
ABSTRACT
Macroautophagy/autophagy is the process by which cellular components are degraded and recycled within the lysosome. These components include mitochondria, the selective degradation of which is known as mitophagy. Mitochondria are dynamic organelles that constantly adapt their morphology, function, and number to accommodate the metabolic needs of the cell. Extensive metabolic reconfiguration occurs during cell differentiation, when mitochondrial activity increases in most cell types. However, our data demonstrate that during physiologic retinal ganglion cell (RGC) development, mitophagy-dependent metabolic reprogramming toward glycolysis regulates numbers of RGCs, which are the first neurons to differentiate in the retina and whose axons form the optic nerve. We show that during retinal development tissue hypoxia triggers HIF1A/HIF-1 stabilization, resulting in increased expression of the mitophagy receptor BNIP3L/NIX. BNIP3L-dependent mitophagy results in a metabolic shift toward glycolysis essential for RGC neurogenesis. Moreover, we demonstrate that BNIP3L-dependent mitophagy also regulates the polarization of proinflammatory/M1 macrophages, which undergo glycolysis-dependent differentiation during the inflammatory response. Our results uncover a new link between hypoxia, mitophagy, and metabolic reprogramming in the differentiation of several cell types in vivo. These findings may have important implications for neurodegenerative, metabolic and other diseases in which mitochondrial dysfunction and metabolic alterations play a prominent role.
Mitophagy is a mechanism by which damaged mitochondria are selectively removed from the cell via autophagy. Mitochondria are eliminated in the context of cell development, for example during erythrocyte maturation. In red blood cells the removal of mitochondria is regulated by BNIP3L/NIX. This mitophagy receptor binds to mitochondria and to the phagophore membrane protein MAP1LC3, facilitating the envelopment of mitochondria within autophagosomes for subsequent delivery to lysosomes for degradation. We show that erythrocytes are not the only cell types to eliminate mitochondria during cell differentiation. Our data demonstrate that BNIP3L/NIX-dependent mitophagy is also essential for the differentiation of other cells including retinal ganglion cells (RGCs) and proinflammatory macrophages.
Programmed mitophagy regulates the neurogenesis of RGCs, as evidenced by increases and decreases in the numbers of mitochondria and RGCs, respectively, in BNIP3L/NIX-deficient retinas. A similar phenotype is observed in ATG5-deficient retinas. Conversely, we found that ex vivo rapamycin treatment increases mitophagy and RGC neurogenesis, further supporting a link between neuronal differentiation and selective autophagy-dependent removal of mitochondria (). Mature red blood cells lack mitochondria, indicating obligatory mitochondrial elimination in these cells. But why would a recently differentiated neuron degrade its own mitochondria? Our data show that mitophagy is essential to promote a metabolic shift toward glycolysis that is required for cell differentiation. RNA sequencing data demonstrate an increase in the expression of many glycolytic enzymes, and metabolic profiling reveals increased levels of glycolytic intermediates coinciding with peak RGC differentiation. Furthermore, we observed a decrease in RGC number in retinas treated with the non-metabolizable glucose analog 2DG, which blocks the initial steps of glycolysis, and an increase in RCG number increasing glycolysis following pharmacological inhibition of the mitochondrial pyruvate carrier. Taken together, these observations indicate that a metabolic shift toward glycolysis is essential for RGC differentiation.
Figure 1. Mitophagy-dependent metabolic reprogramming during cell differentiation. Retinal neuroblasts and undifferentiated macrophages contain an increased number of mitochondria and display an oxidative phosphorylation metabolic profile, which enables the production of high levels of ATP via glucose oxidation. High levels of respiration in the developing retina and in inflamed tissue lead to decreased oxygen availability, triggering a hypoxia response coordinated by the transcription factor HIF1A. One target of HIF1A is the mitophagy receptor BNIP3L/NIX, which mediates the formation of phagophores and mitochondria engulfment within autophagosomes for subsequent degradation in the lysosome. This reduction in mitochondrial number triggers a metabolic switch whereby glucose is metabolized to lactate, which is required for RGC neurogenesis and M1 macrophage polarization.
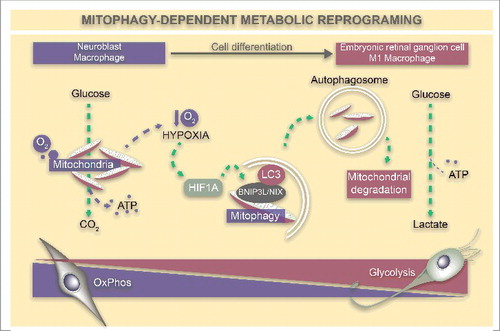
What triggers mitophagy during retinal neurogenesis? Our data show that hypoxia, which induces increased mRNA expression of BNIP3L/NIX, occurs during early retinal development. Interestingly, stabilization of HIF1A, a transcription factor that coordinates the hypoxia response, increases mitophagy and neuronal differentiation. HIF1A also enhances the glycolytic response by upregulating mRNA expression of glycolytic enzymes and glucose transporters. Our data thus suggest that hypoxia triggers a coordinated response in which mitophagy and glycolysis are coupled to promote RGC differentiation.
The observations described above contrast with those described during the development of most cell types, in which differentiation is associated with increased mitochondrial activity, which is required to generate ATP. This suggests that there are certain special metabolic requirements for RGC differentiation. The metabolic shift toward glycolysis in these cells could be similar to that observed in cancer cells, in which metabolic reprogramming is used to generate specific metabolites fueling anabolic reactions to sustain cell growth. In the embryonic stages during which this glycolytic metabolism is observed, RGCs begin to generate their axons, which must travel very long distances to form synapses with their targets in the brain. It is thus plausible that this special metabolic profile fuels axon growth in young RGCs. Alternatively, mitophagic degradation of damaged mitochondria may constitute a quality control mechanism. We observed an increase in mitochondrial biogenesis at postnatal developmental stages, in agreement with the increased number of mitochondria in mature RGCs, which are highly dependent on oxidative phosphorylation. Degradation of damaged mitochondria in RGCs may serve as a means of selecting functional mitochondria, which then divide to generate a pool of healthy mitochondria for mature RGCs. A similar coupling of mitophagy and mitochondrial biogenesis has been observed as a stress response in worms, and alterations in this homeostatic mechanism may be implicated in aging and degenerative diseases.
We found that this mitophagy-dependent metabolic shift also occurs during M1 macrophage polarization, a process that is dependent on metabolic reprograming. Proinflammatory or M1 macrophages rely on glycolysis for ATP production to fuel a rapid inflammatory response. We found that mitophagy inhibition increases mitochondrial mass and reduces the expression of glycolytic genes and proinflammatory cytokines. Therefore, at least in macrophages and RGCs, BNIP3L/NIX-mediated mitophagy can control metabolism and cell differentiation. It remains to be determined whether this also holds true for other cell types in which a shift toward glycolysis is also required for differentiation or other functions.
The implications of these findings are manifold. The generation of RGCs from induced pluripotent stem cells is a promising strategy for the treatment of glaucoma, a disease characterized by ganglion cell death, which is the world's leading cause of irreversible blindness and currently affects over 70 million people. Mitochondrial impairment is a well-established cause of RGC loss, and mitochondrial alterations precede RGC death. Increasing mitophagy and metabolic reprogramming could therefore enhance RGC differentiation while simultaneously improving the quality of the mitochondrial pool in young RGCs. Moreover, a very recent report shows that vision loss in a mouse model of glaucoma can be restored by targeting metabolism using dietary supplementation with vitamin B3, a precursor of NAD+. While neither mitochondrial function nor mitophagy were assessed in that study, it is tempting to speculate that the impressive neuroprotective effects of NAD+ restoration could be mediated via mitophagy. Indeed, we recently demonstrated that NAM (a precursor of NAD+) induces mitophagy in neuronal cells, although further studies are required to confirm this hypothesis.
Given the ubiquity of mitochondrial dysfunction in neurodegenerative and aging-associated diseases, new therapies targeting mitophagy and metabolic reprogramming could hold significant promise. Our findings support the view that metabolic interventions targeting mitophagy could also be applied to reduce inflammatory responses in diseases such as diabetes and atherosclerosis, in which aberrant and chronic inflammation exacerbate the pathology. In summary, we show that selective and programmed autophagy-mediated mitochondrial elimination in physiologic conditions promotes a metabolic switch that regulates cell differentiation in several cell types. These findings suggest that mitophagy constitutes an important target for interventions seeking to manipulate cell fate.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.