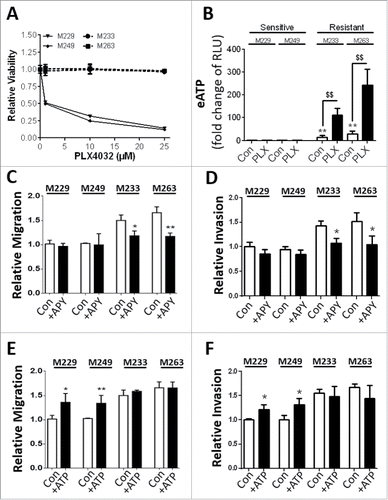
Notably, only PLX-resistant M233 and M263 cells showed a significant secretion of ATP, an ability that could be significantly reinforced in response to PLX (). Interestingly, eATP was not detected in the media of the PLX-sensitive M229 and M249 cells, either under untreated conditions or upon PLX treatment eliciting cell death (). This further indicates that secretion of ATP is an active process associated with the surviving PLX-resistant melanoma cells. Moreover, as shown in the case of melanoma cell lines of acquired PLX resistance (, ), only the primarily PLX-resistant melanoma cells exhibited an increased migration and invasion capacity that was APY-sensitive (). Moreover, as in the case of the isogenic 451-LU and A375 cell lines, exogenously added ATP fostered migration and to a lesser extent, invasion, of the PLX-sensitive cells ( E-F). Finally, assessment of their proliferation capacity indicated no ATP dependent alterations, yet PLX-resistant variants proliferated to a lesser extent (Fig. S2J).
Thus, our data show that patient-derived melanoma cells harboring primary PLX-resistance, exhibit a PLX-stimulated eATP-mediated autocrine-paracrine signaling supporting their migration and invasion.
ATP secreted by the PLX-acquired resistant cells contributes to the maintenance of a melanoma drug-resistant cellular population
Resistance to PLX correlates with increased expression of ABC transporters,Citation30 key markers of drug resistance, proliferation and clonogenicity,Citation31,32 and essential components of a cancer cell subset displaying increased drug efflux capacity, also known as side population (SP).Citation31 The detection of a Hoechstlow population that is verapamil-sensitive,Citation33 is indicative of this SP with heightened ABC transporter-mediated efflux capacity.Citation31,32,34
Since we initially identified an enrichment in the protein expression of ABCB1 (Fig. S1B), we then wondered if eATP could contribute to an increase in a melanoma SP subset. To minimize effects linked to the different genetic background of the patient-derived melanoma cell lines,Citation28,29 we decided to further explore this relevant connection using the 451-Lu and A375 isogenic cellular models. Initially, we observed an increased fraction of melanoma cells with increased efflux capacity in the PLX-resistant melanoma cells (Fig. S3A). Most strikingly, we found that this PLX-resistant melanoma cell subset could be significantly reduced by lowering the concentration of eATP ( and Fig. S3B). Conversely, the addition of exogenous ATP ( and Fig. S3C) or PLX (Fig. S3D) stimulated the SP fraction characteristic of the PLX-resistant cells.
Figure 3. PLX-acquired resistance promotes an aggressive, drug-resistant signature in vitro. 451-Lu isogenic models were assessed, after a 72-h incubation with either extracellular APY (A, 2 U/ml) or ATP (B, 50 µM), for adaptations in side population (SP) subsets. The 451-Lu isogenic cell models were screened for a panel of drug resistance, stemness and tumor aggression markers by q-RTpcr (C-I, including; ABCB1, ABCG2, KDM5B/JARID1B, SOX10, TERT, NGFR, MYC normalized to GAPDH). All experiments are representative of 3 independent experiments and expressed as mean ± SD. */$ = P < 0.05, **/$$ = P < 0.01, ***/$$$ = P < 0.001, **** P < 0.0001.
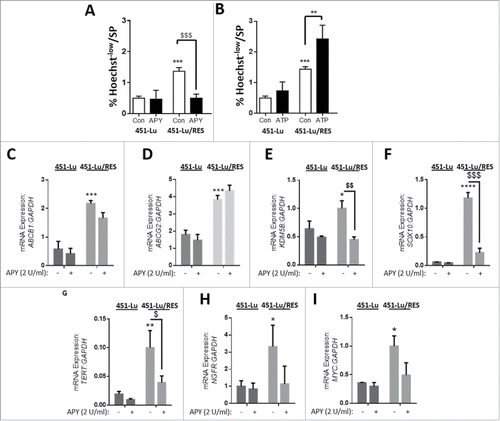
Next, to unravel the aggressive nature of PLX-resistant melanoma cells and confirm the involvement of eATP in vitro, we assessed the mRNA expression levels of known markers of melanoma aggressive phenotype,Citation35-37 SPCitation33 and drug resistanceCitation34 and their dependence on the presence of eATP. We found that acquired PLX-resistance was associated with a marked increase in the expression of the drug resistance pump-coding genes ABCB1 and ABCG2, KDM5B/JARIDB1, SOX10, TERT, NGFR/CD271 and MYC ( and Fig. S3E-K). Interestingly, while APY did not alter mRNA expression of these markers in PLX-sensitive cells, reducing eATP by APY treatment led to a statistically significant reduction in the mRNA expression of KDM5B/JARID1B, SOX10 and TERT ( and Fig. S3G-I), while NGFR and MYC expression showed a similar, albeit not statistically significant, trend ( H, I and Fig. S3J, K).
These results reveal that upon acquired resistance to PLX, eATP enables melanoma cells to maintain a more aggressive and PLX-based drug-resistant signature.
ATP secretion is mediated by heightened autophagy in PLX-resistant melanoma cells
Based on our results implicating ATP release from melanoma cells with acquired or primary PLX-resistance as a mechanism supporting their aggressive and invasive phenotype, we next set out to investigate the mechanism underlying ATP secretion. Recent studies have implicated autophagy as a major mechanism for ATP secretion from dying cancer cells following chemotherapy.Citation22,38 However, little is known about the role of autophagy in ATP secretion from actively proliferating, or therapy-resistant cancer cells. We have recently shown that autophagy is increased following the acquisition of resistance to PLX therapy.Citation8 Thus, we wondered if the stimulated autophagy in PLX-resistant melanoma cells was causally linked to the increased ATP secretion by these cells.
We initially confirmed that upon acquired PLX-resistance (both human and mouse) as well as for primary PLX-resistant patient-derived cell lines (Fig. S4A-D)Citation8,39 the autophagic flux was increased as compared with the parental cells. Indeed, in the presence of the autophagic flux blocker bafilomycin A1 (Baf A1), the accumulation of the autophagic substrates MAP1LC3B/LC3B-II and SQSTM1/p62, as judged by immunoblotting, increased to a greater extent in all the PLX-resistant cells as compared with their respective PLX-sensitive counterparts (Fig. S4A, C, D). Moreover, this pattern of increased autophagic flux was confirmed by immunofluorescence-based imaging of LC3 redistribution in a punctate pattern (Fig. S4B). We also observed that treatment of the PLX-resistant 451-LU and A375 cells with exogenously added ATP could further stimulate the accumulation of LC3-II (Fig. S4E).
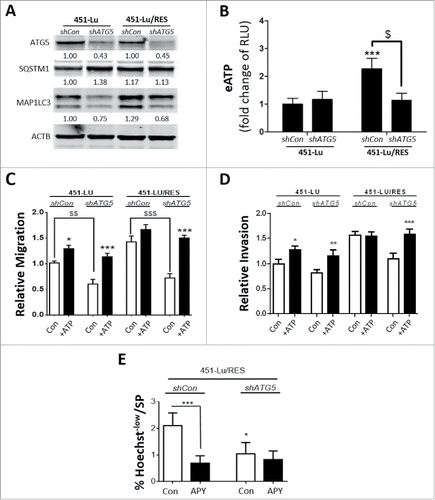
Next, to better understand the role of autophagy in ATP secretion, we stably knocked down ATG5 by shRNA-mediated transduction, in both 451-Lu and 451-Lu/RES cells and assessed whether attenuating basal autophagy () could affect the capacity of PLX-sensitive and -resistant melanoma cells to secrete ATP (). We found that while mock-shRNA transduced PLX-resistant cells (shCon) still exhibited enhanced capacity to export ATP, the silencing of ATG5 in these PLX-resistant cells reverted their ability to secrete ATP back to the levels displayed by their PLX-sensitive counterparts (). Conversely, ATG5 knockdown had no significant effect on the levels of eATP in the media derived from PLX-sensitive cells (). Along with their reduced ability to export ATP, autophagy-compromised PLX-resistant cells, but not their isogenic counterparts, also exhibited a diminished migration and invasion potential (, Fig. S4F). This ATG5-regulated effect was eATP dependent, since exogenous addition of ATP rescued the migratory capacity of the ATG5-silenced PLX-resistant cells to that of their ATG5-proficient counterparts (, Fig. S4G). Furthermore, autophagy was also implicated in maintaining the higher ATP-stimulated SP potential of the PLX-resistant cells, since compromising autophagy in PLX-resistant 451-Lu cells decreased the SP subset and abrogated the susceptibility to APY treatment in these cells (). APY instead was clearly effective in reducing the SP potential of the PLX-resistant shCon cells ().
Figure 4. Elevated secretion of ATP by PLX-resistant melanoma cells is an autophagy-dependent process. 451-Lu PLX isogenic cell models were stably knocked down in ATG5 expression, in comparison to control (shCon) by shRNA and confirmed by western blot analysis of ATG5, SQSTM1 and MAP1LC3B/LC3B-II, normalized to ACTB (A). Following stable ATG5 knockdown, eATP was stained and assessed using a FlexStation 3 microplate reader; RLU, relative luciferase units (B). The effects of ATG5 knockdown on the cell migration or invasion potential were characterized by transwell assays (C, D). Hoechst 33342-based flow cytometry was performed on 451-Lu/RES cells, stably transduced with shCon vs. shATG5, and the percentage of Hoechstlow cells determined (E). All experiments are representative of 3 independent experiments ± SD. */$ = P < 0.05, **/$$ = P < 0.01, ***/$$$ = P < 0.001.
To corroborate that this autocrine/paracrine signaling loop was mediated by bona fide autophagy, we evaluated the effects of the transient knockdown of 2 additional autophagy proteins regulating the formation of autophagosomes, i.e. ATG7 and ULK1, in the human 451-Lu-based PLX isogenic melanoma cell model, which resulted in an autophagy blockade (Fig. S5A, B). Consistent with the observations obtained for ATG5-silenced cells, transient knockdown of either ATG7 or ULK1 blunted the increased ability of the PLX-resistant melanoma cells to secrete ATP () and to migrate faster (; Fig. S5C, D), a process that could be rescued by the addition of exogenous ATP (). Of note, the transient knockdown either of ATG5, ATG7 or ULK1 in the A375 isogenic models recapitulated the migratory phenotypes documented for the 451-Lu cells (Fig. S6), strengthening the significance of autophagy in eATP-mediated migration of PLX-resistant cells.
Figure 5. Autophagy governs ATP secretion of the PLX-resistant melanoma cells. Following knockdown of either ULK1 (A, C) or ATG7 (B, D) in 451-Lu or 451-Lu/RES melanoma cells, eATP (A, B), or migration by transwell assays (C, D) were assessed; RLU, relative luciferase units. The capacity of exogenously added ATP (50 µM) to restore migration was assessed by transwell migration assays (C, D). 451-Lu and 451-Lu/Res isogenic melanoma models were treated with 1 µM quinacrine (green) for 2 h at 37°C alone or in combination with Baf A1 (10 nM, 1 h pre-treatment), before colocalization with autophagosomes was assessed with MAP1LC3B/LC3B-II (red) (E), light microscopy overlays (differential interference contrast/DIC) are provided. Colocalization was calculated using imageJ (F). All experiments are representative of 3 independent experiments and expressed as mean ± SD. Scale bar: 10 µM. */$ = P < 0.05, **/$$ = P < 0.01, *** = P < 0.001.
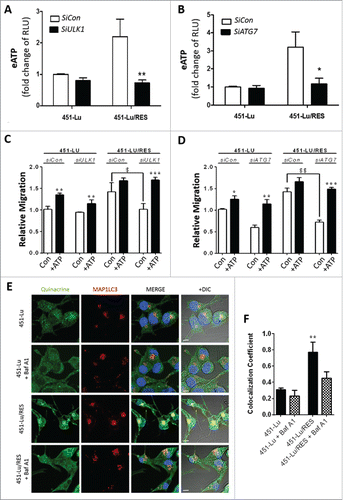
Next, to encapsulate the significance of autophagy in ATP secretion, PLX-sensitive and -resistant 451-Lu cells were treated with quinacrine to stain putative ATP-rich stores, alone or in the presence of Baf A138 and then costained for LC3-II (). Colocalization studies revealed that upon acquisition of PLX resistance, which is accompanied by increased autophagic flux (Fig. S4A, B), the colocalization of intracellular ATP with the pro-autophagy marker LC3 increased. Conversely, the quinacrine staining and LC3 colocalization were significantly reduced in the presence of the vacuolar-type H+-translocating ATPase inhibitor Baf A1, thus suggesting that ATP is mainly trafficked in LC3-decorated acidic vesicles ().
Thus, PLX-resistant melanoma cells rely on their stimulated autophagic machinery to mediate ATP secretion, thereby fostering an eATP-driven autocrine loop that favors migration and the expansion of a drug-resistant melanoma SP subset.
Secreted ATP stimulates PLX-resistant melanoma cell migration by activating purinergic receptors
Interaction between eATP and purinergic receptors accounts for most of the signaling effects of eATP.Citation27,40 To understand the significance of purinergic receptors in the modulation of cell migration of PLX-resistant melanoma cells, we then investigated the effect of the pan-purinergic receptor inhibitor suramin,Citation40 and the P2RX7/P2X7 receptor-specific inhibitor A740003 (A74),Citation40 on ATP secretion and migration of melanoma cells. Treatment with Suramin or A74, did not affect eATP levels wherever applied ( and Fig. S7A). Remarkably, however, addition of either Suramin or A74, reduced the migration/invasion capacity of the PLX-resistant cells, but had no influence on their PLX-sensitive isogenic counterparts (, and Fig. S7B, C). Moreover, autophagy inhibition (shATG5) also abolished the P2RX7 receptor sensitivity of the PLX-resistant melanoma cell's SP subset, without affecting that of the mock-shRNA controls (shCon, ). This finding further reveals that in PLX-resistant cells, an autophagy-mediated eATP-P2RX7 autocrine pathway supports the presence of the drug effluxing SP subset.
Figure 6. Extracellular ATP promotes PLX-resistant melanoma cell migration and invasion in a purinergic receptor-dependent manner. eATP was analyzed in the 451-Lu isogenic models following treatment with either 5 µM suramin or 10 µM A740003; RLU, relative luciferase units. (A). 451-Lu and 451-Lu/RES cell migration (B) and invasion (C) was assessed by scratch and transwell invasion assays following treatment with either 5 µM suramin or 10 µM A740003. 451-Lu/RES cells (shCon vs. shATG5) were analyzed by Hoechst 33342-based flow cytometry and changes in the percentage of Hoechstlow cells determined (D). Data are the mean ± SD of 3 independent experiments. ** = P < 0.01, *** = P < 0.001, **** P < 0.0001.
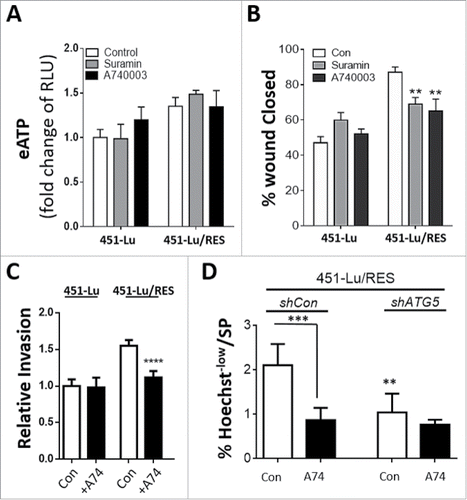
To further expand the importance of the purinergic receptors in our observed eATP-mediated phenotypes, we also treated our primary PLX-resistant melanoma cell lines and the mouse isogenic cell lines with A74 and observed a significant reduction in the invasion potential of the resistant melanoma cells (Fig. S7D, E), with no observable effects on their PLX-sensitive controls.
It has been previously shown that binding of ATP to P2RX7 results in pore opening and increased influx/uptake of small molecules and ions.Citation41 Therefore, to establish eATP-based stimulation of P2RX7, we measured rhodamine 123 (Rh123, 0.38 kDa) uptake by both PLX isogenic melanoma cells. Interestingly, both PLX-resistant melanoma cell models exhibited a significant increase in Rh123 uptake as compared with their isogenic controls (Fig. S8A). Furthermore, when exposed to exogenously added ATP, Rh123 uptake was potentiated only in the PLX-resistant melanoma cell lines ( and Fig. S8B). When PLX-resistant cells were co-incubated with exogenous ATP and the P2RX7 inhibitor A74, the increased influx of Rh123 driven by eATP was significantly dampened ( and Fig. S8B).
Figure 7. Extracellular ATP promotes cellular uptake via P2RX7. 451Lu and 451-Lu/Res were treated with 50 µM ATP for 24 h alone or in combination with 10 µM A740003 (A74) and Rh123 (2 µM for 1 h) positivity assessed by flow cytometry (A). 451-Lu isogenic cells were exposed to 2 µM Rh123 alone or in combination with APY (2 U/ml, overnight pre-incubation) for 1 h and uptake assessed (B). 451Lu and 451-Lu/Res cells were treated with 50 µM ATP for 24 h alone or in combination with 10 µM A740003 (A74) and FITC-dextran (C, 40 µg/ml for 2 h) uptake assessed by flow cytometry. 451-Lu/Res cells harboring shCon compared with shATG5 were analyzed for Rh123 uptake potential at basal conditions (D) or following exposure to 50 µM ATP alone or combined with 10 µM A74 (E). Data are the mean ± SD of 3 independent experiments. * = P < 0.05, ** = P < 0.01, *** = P < 0.001. MFI, mean fluorescence intensity.
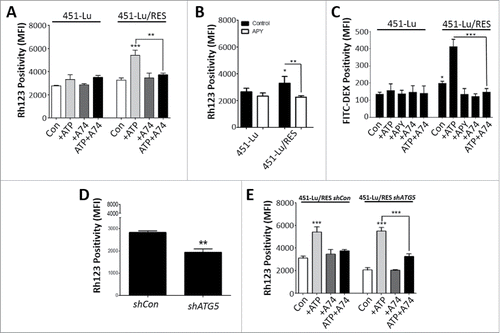
Furthermore, addition of APY blunted the Rh123 uptake by PLX-resistant melanoma cell line ( and Fig. S8C), thus indicating that ATP is indeed the extracellular factor instigating increased uptake of Rh123 in a P2RX7-dependent fashion. However, to differentiate between the influx capacity of the P2RX7 ion channel and its potential to mediate membrane internalization or endocytosis,Citation43,44 we then monitored the uptake of the larger FITC-Dextran (70 kDa, Fig. S8D) by these melanoma cells. In line with reports showing that eATP stimulates membrane internalization and uptake of extracellular material,Citation43,44 both PLX-resistant isogenic melanoma models also displayed an eATP-P2RX7-regulated uptake of FITC-dextran in comparison with their isogenic counterparts ( and Fig. S8E). Notably, a similar pattern of eATP-dependent and A74-sensitive increased FITC-dextran internalization was found in the primary PLX-resistant M233 and M263 human melanoma cells (Fig. S8F), further validating the assumption that this is not an effect limited to, or caused by, the in vitro acquired drug resistance, but a general event associated with an increased melanoma aggressive behavior.
Finally, to further understand the role of autophagy in this autocrine-paracrine response mechanism, we tested Rh123 uptake in ATG5-proficient or ATG5-silenced PLX-resistant 451-Lu cells (). The uptake of Rh123 was significantly blunted in PLX-resistant cells with reduced ATG5 expression (shATG5) in comparison to the control (shCon) cells (). These findings correlated well with the diminished ATP secretion observed in ATG5-attenuated PLX-resistant cells () and in line with this, exogenous addition of ATP was sufficient to re-incite their Rh123 uptake (). However, this exogenous ATP-induced Rh123 uptake remained A74-sensitive ().
Altogether, these results indicate that in PLX-resistant melanoma cells, autophagy-mediated ATP secretion elicits a P2RX7-mediated autocrine-paracrine signaling, which ultimately fuels an aggressive and drug-resistant melanoma cell phenotype.
Discussion
Our study shows that PLX-resistance, either primary or acquired, in melanoma cells potentiates melanoma cell migration and invasion in vitro through an autocrine-paracrine signaling loop. Moreover, this loop is dependent on the ability of autophagy to enhance the secretion of ATP from these drug-resistant melanoma cells. We further demonstrate that PLX-resistant melanoma cells utilize P2RX7 signaling, to stimulate a more aggressive melanoma cell phenotype.
The inhibition of autophagy in PLX-resistant cells sensitizes them to cell death induced by MAP2K inhibitors.Citation38 However, beyond cell death modulation,Citation6,38,39 whether and how autophagy supports the PLX-resistant phenotype of melanoma cells remains poorly understood. Within the tumor microenviroment, extracellular ATP is present at >103 times the concentration found in normal tissues.Citation42 This is due to various reasons ranging from micro-environmental stressors to the presence of intratumor necrosis.Citation43 Thus, it is not surprising that in certain contexts or settings, cancer cells may start exploiting eATP for facilitating their growth and/or metastatic potential.Citation16,20,21,47 However the role of eATP in therapy resistance remains poorly defined.Citation26
Here, we show that the heightened autophagy machinery exhibited by the PLX-resistant melanoma cells is recruited to foster the secretion of ATP, which further promotes their more migratory/invasive phenotype. Autophagosomes have been identified as important traffickers of intracellular ATP into the extracellular environment, as demonstrated convincingly by Fader et al.Citation12 In line with this observation, we found in PLX-resistant melanoma cells the colocalization of ATP-rich stores with a key component of the autophagic machinery, which was significantly higher compared with PLX-sensitive melanoma cells. Moreover, when autophagosome formation was blunted by the silencing of either ATG5, ATG7 or ULK1, PLX-resistant melanoma cells lost their increased ability to secrete ATP, thus causally linking the heightened autophagic capacities of these drug-resistant melanoma cells with their increased ATP-secretory phenotype. Of note, eATP has been shown previously to function as a chemoattractant moleculeCitation26 and to facilitate migratory or invasive phenotypes in glioma cells, breast cancer cells and prostate cancer cells.Citation44 Moreover, vesicular exocytosis of ATP, followed by binding to and activation of purinergic receptors (P2RX7) promotes through an autocrine pathway, lung cancer cell migration.Citation45
Thus, our study not only extends these pro-tumorigenic effects of eATP to BRAF-inhibitor resistant melanoma cells, but demonstrates the key role of autophagy in exporting ATP extracellularly, thus initiating an autocrine loop fostering melanoma cells' metastastic features. Moreover, these findings are in line with the emerging tumor-promoting roles of hyperactivated lysosomal-dependent vesicular traffickingCitation46-48 and of key autophagy genes, such as ATG7,Citation49 in melanoma invasion and progression in vivo.
It is not immediately clear why PLX-resistant metastatic melanoma cells, but not metastatic melanoma cells in general, become more proficient in exploiting eATP as a facilitator of their migration and invasion. However, here we show that PLX4032 and U0126 were capable of increasing or decreasing, respectively, the secretion of ATP and consequently affecting the elevated migratory potential of the PLX-resistant melanoma cell lines. This points out that eATP may potentiate the pro-invasive/metastatic behavior of mainly PLX-resistant melanoma cells possibly through their abberant MAPK signaling system,Citation8 which results in enhanced MAP2K activation.Citation8,12 To that end, an EMT-like transition to a more mesenchymal-like signature leads, in the case of B16BL6 melanoma cells, to an increased invasive nature.Citation50 Here within this study, we documented an EMT-like shift in human melanoma cells following acquired PLX-resistance that correlated with enhanced invasion and greater migratory capacities. Interestingly, research using in silico-based modeling has demonstrated that a cancer cell population may only require a subpopulation of mesenchymal-like cancer cells to mediate invasion and downstream metastasis.Citation51 Aided by their capacity to adapt size and shape, mesenchymal-like cancer cells use cell parturitions to mediate space, allowing migration in a densely packed tumor environment and these ‘scouts’ leave in their wake a predefined path for the mass population to follow.Citation51 PLX-resistance is associated with EMT, in particular the MAP2K-dependent upregulation of transcription factors such as SNAI/SNAIL,Citation52 which promote melanoma cell invasionCitation53 and the capacity of cells to colonize distant sites.Citation54
We also observed that the eATP-driven increase in cell migration is significantly decreased following the inhibition of the purinergic receptors, more specifically, P2RX7 in line with a previous study in lung cancer cells.Citation45 P2RX7 is an ATP-gated ion channel also expressed in melanoma cells,Citation55 with the potential to mediate endocytosis. Interestingly, P2RX7 has a well-established role in facilitating cancer migration and invasion.Citation15 Previously it was shown that the open channel of P2RX7 can allow passage of small molecules such as rhodamine.Citation41 Along these lines, here we found that PLX-resistant melanoma cells displayed an eATP-dependent increased uptake of both FITC-dextran and Rh123, suggesting that eATP mediates endocytosis rather than channel opening-mediated intake, that may ultimately support the enhanced endocytic activity of melanoma and influence melanoma cell migration.
Furthermore, a direct link might exist between eATP-mediated purinergic receptor signaling and MAPK activation such that eATP can activate the phosphoinositide 3-kinase-AKT and MAPK1/ERK2-MAPK3/ERK1 signaling pathways downstream of P2RX7, which in turn can account for increased migration/invasion.Citation16,20 Indeed, PLX-resistant isogenic melanoma cell lines stimulated by PLX, exhibited an increase in MAP2K phosphorylation, indicating an increase in MAPK/ERK activity (as previously documented).Citation8 Hence, these observations suggest the existence of an autophagy-modulated autocrine signal, whereby eATP-P2RX7 interaction causes increased migration of PLX-resistant melanoma cells, ultimately by fuelling their exacerbated MAP2K-MAPK/ERK pathway.Citation15
Intriguingly, we found that this autocrine signal also contributes to the maintenance of a subset of melanoma cells with increased ability to minimize intracellular concentrations of xenobiotics, characteristic of a melanoma drug-resistant SP subset,Citation34 thus increasing the probability of cell survival and modulating the acquisition of drug-resistance.Citation56,57 Previously, in the context of melanoma, PLX-resistance has been linked with ABC transporter-based efflux, with evidence indicating PLX as a direct substrate of ABC transporters, such as ABCB1.Citation58 Indeed, our results show that acquisition of PLX-resistance increased the population of melanoma cells with increased drug-efflux ability in an eATP-dependent fashion, which could be further modulated by the addition of PLX4032 itself. Moreover, the direct modulation of this melanoma cell population by autophagy as shown here, suggests a certain degree of synergy between purinergic receptors and ABC transporter functionality in the migration or invasion of PLX-resistant melanomas. Furthermore, antagonizing the P2RX7 signal with A740003 abolished the enhanced drug effluxing nature of this melanoma cell population. Because as a consequence of tumor heterogeneity and microenviromental stress (i.e., hypoxia), it is unlikely that all cells contribute equally to the increased eATP in the tumor microenvironment,Citation42 it would be interesting in future studies to understand the full tumor-promoting potential of the drug-resistant SP fraction of PLX-resistant melanoma cells and their overall contribution to ATP secretion.
Moreover, our expression studies showed that increased mRNA expression of the melanocytic-lineage specification factor SOX10 and to a lesser extent the oncogene MYC is modulated by the heightened secretion of ATP of the PLX-resistant melanoma cells. Because SOX10 and MYC regulate the levels of RAB7 in melanoma cells, a key factor controlling endosomal cargo fate within the endo-/lyososomal/autophagy pathways and the proliferative or invasive phenotype of melanoma cells,Citation47 it would be interesting to further elucidate the role of autophagy-mediated eATP in modulating RAB7 functions in PLX-resistant melanoma cells.
Development of PLX-resistance is an important concern that mandates the better understanding of mechanisms behind such resistance to enable the design of smart combinational therapies. Our in vitro study suggests that autophagy inhibition in advanced melanoma could be a potent combinatorial strategy as it will not only overcome the cell-autonomous prosurvival function of autophagy (previously demonstrated in refs.Citation6,8,39,59) but potentially also its autocrine/paracrine pro-migratory and pro-invasion effects exerted via eATP-P2RX7 interaction.Citation15,19,55 However, further in vivo studies need to be performed to evaluate the effects of the inhibition of PLX-resistant melanoma cell-associated autophagy on their metastatic potential, because eATP and its degradation products influence key features of the tumor microenvironment, including immuno-surveillance mechanisms and therapeutic responses.Citation40,63 Irrespective of these interesting conjectures, our study unravels a pro-invasive role for melanoma cell-associated autophagy following both in vitro acquired, as well as primary resistance to, PLX, whereby the increased autophagy promotes through the trafficking and extracellular secretion of ATP key hallmarks of melanoma's aggressive phenotype.
Materials and methods
Tissue culture
Human A375 (ATCC, Teddington UK; CRL-3224™), 451-Lu and mouse SM1 (both harboring the BRAFV600E mutation) metastatic melanoma cells, as well as PLX-sensitive M229 (BRAFV600E homozygous), M249 and PLX-resistant M233, and M263 (BRAFV600E heterozygous) cells were cultured in DMEM (A375 and 451-Lu; Sigma-Aldrich, D6546) or RPMI culture media containing 1% glutamine and penicillin/streptomycin (Sigma-Aldrich, G7513 and P0781, respectively) supplemented with 10% or 5% (451-Lu models only) fetal bovine serum (FBS; GE Healthcare, SV30160.03).Citation8 Vemurafenib (PLX4032/PLX)-resistant A375/RES, 451Lu/RES and SM1/RES (BRAFV600E) cells were cultured in the same medium as described for parental cell lines, further supplemented with 1 µM PLX4032 to maintain BRAF acquired resistance. All cells were maintained at 37°C, 5% CO2 for a maximum of 10 passages. We would like to thank Prof. M. Herlyn for the use of the 451-LU isogenic cell linesCitation60-62 and Prof. A Ribas for the patient derived (M cell lines) and murine isogenic cell lines.Citation28,29
Drug treatments
All experiments were performed in the appropriate culture media with reduced (2%) FBS and treated for 72 h with ATP (0.05 – 50 µM; Sigma, FLAAS) or apyrase (2 U/ml; Sigma, A6535), in comparison to untreated controls. Involvement of the RAS-RAF pathway was examined by treatment with 10 µM of either PLX or UO126 (MAP2Ki). Purinergic receptor activation was inhibited by 1 h pretreatment with suramin (10 µM, pan-purinergic receptor inhibitor; Sigma, S2671) or A740003 (5 µM, P2RX7/P2X7 receptor-specific inhibitor; Santa Cruz Biotechnology, A740003) before treatment with ATP. Autophagic flux was observed by treatment with bafilomycin A1 (10 nM, Baf A1; Sigma-Aldrich, B1793) for 24 h.
Autophagy inhibition by knockdown
ATG5 expression was reduced by shRNA-based lentiviral technology as described previously.Citation8 Furthermore, ATG5, ATG7 or ULK1 expression was also reduced by siRNA-mediated knockdown (as described in ref.Citation8). Briefly, cells were transiently transfected either with ATG5-, ATG7- or ULK1-specific siRNA (Thermo Scientific, L-004374, L-020112, L-005049, respectively) or scrambled siRNA control (shCon; Qiagen, SI03650318) in FBS-free medium using DharmaFECT (Dharmacon, T-2001) for 4 h, followed by FBS addition and incubation overnight. Cells were then plated and used for experimentation. Following transfection, the reduction in protein expression was verified by immunoblotting.
Fluorescent detection of autophagosomes
LC3-positive puncta were detected on methanol-fixed melanoma cells stained with quinacrine (Sigma, Q3251–25G; 1 µM for 2 h in serum-free DMEM). Briefly, following treatment, media was discarded and cells washed (PBS/T: phosphate-buffered saline (Sigma, D8537) containing 0.01% Tween 20 (Sigma, P1379) and fixed in 100% ice-cold methanol at 4°C for 30 min. After fixation, cells were washed in PBS/T, incubated for 1 h with PBS/T containing 0.1% Triton X-100 (Sigma, T9284), 0.02% SDS (Sigma, L3771) and incubated for 1 h in PBS/T containing 0.1 M glycine (Sigma, G8898). Next, samples were thoroughly washed, then incubated for 30 min in PBS/T containing 10% FBS, 1% BSA (Sigma, A4503), washed again, and incubated for 90 min with LC3 (1:250, PBS/T containing 0.1% BSA, 1% FBS). Further, samples were washed, incubated in 10% serum for 30 min, followed by the addition of Alexa Fluor 488 secondary antibody (Thermo Fisher Scientific, A27034) for 60 min. Finally, samples were washed, fixed with prolong gold (Thermo Fisher, P10144) and imaged using a Leica confocal microscope (as described in ref. Citation63).
Western blotting
Following treatments, cells were scraped, pelleted, lysed in RIPA buffer (Enzo Life Sciences, 80–1284) supplemented with 1 mM PMSF and phosphatase inhibitors (Thermo Scientific, 88667). Protein concentration was determined with a BCA kit (Thermo Scientific, 23228). Primary antibodies: CDH2/N-Cadherin (ab76057), CDH1/E-Cadherin (ab76055), MITF (ab12039), FN1/fibronectin (ab2413, Abcam), TJP1/ZO-1 (Thermo Fisher, ZO1–1A12), ATG5 (Cell Signaling Technology, 12994), ABCB1 (Cell Signaling Technology, 13342), MAP1LC3/LC3B (Cell Signaling Technology, 3868), ATG7 (Cell Signaling Technology, 8558) and p-ULK1 (Cell Signaling Technology, 5869), SQSTM1 (Cell Signaling Technology, P0067) or ACTB (Sigma-Aldrich, A5441) were diluted 1:1000. Fluorescently-labeled secondary antibodies (anti-rabbit-DyLight 800 [Thermo Scientific, 35571] and anti-mouse-DyLight 680 [Thermo Scientific, 35519]) were diluted 1:2000. The detection was done using the Odyssey infrared-imaging system (Li-Cor Biosciences). Densitometric analyses were generated using ImageJ.
Scratch-wound assay
A scratch-wound was produced on the confluent melanoma monolayers using a 200-μl tip. Cultures were further incubated in media containing 0.5 µM mitomycin C (Sigma, M4287) and photographed at the indicated time periods. Wound closure was measured with NIH ImageJ software and plotted as percentage of open wound calculated as wound area at given time point:wound area at T0.
Transwell migration/invasion assays
5000 cells per insert (50 µl) were seeded into 96-well HTS transwells (migration: Corning, CLS3374–2EA; invasion: basement membrane, 8 µm [Millipore, ECM555]), allowed to adhere overnight and treated as indicated. Appropriate media (150 µl) was added to each receiver well (where indicated, the media in donor wells was supplemented with ATP or APY). Following treatment, insert and receiver wells were washed with PBS, then trypsin-staining solution (dissociation solution [Trevigen, 3455–096–05]; calcein AM [Trevigen, 4892–010–01]; 1:1000 for both in water) was added to the receiver well for 1 h at 37°C. Fluorescence was measured using a FlexStation 3 microplate reader (Molecular Devices Inc., 0310–5627) at excitation 485 nm and emission 520 nm. The acquired fluorescence was plotted relative to control cells.
Cell proliferation assay
Cellular proliferation profiles were assessed according to the manufacturer's protocol (Invitrogen, C35007). Briefly, melanoma cells were plated in experimental media, at a density of 5000 cells per well, in 96-well plates and allowed to attach overnight. Adhered cells were then exposed to relevant stress conditions before the media was aspirated, cells washed and 100 µl of 1x CyQUANT® NF dye reagent was added for 1 h. Fluorescence was captured using a Flexstation 3 microplate reader (Molecular Devices Inc., 0310–5627) at excitation 488 nm and emission 530 nm.
Reverse-transcription quantitative PCR (RT-qPCR)
RNA was isolated from APY-treated 451-Lu and A37 PLX isogenic models via the Qiagen RNeasy Mini Kit, according to the manufacturer's protocol (Qiagen Benelux, 74104). cDNA was constructed with the iScript cDNA kit (Bio-Rad, 170–8890). All Primers were previously validated. Primers for genes involved in cancer drug resistance: ABCB1 (AAATTGGCTTGACAAGTTGTATATGG and CACCAGCATCATGAGAGGAAGTC) and ABCG2 (TGACCTGAAGGCATTTACTG and GGTAGAAAGCCACTCTTCAG). Primers for genes involved in cancer stemness and aggression: KDM5B/Jarid1B (AACAACATGCCAGTGATGGA and TACCAGGTTTTTGGCTCACC), SOX10 (TCTTTGTCTGAGAAACAGCC and CATGGAGGTTGTAGTGGAGG), MYC (TGAGGAGACACCGCCCAC and TGAGGAGACACCGCCCAC), TERT (CCACTCCCCACATAGGAATAGTC and TCCTTCTCAGGGTCTCCACCT), NGFR/CD271 (AACCTCATCCCTGTCTATTG and GTTGGCTCCTTGCTTGTT). The housekeeping gene used for normalization was GAPDH. qPCR was performed on a ABI 7500 RT-PCR system (1 cycle 50°C 2 min, 1 cycle 95°C 10 min and 40 cycles of alternating 95°C 15 sec and 60°C 1 min) using a SYBR Green Master Mix (Thermo Scientific, 4472918) in standard conditions according to the manufacturer's instructions. Careful monitoring of negative controls and dissociation curves for each target showed specific amplification and absence of carryovers.
ATP assay
ATP measurements were performed as described previously.Citation64 Briefly, following culture or treatments done in 96-well plates, supernatant was collected and assayed with ATP assay mix (Sigma, FLAAM-1VL). Bioluminescence was assessed via a FlexStation 3 microplate reader (Molecular Devices Inc., 0310–5627).
Cell death
Following treatments, cells were trypsinized, washed twice with PBS and fixed with 70% ice-cold ethanol. The percentage of propidium iodide-stained cells positive for hypodiploid DNA (subG1 peak) was captured using an Attune flow cytometer (Life Technologies, Paisley, UK).
Cell viability
Viability was assessed as described in ref.Citation8 Briefly, cells were plated at a density of 5000 cells per well of a 96-well plate. Following treatments, cells were washed with PBS and incubated with 0.01 mg/mL MUH (4-methylumbelliferylheptanoate; Sigma, St. Louis, MO, USA, M2514; dissolved in PBS) for 30 min at 37°C. Fluorescence was then measured with a FlexStation microplate reader (Molecular Devices, Wokingham, UK) with an excitation of 355nm, emission of 460 nm and a cut off value of 455 nm.
Rhodamine 123 uptake assay
Following treatments, cells were incubated in 2 µM rhodamine 123 (Rh123; Sigma, R8004) for 1 h before trypsinization, subsequently washed twice in PBS and positivity acquired using an Attune flow cytometer.
FITC-dextran endocytosis assay
Endocytic capacity of the cells, following the required treatments, was investigated by exposing the melanoma cells to 40 µg/ml FITC-dextran (Sigma, 46945–100MG-F) for 2 h.Citation65 Subsequently cells were collected by trypsinization and FITC positivity acquired of 10,000 cells using an Attune flow cytometer.
Assessment of melanoma side population
Side population (SP) was identified and characterized as described previously.Citation33 Briefly, cells were trypsinized, collected and density adjusted to 1 × 106 cells/ml in PBS supplemented with 2% FBS and 1% BSA. Control samples were pretreated for 20 min with verapamil (100 μM; Sigma, V4629). All samples were then incubated with 5 μg/ml Hoechst 33342 (Sigma, B2261) for 90 min at 37°C. Subsequently, cells were washed and resuspended in ice-cold PBS with FBS (2%) and propidium iodide (2 μg/ml; Sigma, P4170–25MG). Finally, samples were analyzed by flow cytometry using a FACS Aria III (BD Biosciences, Erembodegem, Belgium). The SP is revealed as a Hoechstlow population (blue emission [filter BP 450/40] versus red emission [filter BP 630/20]) after (near-) UV excitation. The SP phenotype is verified by sensitivity to the efflux blocker verapamil. Analysis was done, and proportions calculated, within the living (propidium iodide negative) cell population.
Statistics
All data are the mean of at least 3 independent experiments ± standard deviation (SD). For individual observations significance was determined by T-test. One-Way Analysis of variance (ANOVA) or Two-Way ANOVA with Bonferroni post hoc correction were used for comparison of tested time points and treatments. All analyses were performed using GraphPad Prism 5. */$ P < = 0.05, **/$$ P < = 0.01, ***/$$$ P < = 0.001, ****/$$$$ P < 0.0001.
Abbreviations
| ATP | = | adenosine triphosphate |
| ABC | = | ATP-binding cassette |
| APY | = | apyrase |
| ATG | = | autophagy related |
| CM | = | culture medium |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MAP2K/MEK | = | mitogen-activated protein kinase kinase |
| MAPK | = | mitogen activated protein kinase |
| MITF | = | microphthalmia-associated transcription factor |
| MYC | = | myelocytomatosis oncogene |
| P2RX7 | = | purinergic receptor class 2 subset X number 7 |
| SOX10 | = | SRY (sex determining region Y)-box 10 |
| SQSTM1 | = | sequestesome 1 |
| SP | = | side population |
| TERT | = | telomerase reverse transcriptase |
| TJP1/ZO-1 | = | tight junction protein 1 |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KAUP_A_1332550_supplementary.zip
Download Zip (3 MB)Acknowledgments
We would like to thank Prof. M. Herlyn for the use of the 451-LU isogenic cell linesCitation60-62 and Prof. A Ribas for both the patient-derived (M cell lines) and murine isogenic cell lines.Citation28,29
Funding
Research was supported by C16/15/073 grant of the KU Leuven to P.A. and P.V., FWO grant G070115N and Stichting tegen Kanker F/2014/222 to P.A. Melanoma research in PA laboratory is supported by the MEL-PLEX ITN-consortium supported by the H2020 Marie Curie Actions. Aleksandra Dudek-Perić was supported by a fellowship of the Vlaamse Liga tegen Kanker (VLK). Abhishek D. Garg is supported by the FWO Postdoctoral (Renewal) Fellowship. This paper represents research results of the IAP7/32 funded by the Interuniversity Attraction Poles Programme, initiated by the Belgian State, Science Policy Office. We would like to thank Kristine Rillaerts and the flow cytometry core facility (KU Leuven) for technical support. We acknowledge the use, help and support of the Cell imaging Core (CIC, Prof. Pieter Vanden Berghe, KU Leuven) for their help and support within this project.
Related Research Data
References
- Davies MA, Liu P, McIntyre S, Kim KB, Papadopoulos N, Hwu WJ, et al. Prognostic factors for survival in melanoma patients with brain metastases. Cancer 2011; 117:1687-96; PMID:20960525; https://doi.org/10.1002/cncr.25634
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov 2012; 11:873-86; PMID:23060265; https://doi.org/10.1038/nrd3847
- Palmieri G, Ombra M, Colombino M, Casula M, Sini M, Manca A, Paliogiannis P, Ascierto PA, Cossu A. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Frontiers in oncology 2015; 5:183; PMID:26322273; https://doi.org/10.3389/fonc.2015.00183
- Swaika A, Crozier JA, Joseph RW. Vemurafenib: an evidence-based review of its clinical utility in the treatment of metastatic melanoma. Drug Des Devel Ther 2014; 8:775-87; PMID:24966667
- Corazzari M, Fimia GM, Lovat P, Piacentini M. Why is autophagy important for melanoma? Molecular mechanisms and therapeutic implications. Semin Cancer Biol 2013; 23:337-43; PMID:23856558; https://doi.org/10.1016/j.semcancer.2013.07.001
- Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, Fimia GM, Lovat PE, Piacentini M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ 2015; 22:946-58; PMID:25361077; https://doi.org/10.1038/cdd.2014.183
- Spagnolo F, Ghiorzo P, Queirolo P. Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget 2014; 5:10206-21; PMID:25344914; https://doi.org/10.18632/oncotarget.2602
- Martin S, Dudek-Peric AM, Maes H, Garg AD, Gabrysiak M, Demirsoy S, Swinnen JV, Agostinis P. Concurrent MEK and autophagy inhibition is required to restore cell death associated danger-signalling in Vemurafenib-resistant melanoma cells. Biochem Pharmacol 2015; 93:290-304; PMID:25529535; https://doi.org/10.1016/j.bcp.2014.12.003
- Cheng Y, Ren X, Hait WN, Yang JM. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev 2013; 65:1162-97; PMID:23943849; https://doi.org/10.1124/pr.112.007120
- Murrow L, Debnath J. Autophagy as a Stress-Response and Quality-Control Mechanism: Implications for Cell Injury and Human Disease. Annu Rev Pathol-Mech 2013; 8:105-37; https://doi.org/10.1146/annurev-pathol-020712-163918
- Gomes LR, Vessoni AT, Menck CF. Microenvironment and autophagy cross-talk: implications in cancer therapy. Pharmacol Res 2016; https://doi.org/10.1016/j.phrs.2016.03.031
- Fader CM, Aguilera MO, Colombo MI. ATP is released from autophagic vesicles to the extracellular space in a VAMP7-dependent manner. Autophagy 2012; 8:1741-56; PMID:22951367; https://doi.org/10.4161/auto.21858
- Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, Breckpot K, Brough D, Chaurio R, Cirone M, et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Fron Immunol 2015; 6:588; PMID:26635802; https://doi.org/10.3389/fimmu.2015.00588
- Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010; 29:5346-58; PMID:20661219; https://doi.org/10.1038/onc.2010.292
- Qiu Y, Li WH, Zhang HQ, Liu Y, Tian XX, Fang WG. P2X7 mediates ATP-driven invasiveness in prostate cancer cells. PloS One 2014; 9:e114371; PMID:25486274; https://doi.org/10.1371/journal.pone.0114371
- Ledur PF, Villodre ES, Paulus R, Cruz LA, Flores DG, Lenz G. Extracellular ATP reduces tumor sphere growth and cancer stem cell population in glioblastoma cells. Purinergic Signal 2012; 8:39-48; PMID:21818572; https://doi.org/10.1007/s11302-011-9252-9
- Zimmerer RM, Korn P, Demougin P, Kampmann A, Kokemuller H, Eckardt AM, Gellrich NC, Tavassol F. Functional features of cancer stem cells in melanoma cell lines. Cancer Cell Int 2013; 13:78; PMID:23915418; https://doi.org/10.1186/1475-2867-13-78
- Buzzi N, Bilbao PS, Boland R, de Boland AR. Extracellular ATP activates MAP kinase cascades through a P2Y purinergic receptor in the human intestinal Caco-2 cell line. Biochim Biophys Acta 2009; 1790:1651-9; PMID:19836435; https://doi.org/10.1016/j.bbagen.2009.10.005
- Chang SJ, Tzeng CR, Lee YH, Tai CJ. Extracellular ATP activates the PLC/PKC/ERK signaling pathway through the P2Y2 purinergic receptor leading to the induction of early growth response 1 expression and the inhibition of viability in human endometrial stromal cells. Cellular Signal 2008; 20:1248-55; PMID:18434089; https://doi.org/10.1016/j.cellsig.2008.02.011
- Hill LM, Gavala ML, Lenertz LY, Bertics PJ. Extracellular ATP may contribute to tissue repair by rapidly stimulating purinergic receptor X7-dependent vascular endothelial growth factor release from primary human monocytes. J Immunol 2010; 185:3028-34; PMID:20668222; https://doi.org/10.4049/jimmunol.1001298
- Trabanelli S, Ocadlikova D, Gulinelli S, Curti A, Salvestrini V, Vieira RP, Idzko M, Di Virgilio F, Ferrari D, Lemoli RM. Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J Immunol 2012; 189:1303-10; PMID:22753942; https://doi.org/10.4049/jimmunol.1103800
- Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334:1573-7; PMID:22174255; https://doi.org/10.1126/science.1208347
- Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014; 3:e955691; PMID:25941621; https://doi.org/10.4161/21624011.2014.955691
- Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, Kong X, Bosenberg MC, Wiesner T, Rosen N, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015; 520:368-72; PMID:25807485; https://doi.org/10.1038/nature14336
- Basu D, Bewley AF, Sperry SM, Montone KT, Gimotty PA, Rasanen K, Facompre ND, Weinstein GS, Nakagawa H, Diehl JA, et al. EGFR inhibition promotes an aggressive invasion pattern mediated by mesenchymal-like tumor cells within squamous cell carcinomas. Molecular Cancer Ther 2013; 12:2176-86; PMID:23939378; https://doi.org/10.1158/1535-7163.MCT-12-1210
- Jiang JX, Riquelme MA, Zhou JZ. ATP, a double-edged sword in cancer. Oncoscience 2015; 2:673-4; PMID:26425653; https://doi.org/10.18632/oncoscience.230
- Da'dara AA, Bhardwaj R, Ali YB, Skelly PJ. Schistosome tegumental ecto-apyrase (SmATPDase1) degrades exogenous pro-inflammatory and pro-thrombotic nucleotides. PeerJ 2014; 2:e316; PMID:24711968; https://doi.org/10.7717/peerj.316
- Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B, et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PloS One 2011; 6:e28973; PMID:22194965; https://doi.org/10.1371/journal.pone.0028973
- Sondergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S, Sazegar H, MacConaill LE, Barretina JG, Kehoe SM, et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med 2010; 8:39; PMID:20406486; https://doi.org/10.1186/1479-5876-8-39
- Michaelis M RF, Nerreter T, van Rikxoort M, Zehner R, Dirks WG, Wiese M, Cinatl J Jr1. Association between acquired resistance to PLX4032 (vemurafenib) and ATP-binding cassette transporter expression. BMC Res Notes 2014; 10:710; https://doi.org/10.1186/1756-0500-7-710
- Dean M. ABC transporters, drug resistance, and cancer stem cells. J Mammary Gland Biol Neoplasia 2009; 14:3-9; PMID:19224345; https://doi.org/10.1007/s10911-009-9109-9
- Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer 2010; 10:147-56; PMID:20075923; https://doi.org/10.1038/nrc2789
- Wouters J, Stas M, Gremeaux L, Govaere O, Van den Broeck A, Maes H, Agostinis P, Roskams T, van den Oord JJ, Vankelecom H. The human melanoma side population displays molecular and functional characteristics of enriched chemoresistance and tumorigenesis. PloS One 2013; 8:e76550; PMID:24098529; https://doi.org/10.1371/journal.pone.0076550
- Chen KG, Valencia JC, Gillet JP, Hearing VJ, Gottesman MM. Involvement of ABC transporters in melanogenesis and the development of multidrug resistance of melanoma. Pigment Cell Melanoma Res 2009; 22:740-9; https://doi.org/10.1111/j.1755-148X.2009.00630.x
- Zhao Y, Liu ZG, Tang J, Zou RF, Chen XY, Jiang GM, Qiu YF, Wang H. High expression of Sox10 correlates with tumor aggressiveness and poor prognosis in human nasopharyngeal carcinoma. Onco Targets Ther 2016; 9:1671-7; PMID:27051302; https://doi.org/10.2147/OTT.S101344
- Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene 1999; 18:3004-16; PMID:10378696; https://doi.org/10.1038/sj.onc.1202746
- Beretti F, Manni P, Longo C, Argenziano G, Farnetani F, Cesinaro AM, Witkowski AM, De Pol A, Pellacani G. CD271 is expressed in melanomas with more aggressive behaviour, with correlation of characteristic morphology by in vivo reflectance confocal microscopy. Br J Dermatol 2015; 172:662-8; PMID:25066225; https://doi.org/10.1111/bjd.13301
- Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Métivier D, Galluzzi L, Perfettini JL, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ 2014; 21:79-91; PMID:23852373; https://doi.org/10.1038/cdd.2013.75
- Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, Hart LS, Levi S, Hu J, Zhang G, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest 2014; 124:1406-17; PMID:24569374; https://doi.org/10.1172/JCI70454
- Hoyle CH, Knight GE, Burnstock G. Suramin antagonizes responses to P2-purinoceptor agonists and purinergic nerve stimulation in the guinea-pig urinary bladder and taenia coli. Br J Pharmacol 1990; 99:617-21; PMID:2331585; https://doi.org/10.1111/j.1476-5381.1990.tb12979.x
- Browne LE, Compan V, Bragg L, North RA. P2X7 receptor channels allow direct permeation of nanometer-sized dyes. J Neurosci 2013; 33:3557-66; PMID:23426683; https://doi.org/10.1523/JNEUROSCI.2235-12.2013
- Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: In Vivo imaging with plasma membrane luciferase. PloS one 2008; 3; PMID:18612415; https://doi.org/10.1371/journal.pone.0002599
- Morrone FB, Oliveira DL, Gamermann P, Stella J, Wofchuk S, Wink MR, Meurer L, Edelweiss MI, Lenz G, Battastini AM. In vivo glioblastoma growth is reduced by apyrase activity in a rat glioma model. Bmc Cancer 2006; 6; PMID:16995949; https://doi.org/10.1186/1471-2407-6-226
- Li WH, Qiu Y, Zhang HQ, Liu Y, You JF, Tian XX, Fang WG. P2Y2 receptor promotes cell invasion and metastasis in prostate cancer cells. Br J Cancer 2013; 109:1666-75; PMID:23969730; https://doi.org/10.1038/bjc.2013.484
- Takai E, Tsukimoto M, Harada H, Sawada K, Moriyama Y, Kojima S. Autocrine regulation of TGF-beta1-induced cell migration by exocytosis of ATP and activation of P2 receptors in human lung cancer cells. J Cell Sci 2012; 125:5051-60; PMID:22946048; https://doi.org/10.1242/jcs.104976
- Alonso-Curbelo D, Soengas MS. Hyperactivated endolysosomal trafficking in melanoma. Oncotarget. 2015 Feb; 6(5):2583-84;PMID:4413600; https://doi.org/10.18632/Foncotarget.3141
- Alonso-Curbelo D, Riveiro-Falkenbach E, Perez-Guijarro E, Cifdaloz M, Karras P, Osterloh L, Megías D, Cañón E, Calvo TG, Olmeda D, et al. RAB7 controls melanoma progression by exploiting a lineage-specific wiring of the endolysosomal pathway. Cancer Cell 2014; 26:61-76; PMID:24981740; https://doi.org/10.1016/j.ccr.2014.04.030
- Demirsoy S, Martin S, Maes H, Agostinis P. Adapt, Recycle, and Move on: Proteostasis and Trafficking Mechanisms in Melanoma. Front Oncol 2016; 6:240; PMID:27896217; https://doi.org/10.3389/fonc.2016.00240
- Xie X, Koh JY, Price S, White E, Mehnert JM. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Dis 2015; 5:410-23; PMID:25673642; https://doi.org/10.1158/2159-8290.CD-14-1473
- Kushiro K, Chu RA, Verma A, Nunez NP. Adipocytes Promote B16BL6 Melanoma Cell Invasion and the Epithelial-to-Mesenchymal Transition. Cancer Microenviron 2012; 5:73-82; PMID:21892698; https://doi.org/10.1007/s12307-011-0087-2
- Hecht I, Bar-El Y, Balmer F, Natan S, Tsarfaty I, Schweitzer F, Ben-Jacob E. Tumor invasion optimization by mesenchymal-amoeboid heterogeneity. Sci Rep 2015; 5:10622; PMID:26013062; https://doi.org/10.1038/srep10622
- Strippoli R, Loureiro J, Moreno V, Benedicto I, Perez Lozano ML, Barreiro O, Pellinen T, Minguet S, Foronda M, Osteso MT, et al. Caveolin-1 deficiency induces a MEK-ERK1/2-Snail-1-dependent epithelial-mesenchymal transition and fibrosis during peritoneal dialysis. EMBO Mol Med 2015; 7:102-23; PMID:25550395; https://doi.org/10.15252/emmm.201404127
- Fenouille N, Tichet M, Dufies M, Pottier A, Mogha A, Soo JK, Rocchi S, Mallavialle A, Galibert MD, Khammari A, et al. The Epithelial-Mesenchymal Transition (EMT) Regulatory Factor SLUG (SNAI2) Is a Downstream Target of SPARC and AKT in Promoting Melanoma Cell Invasion. PloS One 2012; 7; PMID:22911700; https://doi.org/10.1371/journal.pone.0040378
- Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev 2004; 18:1131-43; https://doi.org/10.1101/gad.294104
- Mantel A, Harvey V. P2X7/PANX1 as a new target for melanoma? Exp Dermatol 2015; 24:336-7; PMID:25594260; https://doi.org/10.1111/exd.12633
- McIntosh K, Balch C, Tiwari AK. Tackling multidrug resistance mediated by efflux transporters in tumor-initiating cells. Expert Opin Drug Metab Toxicol 2016; 12:633-44; https://doi.org/10.1080/17425255.2016.1179280
- Hashida S, Yamamoto H, Shien K, Miyoshi Y, Ohtsuka T, Suzawa K, Watanabe M, Maki Y, Soh J, Asano H, et al. Acquisition of cancer stem cell-like properties in non-small cell lung cancer with acquired resistance to afatinib. Cancer Sci 2015; 106:1377-84; PMID:26202045; https://doi.org/10.1111/cas.12749
- Wu CP, S VA. The pharmacological impact of ATP-binding cassette drug transporters on vemurafenib-based therapy. Acta Pharm Sin B 2014; 4:105-11; PMID:26579371; https://doi.org/10.1016/j.apsb.2013.12.001
- Liu H, He ZY, Simon HU. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin Cancer Biol 2013; 23:352-60; PMID:23831275; https://doi.org/10.1016/j.semcancer.2013.06.008
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010; 18:683-95; PMID:21156289; https://doi.org/10.1016/j.ccr.2010.11.023
- Iliopoulos D, Ernst C, Steplewski Z, Jambrosic JA, Rodeck U, Herlyn M, Clark WH Jr, Koprowski H, Herlyn D. Inhibition of metastases of a human melanoma xenograft by monoclonal antibody to the GD2/GD3 gangliosides. J Natl Cancer Inst 1989; 81:440-4; PMID:2918552; https://doi.org/10.1093/jnci/81.6.440
- Herlyn D, Iliopoulos D, Jensen PJ, Parmiter A, Baird J, Hotta H, Adachi K, Ross AH, Jambrosic J, Koprowski H, et al. In vitro properties of human melanoma cells metastatic in nude mice. Cancer Res 1990; 50:2296-302; PMID:2156614
- Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014; 26:190-206; PMID:25117709; https://doi.org/10.1016/j.ccr.2014.06.025
- Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Extracellular ATP and P2X7 receptor exert context-specific immunogenic effects after immunogenic cancer cell death. Cell Death Dis 2016; 7:e2097; https://doi.org/10.1038/cddis.2015.411
- Lencer WI, Weyer P, Verkman AS, Ausiello DA, Brown D. Fitc-Dextran as a Probe for Endosome Function and Localization in Kidney. Am J Physiol 1990; 258:C309-C17; PMID:1689545