
ABSTRACT
Macroautophagy/autophagy is a self-degradation process that combats starvation. Lipids are the main energy source in kidney proximal tubular cells (PTCs). During starvation, PTCs increase fatty acid (FA) uptake, form intracellular lipid droplets (LDs), and hydrolyze them for use. The involvement of autophagy in lipid metabolism in the kidney remains largely unknown. Here, we investigated the autophagy-mediated regulation of renal lipid metabolism during prolonged starvation using PTC-specific Atg5-deficient (atg5-TSKO) mice and an in vitro serum starvation model. Twenty-four h of starvation comparably induced LD formation in the PTCs of control and atg5-TSKO mice; however, additional 24 h of starvation reduced the number of LDs in control mice, whereas increases were observed in atg5-TSKO mice. Autophagic degradation of LDs (lipophagy) in PTCs was demonstrated by electron microscopic observation and biochemical analysis. In vitro pulse-chase assays demonstrated that lipophagy mobilizes FAs from LDs to mitochondria during starvation, whereas impaired LD degradation in autophagy-deficient PTCs led to decreased ATP production and subsequent cell death. In contrast to the in vitro assay, despite impaired LD degradation, kidney ATP content was preserved in 48-h starved atg5-TSKO mice, probably due to increased utilization of ketone bodies. This compensatory mechanism was accompanied by a higher plasma FGF21 (fibroblast growth factor 21) level and its expression in the PTCs; however, this was not essential for the production of ketone bodies in the liver during prolonged starvation. In conclusion, lipophagy combats prolonged starvation in PTCs to avoid cellular energy depletion.
Introduction
Although the kidney comprises only 0.5% of the total body weight, it utilizes approximately 10% of the oxygen consumed by the body.Citation1 The mitochondrial β-oxidation of nonesterified free fatty acids (FAs) is a major source for renal ATP production, particularly in the proximal tubule, which has a high energy demand and relatively little glycolytic capacity.Citation2-4 Under fasting conditions, plasma levels of adipose-derived FAs rise, and the kidney promotes the uptake of FAs from the circulationCitation5 as well as the retrieval of albumin-bound FAs from the glomerular filtrate.Citation6,7 FAs accumulate in the kidney during fasting in excess of levels used for oxidation, leading to their esterification with glycerol and deposition as triglycerides (TGs) in intracellular lipid droplets (LDs), which must be hydrolyzed for use thereafter.Citation7-9 LDs serve as an energy depot and segregate potentially toxic metabolites, as the excess of some intracellular FAs can disrupt phospholipid bilayer membrane integrity, evoke various stress signaling pathways including inflammasome activation,Citation10 and induce apoptosis.Citation11-13 These depots of stored energy can be rapidly degraded via the activity of lipases, as first suggested by ex vivo experiments in canine kidney slices.Citation14 Currently, 3 different neutral hydrolases (pH-optimum at around 7) are described as responsible for the intracellular lipolysis of TGs in LDs: PNPLA2/ATGL (patatin-like phospholipase domain containing 2), LIPE/HSL (lipase, hormone sensitive) and MGLL (monoglyceride lipase), which catalyze TGs, diacylglycerols (DGs), and MGs, respectively.Citation15 Recently another mode of lipolysis has emerged: the selective breakdown of LDs by autophagy (termed lipophagy). Autophagosomes selectively engulf part of the LDs in response to different nutritional cues, which regulate intracellular lipid stores in hepatocytes, enterocytes, and macrophages.Citation16-18 Although the process by which the cells utilize conventional lipolysis or lipophagy remains unclear, lipophagy is preferentially executed during periods of prolonged or acute starvation.Citation19 In addition, specialized cell types might differentially utilize lipases versus lipophagy for their response to starvation. For example, the release of FAs by lipophagy might be particularly important in cell types with low lipase activity, such as hepatocytes.Citation16,20
The observation that PNPLA2 and LIPE mRNA and protein expression are time-dependently increased by fasting in the kidneyCitation21 and that PNPLA2-deficiency leads to excessive lipid accumulation in the kidneyCitation22 suggests a substantial role for neutral lipases in kidney lipolysis; however, little is known about lipophagy in the kidney. We have previously generated kidney proximal tubular cell (PTC)-specific autophagy-deficient mice and demonstrated that enhanced autophagic activity in the proximal tubules has protective roles during pathological conditions and aging.Citation23-27 More recently, we have established methods for monitoring autophagic activity under basal conditions and metabolic stress in vivo by using GFP-MAP1LC3B (green fluorescent protein-microtubule-associated protein 1 light chain 3B) transgenic mice and drug-inducible PTC-specific autophagy-deficient mice.Citation27
Based on this background, we tested the hypothesis that lipophagy could be induced in kidney PTCs during fasting and aimed to elucidate the potential role of lipophagy in lipid metabolism, cellular function and survival in the kidney.
Results
Starvation promotes LD formation in the basolateral side of kidney proximal tubules
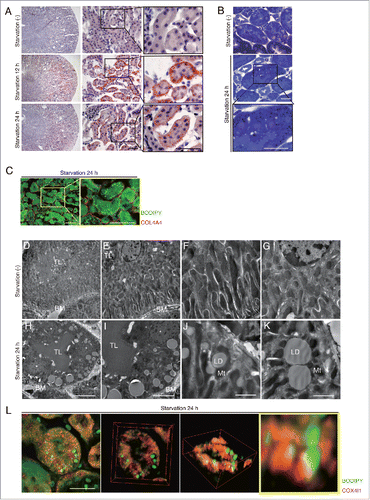
To examine LD formation in PTCs, we subjected 8-wk-old mice to starvation for up to 24 h. Oil Red O and toluidine blue staining, both of which can detect TGs, revealed increased LD formation in the kidney of 12- and 24-h starved mice ( and ). Boron-dipyrromethene (BODIPY) 493/503-labeled LDs were predominantly observed in the LRP2/MEGALIN (low density lipoprotein receptor-related protein 2)-positive PTCs of the starved mice, but not in the -negative (distal) tubular cells or glomeruli (Fig. S1A). Assessment with a confocal quantitative image cytometer, CQ1 (see “Materials and methods”), demonstrated that most LDs (79%) were located at the basolateral region (). Electron microscopy analysis ( to ), COX4I1 (cytochrome c oxidase subunit 4I1) staining (Fig. S1B), and 3-dimensional reconstructed images () revealed that most basolateral LDs exist adjacent to mitochondria. These data indicate that during starvation, free FAs (FFAs) taken up by PTCs are strictly trafficked to the basolateral region adjacent to mitochondria to form LDs.
Figure 1. Starvation promotes LDs formation in the basolateral side of kidney proximal tubules. LDs formation and its localization in the kidney were investigated in wild-type mice. (A and B) Images of Oil Red O- (A) and toluidine blue (B) -stained kidney sections of fed and 12- or 24-h starved mice (n = 6 to 9 in each group). Counterstaining was performed with hematoxylin (A). (C) Images of staining for COL4A4 (red) and BODIPY 493/503 (green) in 24-h starved mice. (D to K) Electron microscopy images of fed (D to G) and 24-h starved mice (H to K) (n = 3 in each group). BM, basement membrane; TL, tubular lumen; Mt, mitochondria; LD, lipid droplet; *, nucleus. (L) Three-dimensional reconstruction images of immunofluorescence staining for COX4I1 (red) and BODIPY 493/503 (green) in 24-h starved mice. Bars: 50 μm (A and C), 20 μm (B and L), 10 μm (H), 5 μm (I), and 2 μm (J and K). All images are representative of multiple experiments.
Starvation-induced autophagic flux peaks at 24 h in PTCs
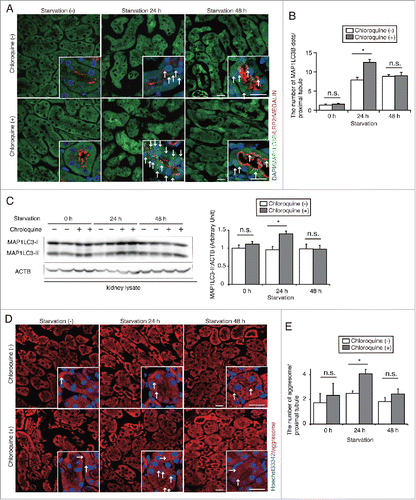
To assess alterations in autophagic flux in the kidney proximal tubules under starvation conditions, 8-wk-old GFP-MAP1LC3B transgenic mice, in which GFP-positive puncta reflect autophagosomes,Citation28 were subjected to starvation for up to 48 h. Some mice were administered chloroquine, a lysosomotropic reagent that inhibits intralysosomal acidification, 6 h before euthanasia.Citation27 GFP-positive puncta were rarely observed in the fed mice, whereas a substantial number of GFP-positive puncta were observed in the LRP2/MEGALIN-positive PTCs of 24- or 48-h starved mice without chloroquine treatment ( and ). Chloroquine administration significantly increased the number of GFP-positive puncta in 24-h starved mice, whereas the number remained unchanged in 48-h starved mice. Western blot analysis for MAP1LC3B using whole kidney lysates confirmed this fluorescence study (). These results demonstrate that starvation stimulates autophagic activity, which peaks at 24 h, in PTCs, followed by the downstream suppression of autophagic flux during prolonged starvation. We also evaluated the time course of protein clearance by autophagy under starvation in vivo. We visualized aggresome accumulation with the ProteoStat dye and counted the number of aggresomes in PTCs of fed and 24- or 48-h starved mice, with or without chloroquine administration ( and ). Chloroquine administration 6 h before euthanasia significantly increased the number of aggresomes in PTCs of 24-h starved mice, whereas the number remained unchanged in PTCs of fed or 48-h starved mice, suggesting that protein clearance peaks after 24 h of starvation.
Figure 2. Starvation-induced autophagic flux peaks at 24 h in PTCs. (A and B) Autophagic flux was assessed in the proximal tubules of GFP-MAP1LC3B transgenic that were either fed or subjected to 24 or 48 h of starvation, with or without chloroquine administration 6 h before euthanasia (n = 6 to 9 in each group). Kidney sections were immunostained for LRP2/MEGALIN, a marker of proximal tubules (red), and counterstained with DAPI (blue). Arrows indicate GFP- MAP1LC3B-positive puncta. (B) The number of GFP-positive puncta per proximal tubule under each condition was counted in at least 10 high-power fields (× 600). (C) Western blot analysis using whole kidney lysates of wild-type mice that were fed or subjected to 24 or 48 h of starvation with or without chloroquine administration (n = 5 to 6 in each group). Quantification by densitometry in protein levels of MAP1LC3-II is shown. Data are expressed as the fold change relative to the mean value of fed control mice without chloroquine administration. (D) Images of staining for protein aggregates detected by ProteoStat dye in the kidney of fed and 24- or 48-h starved mice with or without chloroquine administration 6 h before euthanasia (n = 3 to 4 in each group). Counterstaining was performed with Hoechst 33342 (blue). Arrows indicate protein aggregates. (E) The number of protein aggregates per proximal tubule under each condition was counted in at least 10 high-power fields (× 600). Bars: 20 μm. Images are representative of multiple experiments. Data are provided as mean ± SE. Statistically significant differences (*P < 0.05) are indicated. n.s., not significant.
Prolonged starvation induces lipophagy in PTCs
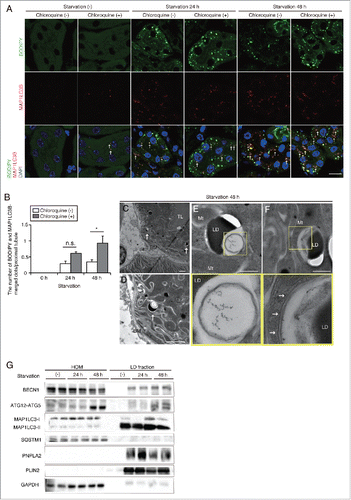
We then investigated whether lipophagy could be induced in PTCs. Starvation promoted colocalization of autophagosomal marker MAP1LC3B-positive dots and BODIPY 493/503-positive LDs in the kidney, hinting that lipophagy is induced during starvation in PTCs ( and ). Chloroquine administration 6 h before euthanasia significantly increased the number of MAP1LC3B- and BODIPY 493/503-merged dots in PTCs of 48-h starved mice, whereas the number remained unchanged in PTCs of 24-h starved mice ( and ). Moreover, colocalization of LAMP1 (lysosomal-associated membrane protein 1) and BODIPY 493/503 was detected in the kidney of 48-h starved mice (Fig. S2). Chloroquine administration increased both the number of BODIPY 493/503-positive dots and colocalization of LAMP1 and BODIPY 493/503-positive dots, indicating impaired LD degradation and subsequent accumulation in autolysosomes (Fig. S2). Electron microscopy analysis revealed the sequestration of regions of LDs in double-membrane organelles, autophagosomes ( to ), and the elongating phagophores along the surface of LDs (). Moreover, autophagosome component proteins such as BECN1/Beclin 1, ATG12–ATG5, and MAP1LC3-II were abundant in the LD fraction isolated from the kidneys of starved mice and were increased under prolonged starvation, whereas the autophagosome cargo protein SQSTM1/p62 was scarcely detected in the LD fraction (). The purity of the isolated LD fraction was confirmed by the enrichment in LD-associated protein PLIN2/ADRP (perilipin 2) and the absence of the cytosolic protein GAPDH (glyceraldehyde-3-phosphate dehydrogenase). Collectively, these results indicate that lipophagy is induced in kidney proximal tubules by prolonged starvation.
Figure 3. Prolonged starvation induces lipophagy in the kidney proximal tubules. Lipophagy in the PTCs was investigated immunohistochemically, morphometrically and biochemically. (A and B) Colocalization of LDs and autophagosomes was assessed in the proximal tubules of fed and 24- or 48-h starved mice, with or without chloroquine administration 6 h before euthanasia (n = 5 in each group). (A) Kidney sections were stained for MAP1LC3B (red) and BODIPY 493/503 (green), and counterstained with DAPI (blue). Arrows indicate MAP1LC3B dots colocalized with BODIPY 493/503. (B) The number of BODIPY and MAP1LC3B-merged dots per proximal tubule under each condition was counted in at least 10 high-power fields (× 600). Data are provided as mean ± SE. Statistically significant differences (*P < 0.05) are indicated. n.s., not significant. (C to F) Electron microscopy analysis of kidney sections in 48-h starved mice. Arrows indicate double-membrane autophagic vacuoles containing only lipids (C to E) and elongating phagophores along the surface of cytosolic LDs (F) (n = 3 in each group). (E and F) Magnified images are presented in the insets. BM, basement membrane; TL, tubular lumen; Mt, mitochondria; LD, lipid droplet. Bars: 10 μm (A), 5 μm (C and D) and 500 nm (E and F). (G) Immunoblots images using homogenates (HOM) and isolated LDs from the kidneys of fed and 24- or 48-h starved mice (n = 5 to 6 in each group). All images are representative of multiple experiments.
Lipophagy mobilizes FAs from LDs to mitochondria during starvation in vitro
We next examined FFA mobilization following lipophagy in an in vitro model using PTCs loaded with oleic acid (OA)-bound albumin to mimic and magnify the LD-forming effect of FFAs. PTCs were initially loaded with OA (250 μM) for 12 h, followed by starvation with serum-limited medium (0.1% fetal bovine serum [FBS]) for 12 h. OA treatment promoted LD formation without an increase in MAP1LC3B dots (Fig. S3A). The subsequent 12 h of starvation decreased the size and number of LDs, and increased the number of MAP1LC3B dots, some of which colocalized with LDs, suggesting the in vitro recapitulation of starvation-induced lipophagy (Fig. S3A, arrow), although juxtalocalization was more likely than true colocalization. Electron microscopy analysis revealed the presence of elongating phagophores along the surface of LDs (Fig. S3B, arrow) and the engulfment of LDs in autolysosome-like structures after 6 h of starvation (Fig. S3B, lower panel).
Next, to explore the trafficking of FFAs during and after lipophagy under a starved condition, we performed a pulse-chase assay by using BODIPY 558/568 C12 (Red C12), a saturated FA analog with a tail composed of 12 carbons and a BODIPY 558/568 fluorophore covalently bound at the hydrophobic end, with an overall length approximately equivalent to that of an 18-carbon FA.Citation19 First, PTCs were loaded with OA (250 μM) and Red C12 (1 μM) for 12 h and were labeled with BODIPY 493/503. The dot-like distribution of Red C12 was almost completely merged with BODIPY 493/503, suggesting that most of the Red C12 was incorporated into LDs (Fig. S3C). Next, cultured PTCs stably expressing GFP-MAP1LC3B were loaded with OA (250 μM) and Red C12 (1 μM) for 12 h, and then chased in serum-limited medium in the absence of Red C12 for up to 24 h, followed by immunostaining for LAMP1 (). In OA-treated PTCs (0 h), little Red C12 signal was observed in autophagosomes or lysosomes (). Six h of starvation induced the autophagosomal localization of Red C12 from 7% at 0 h to 12% and the ratio was maintained largely constant throughout the study. The lysosomal localization of Red C12 increased from 7% at 0 h, to 9% at 6 h and up to 15% at 12 h. Red C12 signal negative for GFP-MAP1LC3B and LAMP1 decreased from 85% at 0 h to 70% at 12 h, and then rebounded to 74% at 24 h. The distribution of Red C12 was dot-like at 0 h, while Red C12 was distributed reticularly throughout the entire cytosol after 24 h of starvation ( and B).
Figure 4. Lipophagy mobilizes FAs from LDs to mitochondria during starvation in vitro. FA mobilization following lipophagy in starved cultured PTCs was investigated by a pulse-chase assay using Red C12, fluorescent FA analog. (A) Schematic representation of the pulse-chase assay. PTCs stably expressing GFP-MAP1LC3B were supplied with 250 μM OA and 1 μM of Red C12 for 12 h, and chased with serum-limited medium (0.1% FBS) for the indicated periods of time and the subcellular localization of Red C12 was determined. Lysosomes were stained with LAMP1 (white) and counterstained with DAPI (blue) (n = 5 in each group). (B) A bar graph of Red C12 localization is shown. The following groups of Red C12 are expressed as percentage of total Red C12 area for each condition: Red C12 negative for GFP-MAP1LC3B and LAMP1, Red C12 positive for GFP-MAP1LC3B or LAMP1, and Red C12 positive for both GFP-MAP1LC3B and LAMP1. (C) OA- and Red C12-treated autophagy-competent or -deficient PTCs with or without 24 h of starvation with serum-limited medium (0.1% FBS) were stained with MitoTracker Green FM (n = 5 in each group). Colocalization of Red C12 and mitochondria was assessed by the Pearson correlation. Bars: 10 μm. All images are representative of multiple experiments. Data are provided as mean ± SE. Statistically significant differences (*P < 0.05) are indicated. n.s., not significant. Atg5+, autophagy-competent PTC; atg5−/−, autophagy-deficient PTC.
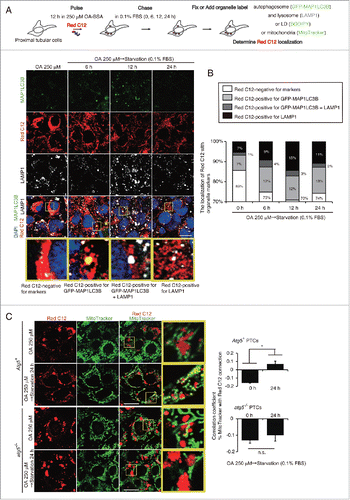
To clarify that autophagy induces the redistribution of FAs from LDs into mitochondria, we subjected 2 cultured cell lines, autophagy-deficient PTCs and genetically reverted autophagy-competent cells, to 24 h of starvation. MitoTracker Green FM staining in OA-treated autophagy-competent PTCs demonstrated that the reticular Red C12 signal was merged with mitochondria after 24 h of starvation (). In contrast, colocalization of mitochondria and Red C12 after starvation was diminished in OA-treated autophagy-deficient PTCs. The Pearson correlation coefficient confirmed this finding (, right). Together, starvation-induced lipophagy leads to the redistribution of FAs from LDs into mitochondria in cultured PTCs for lipid β-oxidation.
Lipophagy produces ATP for cell survival in the proximal tubules during prolonged starvation
To confirm that autophagy is involved in the degradation of LDs, we subjected autophagy-deficient and -competent PTCs to starvation. Both cell lines were initially loaded with OA (250 μM) and Red C12 (1 μM) for 12 h, then cultured in serum-limited medium for 12 or 24 h followed by labeling with BODIPY 493/503. Western blot analysis of MAP1LC3B confirmed that OA treatment and the following starvation by serum-limited medium stimulated autophagic activity, which peaked at 12 h, followed by the suppression of autophagic flux after 24 h of starvation (Fig. S4A). LD content (BODIPY-positive area) significantly decreased after 24 h of starvation in autophagy-competent PTCs, but not in autophagy-deficient PTCs ( and ). Electron microscopy analyses confirmed the impaired LD degradation and an accumulation of enlarged LDs in autophagy-deficient PTCs, whereas lipophagy-like structures in autophagy-competent PTCs was observed (Fig. S4B, arrow).
Figure 5. Lipophagy produces ATP for cell survival in the proximal tubules during starvation in vitro. Effects of starvation-induced lipophagy on cellular energy homeostasis and cell survival were studied. (A and B) Autophagy-competent and -deficient PTCs were supplied with 250 μM OA containing Red C12 for 12 h and then incubated with serum-limited medium (0.1% FBS) for the indicated periods of time. LDs were labeled with BODIPY 493/503. BODIPY 493/503-positive LD content per cell was quantified in the indicated conditions. Counterstaining was performed with DAPI (blue) (n = 5 in each group). (C to E) Autophagy-competent and -deficient PTCs were supplied with 250 μM OA for 12 h and then incubated with HBSS with or without 40 μM etomoxir for the indicated periods of time (n = 3 to 5 in each group). (C) Images of Oil Red O staining. (D and E) Cellular ATP level (D) and cell survival rate (MTS assay) (E) were measured. (B and E) Data are expressed as the fold change relative to the mean value of control PTCs. Bars: 10 μm (A) and 50 μm (C). All images are representative of multiple experiments. Data are provided as mean ± SE. Statistically significant differences (*P < 0.05 vs. autophagy-competent cells of the corresponding condition; #P < 0.05 vs. control PTCs) are indicated. Atg5+, autophagy-competent PTC; atg5−/−, autophagy-deficient PTC.
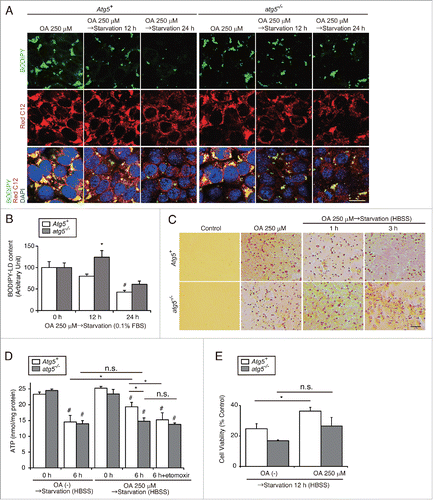
Three-dimensional reconstructed images revealed that the adjacent localization of LDs and mitochondria was disturbed in autophagy-deficient PTCs (Fig. S4C). To examine whether impaired degradation of LDs in autophagy-deficient PTCs could lead to a decrease in cellular ATP levels and cell survival, OA-treated autophagy-deficient and -competent PTCs were subjected to more severe starvation with Hank's balanced salt solution (HBSS). Oil Red O staining revealed that LDs gradually decreased over time in autophagy-competent PTCs, whereas LDs remained unchanged in autophagy-deficient PTCs (). Starvation with HBSS significantly decreased cellular ATP levels in both autophagy-competent and -deficient PTCs in the absence of OA treatment (). Conversely, the starvation-induced decrease in ATP levels was alleviated when autophagy-competent PTCs were pretreated with OA. This alleviating effect of OA was not observed in autophagy-deficient PTCs and was diminished when autophagy-competent PTCs were treated with etomoxir, a specific inhibitor of CPT1A (carnitine palmitoyltransferase 1A) (which leads to an inhibition of lipid β-oxidation), indicating that starvation-induced LD degradation by autophagy contributes to ATP production (). Consistently, starvation with HBSS significantly decreased cell survival in both PTCs, while OA-pretreatment significantly promoted cell survival in autophagy-competent PTCs, while not in autophagy-deficient PTCs (). This trend was confirmed by lactate dehydrogenase (LDH) assay and terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining (Fig. S4D and S4E). Taken together, lipophagy produces ATP for cell survival during starvation.
Autophagy-deficiency impairs LD degradation under prolonged starvation in the proximal tubules in vivo
We next examined the role of autophagy in the kidney during starvation using PTC-specific Atg5-deficient mice (referred to as atg5-tissue specific knockout mice, atg5-TSKO). Eight-wk-old mice were subjected to starvation for up to 48 h. Although Oil Red O staining demonstrated that 24 h of starvation comparably induced LD formation in PTCs of atg5-TSKO and Atg5F/F control (referred to as atg5F/F-CTRL) mice, after 48 h of starvation, the number and size of LDs in atg5F/F-CTRL littermates were decreased, whereas excessive LD accumulation was observed in atg5-TSKO mice ( and ). Although kidney TG content after 24 h of starvation was comparable between atg5-TSKO and atg5F/F-CTRL mice, after a subsequent 24 h of starvation, it was significantly greater in atg5-TSKO mice than atg5F/F-CTRL littermates (), and Plin2 mRNA levels were in agreement with the LD content (). These data demonstrate that autophagy plays an important role in degrading LDs in PTCs during prolonged starvation and confirm the data from mice using chloroquine administration ( and ).
Figure 6. Autophagy-deficiency impairs LD degradation under prolonged starvation in the proximal tubules in vivo. Kidneys of atg5-TSKO and atg5F/F-CTRL mice, that were fed or starved for up to 48 h, were analyzed for the LD degradation (A to C, n = 10 to 14; D, n = 7 to 9; E, n = 5 to 6; F to I, n = 3 in each group). (A) Images of Oil Red O-stained kidney sections. Counterstaining was performed with hematoxylin. (B) Quantification of the Oil Red O staining. (C to E) TGs level per kidney weight (gKW) (C) and mRNA expression level of Plin2 in the kidney (D), immunoblots of the indicated molecules using homogenates (HOM) and isolated LD fraction from the whole kidney (E), and images of toluidine blue-stained kidney sections (F) are shown. (G to I) Transmission electron microscopy images of 48-h starved atg5-TSKO mice. BM, basement membrane. (D) Data are expressed as the fold change relative to the mean value of fed atg5F/F-CTRL mice. Bars: 50 μm (A), 10 μm (F and G), 5 μm (H), and 2 μm (I). All images are representative of multiple experiments. Data are provided as mean ± SE. Statistically significant differences (*P < 0.05 vs. 48-h starved atg5F/F-CTRL mice; #P < 0.05 vs. fed mice) are indicated. CTRL, atg5F/F-CTRL mice; TSKO, atg5-TSKO mice.
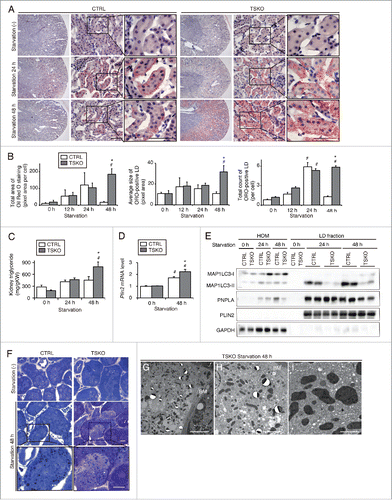
To investigate whether autophagy-deficiency affects cytosolic lipase activity, we next evaluated the protein levels of PNPLA2 as well as MAP1LC3-II in the LD fractions isolated from the kidneys of starved atg5-TSKO and atg5F/F-CTRL mice. Forty-eight-h starved atg5-TSKO mice displayed reductions in protein levels of PNPLA2 as well as MAP1LC3-II in the LD fraction compared with atg5F/F-CTRL littermates (). PNPLA2 localization around the surface of BODIPY 493/503-labeled LDs was disturbed in starved atg5-TSKO mice compared with starved atg5F/F-CTRL littermates (Fig. S5A). Moreover, PNPLA2 mRNA and protein expression were significantly lower in the 48-h starved atg5-TSKO mice compared with atg5F/F-CTRL littermates (Fig. S5B and S5C). We next investigated the effect of cytosolic lipase activity on LD degradation in autophagy-competent and -deficient PTCs in vitro. Treatment with atglistatin, a PNPLA2 inhibitor, significantly impaired LD degradation in autophagy-competent PTCs, whereas it did not affect LD degradation in autophagy-deficient PTCs, indicating that cytosolic lipase contributes LD degradation to some extent during prolonged starvation in autophagy-dependent manner (Fig. S5D). These findings indicate that lipophagy and PNPLA2 cooperatively contribute to lipolysis during starvation.
The characteristic basolateral distribution of LDs in PTCs was disturbed in atg5-TSKO mice, as assessed by toluidine blue staining (), and the percentage of LDs localized in the basolateral region, as quantified by CQ1, was significantly lower in atg5-TSKO mice compared with atg5F/F-CTRL littermates (69% versus 79%; P < 0.05). Similarly, the adjacent localization between LDs and mitochondria in PTCs was disturbed in atg5-TSKO mice, as indicated in electron microscopy analysis ( to ), COX4I1 staining (Fig. S5E), and 3-dimensional reconstructed images (Fig. S5F). These findings show that autophagy-deficiency impairs LD degradation as well as FA trafficking in proximal tubules during prolonged starvation.
Furthermore, we performed comparative lipidomic analysis of whole kidneys from fed or 48-h starved atg5F/F-CTRL and atg5-TSKO mice to analyze the impact of starvation-induced lipophagy on the qualitative and quantitative changes in the phospholipid composition of the kidneys. Total amount of phosphatidylcholine (PC) was significantly higher in 48-h starved atg5-TSKO mice compared with starved atg5F/F-CTRL littermates, suggesting that some kinds of PC are substrates of starvation-induced autophagy including lipophagy (Fig. S6A). To further investigate what kinds of phospholipids undergo lipophagy, we then analyzed the changes in lipid species composition and found that the content of unsaturated PCs such as PC 34:3, 36:5, and 38:2 was significantly lower in atg5F/F-CTRL mice than that in atg5-TSKO mice after 48 h of starvation (Fig. S6B and S6C). These results were consistent with the previous report showing that PC, especially with an unsaturated acyl chain, is the most abundant phospholipid present in LDs relative to total membrane.Citation29 Total amount of phosphatidylinositol (PtdIns) and lysophosphatidylcholine (LPC) significantly decreased in atg5-TSKO mice after 48 h of starvation, but not in atg5F/F-CTRL littermates (Fig. S6A and S6B).
Impairment of lipid utilization induced by autophagy-deficiency during prolonged starvation is compensated for by ketone bodies
Next, we examined the impact of failure of LD degradation in autophagy-deficient kidney on whole-body energy metabolism during prolonged starvation. FFAs derived from LDs activate PPARA (peroxisome proliferator activated receptor α) and downstream β-oxidation pathways.Citation22 Immunohistochemical analysis revealed that the intensity of PPARA staining increased in the LRP2/MEGALIN-positive PTCs of the 48-h starved atg5F/F-CTRL littermates, whereas it decreased in the 48-h starved atg5-TSKO mice (). We analyzed mRNA levels of lipid β-oxidation- and mitochondrial oxidative phosphorylation (OXPHOS)-related genes in the whole kidney ( and ). During starvation, mRNA levels of enzymes related to lipid β-oxidation, such as acyl-Coenzyme A oxidase 1 (Acox1/Aox), acyl-Coenzyme A dehydrogenase, C-4 to C-12 straight chain (Acadm/Mcad), were significantly lower in the 48-h starved atg5-TSKO mice compared with atg5F/F-CTRL littermates (). Consistent with these observations, mRNA levels of enzymes related to OXPHOS, such as Ndufb5 (NADH dehydrogenase [ubiquinone] 1 β subcomplex 50), and Uqcrb (ubiquinol-cytochrome c reductase binding protein), were preserved in the 48-h starved atg5F/F-CTRL littermates, whereas they were significantly suppressed in the 48-h starved atg5-TSKO mice (). Unexpectedly, the ATP content in the kidney remained unchanged in the 24- or 48-h starved atg5-TSKO mice compared with starved atg5F/F-CTRL littermates (), which prompted us to investigate possible compensatory mechanisms for impaired FFAs utilization in atg5-TSKO mice.
Figure 7. Impairment of lipid utilization induced by autophagy-deficiency under prolonged starvation is compensated by ketone bodies. Metabolic and energetic changes of atg5F/F-CTRL and atg5-TSKO mice, that were fed and starved for up to 48 h, were investigated using plasma and organs including kidney and liver (n = 7 to 9 in each group). (A) Representative images of immunostaining of PPARA in the kidney cortical regions. Kidney sections were immunostained for the proximal tubule marker LRP2/MEGALIN in blue. (B to E) mRNA expression levels of genes related to lipid β-oxidation (B) and to mitochondrial oxidative phosphorylation (C), ATP content per protein in the kidney (D), and plasma ketone body concentration (E) are shown. (F) mRNA expression level of Hmgcs2 in the liver. (G) Representative images of immunostaining for PPARΑ in the liver. Tissue sections were counterstained with hematoxylin. (H) mRNA expression levels of genes related to lipid β-oxidation in the liver. (B, C, F and H) Data are expressed as the fold change relative to the mean value of fed atg5F/F-CTRL mice. Cpt1a, carnitine palmitoyltransferase 1a, liver; Acads, acyl-Coenzyme A dehydrogenase, short-chain; Acadl, acyl-Coenzyme A dehydrogenase, long-chain; Acadvl, acyl-Coenzyme A dehydrogenase, very long chain. Bars: 50 μm (A) and 100 μm (G). All images are representative of multiple experiments. Data are provided as mean ± SE. Statistically significant differences (*P < 0.05 vs. 48-h starved atg5F/F-CTRL mice; #P < 0.05 vs. fed mice) are indicated. n.s., not significant. CTRL, atg5F/F-CTRL mice; TSKO, atg5-TSKO mice.
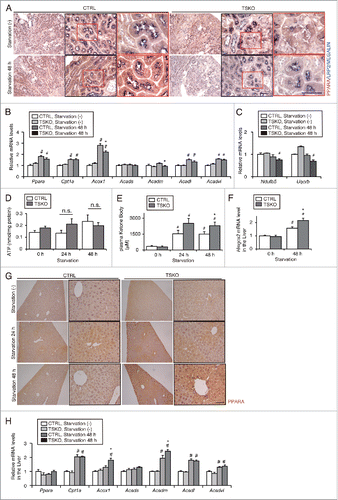
Metabolic profiles in fed or starved atg5-TSKO and atg5F/F-CTRL mice are shown in . Of note, plasma ketone body levels were significantly higher in atg5-TSKO mice under the 48-h starved condition, whereas plasma cholesterol and phospholipid levels were rather lower in 48-h starved atg5-TSKO mice and there were no significant differences in body weight, plasma glucose, FFAs, TGs ( and ). Consistent with the higher plasma ketone body levels in 48-h starved atg5-TSKO mice, the mRNA levels of Hmgcs2 (3-hydroxy-3-methylglutaryl-coenzyme A synthase 2), which is the rate-limiting enzyme for ketogenesis, was significantly higher in the liver of 48-h starved atg5-TSKO mice (). Ketone bodies are produced exclusively in the liver, mainly from β-oxidation of FAs generated by lipolysis in adipose tissue, during prolonged starvation, and are then delivered to peripheral organs, converted to acetyl-CoA and used as an energy source via the tricarboxylic acid (TCA) cycle.Citation30 Immunohistochemical analysis revealed that the intensity of PPARA staining increased in the liver of the 48-h starved atg5-TSKO mice (). mRNA levels of the lipid β-oxidation pathway modulators, such as Acox1 and Acadm were significantly higher in the liver of 48-h starved atg5-TSKO mice (). These findings indicate that impaired FFA utilization due to lipophagy-deficiency in atg5-TSKO mice is compensated for by the production of ketone bodies in the liver.
Table 1. Metabolic profiles in fed or starved atg5-TSKO and atg5F/F-CTRL mice.
Increased secretion of FGF21 (fibroblast growth factor 21) from autophagy-deficient kidney is associated with increased ketone bodies during prolonged starvation
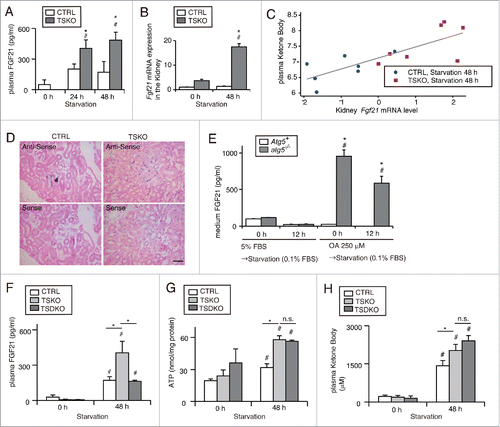
A recent report reveals that mitochondrial dysfunction due to skeletal muscle-specific deletion of Atg7 promotes FGF21 secretion from skeletal muscle, which protects against diet-induced obesity and insulin resistance.Citation31 FGF21 is known to promote lipid β-oxidation and ketogenesis in the liver.Citation32 Inspired by these reports, we postulated that FGF21 secreted from the kidney of atg5-TSKO mice could promote lipid β-oxidation and ketogenesis in the liver. Indeed, the plasma FGF21 concentration was significantly higher in atg5-TSKO mice compared with atg5F/F-CTRL littermates under starved conditions (). To identify the source of plasma FGF21 in atg5-TSKO mice, Fgf21 mRNA levels were determined in each organ. The Fgf21 mRNA levels were significantly higher in the kidney of 48-h starved atg5-TSKO mice compared with atg5F/F-CTRL littermates, whereas no significant differences were observed in the Fgf21 mRNA levels of the liver, adipose tissue, pancreas, and skeletal muscle ( and Fig. S7A). Importantly, the levels of Fgf21 mRNA in the kidney were significantly correlated with plasma ketone body levels (). In situ hybridization corroborated the higher Fgf21 expression in PTCs of atg5-TSKO mice compared with atg5F/F-CTRL littermates under the 48-h starved condition (). In addition, to elucidate whether autophagy-deficiency induces the expression and secretion of FGF21 in a cell-autonomous manner in vitro, we assessed FGF21 concentrations in the medium of autophagy-deficient and -competent PTCs. OA administration with or without starvation caused a significant elevation in FGF21 secretion and protein expression, exclusively in autophagy-deficient PTCs ( and Fig. S7B), and this trend was confirmed in primary cultured PTCs (Fig. S7C). These results confirmed the FGF21 expression and secretion from PTCs in atg5-TSKO mice. Finally, to determine that increased secretion of FGF21 from the kidney of atg5-TSKO mice actually promotes β-oxidation and ketogenesis in the liver, we generated PTC-specific Fgf21- and Atg5-deficient mice (fgf21, atg5-TSKO mice). The increased plasma level of FGF21 after 48-h starvation was attenuated in fgf21, atg5-TSKO mice compared with Fgf21+/+, atg5-TSKO mice (). However, the compensated ATP content in the kidney, the increased plasma ketone body levels, activated β-oxidation, and ketogenesis in the liver of 48-h starved Fgf21+/+, atg5-TSKO mice were not abolished in 48-h starved fgf21, atg5-TSKO mice ( and ; Fig. S7D and S7E). Taken together, these results demonstrate that increased kidney FGF21 expression via autophagy-deficiency is associated with increased ketone bodies, but is not essential for the production of ketone bodies in the liver during prolonged starvation.
Figure 8. Increased secretion of FGF21 from autophagy-deficient kidney is associated with increased ketone bodies under prolonged starvation. FGF21 expression in the PTCs in response to autophagy-deficiency was investigated in vivo and in vitro. (A to C) Plasma concentration (A) and mRNA expression level in the kidney (B) of FGF21 in fed and 24- or 48-h starved atg5-TSKO and atg5F/F-CTRL mice (n = 10 to 14 (A), 7 to 9 (B) in each group). (B) Data are expressed as the fold change relative to the mean value of fed atg5F/F-CTRL mice. (C) Correlation between plasma ketone body concentration and Fgf21 mRNA expression level in the kidney on log-scale (n = 14; R2 = 0.622; P < 0.001, as determined by linear regression analysis). (D) Representative images of in situ hybridization of Fgf21 in the kidney of 48 h starved atg5F/F-CTRL and atg5-TSKO mice. Bars: 50 μm. CTRL, atg5F/F-CTRL mice; TSKO, atg5-TSKO mice. (E) Medium FGF21 concentration in autophagy-competent and -deficient PTCs that were incubated with 5% FBS or 250 μM OA for 12 h followed by starvation with 0.1% FBS for 12 h (n = 5 in each group). Atg5+, autophagy-competent PTC; atg5−/−, autophagy-deficient PTC. (F to H) Plasma FGF21 concentration (F), ATP per protein amount in the kidney (G) and plasma ketone body concentration (H) in atg5F/F or fgf21F/F-CTRL, Fgf21+/+atg5-TSKO and fgf21, atg5-TSKO mice under fed or 48-h starved condition (n = 5 in each group). CTRL, atg5F/F or fgf21F/F-CTRL mice; TSKO, Fgf21+/+atg5-TSKO mice; TSDKO, fgf21, atg5-TSKO mice. Data are provided as mean ± SE. Statistically significant differences (*P < 0.05 vs. atg5F/F-CTRL mice under the corresponding condition (A and B); *P < 0.05 vs. autophagy-competent cells of the corresponding condition (E); #P < 0.05 vs. fed mice (A and B, F to H); #P < 0.05 vs. control PTCs (E)) are indicated. n.s., not significant.
Discussion
In this study, we elucidated the adaptive role of lipophagy in kidney proximal tubules in regulating energy homeostasis during prolonged starvation. The main results are: 1) during starvation, FFAs taken by PTCs are strictly trafficked to the basolateral region adjacent to mitochondria to form LDs, which allows FFAs released from LDs to be used as an energy source readily and safely without leaking out to the cytosol, 2) lipophagy induced by prolonged starvation in PTCs mobilizes FFAs from LDs to mitochondria for lipid β-oxidation, 3) lipophagy produces ATP for cell survival by breaking down LDs for β-oxidation, 4) impaired FFA utilization due to lipophagy-deficiency is compensated for by the production of ketone bodies in the liver.
We found that conventional autophagy and lipophagy have different cycles of phases: overall autophagic flux has its peak at 24 h during starvation, and prolonged starvation leads to downstream suppression. In contrast, the activity of lipophagy continues even after the downstream suppression of overall autophagy, which is in agreement with the previous observation that lipophagy is activated during longer periods of starvation.Citation16 It is reasonable that lipophagy can provide FFAs that are used by PTCs as a more efficient and essential energy fuel compared with amino acids. Although what regulates this “class switch of autophagy” is unclear, transcription factors such as PPARA, CREB (cAMP responsive element binding protein), or TFEB (transcription factor EB) are responsible for the coordination of this process.Citation33-35 To examine whether lipophagy involves MTORC1 (mechanistic target of rapamycin [serine/threonine kinase] complex 1)-mediated conventional autophagy induction, we assessed the time dependent change in the activity of MTORC1 and the nuclear translocation of TFEB during starvation. Suppression of MTORC1 activity (assessed by phosphorylated-[p-]RPS6 [ribosomal protein S6] and p-RPS6KB/p70S6K [ribosomal protein S6 kinase]) and the nuclear translocation of TFEB were observed after 24 h of starvation and persisted for an additional 24 h (Fig. S8). The mechanisms underlying the molecular switch between conventional autophagy and lipophagy remain to be elucidated.
The previous observation that PNPLA2 and LIPE expression are time-dependently increased by fasting in the kidneyCitation21 and that genetic ablation of PNPLA2 leads to excessive lipid accumulation in the kidney,Citation22 suggests a substantial role for neutral lipases in kidney lipolysis. Indeed, PNPLA2 inhibition significantly impaired LD degradation in autophagy-competent PTCs (Fig. S5D). It remains to be elucidated how cells utilize 2 modes of lipolysis (neutral lipase and lipophagy) depending on the condition. It is easy to infer that lipophagy, which will allow cells to obtain energy in bulk, will be preferred during prolonged starvation. In addition, there seems to be crosstalk between neutral lipase and lipophagy. For example, MAP1LC3-II supports PNPLA2-mediated lipolysis, possibly by serving as scaffolds regulating the recruitment and activity of PNPLA2.Citation36 Conversely, neutral lipase PNPLA5 (patatin-like phospholipase domain containing 5), which localizes to LDs, is required for optimal initiation of autophagy.Citation37 Indeed, the association of PNPLA2 around the surface of LDs was disturbed in starved atg5-TSKO mice in our model (Fig. S5A). In addition, disarranged (loss of basolateral polarity and adjacency to mitochondria) and enlarged LDs (; Fig. S4B and S4C) may suggest that autophagy-deficiency leads to the formation of altered “bad” LDs that neutral lipase could not act on effectively. Further studies are warranted to test this hypothesis.
Another important finding is that lipid-containing autophagosomes and autolysosomes are mainly located in the perinuclear or apical region of PTCs despite that LDs are localized in the basolateral region and adjacent to mitochondria. Although the mechanism underlying characteristic localization of these cytoplasmic organelles is largely unknown, it is generally supposed that FFAs released by lipophagy-mediated lipolysis bind to the chaperon proteins such as fatty acid binding proteins and are translocated to mitochondria. Interestingly, certain FFAs degraded by lipophagy are known to translocate into nuclei and activate PPARA to stimulate β-oxidation.Citation38 This concept is consistent with the perinuclear or apical localization of lipid-containing autophagosomes. Another possibility is that lipophagy takes place in the apical region to efflux excessive lipids efficiently when PTCs are exposed to massive FFAs as previously reported in macrophages.Citation18
There is accumulating evidence of the pathophysiological roles of lipid metabolism in the progression of acute kidney injury (AKI) and chronic kidney disease (CKD). Diverse forms of AKI, including endotoxic, toxic, and ischemic injury, evoke dramatic TG accumulation in proximal tubules, which may predispose tubules to concomitant ATP depletion or oxidant attack.Citation39 Furthermore, during the acute phase of ischemia reperfusion injury, there is a notable switch of energy source from glucose to lipids.Citation40 Another report has demonstrated that humans and mouse models with tubulointerstitial fibrosis have decreased expression of key enzymes and regulators of β-oxidation and increased intracellular lipid deposition and that the inhibition of β-oxidation in tubule epithelial cells causes ATP depletion, cell death, dedifferentiation and intracellular lipid deposition, which are phenotypes observed in fibrosis.Citation41 These findings led us to speculate that lipophagy may be involved in the progression of AKI and CKD. Lipophagy may be upregulated to attenuate TG overload in proximal tubules during AKI. Alternatively, stagnation of lipophagy could cause increased TG accumulation. In the latter case, restoration of lipophagy (and thereby an activation of β-oxidation) could have a therapeutic effect on the progression of AKI and CKD.
In this study, we demonstrated a novel example of kidney-other organ communication, namely that autophagy deficiency in the kidney is accompanied by increased secretion of ketone bodies in the liver during prolonged starvation, which serves in energy homeostasis. Since a previous report demonstrates that genetic manipulation of autophagy deficiency in skeletal muscle or liver promotes FGF21 expression in each organ, which promotes protection from diet-induced obesity and insulin resistance,Citation31 we hypothesized that FGF21 is a mediator of ketogenesis. Indeed, we, for the first time, found that FGF21 is secreted by kidney PTCs; however, the increased levels of plasma ketone bodies and liver β-oxidation were not abolished in starved fgf21, atg5-TSKO mice, suggesting that together with FGF21 another mediator may be involved in the adaptive response. One possible candidate is the plasma FFAs themselves, which could be elevated due to the decreased uptake into the PTCs during autophagy-deficiency. Indeed, atg5-TSKO mice tended to exhibit higher concentrations of plasma FFA levels during prolonged starvation ().
In conclusion, lipophagy is induced in kidney PTCs during prolonged starvation to break down LDs, which serves to maintain the quality and quantity of LDs and contributes to energy homeostasis by mobilizing FFAs to the mitochondria.
Materials and methods
Mice
GFP-MAP1LC3B transgenic mice and atg5-TSKO with C57BL/6 background have been described previously.Citation23,28 To generate PTC-specific Fgf21- and Atg5-deficient mice (fgf21, atg5-TSKO mice), Fgf21-floxed mice were crossed with atg5-TSKO mice. Eight-wk-old mice were housed in box cages, maintained on a 12-h light/12-h dark cycle, and fed a normal fat chow diet (Oriental Yeast, MF). To assess the autophagic flux and the LD degradation by the autophagy-lysosome pathway, mice were injected intraperitoneally with chloroquine (Sigma-Aldrich, C6628; 50 μg/g body weight) and were killed 6 h following the injection. For studying the effects of starvation, mice were completely deprived of food for 24 or 48 h (9 a.m. to 9 a.m.) with having free access to drinking water. All animal experiments were approved by the institutional committee of the Animal Research Committee of Osaka University and the Japanese Animal Protection and Management Law (No. 25).
Antibodies
We used the following antibodies: antibodies for LRP2/MEGALIN (a gift from T. Michigami, Department of Bone and Mineral Research, Osaka Medical Center and Research Institute for Maternal and Child Health, Japan), MAP1LC3B (Cell Signaling Technology, 2755 for western blotting, Medical and Biological Laboratory [MBL], PM036 for immunostaining), LAMP1 (BD Biosciences, 553792), COL4A4 (collagen, type IV, α 4; Cosmo Bio, LSL-LB-1403), COX4I1 (MBL, PM063), ATG5 (MBL, PM050), BECN1 (MBL, PD017), SQSTM1 (MBL, PM045), PLIN2 (Progen Biotechnics, GP40), PNPLA2 (Cell Signaling Technology, 2439), GAPDH (Gene Tex, GTX100118), PPARA (abcam, ab2779), FGF21 (abcam, ab64857), ACTB (Sigma-Aldrich, A5316), phosphorylated RPS6 (Ser235/236; Cell Signaling Technology, 2211), phosphorylated RPS6KB (Thr389; Cell Signaling Technology, 9234), TFEB (Invitrogen, PA1–31552), biotinylated secondary antibodies (Vector Laboratories, BA-1000 [anti-rabbit IgG], BA-2001 [anti-mouse IgG]), horseradish peroxidase-conjugated secondary antibodies (DAKO, P0448 [anti-rabbit IgG], P0447 [anti-mouse IgG]), and Alexa Fluor-conjugated secondary antibody (Invitrogen, A31572 [anti-rabbit Alexa Fluor 555], A21432 [anti-goat Alexa Fluor 555], A21434 [anti-rat Alexa Fluor 555], and A21247 [anti-rat Alexa Fluor 647]).
Histological analysis
Histological analysis was performed as described previously with modification.Citation27 To investigate the distribution of LDs, postfixed frozen kidney sections were immunostained for COL4A4 (tubular basement membrane marker), and LDs were labeled with BODIPY 493/503 (Life Technologies, D3922) at 200 ng/ml immediately before imaging. Tubular cells were divided into 2 regions determined by the distance from COL4A4; less than 5 μm in basolateral region and more than 5 μm in apical region using a confocal quantitative image cytometer, CQ1 (Yokogawa, Kanazawa, Japan), and the number of LDs in each region was counted. Postfixed frozen kidney was immunostained for COX4I1, labeled with BODIPY 493/503 to elucidate the relationship between mitochondria and LDs. Three-dimensional reconstruction images were acquired using CQ1. To examine the number of GFP-positive dots, postfixed frozen kidney was sectioned and immunostained for LRP2/MEGALIN (proximal tubular cell maker). GFP-MAP1LC3B dots per proximal tubule were counted. To access the number of LDs colocalized with MAP1LC3B or LAMP1 dots, postfixed frozen kidney was immunostained for MAP1LC3B or LAMP1, and LDs were labeled with BODIPY 493/503 followed by imaging and LDs colocalized with MAP1LC3B or LAMP1 dots per proximal tubule were counted. To assess the levels of the post-translational modifications of MTORC1 (p-RPS6), post fixed frozen tissue was immunostained for p-RPS6. The intensity of the positive staining area was measured using ImageJ. Post-fixed frozen tissue was immunostained for TFEB to evaluate the localization of TFEB. Cells were visually scored as having nuclear localization if the nuclear levels of TFEB exceeded those in the cytoplasm. The percentage of cells showing nuclear localization was quantified. At least 10 high-power fields (× 600) were evaluated for each quantitative assessments. The fluorescence images were collected using confocal microscopy (FV1000-D [Olympus, Tokyo, Japan]). Immunohistochemical staining for PPARA was performed on paraffin-embedded sections after antigen retrieval via autoclaving in 0.01 mM citrate buffer (pH 6.0) for 10 min at 120°C and blocking with 1.5% bovine serum albumin (BSA; Sigma-Aldrich, A3059–100G) in phosphate-buffered saline (PBS, composed of 137 mM NaCl [Nacalai, 31320–05], 8 mM Na2HPO4 [Nacalai, 31723–35], 2.7 mM KCl [Nacalai, 2851475], 1.5 mM KH2PO4 [Wako, 169–04245], pH 7.4) for 60 min. For double staining for PPARA and LRP2/MEGALIN, each molecule was first visualized using a horseradish peroxidase-diaminobenzidine (DAB) compound (Nichirei, 415172), followed by detection of LRP2/MEGALIN using alkaline phosphatase and nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate (NBT-BCIP) Stock Solution (Roche, 11681451001).
Electron microscopy
For electron microscopy, kidney specimens were fixed with 2.5% glutaraldehyde (Wako, 071–01931) and then in a 2% OsO4 solution in distilled water. They were embedded in Quetol812 (Nisshin EM) and ultrathin sections (80 nm) were obtained on a Ultracut E ultramicrotome (Reichert-Jung, Vienna, Austria), stained with a uranyl acetate and lead solution and observed using a Hitachi H-7650 transmission electron microscope (Hitachi, Tokyo, Japan).
Toluidine blue staining
After fixing specimens and embedding them, as described above, 1-μm-thick semithin sections mounted on glass slides were stained with a 0.25% toluidine blue dye solution on a slide warmer at 100 to 120°C for 10 sec.
Oil Red O staining
Frozen, 4% paraformaldehyde (PFA) embedded tissue sections (5 μm thick) or 4% PFA-fixed cultured cells were rinsed in PBS, and placed in 60% isopropylene for 1 min. They were stained in 60% Oil Red O solution (100% solution: 0.3 g of Oil Red O [Sigma-Aldrich, O0625] dissolved in 100 mL isopropylene) for 20 min at 37°C, placed in 60% isopropylene for 1 min, washed with PBS 3 times. Tissue sections were counterstained with hematoxylin. The mean size, the average number and total area of the Oil Red O positive LDs per tubular cell were calculated using a digital image-analyzing software, ImageJ (available at http://rsbweb.nih.gov/ij/index.html; National Institutes of Health, Bethesda, MD, USA). For each kidney, at least 10 high-power fields (× 400) were analyzed. Total area of LDs was calculated by multiplying the mean size by the average number of LDs.
Aggresome assay
Aggresomes were detected using the Proteostat Aggresome Detection Kit (Enzo Life Sciences, ENZ-51035–0025) according to the manufacturer's instructions. Briefly, postfixed frozen kidney was sectioned, rinsed in PBS, stained with ProteoStat dye for 30 min at room temperature and washed with PBS 3 times. Aggresomes per proximal tubule in at least 10 high-power fields (× 600) were counted. The fluorescence images were collected using confocal microscopy (FV1000-D [Olympus, Tokyo, Japan]).
Biochemical parameters
Blood samples were collected from mice under anesthesia. Plasma was obtained after centrifugation (15 min, 845 g, 4°C) and concentrations of glucose, total protein, albumin, TGs, FFAs, cholesterol, phospholipids, ketone bodies and FGF21 were measured using the Glucose CII-test (Wako, 439–90901), the A/G B-test (Wako, 274–24301), the Triglyceride E-test (Wako, 432–40201), the NEFA C-test (Wako, 279–75401), the Cholesterol E-test (Wako, 439–17501), the Phospholipid C-test (Wako, 433–36201), the Total Ketone Bodies kit (Wako, 415–73301, 411–73401) and the Mouse/Rat FGF21 Quantikine ELISA Kit (R&D Systems, MF2100). All kits were used in accordance with the manufacturer's protocols.
Cell fractionation
LDs were isolated from mouse kidneys by density gradient centrifugation following a modification of a method described previously.Citation42 Kidneys were homogenized followed by ultrasonic disruption, and were centrifuged at 3000 g for 10 min at 4°C. The supernatant was adjusted to 20% sucrose in hypotonic lysis buffer (HLM; 20 mM Tris, 1 mM EDTA, pH 7.4), placed in a centrifuge tube and sequentially overlaid with 4 ml of 5% sucrose in HLM and HLM. After centrifugation at 100,000 g for 60 min at 4°C, the LD fraction was collected from the top of the tube. LD fractions were washed 3 times by HLM and delipidated with acetone. The protein pellets were solubilized in sodium dodecyl sulfate (Wako, 191–07145) and an equal protein amount was analyzed by western blotting. Each sample was prepared from 8 kidneys of 4 mice to obtain enough LD fraction.
Tissue triglyceride measurement
Samples were weighed and homogenized by polytron, and total lipids were extracted by the Folch method.Citation43 Total tissue TGs were assayed using the Triglyceride E-test (Wako, 432–40201).
Measurement of ATP
ATP was measured using the ATP Colorimetric/Fluorometric Assay Kit (BioVision, K354–100) according to the manufacturer's instructions.
In situ hybridization
Mouse cDNA was obtained by reverse transcription polymerase chain reaction (RT-PCR) using mRNA extracted from mouse embryonic kidney. DNA fragments corresponding to positions 340 through 786 of Fgf21 mRNA (accession no. NM_020013.4) were amplified by PCR (GeneAmp PCR System 2700 [Applied Biosystems, Foster City, CA, USA]) and subcloned into pGEM-T vector (Promega Corporation, A3600). The digoxigenin labeled RNA probe was prepared by in vitro transcription using a Digoxigenin RNA labeling kit (Roche Diagnostics, 11175025910) according to the manufacturer's protocol. Kidneys were dissected from 48-h starved mice and fixed in tissue fixative (Genostaff, STF-01), embedded in paraffin, and sectioned at 8-μm thickness. Tissue sections were processed as described previously.Citation44
Lipidomic analysis
Lipid extraction from mice kidney tissue was performed using a modified Folch method.Citation43 The levels of PtdIns, PC, and LPC were quantified using supercritical fluid chromatography triple quadrupole mass spectrometry (SFC/MS/MS) in multiple reaction monitoring mode.Citation45,46 The SFC/MS/MS system is composed of a SFC (ACQUITY UPC2 [Waters Corporation, Milford, MA, USA]) and a triple quadrupole mass spectrometer (Xevo TQ-S micro [Waters Corporation, Milford, MA, USA]). Phosphatidylserine 17:0/17:0 (25 μM), PC 17:0/17:0 (5 μM), and LPC 17:0 (0.25 μM) were used as internal standard.
Cell culture
Autophagy-deficient and -competent PTC lines were described previously.Citation23 In brief, Atg5-deficient kidney PTCs (atg5-negative PTCs) were isolated from 3-wk-old atg5-TSKO mice and immortalized by using pEF321-T, an SV-40 large T antigen expression vector. As a control, we generated Atg5-positive PTCs by stably transfecting pMRX-IRES-Atg5-bsr (Atg5-expressing retroviral vector cassette) to atg5-negative PTCs. We also isolated PTCs from wild-type mice and immortalized cell lines were established by transformation using pEF321-T, an SV-40 large T antigen expression vector. We transfected the pEGFP-MAP1LC3B vector (a gift from T. Yoshimori, Department of Genetics, Osaka University) to the wild-type PTC lines and then constructed GFP-MAP1LC3B transgenic PTC lines. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Sigma-Aldrich, D5030) containing 5% FBS at 37°C under a humidified atmosphere of 5% CO2 and 95% air.
Lipid-containing medium (OA-BSA) was prepared by conjugation of OA with essentially FA-free, low endotoxin BSA (Sigma, A8806). Sodium OA (Sigma, O7501) were dissolved in 50% ethanol and vigorously mixed with BSA in PBS at a molar ratio of 6.6:1,Citation47 filter-sterilized, and added to culture media at the final FA (OA) concentration of 250 μM. In the current experimental condition, the BSA concentration was 2.5 mg/ml (37.88 μM) when the FA was used at 250 μM. This concentration of FFA was used because the molar ratio of FFA to albumin could reach up to 8.59,Citation48 and the albumin concentration in the proximal tubular lumen could be as high as 2.9 mg/ml (43 μM).Citation49 Twenty-four h after the seeding, the cells were loaded with or without OA-BSA for 12 h and thereafter starved with serum-limited medium (DMEM with 0.1% FBS) or HBSS with or without 40 μM etomoxir (Sigma, E1905) or 40 μM atglistatin (Cayman Chemical, 15284). To assess autophagic activity, PTCs were treated with 200 nM of bafilomycin A1 (BafA1; Wako, 023–11641) for 1 h at 37°C before harvest.
Fluorescence and immunofluorescence microscopy
Cells were cultured on coverslips and fixed with 4% PFA for 10 min, quenched with 50 mM NH4Cl in PBS, permeabilized with 50 mg/ml digitonin (Sigma, D141) in PBS, blocked with 0.2% gelatin (Wako, 074–02761) in PBS, and then incubated with the indicated primary antibodies, followed by subsequent labeling with secondary antibodies.
Fluorescent FA pulse-chase assay
PTCs stably expressing GFP-MAP1LC3B were incubated with complete medium (DMEM with 5% FBS) containing 250 μM OA-BSA and 1 μM BODIPY 558/568C12 (Red C12; Invitrogen, D3835) for 12 h. Cells were then starved with serum-limited medium (DMEM with 0.1% FBS), and chased for the indicated time periods. The percentage of colocalization area of Red C12 to GFP-MAP1LC3B or LAMP1 was quantified using the ImageJ plug-in.
Measurement of FAs distribution into mitochondria
To access FAs distribution into mitochondria, PTCs incubated with 250 μM OA-BSA and 1 μM Red C12 for 12 h were stained with 100 nM of MitoTracker Green FM (Invitrogen, M7514) for 30 min at 37°C before or after 24 h of starvation by serum-limited medium (DMEM with 0.1% FBS). Three-dimensional reconstruction images were acquired using CQ1. Colocalization of FAs and mitochondria was calculated using ImageJ and represented as the Pearson correlation between fluorescence of Red C12 and MitoTracker Green FM.
LD degradation assay
Atg5-positive and atg5-negative PTCs were loaded with OA-BSA for 12 h and thereafter starved with serum-limited medium (DMEM with 0.1% FBS) with or without 40 μM atglistatin. The LDs content was calculated from the area of BODIPY 493/503 labeled LDs using ImageJ and was adjusted by cell number.
Cell proliferation assay
MTS assays were performed using CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega Corporation, G3580) according to the manufacturer's instructions in a 96-well plate. Each experiment was performed in triplicate and repeated at least 3 times. Cell viability in percent refers to the MTS value of the induced cells compared with the value of control cells.
LDH assay
Cytotoxicity was evaluated by measuring LDH release into the culture medium using Cytotoxicity Detection Kit (Roche, 04744934001) according to the manufacturer's protocol in a 96-well plate. Each experiment was performed in triplicate and repeated at least 3 times. The LDH signal that was associated with 100% cell death was determined by lysing cells with 0.2% Triton X-100 (Calbiochem, 648466).
TUNEL assay
Apoptotic cells were detected using the TUNEL assay with an in situ apoptosis detection kit (Takara, MK-500) according to the manufacturer's instructions. The percentage of TUNEL-stained cells in at least 10 high-power fields (× 600) was quantified using ImageJ.
Quantitative RT-PCR and western blot analysis
Quantitative RT-PCR and western blot analyses were performed as described previously.Citation27 The sequences of the primers used were as follows:
Acadl-F, 5´- gcatcaacatcgcagagaaa -3´; Acadl-R, 5´- acgcttgctcttcccaagta -3´;
Acadm-F, 5´- taatcggtgaaggagcaggttt -3´; Acadm-R, 5´- ggcatacttcgtggcttcgt -3´;
Acads-F, 5´- ttacctggcctactccatcg -3´; Acads-R, 5´- tgatccactgttgcttctgc -3´;
Acadvl-F, 5´- gcatcttgctctatggcaca -3´; Acadvl -R, 5´- cactcgagggctctgttagg -3´;
Acox1-F, 5´- tcaacagcccaactgtgacttccatta -3´; Acox1-R, 5´- tcaggtagccattatccatctcttca -3´;
Cpt1a-F, 5´- accactggccgaatgtcaag -3´; Cpt1a-R, 5´- agcgagtagcgcatggtcat -3´;
Fgf21-F, 5´- tacacagatgacgaccaaga -3´; Fgf21-R, 5´- ggcttcagactggtacacat -3´;
Hmgcs2-F, 5´- gcaccgagaccatcattgac -3´; Hmgcs2-R, 5´- ctcgatgtcagtgttgcctg -3´;
Ndufb5-F, 5´- tcctagactcggagtcggaa -3´; Ndufb5-R, 5´- aacttcctgctcctttaacc -3´;
Plin2-F, 5´- ctgtctaccaagctctgctc -3´; Plin2-R, 5´- cgatgcttctcttccactcc -3´;
Pnpla2-F, 5´- atggtgccctatactctgcc -3´; Pnpla2-R, 5´- tcttcagggacatcaggcag -3´;
Ppara-F, 5´- cgtcacggagctcacagaat -3´; Ppara-R, 5´- actcgcgtgtgataaagcca -3´;
Uqcrb-F, 5´- acttacccagaaggcagcg -3´; Uqcrb-R, 5´- tgcccactcttctctctcct -3´;
Gapdh-F, 5´- aactttggcattgtggaagg -3´; Gapdh-R, 5´- acacattgggggtaggaaca -3´.
Statistics
All results are presented as mean ± SE. Statistical analyses were conducted using JMP software (SAS Institute, Cary, NC). The difference between 2 experimental values was assessed by the Student t test. Multiple group comparisons were performed using one-way analysis of variance (ANOVA) followed by the Dunnett test, to detect inter-group differences. Statistical significance was defined as P < 0.05.
Abbreviations
| ACADL/LCAD | = | acyl-Coenzyme A dehydrogenase, long-chain |
| ACADM/MCAD | = | acyl-Coenzyme A dehydrogenase, C-4 to C-12 straight chain |
| ACADS/SCAD | = | acyl-Coenzyme A dehydrogenase, short-chain |
| ACADVL/VLCAD | = | acyl-Coenzyme A dehydrogenase, very long chain |
| ACTB | = | actin, β |
| AKI | = | acute kidney injury |
| ACOX1/AOX | = | acyl-Coenzyme A oxidase 1 |
| ATG | = | autophagy-related |
| atg5F/F-CTRL | = | floxed control mice |
| atg5-TSKO | = | PTC-specific atg5-deficient mice |
| BafA1 | = | bafilomycin A1 |
| BECN1 | = | Beclin 1, autophagy related |
| BODIPY | = | boron-dipyrromethene |
| BSA | = | bovine serum albumin |
| CKD | = | chronic kidney disease |
| COL4A4/COL4 | = | collagen, type IV, α 4 |
| COX4I1 | = | cytochrome c oxidase subunit 4I1 |
| CPT1A | = | carnitine palmitoyltransferase 1A |
| CREB | = | cAMP responsive element binding protein |
| DG | = | diacylglycerol |
| FA | = | fatty acid |
| FBS | = | fetal bovine serum |
| FFA | = | free fatty acid |
| FGF21 | = | fibroblast growth factor 21 |
| fgf21, atg5-TSKO | = | PTC-specific fgf21- and atg5-deficient mice |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| GFP | = | green fluorescent protein |
| HBSS | = | Hank's balanced salt solution |
| HMGCS2 | = | 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 |
| KAP | = | kidney androgen regulated protein |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| LD | = | lipid droplet |
| LDH | = | lactate dehydrogenase |
| LIPE/HSL | = | lipase E, hormone sensitive |
| LPC | = | lysophosphatidylcholine |
| LRP2 | = | low density lipoprotein receptor-related protein 2 |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 β |
| MG | = | monoacylglycerol |
| MGLL | = | monoglyceride lipase |
| MTORC1 | = | mechanistic target of rapamycin (serine/threonine kinase) complex 1 |
| NDUFB5 | = | NADH dehydrogenase (ubiquinone) 1 β subcomplex, 5 |
| OA | = | oleic acid |
| OXPHOS | = | oxidative phosphorylation |
| PC | = | phosphatidylcholine |
| PLIN2/ADRP | = | perilipin 2 |
| PNPLA2/ATGL | = | patatin-like phospholipase domain containing 2 |
| PNPLA5 | = | patatin-like phospholipase domain containing 5 |
| PPAR | = | peroxisome proliferator activated receptor |
| PTC | = | proximal tubular cell |
| PtdIns | = | phosphatidylinositol |
| RPS6 | = | ribosomal protein S6 |
| RPS6KB/S6K | = | ribosomal protein S6 kinase |
| SFC/MS/MS | = | supercritical fluid chromatography triple quadrupole mass spectrometry |
| SQSTM1 | = | sequestosome 1 |
| TCA | = | tricarboxylic acid |
| TFEB | = | transcription factor EB |
| TG | = | triglyceride |
| TSKO | = | tissue specific knockout |
| TUNEL | = | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| UQCRB | = | ubiquinol-cytochrome c reductase binding protein |
Disclosure of potential conflicts of interest
The authors declare that there are no conflicts of interest.
KAUP_A_1341464_Supplemental.pdf
Download PDF (56.9 MB)Acknowledgments
We thank N. Mizushima, University of Tokyo, for the atg5F/F-CTRL and GFP-MAP1LC3B mice; T. Michigami, Osaka Medical Center and Research Institute, for the LRP2/MEGALIN antibody; and N. Horimoto for the technical assistance.
Funding
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan (JP15H06371 [to T.Y.], JP24591196 and JP15K09260 [to Y.T.], and JP24659416 [to Y.I.]), Ono Medical Research Foundation (to A.T.), Japan Foundation for Applied Enzymology (to A.T.), Takeda Medical Research Foundation [to Y.T.], and Merck Sharp and Dohme (MSD) (to Y.I.).
Related Research Data
References
- Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu ASL, Brenner BM. Brenner and Rector's The Kidney. ninth ed. Philadelphia: Saunders; 2012
- Gullans SR, Brazy PC, Mandel LJ, Dennis VW. Stimulation of phosphate transport in the proximal tubule by metabolic substrates. Am J Physiol. 1984;247:F582-7
- Balaban RS, Mandel LJ. Metabolic substrate utilization by rabbit proximal tubule. An NADH fluorescence study. Am J Physiol. 1988;254:F407-16
- Uchida S, Endou H. Substrate specificity to maintain cellular ATP along the mouse nephron. Am J Physiol. 1988;255:F977-83
- Hohenegger M, Schuh H. Uptake and fatty acid synthesis by the rat kidney. Int J Biochem. 1980;12:169-72. doi:10.1016/0020-711X(80)90062-2. PMID:7399018
- Kamijo A, Kimura K, Sugaya T, Yamanouchi M, Hase H, Kaneko T, Hirata Y, Goto A, Fujita T, Omata M. Urinary free fatty acids bound to albumin aggravate tubulointerstitial damage. Kidney Int. 2002; 62:1628-37. doi:10.1046/j.1523-1755.2002.00618.x. PMID:12371963
- Thomas ME, Schreiner GF. Contribution of proteinuria to progressive renal injury: consequences of tubular uptake of fatty acid bearing albumin. Am J Nephrol. 1993;13:385-98. doi:10.1159/000168653. PMID:8116691
- Krzystanek M, Pedersen TX, Bartels ED, Kjaehr J, Straarup EM, Nielsen LB. Expression of apolipoprotein B in the kidney attenuates renal lipid accumulation. J Biol Chem. 2010;285:10583-90. doi:10.1074/jbc.M109.078006. PMID:20103594
- Ducharme NA, Bickel PE. Lipid droplets in lipogenesis and lipolysis. Endocrinology. 2008;149:942-9. doi:10.1210/en.2007-1713. PMID:18202123
- Legrand-Poels S, Esser N, L'homme L, Scheen A, Paquot N, Piette J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem Pharmacol. 2014;92:131-41. doi:10.1016/j.bcp.2014.08.013. PMID:25175736
- Hardy S, El-Assaad W, Przybytkowski E, Joly E, Prentki M, Langelier Y. Saturated fatty acid-induced apoptosis in MDA-MB-231 breast cancer cells. A role for cardiolipin. J Biol Chem. 2003;278:31861-70
- Mishra R, Simonson MS. Saturated free fatty acids and apoptosis in microvascular mesangial cells: palmitate activates pro-apoptotic signaling involving caspase 9 and mitochondrial release of endonuclease G. Cardiovasc Diabetol. 2005;4:2. doi:10.1186/1475-2840-4-2. PMID:15642122
- Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275-81. doi:10.1152/ajpendo.00644.2005. PMID:16492686
- Weidemann MJ, Krebs HA. The fuel of respiration of rat kidney cortex. Biochem J. 1969; 112:149-66. doi:10.1042/bj1120149. PMID:5805283
- Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F. FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15:279-91. doi:10.1016/j.cmet.2011.12.018. PMID:22405066
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131-5. doi:10.1038/nature07976. PMID:19339967
- Khaldoun SA, Emond-Boisjoly M-A, Chateau D, Carrière V, Lacasa M, Rousset M, Demignot S, Morel E. Autophagosomes contribute to intracellular lipid distribution in enterocytes. Mol Biol Cell. 2014;25:118-32. doi:10.1091/mbc.E13-06-0324. PMID:24173715
- Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13:655-67. doi:10.1016/j.cmet.2011.03.023. PMID:21641547
- Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32:678-92. doi:10.1016/j.devcel.2015.01.029. PMID:25752962
- Walther TC, Farese RV. Lipid droplets and cellular lipid metabolism. Annu Rev Biochem. 2012;81:687-714. doi:10.1146/annurev-biochem-061009-102430. PMID:22524315
- Marvyn PM, Bradley RM, Button EB, Mardian EB, Duncan RE. Fasting upregulates adipose triglyceride lipase and hormone-sensitive lipase levels and phosphorylation in mouse kidney. Biochem Cell Biol. 2015;93:262-7. doi:10.1139/bcb-2014-0150. PMID:25879679
- Haemmerle G, Moustafa T, Woelkart G, Büttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med. 2011;17:1076-85. doi:10.1038/nm.2439
- Kimura T, Takabatake Y, Takahashi A, Kaimori J, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902-13. doi:10.1681/ASN.2010070705. PMID:21493778
- Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol. 2012;180:517-25. doi:10.1016/j.ajpath.2011.11.001. PMID:22265049
- Kimura T, Takahashi A, Takabatake Y, Namba T, Yamamoto T, Kaimori J-Y, Matsui I, Kitamura H, Niimura F, Matsusaka T, et al. Autophagy protects kidney proximal tubule epithelial cells from mitochondrial metabolic stress. Autophagy. 2013;9:1876-86. doi:10.4161/auto.25418. PMID:24128672
- Namba T, Takabatake Y, Kimura T, Takahashi A, Yamamoto T, Matsuda J, Kitamura H, Niimura F, Matsusaka T, Iwatani H, et al. Autophagic clearance of mitochondria in the kidney copes with metabolic acidosis. J Am Soc Nephrol. 2014;25:2254-66. doi:10.1681/ASN.2013090986. PMID:24700866
- Yamamoto T, Takabatake Y, Kimura T, Takahashi A, Namba T, Matsuda J, Minami S, Kaimori J-Y, Matsui I, Kitamura H, et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy. 2016;12:801-13. doi:10.1080/15548627.2016.1159376. PMID: 26986194
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101-11. doi:10.1091/mbc.E03-09-0704. PMID:14699058
- Bartz R, Li W-H, Venables B, Zehmer JK, Roth MR, Welti R, Anderson RGW, Liu P, Chapman KD. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res. 2007;48:837-47. doi:10.1194/jlr.M600413-JLR200. PMID:17210984
- Rui L. Energy metabolism in the liver. Compr Physiol. 2014;4:177-97
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim Y-N, Kim SS, Kim DH, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83-92. doi:10.1038/nm.3014. PMID:23202295
- Zhang F, Yu L, Lin X, Cheng P, He L, Li X, Lu X, Tan Y, Yang H, Cai L, et al. Minireview: Roles of Fibroblast Growth Factors 19 and 21 in Metabolic Regulation and Chronic Diseases. Mol Endocrinol. 2015;29:1400-13. doi:10.1210/me.2015-1155. PMID:26308386
- Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112-5
- Seok S, Fu T, Choi S-E, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108-11
- Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647-58. doi:10.1038/ncb2718
- Martinez-Lopez N, Garcia-Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P, Cecconi F, Schwartz GJ, Singh R. Autophagy in the CNS and Periphery Coordinate Lipophagy and Lipolysis in the Brown Adipose Tissue and Liver. Cell Metab. 2016;23:113-27. doi:10.1016/j.cmet.2015.10.008. PMID:26698918
- Dupont N, Chauhan S, Arko-Mensah J, Castillo EF, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T, Deretic V. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol. 2014;24:609-20. doi:10.1016/j.cub.2014.02.008. PMID:24613307
- Folick A, Oakley HD, Yu Y, Armstrong EH, Kumari M, Sanor L, Moore DD, Ortlund EA, Zechner R, Wang MC. Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science. 2015;347:83-6
- Zager RA, Johnson ACM, Hanson SY. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int. 2005;67:111-21. doi:10.1111/j.1523-1755.2005.00061.x. PMID:15610234
- Wei Q, Xiao X, Fogle P, Dong Z. Changes in metabolic profiles during acute kidney injury and recovery following ischemia/reperfusion. PLoS One 2014;9:e106647. doi:10.1371/journal.pone.0106647. PMID:25191961
- Kang HM, Ahn SH, Choi P, Ko Y-A, Han SH, Chinga F, Park ASD, Tao J, Sharma K, Pullman J, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 2015;21:37-46. doi:10.1038/nm.3762
- Ding Y, Zhang S, Yang L, Na H, Zhang P, Zhang H, Wang Y, Chen Y, Yu J, Huo C, et al. Isolating lipid droplets from multiple species. Nat Protoc. 2013;8:43-51. doi:10.1038/nprot.2012.142. PMID:23222457
- Folch J, Lees M SSG. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497-509
- Suzuki A, Ito T, Imai E, Yamato M, Iwatani H, Kawachi H, Hori M. Retinoids regulate the repairing process of the podocytes in puromycin aminonucleoside-induced nephrotic rats. J Am Soc Nephrol. 2003;14:981-91. doi:10.1097/01.ASN.0000057857.66268.8F. PMID:12660332
- Takeda H, Koike T, Izumi Y, Yamada T, Yoshida M, Shiomi M, Fukusaki E, Bamba T. Lipidomic analysis of plasma lipoprotein fractions in myocardial infarction-prone rabbits. J Biosci Bioeng. 2015; 120:476-82. doi:10.1016/j.jbiosc.2015.02.015. PMID:26162515
- Tsugawa H, Ohta E, Izumi Y, Ogiwara A, Yukihira D, Bamba T, Fukusaki E, Arita M. MRM-DIFF: data processing strategy for differential analysis in large scale MRM-based lipidomics studies. Front Genet. 2014; 5:471
- Brasaemle DL, Barber T, Kimmel AR, Londos C. Post-translational regulation of perilipin expression. Stabilization by stored intracellular neutral lipids. J Biol Chem. 1997;272:9378-87
- Ghiggeri GM, Ginevri F, Candiano G, Oleggini R, Perfumo F, Queirolo C, Gusmano R. Characterization of cationic albumin in minimal change nephropathy. Kidney Int. 1987;32:547-53. doi:10.1038/ki.1987.243. PMID:3430951
- Lewy JE, Pesce A. Micropuncture study of albumin transfer in aminonucleoside nephrosis in the rat. Pediatr Res. 1973; 7:553-9. doi:10.1203/00006450-197306000-00002. PMID:4708973