
ABSTRACT
The orphan nuclear receptor ESRRA (estrogen-related receptor α) is a key regulator of energy homeostasis and mitochondrial function. Macroautophagy/autophagy, an intracellular degradation process, is a critical innate effector against intracellular microbes. Here, we demonstrate that ESRRA is required for the activation of autophagy to promote innate antimicrobial defense against mycobacterial infection. AMP-activated protein kinase pathway and SIRT1 (sirtuin 1) activation led to induction of ESRRA, which is essential for autophagosome formation, in bone marrow-derived macrophages. ESRRA enhanced the transcriptional activation of numerous autophagy-related (Atg) genes containing ERR response elements in their promoter regions. Furthermore, ESRRA, operating in a feed-forward loop with SIRT1, was required for autophagy activation through deacetylation of ATG5, BECN1, and ATG7. Importantly, ESRRA deficiency resulted in a decrease of phagosomal maturation and antimicrobial responses against mycobacterial infection. Thus, we identify ESRRA as a critical activator of autophagy via both transcriptional and post-translational control to promote antimicrobial host responses.
Introduction
The nuclear receptors (NRs) are a family of receptors that bind to various hormones and ligands, and play a crucial role in the transcriptional regulation of genes involved in a variety of physiological processes.Citation1 The orphan NRs are categorized as a group of receptors whose endogenous ligands have not been identified. ESRRA (estrogen-related receptor α) is one of the first identified orphan NR family membersCitation2 and belongs to the ESRR family, consisting of ESRRA/NR3B1, ESRRB/NR3B2, and ESRRG/NR3B3.Citation3 ESRRA has been first identified on the basis of the structural similarity of its DNA-binding domain to that of human ESR1/estrogen receptor α,Citation2,3 but it does not bind natural estrogens or estrogen-like molecules. Numerous studies have reported that ESRRA functions as a key transcriptional regulator of energy metabolism, including glucose and lipid metabolism, mitochondrial biogenesis, and metabolic adaptation in various cells, tissues, and organs.Citation3,4 An earlier report has provided insights into an immune function of ESRRA that contributes to an innate host defense during intracellular infection.Citation5 However, the molecular mechanisms by which ESRRA mediates its host defense during infection remain largely unknown.
Mycobacterium tuberculosis (Mtb) is a major etiologic pathogen of tuberculosis (TB) and is well known for its successful ability to inhibit phagosome-lysosome fusion and to promote its survival inside host macrophages.Citation6,7 Autophagy is a constitutive process for targeting intracellular damaged organelles, long-lived proteins and aggregates to lysosomal degradation, thus contributing to the maintenance of intracellular homeostasis.Citation8,9 Increasing autophagy promotes innate host defense in response to diverse intracellular pathogens, especially Mtb infection.Citation8,10-15 We have previously shown that activation of AMPK-activated protein kinase (AMPK) leads to autophagy activation and phagosomal maturation against Mtb in macrophages through PPARGC1A (peroxisome proliferative activated receptor gamma coactivator 1 α).Citation16 Importantly, the ability of ESRRA to induce transcriptional activation of its numerous target genes is dependent on its coexpression with PPARGC1 coactivators, which often act as protein ligands for the ESRRs.Citation17,18 In our previous studies, ESRRA is not only found to transcriptionally activate the NAD+-dependent deacetylase SIRT1 (sirtuin 1) in macrophages,Citation19 but SIRT1 is shown to act as a coactivator of ESRRA transcriptional activity.Citation20 Indeed, SIRT1 plays a critical role in autophagy activation through deacetylation of essential autophagy proteins.Citation21-Citation23 These studies led us to hypothesize that ESRRA may play a role in AMPK- or SIRT1-mediated autophagy activation to enhance antimicrobial host defense during Mtb infection.
In this study, we aimed to determine whether and how ESRRA contributes to autophagy activation and host defense against Mtb. Our results revealed a critical role of ESRRA in driving autophagy activation in macrophages in response to autophagy inducers mediating AMPK and SIRT1 activation. First, ESRRA is required for the transcriptional activation of the autophagy-related genes Atg5, Atg16l1, and Becn1 (Beclin 1, autophagy related), in which the promoter regions contain ERR response elements (ERREs). Second, ESRRA promotes post-translational activation of autophagy through deacetylation of ATG5, BECN1, and ATG7 via activation of SIRT1. In addition, ESRRA increases the expression of SIRT1 in response to autophagy activation, and vice versa, driving a positively activating feed-forward circuit of phagosomal maturation in the presence of Mtb. Furthermore, we demonstrate that ESRRA is essential for antimicrobial host defense against Mtb infection. Together, our work highlights a previously unrecognized role of ESRRA in autophagy activation and the mechanisms by which it regulates autophagy leading to host defenses during mycobacterial infection.
Results
ESRRA is required for autophagy induction in macrophages in response to AMPK and SIRT1 activation
We first found that either AMPK or SIRT1 activation led to robust induction of ESRRA-encoding Esrra mRNA expression in bone marrow-derived macrophages (BMDMs) (data not shown). We thus treated BMDMs with the autophagy activators, including rapamycin; the AMP-mimetic 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), an AMPK activator;Citation16 and resveratrol, a SIRT1 activator.Citation24 Upon autophagy induction, LC3A/B processing, i.e., the increase in the LC3-II to LC3-I ratio, was significantly enhanced in Esrra+/+ BMDMs in a time-dependent manner, compared with in esrra−/− BMDMs ( and S1A). ESRRA protein was induced during autophagy activation in Esrra+/+ BMDMs after application of autophagy activators ( and S1A). It was noted that there are temporal differences in rapamycin-induced ESRRA protein expression between macrophages and hepatocytes.Citation25 In addition, the formation of LC3A/B-positive autophagic punctate structures was markedly decreased in esrra−/− BMDMs in response to autophagy inducers, compared with in Esrra+/+ BMDMs ( and S1B). Time-dependent LC3A/B puncta formation was increased in WT BMDMs in response to AICAR, resveratrol, or rapamycin, compared with those in esrra−/− BMDMs ( and S1C).
Figure 1. ESRRA is required for autophagy activation in macrophages in response to various autophagy inducers. Esrra+/+ and esrra−/− BMDMs were stimulated with AICAR (0.5 mM) or resveratrol (10 µM) for the indicated times. (A) BMDMs were harvested and subjected to immunoblot analysis of LC3A/B, ESRRA, and ACTB. The densitometry values for LC3-II were normalized to ACTB in each right panel. (B, D and E) Esrra+/+ and esrra−/− BMDMs were stimulated with AICAR (B to E) or resveratrol (B to D) for 24 h. (B) Immunofluorescence microscopy analysis of LC3A/B puncta formation. Merged signals of LC3A/B (Alexa Fluor 488-conjugated goat anti-rabbit IgG, green) and DAPI (nuclei, blue). Scale bar: 10 μm. (C) Quantification of LC3A/B punctate foci per cell. (D) Quantitative LC3B analysis by flow cytometry. (E) Representative transmission electron microscopy images. Scale bar: 1 and 0.2 μm. (F) Quantification of 200 cells with autophagic vacuoles per experimental condition by transmission electron microscopy. (G and H) Esrra+/+ and esrra−/− BMDMs were transduced with retroviruses expressing a tandem LC3B plasmid (mCherry-EGFP-LC3B) for 24 h and stimulated with AICAR or resveratrol for 24 h. (G) Representative immunofluorescence images. Scale bar: 10 µm. (H) Quantification of the number of red puncta per cell. Experiments were repeated at least 3 times. (B and G) Immunofluorescence microscopy images are from one representative of 7 independent samples, with each experiment including at least 200 cells scored in 7 random fields. Graphs show mean ± SD. **P < 0.01 (two-tailed unpaired Mann–Whitney). U, untreated; A, AICAR; R or RSV, resveratrol.
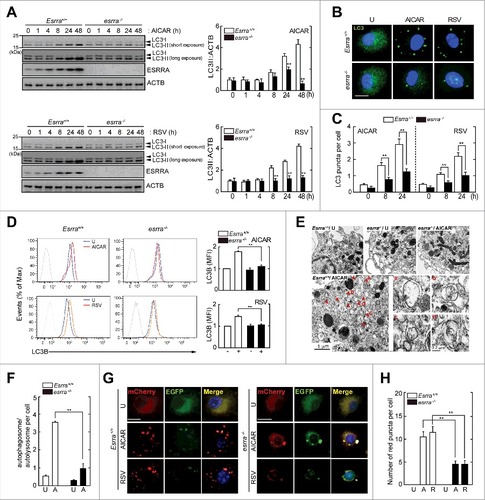
Further analysis showed that the AMPK-PPARGC1A pathway was required for ESRRA mRNA and protein expression in BMDMs after treatment with AMPK and SIRT1 stimulation (Fig. S2). Flow cytometric analysis using an anti-LC3B antibody showed that autophagy induction was significantly decreased in esrra−/− BMDMs, compared were in Esrra+/+ BMDMs, in response to AMPK or SIRT1 activation (). In addition, ultrastructural analysis revealed that the numbers of autophagosomal and autolysosomal structures were dramatically decreased in esrra−/− BMDMs in response to AMPK activation compared with those in Esrra+/+ BMDMs ( and ).
We further assessed the involvement of ESRRA in the increased autophagic flux in macrophages. To this end, we transduced Esrra+/+ and esrra−/− BMDMs with a retroviral vector containing a mCherry-enhanced green fluorescent protein EGFP-LC3B before autophagic stimulation. The abundance or amount of red (mCherry; acid stable) punctate structures, which indicate the positions of autolysosomes (acidic pH quenches GFP fluorescence), by either AMPK or SIRT1 activation, was markedly increased in Esrra+/+ BMDMs, when compared with those in esrra−/− BMDMs ( and ). These data suggest that ESRRA is required for autophagy induction and the increase in autophagic flux in macrophages.
ESRRA is required for the transcriptional activation of autophagy genes in macrophages in response to autophagy inducers
Given the potential role of ESRRA in autophagy activation in macrophages, we revisited the microarray analysis data from our previous studyCitation19 to assess a differential expression of key autophagy genes, which are involved in the essential phases of the autophagic process between Esrra+/+ and esrra−/− BMDMs exposed to LPS ( and S3). The gene expression heatmap analysis showed that 39 autophagy-related genes were differentially expressed between Esrra+/+ and esrra−/− BMDMs (). Among them, we selected 15 autophagy genes that contained at least 3 ESRRA consensus motifs (5′-TNAAGGTCA-3′) in their promoter regions (within 5 kb upstream of the transcription start sites), which mediate regulation of the transcription of target genes through ESRRA binding (Fig. S4).Citation26 In addition, Atg7, which is essential for autophagy initiation and has a single binding site for ESRRA, was included in the qPCR analysis. Among the 16 candidate autophagy genes, the transcriptional activities of 7 Atgs (red-colored) were significantly downregulated in either AICAR- or resveratrol-stimulated esrra−/− BMDMs, when compared with Esrra+/+ BMDMs ().
Figure 2. ESRRA is required for the transcriptional activation of autophagy-related genes in macrophages in response to various autophagy inducers. (A) Heatmap analysis from gene array data of Esrra+/+ and esrra−/− BMDMs. Differential gene expression patterns were observed in 39 autophagy-related genes between Esrra+/+ and esrra−/− BMDMs before and after LPS (100 ng/ml, for 6 h) stimulation (gene accession number: GSE58515). The genes investigated in this study are indicated by red color. (B) Esrra+/+ and esrra−/− BMDMs were stimulated with AICAR (0.5 mM) or resveratrol (10 µM) for 6 h. (C) BMDMs were transduced with lentiviruses expressing nonspecific shRNA (shNS) or shRNA specific for Esrra (shEsrra), and then stimulated with AICAR or resveratrol for 6 h. (D and E) BMDMs were incubated with or without XCT790 (1, 5, and 10 µM) for 48 h and followed by AICAR or resveratrol for 24 h (D) and 6 h (E). (B and C) Quantitative PCR analysis of the mRNA levels of 16 essential autophagy genes for which 15 contain > 3 ESRRA binding sites and ATG7 contains 1 motif (B) or Atg5, Becn1, Maplc13b, Atg16l1 and Ambra1 mRNA expression (C). (D) Cell lysates were subjected to immunoblot analysis using antibodies against ESRRA and ACTB. (E) Quantitative PCR analysis of Atg5, Becn1, and Map1lc3b mRNA expression. Experiments were repeated at least 3 times. Graphs show mean ± SD. *P < 0.05 and **P < 0.01 (2-tailed unpaired Mann–Whitney). U, untreated; SC, solvent control; RSV, resveratrol.
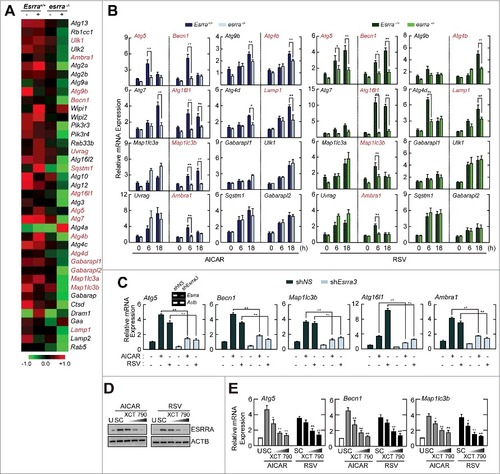
We chose 5 candidate Atg genes (Atg5, Becn1, Atg16l1, Map1lc3b [microtubule-associated protein 1 light chain 3 β], and Ambra1 [autophagy Beclin 1 regulator 1]) that were significantly decreased in both AICAR- and resveratrol-stimulated esrra−/− BMDMs, when compared with Esrra+/+ BMDMs. The gene expression of the candidate Atg genes was further confirmed by transduction of BMDMs with lentiviral short hairpin RNA (shRNA) specific to Esrra (shEsrra). We examined the knockdown efficiencies of several lentiviral shRNA constructs for Esrra expression in BMDMs. Three different shRNA constructs designed for silencing of Esrra were tested and 2 of them showed potent inhibitory effects upon Esrra mRNA expression (Fig. S5A; shEsrra2, > 75%; shEsrra3, > 85%). Silencing of Esrra using shEsrra2 or shEsrra3 led to a significant reduction of mRNA expression of 5 candidate Atgs in BMDMs in response to either AICAR or resveratrol stimulation ( and S5B). In addition, we examined whether the knockdown effects of shEsrra for the regulation of autophagy gene transcription were rescued by re-expression of ESRRA. As shown in Figure S6, we infected shNS- or shEsrra-transduced BMDMs with AdEsrra, and determined the mRNA expression of several autophagy genes including Atg5, Becn1, and Map1lc3b. When Esrra expression was rescued in shEsrra-transduced BMDMs (Fig. S6A), the decreased transcriptional expression of autophagy genes was nearly completely recovered to the levels observed in shNS-transduced BMDMs (Fig. S6B). These data demonstrate that ESRRA is essentially involved in the transcriptional activation of several autophagy genes in BMDMs.
Moreover, we tried to examine the effects of an ESRRA inverse agonist upon the regulation of autophagy genes expression. To investigate this, we pretreated BMDMs with the ESRRA inverse agonist XCT790,Citation 27 and observed that ESRRA protein was degraded in a dose-dependent manner (). When XCT790-pretreated BMDMs were stimulated with AICAR or resveratrol, autophagy gene expression (Atg5, Becn1, and Map1lc3b) was inhibited in a dose-dependent manner (). Together, these results indicate that ESRRA promotes transcriptional activation of numerous Atgs which contain ESRRA consensus motifs in their promoters.
ESRRA is recruited to the promoter regions of Atg5, Becn1, and Atg16l1 to promote transcriptional activation of these genes
Next, we focused on the mechanistic role of ESRRA in the transcriptional activation of Atg5, Becn1, and Atg16l1, which were the most differentially expressed transcripts between Esrra+/+ and esrra−/− BMDMs (). We thus performed ChIP assays to assess the binding of ESRRA to the promoters of these genes. We found a strong interaction between ESRRA and the Atg5 (−841/−1018 and −931/−1143), Becn1 (−173/−362 and −1060/−1239), and Atg16l1 (−3222/−3372) promoter regions in BMDMs after AICAR or resveratrol stimulation ( to ). We next determined whether the differential occupancy of ESRRA at the promoters of Atg5, Becn1, and Atg16l1 correlates with significant differences in promoter activities of these genes. The consensus ESRRA motifs in the promoter regions of Atg5, Becn1, and Atg16l1 are shown (4 in Atg5 and Becn1; 3 in Atg16l1) (, left). Cell-based luciferase reporter gene assays using a 5-kb DNA fragment of the promoter regions of these genes containing either wild-type (WT) or mutant ERREs (, right) were conducted ( to ).
Figure 3. ESRRA upregulates the transcriptional activation of Atg5, Becn1, and Atg16l1, through direct binding to their promoter regions. (A to C) ESRRA ChIP analyses of the 4 proximal ERREs within Atg5 and Becn1, the 3 proximal ERREs within Atg16l1 and a distal region of the Atg5, Becn1, and Atg16l1 promoters. BMDMs were stimulated with AICAR (0.5 mM) or resveratrol (10 µM) for 6 h and subjected to chromatin immunoprecipitation assays using Abs specific for ESRRA, and semi-quantitative-PCR analysis using primers specific for the different Atg5, Becn1, and Atg16l1 ERREs. (D) Schematic diagram of Esrra consensus motifs in the Atg5, Becn1, and Atg16l1 promoters (left), and the wild-type (WT) and mutant construct sequences (right). (E to G) RAW264.7 cells were transfected with Atg5, Becn1, and Atg16l1-luciferase reporter constructs (ATG5–3 or ATG5–4 mutant; BECN1–1 or BECN1–4 mutant; ATG16L1–3 mutant carrying point mutations in the critical ESRRA-binding site) in the presence or absence of plasmid encoding FLAG-tagged murine ESRRA (pcDNA-Esrra). (H) RAW264.7 cells were transfected with Atg5-, Becn1- or Atg16l1-luciferase reporter constructs in the presence or absence of pcDNA-Esrra and a plasmid encoding FLAG-tagged human PPARGC1A (pcDNA-PPARGC1A). (E to H) Cells were stimulated with AICAR or resveratrol for 6 h and subjected to luciferase activity. Experiments were repeated at least 3 times. Graphs show mean ± SD. *P < 0.05, **P < 0.01 and ***P < 0.001 (2-tailed unpaired Welch t test). A5–1, A5–2, A5–3, and A5–4, for Atg5 promoter regions −3092/−2842, −2646/−2436, −1143/−931, and −1018/−841, respectively; B1–1, B1–2, B1–3 and B1–4, for Becn1 promoter regions −1239/−1060, −1131/−911, −819/−609, and −362/−173, respectively; A16L1-1, A16L1-2, and A16L1-3, for Atg16l1 promoter regions −4493/−4224, −3447/−3233 and −3372/−3222, respectively; NC, negative control; SC, solvent control; RSV, resveratrol.
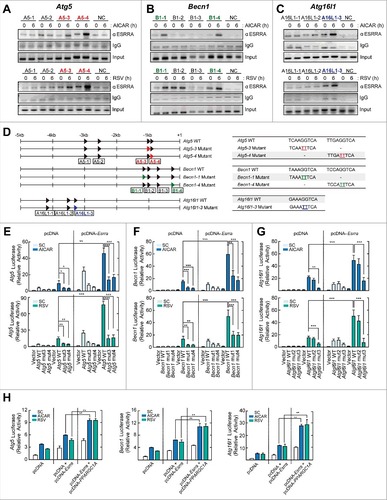
AICAR and resveratrol treatment significantly increased the activity of WT promoter constructs of Atg5, Becn1, and Atg16l1 in RAW264.7 cells ( to ). Mutation of the ESRRA DNA binding motifs within the promoters of all 3 genes significantly decreased the reporter gene activity in RAW264.7 cells and abrogated the response to AICAR or resveratrol treatment ( to ). In addition, overexpression of ESRRA significantly increased Atg5, Becn1, and Atg16l1 promoter activities before and after stimulation with AICAR or resveratrol (E to G). Next, we examined whether PPARGC1A is required for ESRRA-mediated enhancement of Atg transcriptional activities in macrophages. As shown in , the increased promoter activities of Atg5, Becn1, and Atg16l1 by ESRRA overexpression were significantly amplified by cotransfection with PPARGC1A, in RAW264.7 cells. Together, these results confirmed the direct interactions of ESRRA with the Atg promoter regions important for augmenting the transcriptional activation of Atg5, Becn1, and Atg16l1. These results also indicate that PPARGC1A can boost ESRRA-mediated transcriptional activation of Atg5, Becn1, and Atg16l1, as expected from its well-known function as an ESRRA coactivator.Citation17,18
Mutual induction of ESRRA and SIRT1 expression in macrophages
Our recent study shows that ESRRA overexpression results in the transcriptional activation of SIRT, a NAD+-dependent deacetylase, in macrophages after LPS stimulation.Citation19 We thus determined whether AMPK or SIRT1 activation enhanced ESRRA and SIRT1 expression in macrophages. As shown in and , treatment of BMDMs with AICAR or resveratrol led to a robust increase in SIRT1 mRNA and protein levels in a time-dependent manner. Indeed, AICAR- or resveratrol-induced SIRT1 expression was significantly decreased in esrra−/− BMDMs ( and ). In addition, AMPK or SIRT1 activation via AICAR or resveratrol, respectively, resulted in Sirt1 promoter induction, which was further enhanced by ESRRA overexpression ( and ). However, a mutation within the ESRRA consensus motif of the Sirt1 promoter markedly decreased Sirt1 reporter gene activities in RAW264.7 cells in the absence or presence of AICAR or resveratrol ( and ). Furthermore, pretreatment of BMDMs with an ESRRA inverse agonist, XCT790, suppressed the Sirt1 mRNA expression and reporter gene activities induced by AICAR or resveratrol in a dose-dependent manner ( and ). It was also noted that the induced Sirt1 mRNA expression in response to rapamycin was significantly dampened in esrra−/− BMDMs, compared with Esrra+/+ BMDMs (Fig. S7).
Figure 4. Mutual induction of ESRRA and SIRT1 expression in macrophages. (A and B) Esrra+/+ and esrra−/− BMDMs were stimulated with AICAR (0.5 mM) or resveratrol (10 µM) for the indicated times. (A) Quantitative PCR analysis of Sirt1 mRNA expression. (B) Immunoblot analysis of SIRT1 and ACTB. (C and D) RAW264.7 cells were transfected with a Sirt1-luciferase reporter construct (Sirt1 mut1, SIRT1 carrying point mutations in the ESRRA binding site) in the presence or absence of plasmid encoding FLAG-tagged murine ESRRA (pcDNA-Esrra). Cells were stimulated with AICAR (C) or resveratrol (D) for 6 h and subjected to luciferase activity. (E) BMDMs were incubated with or without XCT790 (1, 5, and 10 µM) for 48 h and followed by AICAR or resveratrol for 6 h. (F) RAW264.7 cells were transfected with pGL3 basic vector or Sirt1-luciferase reporter construct. Cells were incubated with or without XCT790 (1, 5, and 10 µM) for 48 h and followed by AICAR or resveratrol for 6 h. (G and H) BMDMs were transduced with lentiviruses expressing nonspecific shRNA (shNS) or shRNA specific for Sirt1 (shSirt1), followed by AICAR or resveratrol for 4 or 24 h. (I and J) The mock vector or SIRT1-expressed RAW264.7 cells followed by AICAR or resveratrol for 4 or 24 h. Cell lysates were subjected to immunoblot analysis using antibodies against ESRRA, SIRT1, and ACTB. (B and G to J) The densitometry values for SIRT1 or ESRRA were normalized to ACTB in each right panel. Experiments were repeated at least 3 times. Graphs show mean ± SD. *P < 0.01, **P < 0.01, and ***P < 0.001 (2-tailed unpaired Mann–Whitney, A, B, and E to J; 2-tailed unpaired t test, C and D). U, untreated; SC, solvent control; RSV, resveratrol.
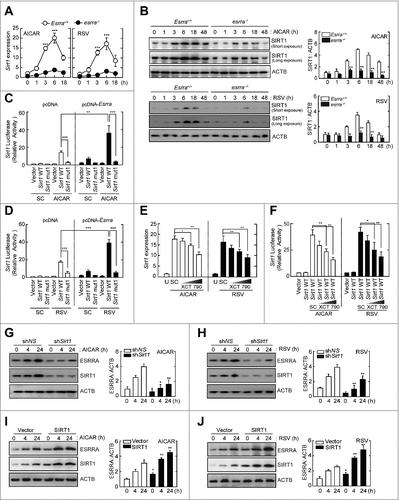
We then questioned whether SIRT1 affected ESRRA expression in macrophages. To this end, we abrogated Sirt1 in BMDMs before treatment with AICAR or resveratrol. As shown in and , the increased protein expression of ESRRA induced by AICAR or resveratrol was significantly inhibited in BMDMs by knockdown of SIRT1 using shRNA specific to Sirt1 (shSirt1). In addition, overexpression of SIRT1 in RAW264.7 cells led to a marked increase in ESRRA protein levels ( and ). Together, these data show that AMPK or SIRT1 activation upregulates ESRRA, which is required for the increased expression of SIRT1 mRNA and protein in macrophages, and vice versa.
ESRRA-mediated SIRT1 induction in macrophages contributes to deacetylation of ATG5, BECN1, and ATG7
SIRT1 is a critical in vivo positive regulator of autophagy that functions through deacetylation of ATG5, ATG7 and ATG8.Citation21,23 Recently, studies have shown that BECN1 is deacetylated by SIRT1 and acetylated by EP300/p300, which is important for regulation of autophagosome maturation and tumor growth.Citation22 We therefore examined the levels of acetylation of the autophagy proteins ATG5, BECN1, and ATG7 under basal and AICAR- or resveratrol-stimulated conditions. We first confirmed acetylation of ATG5, BECN1, and ATG7 in HEK293T cells by using an anti-acetyl-lysine antibody to immunoprecipitate endogenous autophagy proteins. After AICAR or resveratrol treatment, the acetylation levels of ATG5, BECN1, and ATG7 in HEK293T cells were reduced in a time-dependent manner in parallel with increasing SIRT1 levels ( and ). Moreover, ESRRA overexpression, which induces SIRT1 expression, led to a significant decrease in ATG5, BECN1, and ATG7 acetylation ( and ). In contrast, knockdown of Esrra increased the acetylation levels of the autophagy proteins in BMDMs ( and S8A). Similarly, the acetylation levels of ATG5, BECN1, and ATG7 were considerably increased in esrra−/− BMDMs, compared with those in Esrra+/+ BMDMs (Fig. S8B). Of note, the acetylation levels of ATG5, BECN1, and ATG7 were consistently increased in ESRRA-deficient BMDMs after either AICAR or resveratrol treatment ( and S8A).
Figure 5. ESRRA-mediated SIRT1 induction contributes to deacetylation of ATG5, BECN1, and ATG7 in macrophages. (A and B) HEK293T cells were transfected with mock vector (vector) or Esrra-Flag plasmid and stimulated with AICAR (0.5 mM; A) or resveratrol (10 µM; B) for 8 or 24 h. (C) BMDMs were transduced with lentiviruses expressing shRNA (shNS) or shRNA specific for Esrra (shEsrra) and the cells were stimulated with AICAR for 8 or 24 h. (D and E) HEK293T cells transfected with mock vector (vector) or a FLAG-tagged BECN1-encoding plasmid (BECN1; D), and RAW264.7 cells transfected with mock vector (vector) or a HA-tagged SIRT1-encoding plasmid (SIRT1; E) were treated with AICAR or resveratrol for 8 or 24 h. (A to E) Cells were immunoprecipitated using Ac-Lys, FLAG, or SIRT1 antibody, followed by immunoblotting with indicated proteins. Protein levels in nonprecipitated cell lysates were determined by immunoblot analysis. Experiments were repeated at least 3 times. RSV, resveratrol.
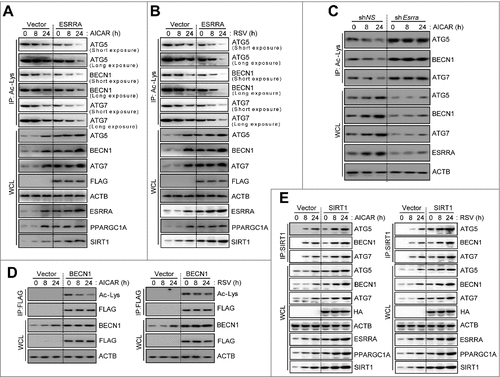
Next, we overexpressed BECN1 in HEK293T cells and assessed whether treatment with AICAR or resveratrol led to the inhibition of BECN1 lysine acetylation. By performing CoIP with an anti-FLAG antibody (BECN1-FLAG) and then probing with an anti-acetyl-lysine antibody, a significant decrease in BECN1 acetylation was confirmed in AICAR- and resveratrol-treated cells (). Finally, we examined whether SIRT1 immunoprecipitated with ATG5, BECN1, and ATG7 in RAW264.7 cells before and after AMPK and SIRT1 activation. RAW264.7 cells were transfected with an HA-SIRT1 DNA expression plasmid. The lysates were then coimmunoprecipitated with anti-SIRT1 antibody, followed by western blot analysis with anti-ATG5, BECN1, and ATG7 antibodies, which demonstrated their strong association (). It was also noted that the interaction between SIRT1 and ATGs was increased after AICAR treatment in both vector control- and SIRT1-overexpressed RAW264.7 cells (). These results were like those obtained with the SIRT1 activator, resveratrol (). Moreover, we overexpressed BECN1 in HEK293T cells and assessed whether treatment with rapamycin led to the inhibition of BECN1 lysine acetylation. By performing CoIP with an anti-FLAG antibody and then probing with an anti-acetyl-lysine antibody, a significant decrease in BECN1 acetylation was confirmed in rapamycin-treated cells (Fig. S9). These data collectively indicate that the ESRRA-mediated SIRT1 deacetylase is a critical activator of autophagy that functions via interacting with and deacetylating ATG5, BECN1, and ATG7.
SIRT1 deficiency affects autophagy induction and Mtb phagosomal maturation in macrophages in response to AICAR or resveratrol
SIRT1 is required for autophagy activation.Citation21-Citation24 However, whether SIRT1 is required for phagosomal maturation of Mtb in macrophages is unclear. We first examined whether SIRT1 contributes to autophagy activation in macrophages in response to AICAR or resveratrol treatment. As shown in , knockdown of Sirt1 by shSirt1 dramatically inhibited LC3A/B-positive autophagosome formation, which was enhanced by treatment with AICAR or resveratrol in BMDMs, when compared with control conditions. In addition, Sirt1-silencing led to a decrease in LC3-II accumulation and the LC3-II:LC3-I ratio in BMDMs after AICAR or resveratrol treatment (). In addition, the colocalization between LC3A/B autophagosomes and LAMP1 (lysosomal-associated membrane protein 1) was significantly downregulated in shSirt1-transduced BMDMs, after AICAR or resveratrol treatment, when compared with shNS-transduced BMDMs ().
Figure 6. SIRT1 deficiency affects autophagy induction and Mtb phagosomal maturation in macrophages in response to AICAR and resveratrol. (A and B) BMDMs were transduced with lentiviruses expressing nonspecific shRNA (shNS) or shRNA specific for Sirt1 (shSirt1). Cells were stimulated with AICAR (0.5 mM) or resveratrol (10 µM) for 24 h. (A) Immunofluorescence microscopy analysis of LC3A/B puncta formation (left). Quantification of LC3A/B punctate foci per cell (right). Scale bar: 10 µm. (B) Cell lysates were subjected to immunoblot analysis using antibodies against LC3A/B, SIRT1, and ACTB. The densitometry values for LC3-II were normalized to ACTB in each right panel. (C and D) BMDMs were transduced with shSirt1 and infected with Mtb-ERFP (MOI = 10, for 4 h), and then stimulated with AICAR or resveratrol for 24 h. (C) Cells were immunostained using antibodies for LC3A/B (Alexa Fluor 488-conjugated goat anti-rabbit IgG, green), LAMP1 (Alexa Fluor 594-conjugated goat anti-rat IgG, red) and DAPI to visualize nuclei. Quantitative analysis of cells showing LC3A/B and LAMP1 colocalization (right). Scale bar: 10 µm. (D) Cells were immunostained using an anti-LC3A/B antibody (Alexa Fluor 488-conjugated goat anti-rabbit IgG, green) and DAPI to visualize nuclei (blue). Quantification of LC3A/B and Mtb-ERFP colocalization per cell with 200 internalized mycobacteria (right). Scale bar: 10 µm. (E) BMDMs were transduced with shRNA specific for Sirt1 (shSirt1) and infected Mtb (MOI = 10, for 4 h), and then stimulated with AICAR or resveratrol for 3 d after which the cells were lysed to determine intracellular bacterial loads. (E, inset) RT-PCR analysis of transduction efficiency. Experiments were repeated at least 3 times. Immunofluorescence microscopy images are from one representative of 7 independent samples, with each experiment including at least 200 cells scored in 7 random fields (A, C, and D). Experiments were repeated at least 3 times. Graphs show mean ± SD. **P < 0.01 (2-tailed unpaired Mann–Whitney). U, untreated; RSV, resveratrol.
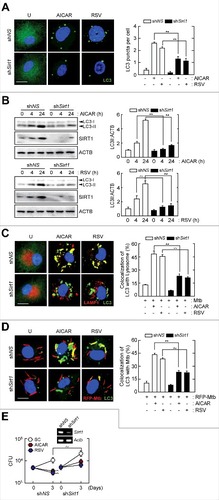
We next examined the effects of SIRT1 on phagosome maturation in Mtb-infected macrophages. To determine whether SIRT1 affects the colocalization of Mtb and the autophagy marker LC3, we transduced BMDMs with shSirt1 or shNS and infected them with RFP-expressing Mtb, followed by treatment with AICAR or resveratrol for 24 h. Merged images showed that AICAR or resveratrol treatment increased colocalization between Mtb (red) and LC3A/B puncta (green) in large punctate structures (). This indicates that in shNS-transduced BMDMs, Mtb was wrapped in LC3A/B autophagosomal structures after AICAR or resveratrol treatment. However, knockdown of Sirt1 significantly decreased the colocalization of Mtb with LC3A/B autophagosomes after stimulation with AICAR or resveratrol (). Furthermore, silencing of Sirt1 led to a marked attenuation of intracellular killing effects against Mtb in BMDMs, suggesting an essential role of SIRT1 in mediating antimicrobial responses induced by AICAR or resveratrol (). These data suggest that SIRT1 is critical for the increase in the formation of autophagosomes and autolysosomes and Mtb phagosomal maturation in macrophages in response to AMPK or SIRT1 activation.
ESRRA is critical for phagosomal maturation, antimicrobial responses and regulation of excessive inflammation during mycobacterial infection
To confirm the effect of ESRRA on phagosomal maturation and antimicrobial responses against Mtb in macrophages, we next analyzed Esrra+/+ and esrra−/− BMDMs treated with AICAR or resveratrol. When Esrra+/+ BMDMs were treated with AMPK or a SIRT1 activator, colocalization of RFP-expressing Mtb and autophagic vesicles was markedly increased compared with vehicle-treated conditions ( and ). In contrast, Esrra deficiency caused a significant reduction in the percentage of Mtb within autophagosomal structures in BMDMs after stimulation with AICAR or resveratrol, compared with WT BMDMs ( and ). In addition, treatment of Esrra+/+ BMDMs with AICAR or resveratrol significantly increased colocalization of Mtb with lysosomes ( and ). Similar results were obtained in Esrra+/+ and esrra−/− BMDMs after infection with BCG-GFP, followed by treatment with AICAR or resveratrol (data not shown). It was noted that the increased phagosomal maturation in Esrra+/+ BMDMs was significantly downregulated in esrra−/− BMDMs, in response to AICAR or resveratrol ( and ).
Figure 7. ESRRA is critical for the AMPK- or SIRT1-mediated enhancement of Mtb phagosomal maturation and antimicrobial responses in macrophages. (A to D) Esrra+/+ and esrra−/− BMDMs were infected with Mtb-ERFP (MOI = 10, for 4 h), and then stimulated with AICAR (0.5 mM) or resveratrol (10 µM) for 24 h. (A) Immunofluorescence microscopy analysis of LC3A/B colocalization formation. Scale bar: 10 µm. (B) Quantitation of LC3A/B colocalization per cell with 200 internalized mycobacteria. (C) Cells were immunostained using antibodies for LAMP2 (Alexa Fluor 488-conjugated goat anti-rabbit IgG, green) and DAPI to visualize nuclei. Scale bar: 10 µm. (D) Quantitative analysis of cells showing LAMP2 and Mtb-ERFP colocalization per cell with 200 internalized mycobacteria colocalization. (E and F) Esrra+/+ and esrra−/− BMDMs were infected with Mtb (MOI = 1 or 10) for 4 h, stimulated with AICAR (0.1, 0.5, and 1 mM) or resveratrol (0.1, 1, or 10 μM) for 3 d after which the cells were lysed to determine intracellular bacterial loads. (G) Esrra+/+ and esrra−/− mice were infected with Mtb (1 × 106 CFU/mouse, i.v.) for 3 wk and then treated with AICAR (500 mg/kg, i.p.) or vehicle (PBS, i.p.) for 7 consecutive d. Mice were killed 4 wk after mycobacterium infection and bacterial loads in organs of infected mice (n = 4 to 5 per group) were determined by CFU assay. Data are from a representative of 2 independent experiments. (H and I) Three wk after Mtb infection in vivo, Esrra+/+ and esrra−/− mice were treated with resveratrol (20 mg/kg, i.p.) or vehicle (5% EtOH, i.p.) for 7 consecutive d. (H) Lung lysates were harvested and subjected to immunoblot analysis. Data are from a representative of 2 independent experiments. The densitometry values for LC3-II were normalized to ACTB in the right panel. (I) H&E staining in lung tissue (n = 3). Scale bar: 100 μm. Experiments were repeated at least 3 times. Immunofluorescence microscopy images are from one representative of 7 independent samples, with each experiment including at least 200 cells scored in 7 random fields (A and C). Graphs show mean ± SD. *P < 0.05 and **P < 0.01 (2-tailed unpaired Mann–Whitney). ns, not significant; U, untreated; SC, solvent control; Vehi, vehicle control; RSV, resveratrol.
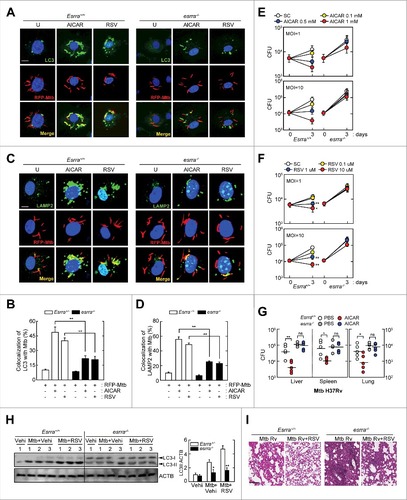
We then examined the survival of intracellular mycobacteria in Esrra+/+ and esrra−/− BMDMs and mice after AICAR or resveratrol treatment. As shown in and , both AICAR and resveratrol inhibited the intracellular survival of Mtb in Esrra+/+ BMDMs in a dose-dependent manner. However, AICAR- and resveratrol-induced antimicrobial responses against Mtb were significantly attenuated in esrra−/− BMDMs ( and ). Moreover, we monitored Mtb survival in Esrra+/+ and esrra−/− mice after treatment with AICAR. As shown in , in vivo administration of AICAR (500 mg/kg) to Mtb-infected Esrra+/+ mice significantly inhibited in vivo bacterial growth in the liver, spleen, and lung, whereas this effect was lost in esrra−/− mice. Moreover, in vivo LC3-II induction was significantly increased in lungs from Esrra+/+ mice compared with esrra−/− mice after administration of resveratrol ().
Since ESRRA is critical in the regulation of inflammatory responses,Citation19 we investigated its role in immunopathological responses during Mtb infection in vivo. As shown in , histopathological changes confirmed that Mtb-infected esrra−/− mice showed more severe pathological responses and granulomatous lesions in the lungs than those from Esrra+/+ mice. Administration of resveratrol considerably attenuated the pathological responses in Mtb-infected Esrra+/+ mice, but did not in esrra−/− mice (). We further examined proinflammatory cytokine generation in response to Mtb or BCG in Esrra+/+ and esrra−/− BMDMs. Following Mtb or BCG infection, TNF (tumor necrosis factor), IL6 (interleukin 6), and IL1B (interleukin 1 β) mRNA and protein levels were increased in esrra−/− BMDMs compared with esrra+/+ BMDMs (Fig. S10). Collectively, these data indicate that ESRRA is critical for phagosomal maturation, antimicrobial responses, and regulation of inflammatory responses during mycobacterial infection.
Discussion
Maintaining homeostasis during infection and/or other stresses is crucial for a variety of physiological functions, including immune regulation. NRs and the autophagy pathway are important in the maintenance of intracellular homeostasis and could be subject to multiple layers of crosstalk, most of which are poorly understood. In particular, the orphan NR, ESRRA, is increasingly recognized as a crucial regulator of immune and inflammatory pathways,Citation19,28,29 beyond its known function in mitochondrial biogenesis and metabolic pathways.Citation3,30 Here, we identified ESRRA as a crucial activator of the antibacterial autophagy response, which protects against mycobacterial infection in vitro and in vivo. In particular, loss of ESRRA resulted in defective autophagy activation and phagosomal maturation after mycobacterial infection. Although the ligand for ESRRA is unknown, we found that autophagic signals robustly induced ESRRA mRNA and protein expression through the AMPK-PPARGC1A pathway in macrophages. Ablation of ESRRA expression revealed defects in autophagy induction, flux, and control of intracellular mycobacterial survival. At the molecular level, ESRRA controlled the transcriptional and post-translational modification of members of the ATG family, primarily via SIRT1 activity. Our findings, therefore, establish that ESRRA acts as a central regulator of autophagy activation and host defense against mycobacterial infection.
Our findings demonstrate that ESRRA plays a key role in autophagy activation, an effect mediated by regulation of the transcription of numerous autophagy genes in innate immune cells. It is well known that autophagy-related genes and proteins serve essential roles in different steps of autophagy, thus being involved in a variety of biological processes.Citation31 Recent studies have highlighted the roles of several NRs—VDR/vitamin D receptor,Citation15 ESR1,Citation32 PPARA (peroxisome proliferator activated receptor α),Citation33 and PPARG,Citation34 involved in the regulation of autophagy, through mainly transcriptional activation of autophagy-related genes in various cell types. Transcriptional regulation of hepatic autophagy genes by 2 NRs, PPARA, and NR1H4 (nuclear receptor subfamily 1, group H, member 4), which are activated in the fasted and fed liver, respectively, has been reported.Citation33 PPARA and NR1H4 are required for autophagic regulation and function via competitive binding to shared sites in the promoter regions of genes related to autophagy during nutrient-sensing signaling.Citation33 Our data showed that ESRRA directly binds to the promoter regions of the autophagy genes Atg5, Becn1, and Atg16l1, required for the upregulation of their promoter activities, especially in response to activators of AMPK or SIRT1. ESRRA transcriptional activation of target genes is dependent on its coexpression with PPARGC1 coactivators.Citation17,18 We have previously shown that the AMPK-PPARGC1A pathway is essential in upregulation of the expression of autophagy-related genes in macrophages during Mtb infection.Citation16 Combined with the present study, these findings suggest that AMPK-dependent autophagy activation requires the coexpression of ESRRA and PPARGC1A, both of which function by promoting the transcriptional activation of autophagy gene sets in macrophages.
Our findings establish ESRRA and SIRT1 signaling as an essential coordinator of host innate defenses through enhancing autophagy activation in macrophages. As a NAD+-dependent deacetylase, SIRT1 is induced by caloric restriction in various cells and tissuesCitation35 and is required for AMPK activation and increased mitochondrial biogenesis and functionality.Citation36 Although the SIRT1 activator resveratrol can activate autophagy,Citation24 its function in the antimicrobial response to intracellular microbial infection is unclear. Here, we establish that resveratrol activates autophagy and increases phagosomal maturation of Mtb in macrophages. SIRT1 is crucial for deacetylation of multiple non-histone protein targets, including the FOXO family of Forkhead transcription factors FOXO1/3/4Citation37-Citation39 and PPARGC1A.Citation40 The FOXO1-dependent pathway contributes to the activation of autophagic flux by increasing the expression of ATG-family genes including ATG4B, ATG12, BECN1, and PIK3C3 and suppressing MTOR (mechanistic target of rapamycin) activity.Citation39,41 Moreover, resveratrol treatment increases autophagic flux by activating the SIRT1-FOXO1-RAB7A axis and improved cardiac function in diabetic hearts.Citation42 We cannot rule out a role for FOXO-family members in resveratrol-induced autophagy activation during mycobacterial infection. Future studies will evaluate crosstalk between ESRRA and FOXO1 in SIRT1-activated autophagy in macrophages.
These findings also provide evidence of a novel role of ESRRA in the post-translational modification of several autophagy proteins (ATG5, BECN1, and ATG7), all of which are deacetylated by SIRT1. SIRT1 is essential in the regulation of autophagy induction and maturation through deacetylation of several components of the autophagic machinery. It has been noted that SIRT1 activity is required for autophagy induction under basal and starved conditions through interaction and deacetylation of ATG5, ATG7, and MAP1LC3A.Citation21,22 Recent studies have shown that acetylation and deacetylation of ATG6/BECN1 are modulated by EP300 and SIRT1, respectively, which is important for the fine-tuning of the autophagic response.Citation22 In addition, the LC3 protein, which shuttles between the nucleus and cytoplasm, is activated in the nucleus via deacetylation by SIRT1, where it associates with autophagy factors, and is returned to the cytosol to initiate autophagy.Citation23 Moreover, overexpression of ESRRA resulted in increased SIRT1 mRNA and protein levels in AMPK- and SIRT1-activated macrophages, and vice versa. Thus, our data strongly indicate that ESRRA, acting in cooperation with SIRT1, contributes to the deacetylation required for induction of autophagy.
An increasing number of adjunctive host-directed therapies are being explored to improve the treatment response and to facilitate the development of new strategies in therapeutics against Mtb infection.Citation43 Understanding how ESRRA-mediated autophagy activation works in the context of host defense against mycobacterial infection in vivo is of great interest and may lead to the design of beneficial therapeutics against TB. Our study is important in demonstrating the role of ESRRA in host defense against mycobacterial infection through acting as a positive regulator of autophagy in vivo. In addition, our data show that ESRRA may prevent excessive immune-related pathological responses during mycobacterial infection. Our data are, in part, in agreement with previous reports that ESRRA is essential for IFNG (interferon gamma)–mediated clearance of listerial infection,Citation5 suggesting a crucial role for ESRRA in host defense against infection with intracellular bacteria. Notably, ESRRA is involved in modulation of TLR4 (toll-like receptor 4)-mediated signaling through TNFAIP3/A20 induction and metabolic reprogramming,Citation19 highlighting its role in the regulation of inflammation. Approaches based on augmentation of antimicrobial mechanisms and/or directly reducing inflammation may provide promising new adjunctive host-directed therapeutics for the treatment of TB.Citation43-Citation45 Indeed, several candidates which are involved in autophagy activation and in reducing inflammatory damage and tissue destruction are increasingly considered as promising TB therapies.Citation43-Citation45 Taken together, ESRRA could play a key fine-tuning role during microbial infection by promoting bacterial clearance through autophagy induction and preventing severe tissue damage. Approaches based on targeting ESRRA may provide a promising new approach for the treatment of intracellular bacterial infection and amelioration of pathologic inflammatory conditions.
Materials and methods
Mice and cell culture
Esrra+/+ and esrra−/− mice were 6 to 8 wk of age and sex-matched. All mice were maintained under specific pathogen-free conditions. Animal-related procedures were reviewed and approved by the Institutional Animal Care and Use Committee, Chungnam National University School of Medicine (CNUH-014-A0006; Daejeon, Korea). BMDMs were isolated and differentiated after culture for 4 to 5 days in medium containing 25 ng/ml CSF1/macrophage colony-stimulating factor (R&D Systems, 416-ML), as described previously.Citation19 The culture medium consisted of Dulbecco's modified Eagle's medium (DMEM; Lonza, 12–604F) supplemented with 10% fetal bovine serum (FBS; Lonza, BW14–503E), and penicillin–streptomycin-amphotericin B (Lonza, 17–745E). RAW264.7 (ATCC, TIB-71), HEK293T (ATCC, 11268), and human Phoenix AMPHO (ATCC, CRL-3213) cells were purchased from the American Type Culture Collection and maintained in DMEM supplemented with 10% heat-inactivated FBS and penicillin–streptomycin-amphotericin B.
Mycobacterial infection in vivo
Mycobacterial infection in vivo was performed as described previously.Citation16 Esrra+/+ and esrra−/− mice were intravenously injected with Mtb (1 × 106 colony-forming unit [CFU]/mouse). After 3 wk, mice were intraperitoneally treated with AICAR (500 mg/kg) or resveratrol (20 mg/kg) for 7 consecutive d. At 4 wk, mice were killed and the lungs, spleens, and livers harvested for CFU assay and immunoblot analysis. Mice were maintained in biosafety level (BSL)-3 laboratory facilities. The experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Bioleaders Corporation (Daejeon, Korea, protocol number: BLS-ABSL3–13–11). For immunohistochemistry (IHC), lungs were fixed in 10% formalin (Sigma, HT501128) and embedded in paraffin wax. Then, paraffin sections (4 µm) were cut and stained with hematoxylin and eosin. IHC tissue slides were imaged using an Aperio digital pathology slide scanner (Leica, Nussloch, Germany). The IHC images used for this study were provided by the Biobank of Chungnam University Hospital, a member of the Korea Biobank Network.
Mycobacterial culture
Mycobacterium tuberculosis H37Rv (Mtb; provided by R L Friedmann, University of Arizona, Tucson, AZ USA), and enhanced Mtb-enhanced red fluorescent protein (ERFP) were grown in single cell suspensions, as described previously.Citation16 Briefly, Mtb was grown in Middlebrook 7H9 medium (Difco, 271310) supplemented with 10% OADC (BD Biosciences, 212240), 5% glycerol (Sigma, G5516), and 0.05% Tween-80 (Sigma, P1754). Mtb-ERFP strains were grown in Middlebrook 7H9 medium supplemented with 10% OADC and selected kanamycin (50 μg/ml; Sigma, 60615). All mycobacterial suspensions were aliquoted (1 ml) and stored at −80°C. Mid-logarithmic-phase bacteria (optical density 0.6) were used in all assays. CFUs were enumerated on Middlebrook 7H10 agar (Difco, 271310).
Reagents and antibodies
AICAR (A9978), resveratrol (R5010), and XCT790 (X4753) were purchased from sigma. Rapamycin (553211) was purchased from Calbiochem. For immunofluorescence analysis, anti-LC3A/B (PM036) was purchased from Medical & Biological Laboratories International. The antibodies against LAMP1 (sc-20011) and LAMP2 (sc-18822) were purchased from Santa Cruz Biotechnology. Alexa Fluor 488-conjugated anti-rabbit IgG (A17041) and Alexa Fluor 594-conjugated anti-rat IgG (A-11007) were purchased from Molecular Probes. The antibody against LC3A/B (L8918) for immunoblotting was purchased from Sigma. Antibodies against ESRRA (ab76228), PPARGC1A (ab54481), and SIRT1 (ab7343) were purchased from Abcam. Antibodies against HA (sc-805), FLAG (sc-807), and ACTB (sc-1616) were purchased from Santa Cruz Biotechnology. Anti-ATG5 (12994), anti-BECN1 (3495), anti-ATG7 (8558), and anti-acetylated-lysine (9441) were purchased from Cell Signaling Technology.
Plasmid construction
The pcDNA3-Flag-Esrra was generated as described previously.Citation19 The pcDNA3-Flag PPARGC1A was kindly provided by Dr. S-H. Koo (Korea University, Seoul, Korea). The DNA fragment corresponding to the coding sequences of the Becn1 gene was amplified by PCR and subcloned into pcDNA3-Flag vector (Addgene, 20011; deposited by Stephen Smale). The pCruzHA-Sirt1 (10962) was purchased from Addgene (deposited by Toren Finkel). All plasmid constructs were sequenced using the ABI PRISM 377 automatic DNA sequencer (Applied Biosystems, Inc., Carlsbad, CA, USA) to verify 100% correspondence with the original sequence.
Immunofluorescence analysis
Immunofluorescence analysis of endogenous and colocalized LC3A/B was performed as described previously.Citation16,46 Briefly, cells were fixed with 4% paraformaldehyde (Santa Cruz Biotechnology, sc-281692) for 15 min, and permeabilized with 0.25% Triton X-100 (Sigma, T8787) for 10 min. Cells were incubated with primary antibodies anti-LC3A/B, anti-LAMP1, and anti-LAMP2 for 2 h at room temperature. After 2 h, primary antibodies were removed and washed with phosphate-buffered saline (PBS; Sigma, P4417), and then cells were incubated with a secondary anti-rabbit IgG-Alexa Fluor 488 (Molecular Probes, A17041) and anti-rat IgG-Alexa Fluor 594 (Molecular Probes, A11007) for 1 h at room temperature. Nuclei were stained by incubation with 4′,6-diamidino-2-phenylindole (DAPI; Sigma, D9542) for 1 min. Slides were examined using a confocal laser-scanning microscope (LSM 710; Zeiss, CLSM, Jena, Germany). To analyze endogenous LC3A/B puncta, we used the ImageJ software. Slides were assayed in triplicate, and at least 200 cells per well were counted. To quantify the percentage of mycobacterial phagosomes, autophagosomes (LC3A/B), lysosomes (LAMP1 or LAMP2), Mtb-infected cells were visualized using confocal laser scanning microscopy, and the images were captured using the ZEN 2009 software (version 5.5 SP1; Zeiss). Each experiment was completed on triplicate coverslips and expressed as the average and standard deviation, and at least 200 bacterial phagosomes or autophagosome and lysosome markers were analyzed per coverslip for each experiment.
Chromatin immunoprecipitation assay
A chromatin immunoprecipitation assay was performed according to the manufacturer's protocol (Upstate Biotechnology, 17–295). Cells were fixed with 37.5% formaldehyde (1% final; Sigma F8775) and cross-linked chromatin was sheared by sonication. Soluble chromatin was immunoprecipitated using a goat polyclonal anti-ESRRA antibody (Santa Cruz Biotechnology, sc-32972) and anti-goat IgG control antibody (Santa Cruz Biotechnology, 2028). After recovery of DNA, enrichment of ESRRA in Atg5, Becn1, and Atg16l1 was assessed by semiquantitative-PCR with specific primers. The sequences of the primers used for semiquantitative-PCR are shown in Table S1. Amplified products were resolved by electrophoresis in 2% agarose gels and visualized by ethidium bromide (Sigma, E1510) staining.
Luciferase reporter gene assays
RAW264.7 cells were transiently transfected with a luciferase gene under the control of the pGL3 basic promoter (Promega, E1751), or the ESRRA transcription recognition sequences were used. For luciferase reporter gene assays, Atg5, Becn1, Atg16l1 and Sirt1 promoter constructs were used. The Sirt1 promoter constructs was generated as described previously.Citation19 To produce the Atg5, Becn1, and Atg16l1 promoter constructs, a 5000-bp fragment upstream of the predicted transcription start site of each mouse gene was generated by PCR amplification from mouse genomic DNA, using the following primers: Atg5 (sense: 5′-AGGTTTAAGCGTGTTTCATAAAGTAA-3′ and antisense: 5′-GGCGCCCGGAGATGACGTCACCAAGA-3′), Becn1 (sense: 5′-CCTAGGACAGTGGTTCTCAATTTTCC-3′ and antisense: 5′-ACCAGAAGTGACGTCACGACGGGAG-3′), Atg16l1 (sense: 5′-ACTATTCCTCATCCCATACCTTCCCC-3′ and antisense: 5′-CTTCCCACACAGGGCACCCGGAGGCG-3′), and Searches for putative DNA-binding sites for transcription factors in the mouse Atg5, Becn1, and Atg16l1 5′-flanking region sequences were conducted using TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH.html) and MOTIF Search (http://motif.genome.jp). The reporter gene assay was performed as described previously. RAW264.7 cells were plated in 6-well culture plates and transfected with a firefly reporter vector (4 μg) and pRL vector expressing the Renilla luciferase gene (0.1 μg; Promega, E2241), together with the indicated expression plasmids (4 μg) using the Lipofectamine 2000 (Invitrogen, 11668027) according to the manufacturer's instructions. After 2 days of transfection, cells were lysed in Passive Reporter Lysis Buffer (Promega, E1910), and luciferase activity was measured. Firefly luciferase activity was normalized to that of Renilla luciferase.
Transmission electron microscopy
For transmission electron microscopy, BMDMs were washed with PBS, fixed with 3% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) containing 0.1% CaCl2 for 3 h, and post-fixed with 1% osmium tetroxide in 0.1 M sodium cacodylate buffer containing 0.1% CaCl2 for 2 h. After rinsing with cold distilled water, the cells were dehydrated slowly with ethanol series and propylene oxide at 4°C. The samples were embedded in Embed-812 and cured at 60°C for 36 h. Ultrathin sections (70 to 80 nm) were cut with a diamond knife and an ULTRACUT UC7 ultramicrotome (Leica, Austria, Wien), and mounted on formvar-coated slot grids. Sections were stained with 4% uranyl acetate for 10 min and lead citrate for 7 min. Sections were observed using a Tecnai G2 Spirit Twin transmission electron microscope (FEI Company, USA).
Flow cytometry analysis
Flow cytometry analysis of LC3B was performed as described previously.Citation16 AICAR or resveratrol-stimulated Esrra+/+ and esrra−/− BMDMs were washed in fixation and permeabilization solution (BD Biosciences, 554722) for 10 min. Cells were incubated with an anti-LC3B (Cell Signaling Technology, 2775) for 1 h and a secondary anti-rabbit IgG-Alexa Fluor 488 (Molecular Probes, A17041) for 30 min on ice. Finally, cells were washed in PBS, resuspended in FACS buffer and analyzed immediately. The samples were examined using a FACSCanto II flow cytometer (Becton Dickinson, San Jose, CA, USA). Flow cytometry data of 10,000 cells were collected and analyzed using the FlowJo software (Tree Star, Ashland, OR, USA).
Lentiviral shRNA generation and transduction
For silencing of target genes in BMDMs, the following pLKO.1-based lentiviral shRNA constructs were used: Esrra (GE Healthcare Dharmacon, TRCN0000026146, TRCN0000026187, and TRCN000002620), and Sirt1 (Santa Cruz Biotechnology, sc-40987-SH). Briefly, lentiviruses were produced by transient transfection using 3 packaging plasmids (pMDLg/pRRE [Addgene, 12251; deposited by Didier Trono], pRSV-Rev [Addgene, 12253; deposited by Didier Trono], and pMD2.G [Addgene, 8454; deposited by Bob Weinberg]) after Lipofectamine 2000-mediated transient transfection into HEK293T cells, as described previously.Citation19 After 72 h, lentivirus-containing supernatants were collected and several lentiviral vectors titrated, as described previously.Citation19
Lentivirus transduction was performed as described previously.Citation19 Briefly, BMDMs in medium containing 10% FBS were infected with lentiviral vectors in the presence of 8 µg/ml polybrene (Sigma, H9268). After 2 d, cells were harvested and target gene knockdown analyzed.
Tandem-tagged mCherry-EGFP-LC3B retrovirus generation and autophgic flux analysis
Generation of retroviruses encoding tandem-tagged mCherry-EGFP-LC3B were prepared, as described previously.Citation47 Human Phoenix AMPHO cells were seeded at 70% confluence into a 6-well plate and transfected with 1 μg of pBABE-puro mCherry-EGFP-LC3 (Addgene, 22418; deposited by Jayanta Debnath), 0.75 μg of pCL-Eco (Addgene, 12371; deposited by Inder Verma), and 0.25 μg of pDM2.G (Addgene, 12259; deposited by Didier Trono) using Lipofectamine 2000. After 18 h, transfected cell culture medium was removed and replaced with fresh medium. The retrovirus-containing medium was harvested at 60 h post-transfection and filtered through a 0.45 μm syringe filter. Autophagic flux analysis was performed as described previously.Citation47-Citation49 BMDMs were transduced with mCherry-EGFP-LC3B retrovirus for 24 h and stimulated with AICAR or resveratrol. The stimulated cells on coverslips were washed twice with PBS, fixed with 4% paraformaldehyde for 15 min. After mounting, fluorescence images were acquired using a confocal laser scanning microscope. To analyze endogenous autophgic flux, we used the ImageJ software. Slides were assayed in triplicate, and at least 200 cells per well were counted.
Transfection of plasmid DNA into RAW264.7 cells
RAW264.7 cell transfections were performed, as described previously.Citation50-Citation53 Prior to transfection, RAW 264.7 cells at 50% to 60% confluence were plated per 10-cm cell culture plate in DMEM medium supplemented with 10% FBS (Lonza, BW14–503E). One h before transfection, cells were washed with Opti-MEM (Invitrogen, 51985091). For RAW264.7 cell transfections, OptiMEM (500 μl) were mixed with each plasmid DNA (10 μg: pCruzHA-Sirt1) or Lipofectamine 2000 (25 μl; Invitrogen, 12566014), according to the manufacturer's protocol. After 5 min, the plasmid DNA mixture and Lipofectamine 2000 mixture were mixed gently and incubated for 25 min at room temperature. RAW264.7 cells were then transfected with plasmid DNA and Lipofectamine complex in each plate. After 4 to 6 h, transfected cell culture medium was removed and replaced with fresh medium.
Immunoprecipitation and immunoblotting
Immunoprecipitation (IP) was performed as described previously.Citation19 Briefly, cells were collected and lysed in RIPA buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 140 mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100; Sigma, R0278) containing a protease inhibitor cocktail (PI; Roche, REF11697498001). After preclearing with protein A agarose beads (GE Healthcare, 17–0780–01) for 30 min at 4°C, whole-cell lysates were subjected to IP with the antibodies: anti-acetylated-lysine, anti-FLAG, anti-HA and anti-SIRT1. Generally, 1 µg of antibody was added to 500 µg of cell lysate and incubated for 18 h at 4°C. After incubation with protein A agarose beads for 6 h, immunoprecipitates were extensively washed with lysis buffer and eluted with SDS loading buffer by boiling for 10 min. The immunoprecipitated proteins were subjected to immunoblotting analysis.
For immunoblotting, cells were lysed in RIPA buffer with PI cocktail at 4°C for 10 min, as described previously.Citation19 The protein extracts were boiled in SDS sample buffer, loaded onto 10% or 12% SDS-polyacrylamide gels for electrophoresis, and then transferred to polyvinylidene difluoride membranes (PVDF; Millipore, IPVH0001). Membranes were blocked in 5% nonfat milk in PBST (3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl, 135 mM NaCl, 0.05% Tween 20 [Sigma, P9416], pH 7.4) for 1 h and incubated with the following primary antibodies: anti-LC3A/B, anti-ATG5, anti-BECN1, anti-ATG7, anti-PPARGC1A, anti-ESRRA, anti-SIRT1, anti-FLAG, anti-HA, and anti-ACTB. These proteins were visualized by enhanced chemiluminescence using an ECL reagent (Millipore, WBKLS0500), and bands were detected using a Vilber chemiluminescence analyzer (Vilber Lourmat, Fusion SL 3, Eberhardzell, Germany).
RNA extraction, quantitative PCR, and enzyme-linked immunosorbent assays
RNA extraction, quantitative PCR (qPCR), and semiquantitative RT-PCR were performed as described previously.Citation16 Briefly, total RNA from cells was isolated using TRIzol reagent (Invitrogen, 15596026) and used for synthesis of cDNA with Superscript II reverse transcriptase (Invitrogen, 18064). qPCRs were performed using cDNA, primers, and SYBR Green PCR Kits (Qiagen, 204074) using the real-time PCR cycler Rotor-Gene Q 2plex system (Qiagen GmbH, 9001620, Hilden, Germany). The samples were amplified for 40 cycles as follows: 95°C for 10 sec and 60°C for 30 sec. To analyze qPCR data, we performed relative quantification using the 2ΔΔCt method with Gapdh as an internal control gene; data were expressed as relative fold changes. Primer sequences are provided in Supplemental materials Table S2.
TNF (558534), IL6 (555240), and IL1B (559603) protein levels were measured by sandwich ELISA using Duoset Ab pairs (BD PharMingen), according to the manufacturer's protocol.
Colony-forming unit assays
For quantification of intracellular bacteria, CFU assays were performed.Citation16 After Mtb infection, BMDMs or in vivo tissues were washed 3 times with PBS, and fresh medium containing 200 μg/ml amikacin (Sigma, A2324) was added. After incubation for various periods, cells were lysed with 0.3% saponin (Sigma, 47036) to release intracellular bacteria, and the lysates of infected samples were resuspended vigorously, transferred to screw-capped tubes, and sonicated in a preheated 37°C water bath sonicator (Elma, SH250EL, Germany, Singen) for 5 min. Aliquots of the sonicates were diluted 10-fold in 7H9 medium. Four dilutions of each sample were plated separately on 7H10 agar plates and incubated at 37°C in 5% CO2 for 14 to 21 d.
Statistical analysis
Data are presented as means ± standard deviation (SD). As a Shapiro-Wilk test indicated that the data were not normally distributed, nonparametric statistics were used. The Kruskal-Wallis test in SPSS for Windows (ver. 21.0; SPSS Inc.) was used to test for significant differences. Group-wise post-hoc comparisons were performed using the Mann-Whitney U-test and the Welch t test in SPSS if significant differences were evident. P < 0.05 was considered to indicate statistical significance.
Abbreviations
| AICAR | = | AMP-mimetic 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside |
| AMPK | = | AMP-activated protein kinase |
| ATG/Atg | = | autophagy-related |
| BCG | = | Mycobacterium bovis-Bacillus Calmette-Guérin |
| BECN1 | = | Beclin 1, autophagy related |
| BMDMs | = | bone marrow-derived macrophages |
| CFU | = | colony-forming unit |
| ERREs | = | ERR response elements |
| ESRRA | = | estrogen-related receptor α |
| Esrra+/+ | = | Esrra wild type |
| esrra−/− | = | Esrra homozygous knockout |
| GFP | = | green fluorescent protein |
| IL | = | interleukin |
| LAMP | = | lysosomal-associated membrane protein |
| MOI | = | multiplicity of infection |
| MAP1LC3/LC3 | = | microtubuleassociated protein 1 lightchain 3 |
| MAP1LC3B | = | microtubule-associated protein 1 light chain 3 β |
| Mtb | = | Mycobacterium tuberculosis |
| NR1H4 | = | nuclear receptor subfamily 1, group H, member 4 |
| NRs | = | nuclear receptors |
| PCR | = | polymerase chain reaction |
| PPAR | = | peroxisome proliferator activated receptor |
| PPARGC1A | = | peroxisome proliferative activated receptor, gamma, coactivator 1 α |
| SIRT1 | = | sirtuin 1 |
| TB | = | tuberculosis |
| TNF | = | tumor necrosis factor |
Disclosure of potential conflicts of interest
The authors declare that they have no competing financial interests.
KAUP_A_1339001_supplemental.doc
Download MS Word (2.4 MB)Acknowledgments
We thank Dr R. L. Friedman (Arizona University) for providing mycobacterial strains; Dr J. C. Jang (GyeongSang University) for providing ERFP-Mtb; Dr S.H. Koo (Korea University) for providing adenoviruses encoding PPARGC1A and plasmid; Dr T. Bae (Indiana University) for providing BABE-puro mCherry-EGFP-LC3 plasmid; Drs. D. M. Shin and H. S. Jin (Chungnam National University) for helpful discussions and critical reading of the manuscript. We also thank members of E-K Jo's laboratory for their helpful discussions and technical assistance.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NRF-2015M3C9A2054326 and 2017R1A5A2015385) at Chungnam National University and No. NRF-2016R1D1A1A02937312 at Hanyang University, by a grant of the Korea Healthcare Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant HI15C0395 and HI16C1653). This work was supported by National Creative Research Initiatives Grant (20110018305) through the National Research Foundation of Korea (NRF) funded by the Korean government (Ministry of Science, ICT & Future Planning) to HSC. This work was supported under the framework of international cooperation program managed by National Research Foundation of Korea 2015K2A2A6002008. This work was funded by the Canadian Institutes of Health Research (MOP-125885) to VG.
Related Research Data
References
- Evans RM, Mangelsdorf DJ. Nuclear receptors, RXR, and the Big Bang. Cell 2014; 157:255-66; PMID:24679540; https://doi.org/10.1016/j.cell.2014.03.012
- Giguère V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature 1988; 331:91-4; PMID:3267207; https://doi.org/10.1038/331091a0
- Deblois G, Giguère V. Functional and physiological genomics of estrogen-related receptors (ERRs) in health and disease. Biochim Biophys Acta 2011; 1812:1032-40; PMID:21172432; https://doi.org/10.1016/j.bbadis.2010.12.009
- Villena JA, Kralli A. ERRalpha: a metabolic function for the oldest orphan. Trends Endocrinol Metab 2008; 19:269-76; PMID:18778951; https://doi.org/10.1016/j.tem.2008.07.005
- Sonoda J, Laganière J, Mehl IR, Barish GD, Chong LW, Li X, Scheffler IE, Mock DC, Bataille AR, Robert F, et al. Nuclear receptor ERR alpha and coactivator PGC-1 beta are effectors of IFN-gamma-induced host defense. Genes Dev 2007; 21:1909-20; PMID:17671090; https://doi.org/10.1101/gad.1553007
- Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, Allen RD, Gluck SL, Heuser J, Russell DG. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 1994; 263:678-81; PMID:8303277; https://doi.org/10.1126/science.8303277
- Vergne I, Fratti RA, Hill PJ, Chua J, Belisle J, Deretic V. Mycobacterium tuberculosis phagosome maturation arrest: mycobacterial phosphatidylinositol analog phosphatidylinositol mannoside stimulates early endosomal fusion. Mol Biol Cell 2004; 15:751-60; PMID:14617817; https://doi.org/10.1091/mbc.E03-05-0307
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; https://doi.org/10.1038/nature09782
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; https://doi.org/10.1038/nature06639
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753-66; PMID:15607973; https://doi.org/10.1016/j.cell.2004.11.038
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253-7; PMID:18097414; https://doi.org/10.1038/nature06421
- Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee HM, Krutzik SR, Schenk M, Sieling PA, et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med 2011; 3:104ra2; https://doi.org/10.1126/scitranslmed.3003045
- Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012; 150:803-15; PMID:22901810; https://doi.org/10.1016/j.cell.2012.06.040
- Chandra V, Bhagyaraj E, Nanduri R, Ahuja N, Gupta P. NR1D1 ameliorates Mycobacterium tuberculosis clearance through regulation of autophagy. Autophagy 2015; 11:1987-97; PMID:26390081; https://doi.org/10.1080/15548627.2015.1091140
- Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, Lee ZW, Lee SH, Kim J M, Jo EK. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009; 6:231-43; PMID:19748465; https://doi.org/10.1016/j.chom.2009.08.004
- Yang CS, Kim JJ, Lee HM, Jin HS, Lee SH, Park JH, Kim SJ, Kim JM, Han YM, Lee MS, et al. The AMPK-PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy 2014; 10:785-802; PMID:24598403; https://doi.org/10.4161/auto.28072
- Huss JM, Torra IP, Staels B, Giguère V, Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol 2004; 24:9079-91; PMID:15456881; https://doi.org/10.1128/MCB.24.20.9079-9091.2004
- Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, Fernández-Ruiz S, Morciano G, Caro-Maldonado A, Guiu M, et al. The metabolic co-regulator PGC1α suppresses prostate cancer metastasis. Nat Cell Biol 2016; 18:645-56; PMID:27214280; https://doi.org/10.1038/ncb3357
- Yuk JM, Kim TS, Kim SY, Lee HM, Han J, Dufour CR, Kim JK, Jin HS, Yang C S, Park KS, et al. Orphan nuclear receptor ERRα controls macrophage metabolic signaling and A20 expression to negatively regulate TLR-induced inflammation. Immunity 2015; 43:80-91; PMID:26200012; https://doi.org/10.1016/j.immuni.2015.07.003
- Wilson BJ, Tremblay AM, Deblois G, Sylvain-Drolet G, Giguere V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol Endocrinol 2010; 24:1349-58; PMID:20484414; https://doi.org/10.1210/me.2009-0441
- Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt F W, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 2008; 105:3374-9; PMID:18296641; https://doi.org/10.1073/pnas.0712145105
- Sun T, Li X, Zhang P, Chen WD, Zhang HL, Li DD, Deng R, Qian XJ, Jiao L, Ji J, et al. Acetylation of beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun 2015; 6:7215; PMID:26008601; https://doi.org/10.1038/ncomms8215
- Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, Liu B, Chang C, Zhou T, Lippin cott-Schwartz J, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell 2015; 57:456-66; PMID:25601754; https://doi.org/10.1016/j.molcel.2014.12.013
- Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I, et al. The life span-prolonging effect of sirtuin-1 is mediated by autophagy. Autophagy 2010; 6:186-8; PMID:20023410; https://doi.org/10.4161/auto.6.1.10817
- Chaveroux C, Eichner LJ, Dufour CR, Shatnawi A, Khoutorsky A, Bourque G, So-nenberg N, Giguere V. Molecular and genetic crosstalks between mTOR and ERRalpha are key determinants of rapamycin-induced nonalcoholic fatty liver. Cell Metab 2013; 17:586-98; https://doi.org/10.1016/j.cmet.2013.03.003
- Sladek R, Beatty B, Squire J, Copeland NG, Gilbert DJ, Jenkins NA, Giguère V. Chromosomal mapping of the human and murine orphan receptors ERRalpha (ESRRA) and ERRbeta (ESRRB) and identification of a novel human ERRalpha-related pseudogene. Genomics 1997; 45:320-6; PMID:9344655; https://doi.org/10.1006/geno.1997.4939
- Lanvin O, Bianco S, Kersual N, Chalbos D, Vanacker JM. Potentiation of ICI182, 780 (Fulvestrant)-induced estrogen receptor-alpha degradation by the estrogen receptor-related receptor-alpha inverse agonist XCT790. J Biol Chem 2007; 282:28-34; https://doi.org/10.1074/jbc.M704295200
- Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, Dwyer M A, Nelson ER, Pollizzi KN, Ilkayeva O, et al. Estrogen-related receptor-α is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci U S A 2011; 108:18348-53; PMID:22042850; https://doi.org/10.1073/pnas.1108856108
- Hong EJ, Levasseur MP, Dufour CR, Perry MC, Giguere V. Loss of estrogen-related receptor alpha promotes hepatocarcinogenesis development via metabolic and inflammatory disturbances. Proc Natl Acad Sci U S A 2013; 110:17975-80; PMID:24127579; https://doi.org/10.1073/pnas.1315319110
- Huss JM, Garbacz WG, Xie W. Constitutive activities of estrogen-related receptors: Transcriptional regulation of metabolism by the ERR pathways in health and disease. Biochim Biophys Acta 2015; 1852:1912-27; PMID:26115970; https://doi.org/10.1016/j.bbadis.2015.06.016
- Shibutani ST, Saitoh T, Nowag H, Münz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol 2015; 16:1014-24; PMID:26382870; https://doi.org/10.1038/ni.3273
- Fan D, Liu SY, van Hasselt CA, Vlantis AC, Ng EK, Zhang H, Dong Y, Ng SK, Chu R, Chan AB, et al. Estrogen receptor α induces prosurvival autophagy in papillary thyroid cancer via stimulating reactive oxygen species and extracellular signal regulated kinases. J Clin Endocrinol Metab 2015; 100:E561-71; PMID:25594859; https://doi.org/10.1210/jc.2014-3257
- Lee JM, Wagner M, Xiao R1, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014; 516:112-5; PMID:25383539
- Zhou J, Zhang W, Liang B, Casimiro MC, Whitaker-Menezes D, Wang M, Lisanti MP, Lanza-Jacoby S, Pestell RG, Wang C. PPARgamma activation induces autophagy in breast cancer cells. Int J Biochem Cell Biol 2009; 41:2334-42; PMID:19563910; https://doi.org/10.1016/j.biocel.2009.06.007
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004; 305:390-2; PMID:15205477; https://doi.org/10.1126/science.1099196
- Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 2012; 15:675-90; PMID:22560220; https://doi.org/10.1016/j.cmet.2012.04.003
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004; 303:2011-5; PMID:14976264; https://doi.org/10.1126/science.1094637
- Giannakou ME, Partridge L. The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends Cell Biol 2004; 14:408-12; PMID:15308206; https://doi.org/10.1016/j.tcb.2004.07.006
- Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of foxo by sirt1 Plays an essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res 2010; 107:1470-82; PMID:20947830; https://doi.org/10.1161/CIRCRESAHA.110.227371
- Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem 2005; 280:16456-60; PMID:15716268; https://doi.org/10.1074/jbc.M501485200
- Zhang J, Ng S, Wang J, Zhou J, Tan SH, Yang N, Lin Q, Xia D, Shen HM. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015; 11:629-42; PMID:25919885; https://doi.org/10.1080/15548627.2015.1023981
- Wang B YQ, Sun YY, Xing YF, Wang YB, Lu XT, Bai WW, Liu XQ, Zhao YX. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med 2014; 18:1599-611; PMID:24889822; https://doi.org/10.1111/jcmm.12312
- Zumla A, Rao M, Wallis RS, Kaufmann SH, Rustomjee R, Mwaba P, Vilaplana C, Yeboah-Manu D, Chakaya J, Ippolito G, et al. Host-Directed Therapies Network consortium. Host-directed therapies for infectious diseases: current status, recent progress, and future prospects. Lancet Infect Dis 2016; 16:e47-63; PMID:27036359; https://doi.org/10.1016/S1473-3099(16)00078-5
- Wallis RS, Maeurer M, Mwaba P, Chakaya J, Rustomjee R, Migliori GB, Marais B, Schito M, Churchyard G, Swaminathan S, et al. Tuberculosis-advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect Dis 2016; 16:e34-46; PMID:27036358; https://doi.org/10.1016/S1473-3099(16)00070-0
- Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, Ko DC, Zou Y, Bang ND, Chau TT, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell 2012; 148:434-46; PMID:22304914; https://doi.org/10.1016/j.cell.2011.12.023
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; https://doi.org/10.1080/15548627.2015.1100356
- Kim JK, Lee HM, Park KS, Shin DM, Kim TS, Kim YS, Suh HW, Kim SY, Kim IS, Kim JM, et al. MIR144* inhibits antimicrobial responses against Mycobacterium tuberculosis in human monocytes and macrophages by targeting the autophagy protein DRAM2. Autophagy 2016; 20:1-19.
- Yoo BH, Zagryazhskaya A, Li Y, Koomson A, Khan IA, Sasazuki T, Shirasawa S, Rosen KV. Upregulation of ATG3 contributes to autophagy induced by the detachment of intestinal epithelial cells from the extracellular matrix, but promotes autophagy-independent apoptosis of the attached cells. Autophagy 2015; 11:1230-46; PMID:26061804; https://doi.org/10.1080/15548627.2015.1056968
- Sidjanin DJ, Park AK, Ronchetti A, Martins J, Jackson WT. TBC1D20 mediates autophagy as a key regulator of autophagosome maturation. Autophagy 2016; 12:1759-75; PMID:27487390; https://doi.org/10.1080/15548627.2016.1199300
- Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell 2008; 29:69-80; PMID:18206970; https://doi.org/10.1016/j.molcel.2007.11.002
- Ho PC, Tsui YC, Feng X, Greaves DR, Wei LN. NF-kappaB-mediated degradation of the coactivator RIP140 regulates inflammatory responses and contributes to endotoxin tolerance. Nat Immunol 2012; 13:379-86; PMID:22388040; https://doi.org/10.1038/ni.2238
- Hu D, Wu J, Xu L, Zhang R, Chen L. A method for the establishment of a cell line with stable expression of the GFP-LC3 reporter protein. Mol Med Rep 2012; 6:783-6; PMID:22825056
- Zhu GF, Yang LX, Guo RW, Liu H, Shi YK, Wang H, Ye JS, Yang ZH, Liang X. miR-155 inhibits oxidized low-density lipoprotein-induced apoptosis of RAW264.7 cells. Mol Cell Biochem 2013; 382:253-61; PMID:23797321; https://doi.org/10.1007/s11010-013-1741-4