
ABSTRACT
Inflammation is crucial in the defense against infections but must be tightly controlled to limit detrimental hyperactivation. Our diet influences inflammatory processes and omega-3 polyunsaturated fatty acids (n-3 PUFAs) have known anti-inflammatory effects. The balance of pro- and anti-inflammatory processes is coordinated by macrophages and macroautophagy/autophagy has recently emerged as a cellular process that dampens inflammation. Here we report that the n-3 PUFA docosahexaenoic acid (DHA) transiently induces cytosolic speckles of the autophagic receptor SQSTM1/p62 (sequestosome 1) (described as SQSTM1/p62-bodies) in macrophages. We suggest that the formation of SQSTM1/p62-bodies represents a fast mechanism of NFE2L2/Nrf2 (nuclear factor, erythroid 2 like 2) activation by recruitment of KEAP1 (kelch like ECH associated protein 1). Further, the autophagy receptor TAX1BP1 (Tax1 binding protein 1) and ubiquitin-editing enzyme TNFAIP3/A20 (TNF α induced protein 3) could be identified in DHA-induced SQSTM1/p62-bodies. Simultaneously, DHA strongly dampened the induction of pro-inflammatory genes including CXCL10 (C-X-C motif chemokine ligand 10) and we suggest that formation of SQSTM1/p62-bodies and activation of NFE2L2 leads to tolerance towards selective inflammatory stimuli. Finally, reduced CXCL10 levels were related to the improved clinical outcome in n-3 PUFA-supplemented heart-transplant patients and we propose CXCL10 as a robust marker for the clinical benefits mobilized by n-3 PUFA supplementation.
Introduction
Low-grade persistent inflammation is involved in the initiation and progression of age-related diseases.Citation1 The individual risk of developing inflammation-related diseases clearly varies due to genetic and environmental differences. The diet is an important factor influencing the immune system, and a diet rich in marine n-3 PUFAs possesses anti-inflammatory properties. Beneficial effects of n-3 PUFAs involve a changed membrane lipid composition and the promoted synthesis of anti-inflammatory and pro-resolving eicosanoids.Citation2 Also, regulatory signaling by n-3 PUFAs has been reported via, among others, the selective FFAR4/GPR120 (free fatty acid receptor 4) protein leading to reduced activity of the NFKB (nuclear factor kappa B) complex and the inflammasome.Citation3-5
Macroautophagy (hereafter referred to as autophagy) is a catabolic process that eliminates damaged and excessive intracellular components by lysosomal degradation.Citation6 Cytoplasmic proteins and organelles are sequestered by a growing double membrane that forms an autophagosomal vesicle where the content is degraded after fusion with a lysosome.Citation7 The autophagic receptor SQSTM1 binds to ubiquitinated cargos and interacts with mammalian orthologs of yeast ATG8 (autophagy-related 8) on the growing phagophore membrane leading to selective degradation.Citation8
Homopolymerization of SQSTM1 is important for efficient sequestration and turnover of cargo.Citation9 In the case of altered protein degradation or in response to cellular stresses, the accumulation of SQSTM1 in ubiquitin (Ub)-positive so-called SQSTM1/p62-bodies (also known as aggresome-like induced structures [ALIS]) has been described and might provide a temporary form of storage for unfolded proteins to be destroyed.Citation10 Autophagy is important to prevent inflammation-prone conditions by maintaining cellular homeostasis but also directly by selective elimination of invading pathogens (xenophagy)Citation11-13 and the removal of activated inflammasomes.Citation14-16
As for autophagic removal of cargo, several inflammatory responses depend on ubiquitination for complex formation, activation or degradation of the signaling proteins.Citation17 Interestingly, SQSTM1 can also have a pro-inflammatory role as its ubiquitin-binding and polymerizing abilities are necessary for NF-kB complex activation, suggesting a scaffold function in the formation of signaling complexes.Citation18,Citation19
We recently reported that physiologically relevant doses of the n-3 PUFA DHA elevates levels and cytosolic structures of SQSTM1 and increases turnover of polyubiquitinated proteins in retinal pigment epithelium cells (ARPE-19), possibly reducing the risk of age-related macular degeneration.Citation20 We thus hypothesized that n-3 PUFAs affect autophagy or ubiquitination to mobilize an anti-inflammatory effect in macrophages. We here report that human primary monocyte-derived macrophages (MDM) and RAW264.7 mouse macrophages respond to n-3 PUFAs by induction of SQSTM1 mRNA and protein levels followed by a transient rise in the number of ALIS like SQSTM1/p62-bodies. While SQSTM1/p62-bodies are present, there is a clearly dampened response to lipopolysaccharide (LPS), particularly by a reduced IFN (interferon) type-I response. Consistently, the reduced secretion of the IFN response protein CXCL10 is evident both in macrophage cell cultures and in blood samples from patients on n-3 PUFA supplements.
Results
The n-3 PUFA DHA induces SQSTM1/p62-bodies in macrophages
To determine if n-3 PUFAs affect localization of SQSTM1 and ubiquitinated proteins in macrophages, RAW264.7 cells were supplemented with 70 µM of the n-3 PUFA DHA, the n-6 PUFA arachidonic acid (AA) or the n-9 monounsaturated fatty acid oleic acid (OA). The cells were immunostained for SQSTM1 and conjugated Ub. Confocal inspection showed formation of SQSTM1/p62-bodies in response to DHA in a lipid-selective manner. Further, the DHA-induced \SQSTM1/p62-bodies colocalized more stringently with ubiquitinated proteins () than with MAP1LC3B (microtubule associated protein 1 light chain 3 β; data not shown), thus representing aggregates rather than autophagosomes. The number of SQSTM1/p62-bodies and colocalization with Ub transiently increased up to 16 h and decayed to basal levels after 24 h (). Consistently, the level of SQSTM1 and Ub-conjugated proteins also rose in response to DHA supplementation in RAW264.7 cells in a time- and concentration-dependent manner (, S1A and S1B).
Figure 1. The n-3 PUFA DHA induces SQSTM1/p62-bodies in macrophages. (A) Confocal analysis of RAW264.7 cells treated with DHA, OA or AA (70 µM) for 8 h (scale bars: 10 µm) and (B) automated quantification of SQSTM1- and Ub-positive speckles (< 7000 cells/condition). Data are representative of 3 independent experiments of which 2 were manually counted. (C) Immunoblot (IB) analysis and quantification (below) of RAW264.7 cells treated with DHA, OA and AA (70 µM) for 16 h, n = 4, (repeated measures ANOVA with Dunnett's). (D) Confocal analysis of MDMs treated with DHA, OA or AA (70 µM) for 24 h (scale bars: 10 µm) and (E) automated quantification of SQSTM1/p62-bodies (< 4000 cells/condition), n = 3 except for 48 h where n = 1. (F) IB and quantification (below) of MDMs treated with DHA, OA and AA (70 µM) for 16 h, n = 5, (Friedman test with Dunn's).
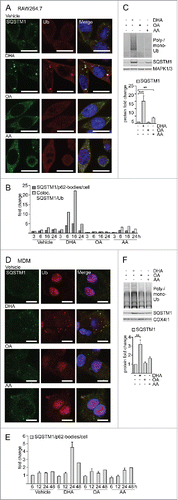
Human primary MDM displayed the same lipid-specific increase in SQSTM1/p62-bodies and colocalized ubiquitin after DHA supplementation (). Automated image analysis demonstrated that the average number of SQSTM1/p62-bodies increased up to 24 h after DHA supplementation and decreased thereafter (), thus showing the same transient rise although at a slightly slower kinetics compared to the mouse macrophages. MDMs similarly demonstrated a selective increase in the protein levels of SQSTM1 and conjugated Ub in response to DHA, whereas no increase was observed after treatment with OA or AA (). Interestingly, the increase in protein level of SQSTM1 was observed already after 6 h treatment with DHA and was further increased with time up to 24 h (Fig. S1C).
DHA induces formation of detergent-resistant SQSTM1/p62-bodies degraded by autophagy and activates NFE2L2
DHA supplementation caused a striking translocation of SQSTM1 from a mainly diffuse cytoplasmic location into cytoplasmic bodies associated with ubiquitinated proteins. Such protein deposits or ALIS tend to be resistant to detergents.Citation21,Citation22 To determine if DHA-induced SQSTM1/p62-bodies were detergent resistant, the pellets that remained after centrifugation in a detergent lysis buffer were resolved in 8 M urea buffer. Interestingly, the protein level of SQSTM1 in the detergent-resistant pellet was clearly elevated after DHA supplementation in RAW264.7 cells () and increased with centrifugation speed (Fig. S1D). Thus, for the remainder of the study, cells were lysed directly in 8 M urea buffer to avoid losing part of the SQSTM1 pool.
Figure 2. DHA induces formation of detergent-resistant SQSTM1/p62-bodies degraded by autophagy and activates NFE2L2. IB of soluble and insoluble fractions of RAW264.7 cells treated with DHA (70 µM) with or without BafA1 (100 nM) for (A) 16 h or (B) 8 h followed by 12 h incubation in fresh medium (Med) with or without BafA1. Hmw SQSTM1 was visualized by long exposure (exp). (C) IB of RAW264.7 cells treated with DHA, OA or AA (70 µM) with or without BafA1 (100 nM) for 8 h. Data are representative of 3 independent experiments. (D) FACS analysis of ROS levels in RAW264.7 cells at different time points after DHA (70 µM) using a fluorescent CM-H2DCFDA probe, n = 3, (paired t-test). (E) Confocal analysis of RAW264.7 cells treated with DHA, OA or AA (70 µM) for 3 h (scale bars: 10 µm). Representative images are shown, n = 3. (F) IB analysis of cytosolic (Cyt) or nuclear (Nuc) fractions after 3 h DHA with or without addition of 5 mM NAC or 6 h DHA and quantification of cytosolic SQSTM1 levels and nuclear NFE2L2 levels (below), n = 3, (repeated-measures ANOVA with Dunnett's). (G) IB analysis and quantification (right) of RAW264.7 cells after 3 h DHA with or without addition of 5 mM NAC, n = 3, (repeated-measures ANOVA with Dunnett's). (H) IB analysis and quantification (right) of MDM after 3 h DHA with or without addition of 5 mM NAC, n = 3. (I) QRT-PCR analysis of NFE2L2 target genes SQSTM1, HMOX1, NQO1 and SLC7A11 in vehicle- and DHA-treated MDMs, n = 5 (Friedman test with Dunn's).
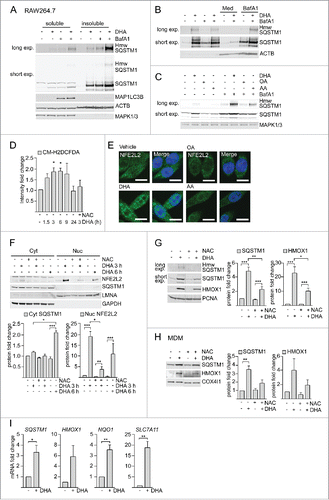
SQSTM1 is continuously turned over by autophagy.Citation21 A combined treatment with DHA and the lysosomal inhibitor bafilomycin A1 (BafA1) caused an additive increase in the protein level of SQSTM1 in the detergent-resistant fraction. However, no concurrent increase in the protein level of MAP1LC3B was observed after BafA1 treatment (). This shows that autophagy can be elevated as measured by the number of SQSTM1 proteins degraded per cell, without affecting the turnover of MAP1LC3B-II. Interestingly, DHA also induced some slower migrating proteins detected by SQSTM1 antibodies in both RAW264.7 cells and MDMs ( and S1E), which were clearly reduced by siRNA targeting SQSTM1 (Fig. S1E). The same migration pattern was detected using 5 different SQSTM1-specific antibodies in ARPE-19 cells (Fig. S1F). The 2 most prominent of these additional SQSTM1 bands migrated at approximately 70,000 and 120,000, of which the latter is indicated as high molecular weight (hmw) SQSTM1. Each of the additional bands accounted for approximately 5% of total SQSTM1 levels in DHA- and BafA1-treated cells. The hmw SQSTM1 showed the same concentration- and time-dependent increase as unmodified SQSTM1 (Fig. S1A and S1B), was especially prominent in the detergent-resistant fraction and was strikingly elevated after cotreatment with DHA and BafA1 (). Importantly, hmw SQSTM1 could be depleted from the soluble fraction and enriched in the detergent-resistant pellet by increasing centrifugation speed (Fig. S1D). The elevated protein levels of SQSTM1 and hmw SQSTM1 were of a transient nature. Removal of DHA and incubation for an additional 12 h in fresh medium clearly reversed the DHA-induced effects (). The reduction in SQSTM1 protein levels could be blocked by adding BafA1 in the recovery medium, suggesting turnover by autophagy. As for the SQSTM1/p62-bodies, hmw SQSTM1 was mainly induced by DHA (). Together, these results demonstrate that DHA induces SQSTM1 protein levels, posttranslational modifications and translocation of the protein into detergent-resistant cytoplasmic bodies in a lipid-selective manner. Furthermore, the formed structures are of a transient nature and degraded by autophagy.
The SQSTM1 gene is induced in response to cellular stress such as reactive oxygen species (ROS) via the transcription factor NFE2L223,24 and ROS are involved in the formation of ALIS.Citation22 We have previously found that DHA induced transient ROS and NFE2L2 nuclear translocation in ARPE-19 cells.Citation20 Also in macrophages, DHA induced ROS at time points preceding the increase in SQSTM1 protein levels as measured by a fluorescent ROS DCF probe (). Nuclear translocation of NFE2L2 could be observed by confocal microscopy () and immunoblotting of cytosolic (Cyt) and nuclear fractions (Nuc) (). Consistent with a NFE2L2-mediated anti-oxidative response, the NFE2L2 target protein HMOX1 (heme oxygenase 1) showed a similar increase as SQSTM1 in response to DHA (). Both the DHA-induced ROS increase, translocation of NFE2L2 and rise in SQSTM1 and HMOX1 protein levels was strongly reduced by a pretreatment with the ROS scavenger N-acetyl cysteine (NAC) (). Similarly, in MDMs, HMOX1 protein levels increased in response to DHA, but clearly less so in the presence of NAC ( and S1C). Importantly, DHA induced the transcriptional activity of NFE2L2 as measured by increased mRNA levels of its target genes SQSTM1, HMOX1, NQO1 (NAD[P]H quinone dehydrogenase 1) and SLC7A11 (solute carrier family 7 member 11) in MDMs ().
DHA strongly reduced expression of CXCL10 by affecting several signaling pathways
Anti-inflammatory effects of n-3 PUFAs have been determined as reduced induction of diverse pro-inflammatory cytokines. To identify putative anti-inflammatory effects of DHA in our system, we determined if macrophages respond differently to LPS when pretreated with DHA for 16 h. This time point was used because the number of SQSTM1/p62-bodies peaked at this time. As an initial screen, NanoString technology was used to quantify the levels of 579 inflammation-related transcripts in DHA-pretreated MDMs from 2 donors that were stimulated with LPS for 3 h. Of these genes, 163 were selected as LPS-responsive transcripts as their mRNA levels increased more than 1.5-fold in each donor. Among those, 148 were downregulated and 15 upregulated by DHA pretreatment ( and Table S1). The clearest reduction was seen for CXCL9, CXCL10 and CXCL11, a subfamily of chemokines that are highly inducible by IFNG/IFNγ and signal through their common receptor CXCR3 (C-X-C motif chemokine receptor 3)Citation25 to attract additional immune cells. CXCL10 is the best studied of these and is implicated in diseases with underlying inflammation.Citation26 Of note, classical NFKB target genes including TNF (tumor necrosis factor), IL1B (interleukin 1 β) and IL6, displayed only minor or opposite changes by DHA pretreatment in this screen. These observations were validated by qRT-PCR as in primary macrophages isolated from 5 different healthy donors, where DHA pretreatment caused an approximately 80% reduction in the LPS-induced expression of CXCL10 in a lipid selective manner (). The lowered mRNA levels of CXCL10 were accompanied by reduced secretion of CXCL10 measured by ELISA (Fig. S2A). Also in RAW264.7 macrophages, we observed a significant decrease in the induction of Cxcl10 but also Tnf mRNA after DHA pretreatment and stimulation with LPS for 3 h (Fig. S2B). We thus asked if this difference could be explained by different kinetics of TNF induction in both systems. Indeed, in MDMs, DHA pretreatment caused reduction in the LPS induction of TNF mRNA levels at earlier time points and was more pronounced at the peak of TNF expression at 2 h of LPS stimulation (Fig. S2C). The DHA-mediated decrease in CXCL10 was not limited to induction by LPS but could also be seen with other inducers of IFN signaling such as TLR3 (toll like receptor 3) ligand poly(p)(I:C) and the synthetic TLR8 ligand pU (Fig. S2D). Overall, these data show that DHA particularly dampens type-I IFN responses, including CXCL10 induction, in a lipid-selective manner in mouse and human macrophages.
Figure 3. The n-3 PUFA DHA strongly reduced expression of CXCL10 by affecting several signaling pathways. (A) MDMs were treated with DHA (70 µM) for 16 h before stimulation with LPS (100 ng/ml) for 3 h. Transcript levels of 579 genes (nCounter GX Human Immunology kit assay) were determined. Transcripts that were more than 1.5-times increased by LPS in both donors were selected and the fold change between vehicle- and DHA-treated cells calculated, mean ± SD, n = 2. (B) QRT-PCR analysis of CXCL10 and (C) TNF in MDM treated with DHA, OA or AA for 16 h and LPS for 3 h, n = 5 (Friedman test with Dunn's). (D) IB analysis and quantification of IRF3 and RELA phosphorylation in MDMs treated with DHA, OA or AA for 16 h and LPS for 1 h, n = 5. (Friedman test with Dunn's), of (E) TBK1, IKBKB, STAT1 and STAT3 phosphorylation in MDMs treated with DHA, OA or AA for 16 h and LPS for 1 or 4 h, n = 5 (Wilcoxon matched-pairs signed rank test, one-tailed) and of (F) IRF1 and RELA in cytosolic (Cyt) and nuclear (Nuc) fractions in MDMs treated with DHA, OA or AA for 16 h and LPS for 2 or 4 h, n = 5 (Wilcoxon matched-pairs signed rank test). (G) IFNB1 mRNA levels after incubation with DHA for 16 h and LPS for 2 h, n = 6 (Wilcoxon matched-pairs signed rank test).
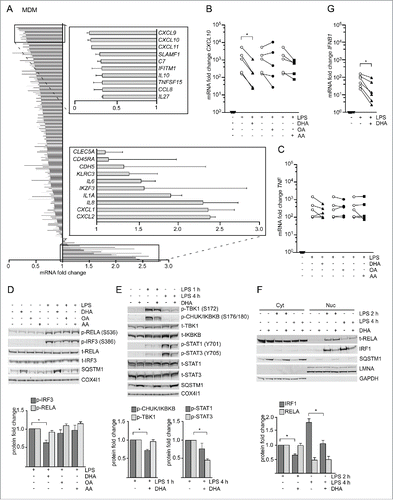
The synthesis of CXCL10 is regulated at the transcriptional level by the transcription factors NFKB complex, IRF3 (interferon regulatory factor 3), IRF1 and STAT1 (signal transducer and activator of transcription 1).Citation27,Citation28 To determine which initial signaling axis DHA affects, phosphorylation of IRF3 (S386) and the NFKB subunit RELA/p65 (S536) was analyzed. DHA pretreatment led to reduced levels of phosphorylated IRF3 in response to LPS in MDMs while no change in phosphorylation of RELA (S536) was observed (). We also checked subunit NFKB1/p105 (S933) of the heterodimeric NFKB complex, which showed a reduced phosphorylation in response to LPS after DHA pretreatment (Fig. S2E). In RAW264.7 cells, DHA similarly reduced the phosphorylation of IRF3 (S396) and NFKB1 (S933) but not RELA (S536) (Fig. S2F). We then analyzed the phosphorylated, active forms of IKBKB/IKKβ (inhibitor of nuclear factor kappa B kinase subunit β) and TBK1 (TANK binding kinase 1) that are upstream kinases of classical NFKB and IRF3 signaling, respectively.Citation29 Surprisingly, in MDMs, DHA pretreatment decreased the LPS-induced levels of phosphorylated IKBKB but not TBK1 ().
Besides direct induction via IRF3 and NFKB, CXCL10 is induced as a secondary response gene in an IFN-dependent manner. After TLR4 stimulation, secreted IFNs bind to IFNAR1 (interferon α and β receptor subunit 1) in an autocrine manner and activate JAK2 (Janus kinase 2)-STAT1 signaling and synthesis of CXCL10.Citation30,Citation31 We found approximately 50% reduced IFNB and IFNG mRNA induction after DHA pretreatment in the NanoString data (Table S1), and could confirm limited induction of IFNB mRNA 2 h after LPS stimulation in DHA-pretreated MDMs (). Moreover, inhibition of IFNAR1 suppressed LPS-mediated CXCL10 induction similarly to DHA and limited further reduction by DHA (Fig. S2G), demonstrating that the n-3 PUFA supplementation also dampened autocrine IFN signaling. Consistently, DHA pretreatment decreased the phosphorylation of both STAT1 and STAT3 when induced by 4 h LPS stimulation; however, this was more pronounced and significant only for STAT3 (). Also in RAW264.7 macrophages DHA but not OA or AA pretreatment dampened the activation of STAT1 (Fig. S2F).
Of note, in the mRNA expression screen DHA pretreatment also reduced the LPS induction of IRF1, IRF4, IRF7 and IRF8 (Table S1). IRF1 is strongly induced in response to IFN and other cytokinesCitation32 and binds to the CXCL10 promotor.Citation33 Therefore, we investigated if nuclear translocation of IRF1 is affected by DHA. Immunoblotting showed a clear increase of nuclear IRF1 levels after 2 h and 4 h of LPS stimulation that was clearly reduced in DHA-pretreated MDMs. RELA could also be detected in nuclear lysates after 2 h of LPS stimulation, but was unchanged by DHA-pretreatment (). In addition, confocal image analysis showed increased intensity of nuclear IRF1 staining at 2 h of LPS stimulation, which was limited by DHA pretreatment while number, area and maximum radius of SQSTM1/p62-bodies were augmented (Fig. S2I).
The DHA-mediated inflammatory tolerance is related to the formation of SQSTM1/p62-bodies
The results presented above demonstrate that DHA selectively induced the formation of SQSTM1/p62-bodies or ALIS and reduced LPS-induced CXCL10 expression. To assess the relation between both observations, MDMs were pretreated with DHA for increasing times and finally stimulated with LPS for 1 h. Interestingly, the decrease in phosphorylated IRF3 and IKBKB was clearest when SQSTM1 levels were most induced ().
Figure 4. The DHA-mediated inflammatory tolerance is related to the formation of SQSTM1/p62-bodies. (A) IB analysis of MDMs treated with DHA (70 μM) for the indicated times followed by LPS for 1 h. The quantification (below) represents mean fold changes ± SEM of DHA + LPS compared to vehicle + LPS on a log scale, n = 6, (Friedman test with Dunn's). (B) QRT-PCR analysis of CXCL10 levels in MDM after 16 h DHA with and without addition of 5 mM NAC followed by 3 h LPS, n = 3, (repeated-measures ANOVA with Dunnett's). (C) QRT-PCR analysis of CXCL10 in SQSTM1 siRNA-transfected MDMs treated with DHA for 16 h and LPS for 3 h, n = 5, (Friedman test with Dunn's). (D) IB analysis of IRF3, RELA and IKBKB phosphorylation in control and SQSTM1 knockdown MDMs, n = 6 (Friedman test with Dunn's). (E) CXCL10 protein levels in Sqstm1+/+ (n = 8) and sqstm1−/− (n = 10) male mice were measured by ELISA. Serum was analyzed at baseline and at 3 and 6 h after challenge with 3.5 µg/g LPS. ND, not detected.
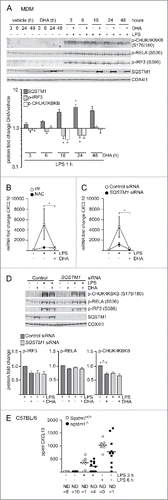
We thus speculated that formation of SQSTM1/p62-bodies could play a role in establishing tolerance towards inflammatory signaling. We first checked if impaired formation of SQSTM1/p62-bodies would impair downregulation of CXCL10 by DHA. As shown above, the induction of SQSTM1 by DHA could be prevented by adding the anti-oxidant NAC. Treatment with NAC before stimulation with LPS limited CXCL10 induction to a similar extent as DHA alone, showing little further effect of the combination of NAC and DHA (). As activation of NFE2L2 might also play a role in this observation, we treated MDMs with L-sulforaphane (L-Sulf), an established activator of NFE2L2. Treatment with L-Sulf abolished LPS-induced CXCL10 expression (Fig. S2H). SQSTM1 is known to be necessary for the formation of ALIS.Citation22 Thus we speculated that knockdown of SQSTM1 would interfere with the formation of SQSTM1/p62-bodies and the downregulation of CXCL10 by DHA. SQSTM1 was shown to have anti-inflammatoryCitation12,Citation16,Citation34 but also pro-inflammatory effects itself.Citation18,Citation19 In line with the pro-inflammatory role of SQSTM1, the SQSTM1-depleted MDMs displayed reduced expression of CXCL10 in response to LPS (). SQSTM1-depleted MDMs, also showed decreased levels of phosphorylated IKBKB and IRF3, although not significant for IRF3, and no further decrease by DHA treatment (). Similarly, a trend of decreased CXCL10 secretion after 3 and 6 h LPS challenge was observed in Sqstm1 depleted (sqstm1−/−) mice (), suggesting that SQSTM1 is involved in the induction of CXCL10 signaling in vivo. Together these data indicate that SQSTM1 is necessary for full induction of CXCL10 expression, whereas the formation of SQSTM1/p62-bodies is important for the LPS tolerance induced by n-3 PUFAs like DHA.
KEAP1 is sequestered in DHA-induced SQSTM1/p62-bodies and contributes to inflammatory tolerance
The obtained results indicated that the DHA-mediated formation of SQSTM1/p62-bodies dampened LPS-induced signaling in macrophages. We therefore aimed to characterize the proteins recruited into the DHA-induced SQSTM1/p62-bodies that mediate the tolerance towards inflammatory stimuli.
The previously established centrifugation protocol was used for partial purification of DHA-induced SQSTM1/p62-bodies from RAW264.7 cells and the isolated proteins were identified by mass spectrometry (MS). Proteins were considered as DHA-specifically enriched in the detergent insoluble fraction if they were more abundant in DHA-treated samples versus vehicle as well as further elevated in samples cotreated with DHA and BafA1 versus BafA1 alone. Using these criteria, SQSTM1 was clearly the most abundant DHA- and BafA1-induced protein in the detergent-resistant fraction (Fig. S3A). Interestingly, TAX1BP1 and KEAP1 were the second and third most prominent proteins recruited into the detergent-resistant fraction in response to DHA and clearly further accumulated by inhibiting lysosomal degradation. However, since this analysis was only based on different detergent solubility, the detected proteins could reside in many different compartments and structures. In order to limit the detection of proteins to DHA-induced partners of SQSTM1, we performed immunoprecipitation (IP) of endogenous SQSTM1 from vehicle treated or DHA-stimulated RAW264.7 cells and identified copurified interaction partners by MS (Fig. S3B). KEAP1 and TAX1BP1 were the only proteins that were found both in the detergent-resistant fraction as well as co-immunoprecipitated with SQSTM1 and were thus considered as located to SQSTM1-containing protein bodies in a DHA-induced manner. These interactions were validated in MDMs where KEAP1 and TAX1BP1 co-immunoprecipitated with SQSTM1 (). Apparently, DHA did not change the affinity of TAX1BP1 or KEAP1 for SQSTM1 but the association increased proportionally with the lipid-induced rise in SQSTM1 protein level.
Figure 5. KEAP1 is sequestered in DHA-induced SQSTM1/p62-bodies and contributes to inflammatory tolerance. (A) IP of SQSTM1 from MDMs treated with DHA for 16 h and IB analysis of KEAP1 and TAX1BP1; INPUT (10%). Data are representative of 3 independent experiments. (B) Immunostaining for SQSTM1 and KEAP1 in MDMs treated with DHA for 24 h (scale bars: 10 µm). (C) IB analysis and quantification (below) of SQSTM1 and KEAP1 in soluble and insoluble fractions from RAW264.7 cells after 16 h DHA, n = 4 (repeated measures ANOVA). (D) QRT-PCR analysis of CXCL10 or (E) CXCL11 in KEAP1 siRNA-transfected MDMs treated with DHA for 16 h and LPS for 3 h, n = 5 (Friedman test with Dunn's). (right) Comparison of the downregulating effect of DHA in control or KEAP1 siRNA-treated MDMs from (D) or (E) respectively, n = 5 (Mann Whitney test).
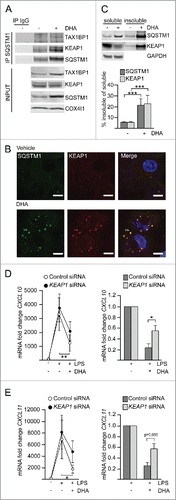
TAX1BP1 is an autophagy receptor involved in xenophagyCitation35 and could bind SQSTM1 indirectly via ubiquitin. KEAP1 mediates ubiquitination and degradation of NFE2L2 under normal unstressed conditionsCitation36 and has been suggested to contribute to degradation of protein aggregates.Citation37 Also, the association of KEAP1 with SQSTM1/p62-bodies has been shown to stabilize and activate NFE2L2.Citation38 NFE2L2 activation, besides its anti-oxidative function, plays an important role in the regulation of inflammation.Citation39-42 In addition, we recently demonstrated a role for KEAP1 in the regulation of inflammatory signaling, via ubiquitination and degradation of IKBKB.Citation43 Intriguingly, TAX1BP1 has been implicated as a negative regulator of NFKB signaling due to the recruitment of the ubiquitin-editing enzyme TNFAIP3.Citation44 Also, IRF3-dependent signaling is negatively regulated by TAX1BP1 and TNFAIP3.Citation45,Citation46 Immunostaining of MDMs demonstrated that both KEAP1 and TAX1BP1 are located in DHA-induced SQSTM1/p62-bodies ( and S3C). In contrast to MS data and IPs, by confocal analysis also TNFAIP3 could be detected in SQSTM1/p62-bodies (Fig. S3C) indicating that this protein interacts with the SQSTM1/p62-bodies in a more transient manner. Importantly, about 20% of KEAP1 was found in insoluble aggregates and corresponded well with the fraction of SQSTM1 that translocated to the detergent-resistant fraction in response to DHA treatment (). As the sequestration of KEAP1 by SQSTM1 has previously been shown to lead to NFE2L2 activation, we analyzed the mRNA level of NQO1 in SQSTM1 knockdown cells treated with LPS and DHA. In line with an important role of SQSTM1 in activation of NFE2L2 in the presence of DHA, NQO1 mRNA (as opposed to CXCL10) was significantly induced by DHA in a SQSTM1-dependent manner (Fig. S3D).
We next asked if recruitment of KEAP1, TAX1BP1 or TNFAIP3 into SQSTM1/p62-bodies has a repressive function on CXCL10 signaling. MDMs were transfected with siRNA targeting KEAP1, TAX1BP1 and TNFAIP3 and decreased protein or mRNA levels were observed, albeit TNFAIP3 was less efficiently reduced (Fig. S3E, S3F). As KEAP1 is the repressor of NFE2L2, the expression of NFE2L2 target genes was analyzed in KEAP1-depleted MDMs and showed at least partial activation of NFE2L2 with induction of NQO1 and SLC7A11 (Fig. S3G). Depletion of TAX1BP1 or TNFAIP3 did not have any significant effect on CXCL10 levels (Fig. S3H and S3I), whereas depletion of KEAP1 significantly limited the ability of DHA to dampen LPS-induced CXCL10 expression (). When directly comparing the reduction by DHA-pretreatment on LPS-induced CXCL10 expression, there was a significant difference in the presence or absence of KEAP1 (76.4% ± 16.4% vs 44.8% ± 22.2% respectively, with less effective DHA pretreatment in KEAP1-depleted cells in all 5 replicates) (, right). The same pattern could be seen for mRNA induction of CXCL11 (), another chemokine strongly affected by DHA. Thus, the formation of SQSTM1/p62-bodies and sequestration of KEAP1 plays a role in the DHA-mediated inflammatory tolerance in macrophages.
The plasma level of CXCL10 is reduced by n-3 PUFA supplementation but increased in the placebo group in hypertensive heart transplant recipients.
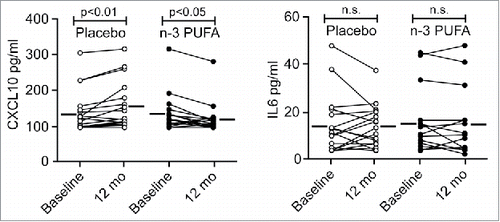
According to our results, we expected n-3 PUFA supplementation to be most effective in conditions with chronically elevated CXCL10 levels. Therefore, we obtained plasma samples of the heart transplant patients in the OmaCor-study, as elevated levels of CXCL9 and CXCL10 are known as causes for transplant rejection.Citation47,Citation48 Plasma samples of these patients were taken at baseline after transplantation and following 12 mo with daily administration of n-3 PUFA (DHA and eicosapentaenoic acid) or placebo (corn oil). This clinical trial documented an improved clinical outcome from intake of n-3 PUFAs compared to the placebo group as measured for example by lowered blood pressure.Citation49 However, a corresponding reduction in any inflammatory mediators in response to the n-3 PUFAs have not been previously found. In fact, TNF was slightly increased by n-3 PUFA treatment in this study.Citation50 We measured CXCL10 by ELISA on those patient plasma samples and found the CXCL10 levels in the placebo group increased after transplantation in line with a nonresolved inflammation in these patients. Conversely, in total agreement with the results from the cell-based assays, n-3 PUFA administration significantly decreased CXCL10 levels after transplantation (, left). In line with our expression analyses, the IL6 levels did not differ between the placebo and the n-3 PUFA group (, right). These data suggest that n-3 PUFA supplementation dampens conditions driven by sterile inflammation and that a reduced type-I interferon response, including lowered CXCL10, represents potent markers for such beneficial effects.
Figure 6. The plasma level of CXCL10 is reduced by n-3 PUFA supplementation but increased in the placebo group in hypertensive heart transplant recipients. The concentration of CXCL10 (left) and IL6 (right) was determined by ELISA in plasma of hypertensive heart transplant recipients isolated at baseline and after 12 mo of daily supplementation of placebo (corn oil, n = 19) or n-3 PUFAs (n = 16). Horizontal lines represent mean plasma levels. p < 0.01, p < 0.05 and nonsignificant (n.s.) were all vs baseline.
To relate this observation to our in vitro mechanism, we wanted to assess NFE2L2 activation in our patient material. IL36G is a direct target gene of NFE2L2 and upregulated by NFE2L2 activation.Citation51 We measured IL36G levels as read-out for NFE2L2 and found an inverse correlation between CXCL10 and IL36G levels at baseline (r = -0.57, p < 0.05), suggesting that NFE2L2 activity contributes to limit CXCL10 levels (data not shown).
In summary, our data are consistent with an important role of DHA-induced SQSTM1 polymerization and resulting NFE2L2 activation in the reduction of serum levels of CXCL10.
Discussion
With increasing evidence of the health benefits and anti-inflammatory effects of n-3 PUFAs, these lipids represent a valuable tool to identify cellular mechanisms of maintenance and homeostasis. In response to polyunsaturated fatty acids, we found that macrophages dampened pro-inflammatory signaling as evident by reduced transcription of a large number of inflammation-related genes after LPS stimulation. The effects were clearest for type-I IFN-related responses with CXCL10 as the most prominent. Importantly, CXCL10 protein levels were reduced both in the media of cultured macrophages and in blood samples of patients supplemented with n-3 PUFAs. We found that SQSTM1/p62-bodies, protein bodies containing the autophagic cargo receptor SQSTM1, are formed in response to the n-3 PUFA DHA. The ROS sensor KEAP1, the autophagy receptor TAX1BP1 and the ubiquitin-editing enzyme TNFAIP3 could be found in SQSTM1/p62-bodies. Of these proteins, depletion of KEAP1 led to limited effects of DHA supplementation. We suggest that the DHA-mediated formation of SQSTM1/p62-bodies and recruitment of KEAP1 represent a fast mechanism for NFE2L2 activation, playing a role in the regulation of inflammatory signaling in macrophages. Thus, the data point to an important balance where dietary lipids influence endogenous and exogenous antioxidants in controlling inflammatory signaling in macrophages.
SQSTM1 is known to form aggregates and to sequester ubiquitinated proteins into speckles also described as ALIS.Citation21 These structures were initially shown in dendritic cells during maturationCitation52 and may occur in response to various cellular stresses such as LPS stimulation, infection, oxidative stress or proteasomal inhibition.Citation53 DHA supplementation led to a transient increase in detergent-resistant SQSTM1/p62-bodies associated with ubiquitinated proteins. Sequestration of potentially harmful and misfolded proteins might help in detoxification of the cytoplasm and could disarm inflammation-prone conditions. Interestingly, Sergin et al. reported a protective role of SQSTM1 in the sequestration of ubiquitinated inclusions in atherosclerosis, as deletion of the gene encoding SQSTM1 caused diffuse distribution of ubiquitinated and misfolded proteins and had worse outcomes.Citation54
ALIS are reported to be degraded via the autophagic-lysosomal pathway.Citation8,Citation22 The increase in SQSTM1/p62-bodies observed after DHA supplementation was transient and reversible in case of functional autophagosome-lysosome fusion, indicating that DHA-induced SQSTM1/p62-bodies are handled via lysosomal degradation. Further, our results suggest that the autophagic clearance of tightly packed protein speckles of SQSTM1 and associating proteins can be elevated without any major changes in basal autophagy as measured by the turnover of MAP1LC3B-II. SQSTM1 can homopolymerizeCitation55 and 1 molcule of MAP1LC3B likely intereacts with a chain of SQSTM1.Citation8 Thus, the autophagic turnover of SQSTM1 may increase by stimuli that increase polymerization without any detectable change in MAP1LC3B degradation. How homopolymerization of SQSTM1 is regulated represents an interesting question as the ability of cargo receptors to polymerize may be an important regulatory step for autophagy. In this regard, we observed posttranslational modifications of SQSTM1, as seen by slower migrating protein forms in response to n-3 PUFA supplementation. SQSTM1 can form covalently crosslinked oligomers in response to ROS-inducing drugs in a PB1-dependent manner.Citation56 We find that DHA stimulates the formation of hmw SQSTM1, which might be explained by the generation of ROS or direct crosslinking of SQSTM1 as DHA is a highly reactive and easily oxidized molecule.Citation57 Also, a transient increase in hmw SQSTM1 as well as activation of NFE2L2 has been reported for other PUFAs.Citation58 Thus, covalent crosslinking of SQSTM1 dimers can be an important mode of regulation of protein turnover after oxidative stress also under noncytotoxic conditions. Also our data indicate that crosslinked SQSTM1 in protein aggregates mediates a transient storage of damaged proteins that are degraded by autophagy after new synthesis of unmodified SQSTM1. On the contrary, diets containing already oxidized n-3 PUFAs increase inflammatory signaling,Citation59 suggesting that the original reactivity of the molecule might be needed for beneficial effects. In this regard, a balanced level of exogenous anti-oxidants in the n-3 PUFA formulation seems important to prevent oxidation of the lipid but also allow activation of endogenous anti-oxidative responses.
We observed activation of NFE2L2 with coinciding induction of its target genes SQSTM1, HMOX1, NQO1 and SLC7A11 during DHA treatment in total agreement with what we recently reported also in nontransformed, diploid epithelial cells.Citation20 The interaction of SQSTM1 and KEAP1 and the sequestration of KEAP1 into SQSTM1/p62-bodies has been shown as an alternative way to liberate and activate NFE2L2.Citation60,Citation61 We here describe the formation of SQSTM1/p62-bodies with sequestration of a substantial fraction of KEAP1 in response to the physiological stimuli DHA as a fast mechanism for NFE2L2 activation. In this line, depletion of SQSTM1 or KEAP1 modulated NFE2L2 activation and impaired the DHA-mediated anti-inflammatory effects. NFE2L2 has been firmly established as a regulator of inflammation.Citation40,Citation62 We found that activation of NFE2L2 by an established inducer strongly limited LPS-induced CXCL10 expression. nfe2l2 knockout mice show increased mortality and inflammatory markers such as phosphorylated IRF3 during septic shock,Citation41 including augmented expression of CXCL10.Citation39 In humans, NFE2L2 polymorphisms are associated with the development of gastric mucosal inflammationCitation63 and increased risk for acute lung injury characterized by inflammation.Citation64 Also, NFE2L2 and expression of HMOX1 have been implicated in the development of a macrophage phenotype with lower inflammatory signature.Citation42,Citation65,Citation66 Finally, in nfe2l2 knockout mice, the anti-inflammatory effects of DHA have been shown to be NFE2L2-dependent.Citation67
Interestingly, exposure to LPS has not only been found to induce ALIS, but also to establish endotoxin tolerance, limiting further inflammatory signaling in septic patientsCitation68 and monocytes.Citation69 In addition, Vasconcellos et al. recently demonstrated that free heme and iron, as a result of hemolysis, induce ALIS as a way to maintain homeostasis.Citation70 These reports suggest that under physiological conditions transient formation of SQSTM1/p62-bodies could represent a beneficial mechanism of NFE2L2 activation. Of interest, a role of KEAP1 in the clearance of SQSTM1/p62-bodies has been reportedCitation37 while CXCL10 secretion is increased in autophagy-deficient cells.Citation71 Thus, the presence of KEAP1 in SQSTM1/p62-bodies might promote their autophagic degradation, limiting CXCL10 expression. Furthermore, we have previously shown that KEAP1 dampens inflammatory signaling by degradation of IKBKB in mycobacterial infection,Citation43 thus KEAP1 might have additional NFE2L2-independent ways of regulating inflammation.
Anti-inflammatory effects of n-3 PUFAs have been established on the expression of many cytokines, but differ between model systems. By taking a broad approach to screen for quantitative differences of more than 500 inflammation gene products, we could identify CXCL10 as the most clearly dampened cytokine after n-3 PUFA treatment. This cytokine was consistently lowered at both the mRNA level and as a secreted protein in mouse macrophages, primary human macrophages and in heart transplant patients in a lipid-selective manner. Increased intake of DHA has been found to reduce the level of CXCL10 in vivo in a mouse model of stress-induced dry eye.Citation72 Also, n-3 PUFA supplementation decreases the CXCL10 concentration in hepatitis C patients.Citation73 Of note, one clinical trial suggested that n-3 PUFAs from flaxseed augment the expression of NQO1 in cystic fibrosis patients,Citation74 making it possible to speculate that the formation of SQSTM1/p62-bodies and activation of NFE2L2 as we have shown in macrophages might also play a role to limit CXCL10 during clinical DHA supplementation. This is further supported by the inverse correlation that we found in our patient samples between CXCL10 and the NFE2L2-controlled cytokine IL36G.Citation51 Importantly, we here show lowered CXCL10 levels to correlate with the better clinical outcome seen in n-3 PUFA-supplemented heart transplant patients. This study used an n-3 PUFA formulation with the minimum of exogenous anti-oxidants. Our data show that exogenous anti-oxidants can interfere with the activation of the anti-oxidative response by DHA and might thus prevent beneficial health effects. Patients with cardiovascular diseases, inflammatory disorders, microbial pathologies and Alzheimer disease also have increased CXCL10 levels as a consequence of persistent inflammation.Citation26,Citation75 Thus, CXCL10 might be a useful marker for anti-inflammatory effects of n-3 PUFAs in these pathological conditions.
In summary, we identify CXCL10 as a robust marker for the inflammatory tolerance established by n-3 PUFAs in clinical settings with low-grade chronic inflammation. We propose that lipid-induced transient activation of the cellular stress response, as seen by activation of NFE2L2 by formation of SQSTM1/p62-bodies with association of KEAP1, counteracts the signals that drive the type-I interferon response and in particular the synthesis of CXCL10. The identified effects of n-3 PUFAs on interferon signaling contribute to our understanding of how the risk of inflammation- and age-related diseases can be modulated by dietary lipids.
Materials and methods
Cell culture and stimulation
RAW264.7 macrophages (ATCC, TIB71) were cultured in RPMI (Sigma, R8758) with 10% fetal bovine serum (Gibco, 10270–106) and gentamicin (0.05 mg/mL; Gibco, 15710049). ARPE-19 (ATCC, CRL-2302) were cultured in DMEM:F12 (Sigma, D8437). PBMC were isolated from buffy coats (obtained from the blood bank of St. Olavs Hospital, Trondheim, Norway) by a Lymphoprep™ (AXIS-SHIELD PoC AS, 1114547) density gradient. Monocytes were selected by plastic adherence and differentiated into monocyte-derived macrophages (MDM) by culture in RPMI with 30% human serum for 5 d, followed with 10% human serum during experiments. All cells were maintained in 5% CO2 at 37°C.
Docosahexaenoic acid (DHA; Cayman, 90310), oleic acid (OA; Cayman, 90260) and arachidonic acid (AA; Cayman, 90010) were added to prewarmed complete medium (70 µM) before being given to the cells. Absolute ethanol was used as control in all experiments.
Other reagents used were: bafilomycin A1 (BafA1; Sigma, B1793), LPS (InvivoGen, tlrl-3pelps), L-sulforaphane (L-Sulf; Sigma-Aldrich, S6317), pU complexed with pL-Arg (Sigma Aldrich, P7762) at a 1:1 ratio (w:w), p(I:C) transfected with Lipofectamine RNAiMAX (Invitrogen, 13778150), and N-acetyl-cysteine (NAC; Sigma, A9165).
Antibodies used were: ATCB/β actin (Abcam, ab6276); COX4I1/COXIV (Abcam, ab33985); GAPDH (Merck Millipore, CB1001); HMOX1 (Enzo Life Sciences, ADI-OSA-110); IRF3 (Santa Cruz Biotechnology, sc-9082); KEAP1 (Proteintech, 10503–2-AP); KEAP1 (Santa Cruz Biotechnology, E20); LMNA/lamin A/C (Santa Cruz Biotechnology, sc-6215); mono- and polyUb conjugates (FK2; Biomol, PW8810); SQSTM1 (Progen, GP62-C); phospho-IRF3 (S386; Abcam, ab76493); STAT1 (BD Biosciences, 610116); TNFAIP3/A20 (Santa Cruz Biotechnology, sc-166692). The following were from Cell Signaling Technology: IKBKB/IKKβ (8943); IRF1 (4302); IRF3 (4302S), MAP1LC3B (3868); MAPK1/3/p44/42 (9107); RELA/p65 (3034); phospho-CHUK/IKBKB/IKKα/β (S176/180; 2697); phospho-IRF3 (S396; 4302); phospho-NFKB2/p105 (S933; 4806); phospho-RELA/p65 (S536; 3033); phospho-STAT1 (Y701; 7649); phospho-STAT3 (Y705; 9145); phospho-TBK1 (S172; 5483); STAT1 (14994); STAT3 (9139); TAX1BP1 (5105); TBK1 (3013). For IP of SQSTM1 MBL antibody (PM045) was used and as control guinea pig IgG (Santa Cruz Biotechnology, sc-2711). To inhibit IFNAR1 signaling, anti-IFNAR2/interferon-α/β receptor chain 2 (Merck Millipore, MAB1155) was used. SQSTM1 antibodies used in Fig. S1G were from: BD (610832), Santa Cruz Biotechnology (sc-28359), Abnova (H00008878-M03), Abcam (ab31782) and Progen (GP62-C). All secondary antibodies were from Invitrogen (Alexa Fluor conjugates; A32723, A32727, A32732, A32731, A-11073, A-21435), Li-Cor Biotechnology (NIR dye conjugates; 925–68070, 925–68071, 925–68074, 925–32210, 925–32211, 925–32411, 925–68077) or Dako (HRP conjugates; P0448, P0447).
ROS detection
Cell were stained with 0.4 µM of the ROS-sensitive probe CM-H2DCFDA (Invitrogen, C6827) for 30 min at 37°C and mean fluorescence was analyzed by a BD FACS Canto (BD Biosciences, San Jose, CA, USA). Experiments were performed in duplicates with 10,000 cells per condition.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated by using the RNeasy kit (Qiagen, 74106). Total RNA (1 µg) was used for cDNA synthesis with the High-Capacity RNA-to-cDNA Kit (Applied Biosystems, 4387406). Real-time PCR was performed using PerfeCTa qPCR FastMix ROX (Quanta, QUNT95077–012) and TaqMan® Gene Expression Assays (Applied Biosystems). The cycling conditions for the StepOne plus system (Applied Biosystems) were 45°C for 2 min, 95°C for 30 sec, 40 cycles of 95°C for 1 sec, 60°C for 20 sec. The following probes were used: CXCL10 (Hs01124251_g1), Cxcl10 (Mm00445235_m1), CXCL11 (Hs04187682_g1), IFNB1 (Hs01077958_s1), TNF (Hs01113624_g1), Tnf (Mm00443260_g1), GAPDH (Hs99999905_m1), Gapdh (Mm99999915_g1), KEAP1 (Hs00202227_m1), SQSTM1 (Hs00177654_m1), HMOX1 (Hs01110250_m1), NQO1 (Hs00168547_m1) and SLC7A11 (Hs00921938_m1). Relative mRNA levels were transformed into linear form by the 2(-ΔΔCt) method.Citation76 Transcripts were normalized to GAPDH.
Nanostring
Total RNA was extracted and mRNA transcripts were assessed by digital transcript counting (nCounter GX Human Immunology kit assay, NanoString Technologies, Seattle, WA, USA). Total RNA (100 ng) was assayed on nCounter Digital Analyzer (NanoString) according to the manufacturer's instructions. Data were normalized by scaling with the geometric mean of the built-in control gene probes for each sample.
Immunoblotting
For total protein extracts, 8 M urea lysis buffer was used (8 M urea, 0.5% [v:v] Triton X-100 [Sigma-Aldrich, X100], DTT [100 mM], Complete® protease inhibitor [PI; Roche, 11873580001]), long with phosphatase inhibitor cocktail (PIC) 2 and 3 (Sigma, P5726 and P0044). Protein concentration was determined by Bio-Rad protein assay (Bio-Rad 500–0006). Proteins (50 µg) were separated using NuPAGE® Novex® Bis-Tris Gels (Invitrogen, NP0322, NP0321, WG1403, WG1402A), transferred to nitrocellulose membranes using Invitrogen's iBlot system. Bound antibodies were imaged using appropriate near-infrared (NIR) dye conjugates (Li-Cor) and an Odyssey NIR scanner (Li-Cor Biosciences) or using HRP-linked secondary antibodies (Dako) and SuperSignal West Femto substrate (Pierce, 34096) with a Li-Cor Odyssey Fc System. Image studio 3.1 was used for quantification. For statistical assessment, mean intensity values were used, while bar graphs of quantifications show fold changes.
Soluble and insoluble fractions
Cells were lysed in a detergent buffer (NP-40 [1%; Calbiochem, 492018], Triton X-100 [0.25%; Sigma-Aldrich, X100], 0.05 M Tris-HCl, 0.15 M NaCl, 0.001 M EDTA, PI, PIC2 and 3) and lysates cleared by centrifugation (1000 g, 10 min). Supernatants were centrifuged at 10,000 g for 20 min and the new supernatant collected (soluble fraction). The detergent-resistant pellet was washed twice and finally resuspended in 8 M urea lysis buffer (insoluble fraction).
Nuclear extraction
Cells were lysed in a hypotonic buffer containing 10 mM HEPES, pH 7.4, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.1% NP-40, PI, and PIC2 and 3. Lysates were kept on ice, vortexed for 15 min and centrifuged at 3000 g for 10 min. The nuclear pellet was washed once and finally resuspended in 8 M urea lyis buffer. Cytoplasmic extracts were added to 20% 8 M urea buffer.
Immunostaining
The cells were fixed in 4% formaldehyde, permeabilized with methanol, blocked with 3% goat-serum (Sigma-Aldrich, G9023) and stained with primary and fluorescently labeled secondary antibodies (Alexa Fluor conjugates, Invitrogen). Nuclear DNA was stained using Draq5 (5 µM; Biostatus, DR50200) or Hoechst 33342 (5 µg/ml; Life Technologies, C10337). Imaging was done on an Axiovert200 microscope with a 63 × 1.2W objective and the confocal module LSM510 META (Carl Zeiss). Images were processed using the ZEN software (Zeiss) and mounted using Canvas 11 (Deneba).
ScanR automated image acquisition
The microscope-based imaging platform ScanR (Olympus, Hamburg, Germany) was used to image SQSTM1/p62-bodies. Images were taken with a 20x objective, using the excitation filters (wavelength [nm]/width [nm]): FITC (485/20), Alexa Fluor 647 (640/30) and Hoechst (650/13). For emission, a combination filter (440/521/607/700) was used for all fluorophores (Chroma Technology Corp, Bellows Falls, VT, USA). The images were analysed by the ScanR Analysis software (Olympus).
Small interfering RNA
MDMs were transfected at d 5 and d 7 using Lipofectamine RNAiMAX (Invitrogen, 13778150) using 20 nM siRNA or nontargeting oligo. The following smartpool siRNA oligonucleotides were obtained from Dharmacon: nontargeting (D-001210–01), SQSTM1 (J-010230–06), TAX1BP1 (L-016892–00), TNFAIP3 (L-009919–00) and KEAP1 (LQ-012453–00).
ELISA
CXCL10 in supernatants and serum was quantified using an ELISA Duo-Set (DY266 [human] or DY466 [murine], R&D, Abingdon, UK) according to the manufacturer's instruction. IL36G in serum was quantified using a human IL-36G/IL-1F9 ELISA Kit (EHIL36G, Thermo Scientific).
IP and mass spectrometry (MS) analyses
For Santa Cruz Biotechnology, Dynabeads protein A (Life Technologies, 10002D) were incubated with antibodies for 1 h at room temperature and complexes were crosslinked with BSCitation3 (Life Technologies, 21580) according to the manufacturer's protocol. Cells were lysed in a buffer containing 0.25% TX-100, 1% NP-40, 50 mM Tris-HCl, pH 8, 150 mM NaCl, 1 mM EDTA, pH 8, PI, PIC 2 and 3. Lysates were adjusted for concentration and volume and immunoprecipitated at 4°C overnight. Beads were washed 3 times with 400 mM KCl and eluted with LDS sample buffer (Invitrogen, NP0007) at 80°C. For MS, eluates were run into NuPAGE® Gels and stained with SimplyBlue SafeStain (Life Technologies, LC6060). Gel pieces were subjected to in-gel reduction, alkylation, and tryptic digestion using 6 ng/μl trypsin (Promega, V511A).Citation77 OMIX C18 tips (Varian, Inc., Palo Alto, CA, USA) were used for sample clean up and concentration. Peptide mixtures containing 0.1% formic acid were loaded onto a Thermo Fisher Scientific EASY-nLC1000 system and EASY-Spray column (C18, 2 µm, 100 Å, 50 µm, 15 cm). Peptides were fractionated using a 2–100% acetonitrile gradient in 0.1% formic acid over 50 min at a flow rate of 250 nl/min. The separated peptides were analyzed using a Thermo Scientific Q-Exactive mass spectrometer. Data were collected in data-dependent mode using a Top10 method. The raw data were processed using the Proteome Discoverer 1.4 software. The fragmentation spectra were searched against the Swissprot SwissProt_2011_12 database using an in-house Mascot server (Matrix Sciences, UK). Peptide mass tolerances used in the search were 10 ppm, and fragment mass tolerance was 0.02 Da. Peptide ions were filtered using a false discovery rate set to 5% for peptide identifications.
Animals
Protocols were approved by the Washington University Animal Studies Committee. Mice used in this study were in the C57BL/6 background and housed in a specific pathogen-free barrier facility. sqstm1−/− mice were as described previously.Citation54,Citation78
Patients
Thirty-five clinically stable hypertensive heart transplant recipients were studied 1–12 y after transplantation and randomized in a double-blind fashion to receive either 3.4 g of omega-3 fatty acids daily or placebo (corn oil) for one y in the OmaCor-study.Citation49,Citation50 Plasma was sampled for cytokine analyses after overnight fasting at baseline and after 12 mo. The study was approved by the Regional Ethics Committee and all patients gave written informed consent for participation.
Statistical analysis
Statistical analyses were performed in Prism 6 (Graph pad). Values are expressed as mean ± standard error of the mean (SEM) if not otherwise stated. Statistical analyses were performed on mRNA fold changes compared to vehicle or on intensities obtained by densitometry of immunoblots, confocal images or FACS analysis. Data from primary cells was analyzed by nonparametric tests where possible; data from cell lines was logtransformed and analyzed by parametric tests as indicated. Coefficients of correlation (r) were calculated using the Spearman's rank test. P-value < 0.05 was considered statistically significant and is labeled with *, and p < 0.01 is labeled with **, p < 0.001 labeled with ***.
Ethics statement
The Regional Committees for Medical and Health Research Ethics at the Norwegian University of Science and Technology (NTNU) approved the use of PBMCs from healthy adult blood donors after informed consent (identification number 2009/2245–2). All donors provided written informed consent.
Abbreviations
| AA | = | arachidonic acid |
| ACTB | = | actin β |
| ALIS | = | aggresome-like induced structures |
| ATG8 | = | autophagy-related 8 |
| BafA1 | = | bafilomycin A1 |
| CHUK/IKKα | = | conserved helix-loop-helix ubiquitous kinase |
| COX4I1 | = | cytochrome c oxidase subunit 4I1 |
| CXCL10 | = | C-X-C motif chemokine ligand 10 |
| DHA | = | docosahexaenoic acid |
| DUB | = | deubiquitinating enzyme |
| FFAR4/GPR120 | = | free fatty acid receptor 4 |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| HMOX1 | = | heme oxygenase 1 |
| IB | = | immunoblot |
| IFN | = | interferon |
| IFNAR1 | = | interferon α and β receptor subunit 1 |
| IKBKB/IKKβ | = | inhibitor of nuclear factor kappa B kinase subunit β |
| IP | = | immunoprecipitation |
| IRF | = | interferon regulatory factor |
| KEAP1 | = | kelch like ECH associated protein 1 |
| LIR | = | light chain 3 (LC3)-interacting region |
| LMNA | = | lamin A/C |
| LPS | = | lipopolysaccharide |
| MAP1LC3B | = | microtubule associated protein 1 light chain 3 β |
| MAPK1/ERK2 | = | mitogen-activated protein kinase 1 |
| MAPK3/ERK1 | = | mitogen-activated protein kinase 3 |
| MDM | = | monocyte-derived macrophages |
| n-3 PUFA | = | omega-3 polyunsaturated fatty acid |
| NAC | = | N-acetylcysteine |
| NFE2L2/NRF2 | = | nuclear factor, erythroid 2 like 2 |
| NFKB | = | nuclear factor kappa B |
| NFKB2/p105 | = | nuclear factor kappa B subunit 2 |
| NQO1 | = | NAD(P)H quinone dehydrogenase 1 |
| OA | = | oleic acid |
| PCNA | = | proliferating cell nuclear antigen |
| PIC | = | phosphatase inhibitor cocktail |
| RELA/p65 | = | RELA proto-oncogene, NF-kB subunit |
| p(I:C) | = | poly I:C |
| ROS | = | reactive oxygen species |
| RPE | = | retinal pigment epithelium |
| SLC7A11/xCT | = | solute carrier family 7 member 11 |
| SQSTM1/p62 | = | sequestosome 1 |
| STAT1 | = | signal transducer and activator of transcription 1 |
| TAX1BP1 | = | Tax1 binding protein 1 |
| TBK1 | = | TANK binding kinase 1 |
| TLR4 | = | toll like receptor 4 |
| TNFAIP3/A20 | = | TNF α induced protein 3 |
| TNF | = | tumor necrosis factor |
| Ub | = | ubiquitin |
| UBA | = | ubiquitin-associated domain |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed
KAUP_A_1345411_Supplemental.zip
Download Zip (3.1 MB)Acknowledgement
We would like to thank the Cellular and Molecular Imaging Core Facility (CMIC), Faculty of Medicine, NTNU for access to instruments and support. We thank the Proteomics core facility at the Institute of Medical Biology, University of Tromsø for the use of instrumentation and expert assistance. We further thank the OmaCor-study group for providing plasma samples from their study on heart transplant patients and Terje Johansen and Kate Fitzgerald for their advice.
Funding
This work was supported by grants from the Norwegian Cancer Society and from the Research Council of Norway through its Centres of Excellence funding program, project number 223255/F50.
References
- Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4-9. doi:10.1093/gerona/glu057. PMID:24833586.
- Calder PC. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochimica Et Biophysica Acta. 2015;1851:469-84. doi:10.1016/j.bbalip.2014.08.010. PMID:25149823.
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687-98. doi:10.1016/j.cell.2010.07.041. PMID:20813258
- Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, Guarda G, Tian Z, Tschopp J, Zhou R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38:1154-63. doi:10.1016/j.immuni.2013.05.015. PMID:23809162.
- Liu Y, Chen LY, Sokolowska M, Eberlein M, Alsaaty S, Martinez-Anton A, Logun C, Qi HY, Shelhamer JH. The fish oil ingredient, docosahexaenoic acid, activates cytosolic phospholipase A(2) via GPR120 receptor to produce prostaglandin E(2) and plays an anti-inflammatory role in macrophages. Immunology. 2014;143:81-95. doi:10.1111/imm.12296. PMID:24673159.
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nature Cell Biology. 2007;9:1102-9. doi:10.1038/ncb1007-1102. PMID:17909521
- Levine B, Klionsky DJ. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463-77. doi:10.1016/S1534-5807(04)00099-1. PMID:15068787.
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-45. doi:10.1074/jbc.M702824200. PMID:17580304.
- Bjorkoy G, Lamark T, Johansen T. p62/SQSTM1: A missing link between protein aggregates and the autophagy machinery. Autophagy. 2006;2:138-9. doi:10.4161/auto.2.2.2405. PMID:16874037.
- Liu XD, Ko S, Xu Y, Fattah EA, Xiang Q, Jagannath C, Ishii T, Komatsu M, Eissa NT. Transient aggregation of ubiquitinated proteins is a cytosolic unfolded protein response to inflammation and endoplasmic reticulum stress. J Biol Chem.. 2012;287:19687-98. doi:10.1074/jbc.M112.350934. PMID:22518844.
- Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nature Immunology. 2009;10:1215-21. doi:10.1038/ni.1800. PMID:19820708.
- Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909-16. doi:10.4049/jimmunol.0900441.
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228-33. doi:10.1126/science.1205405. PMID:21617041.
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264-8. doi:10.1038/nature07383. PMID:18849965.
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology. 2011;12:222-30. doi:10.1038/ni.1980. PMID:21151103.
- Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1[beta] production by targeting ubiquitinated inflammasomes for destruction. Nature Immunology. 2012;13:255-63. doi:10.1038/ni.2215. PMID:22286270.
- Corn JE, Vucic D. Ubiquitin in inflammation: the right linkage makes all the difference. Nature Structural & Molecular Biology. 2014;21:297-300. doi:10.1038/nsmb.2808.
- Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem.. 2005;280:35625-9. doi:10.1074/jbc.C500237200. PMID:16079148.
- Zotti T, Scudiero I, Settembre P, Ferravante A, Mazzone P, D'Andrea L, Reale C, Vito P, Stilo R. TRAF6-mediated ubiquitination of NEMO requires p62/sequestosome-1. Molecular Immunology. 2014;58:27-31. doi:10.1016/j.molimm.2013.10.015. PMID:24270048.
- Johansson I, Monsen VT, Pettersen K, Mildenberger J, Misund K, Kaarniranta K, Schonberg S, Bjorkoy G. The marine n-3 PUFA DHA evokes cytoprotection against oxidative stress and protein misfolding by inducing autophagy and NFE2L2 in human retinal pigment epithelial cells. Autophagy. 2015;11:1636-51. doi:10.1080/15548627.2015.1061170. PMID:26237736.
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. Journal of Cell Biology. 2005;171:603-14. doi:10.1083/jcb.200507002. PMID:16286508.
- Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108:1427-32. doi:10.1073/pnas.1014156108. PMID:21220332.
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. Journal of Biological Chemistry. 2000;275:16023-9. doi:10.1074/jbc.275.21.16023. PMID:10821856.
- Ishii T, Yanagawa T, Yuki K, Kawane T, Yoshida H, Bannai S. Low micromolar levels of hydrogen peroxide and proteasome inhibitors induce the 60-kDa A170 stress protein in murine peritoneal macrophages. Biochem Biophys Res Commun. 1997;232:33-7. doi:10.1006/bbrc.1997.6221. PMID:9125146.
- Muller M, Carter S, Hofer MJ, Campbell IL. Review: The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity–a tale of conflict and conundrum. Neuropathol Appl Neurobiol. 2010;36:368-87. doi:10.1111/j.1365-2990.2010.01089.x. PMID:20487305.
- van den Borne P, Quax PH, Hoefer IE, Pasterkamp G. The multifaceted functions of CXCL10 in cardiovascular disease. BioMed Res Int. 2014;2014:893106. doi:10.1155/2014/893106. PMID:24868552.
- Ohmori Y, Hamilton TA. The interferon-stimulated response element and a kappa B site mediate synergistic induction of murine IP-10 gene transcription by IFN-gamma and TNF-alpha. J Immunol. 1995;154:5235-44..
- Brownell J, Bruckner J, Wagoner J, Thomas E, Loo YM, Gale M, Jr, Liang TJ, Polyak SJ. Direct, interferon-independent activation of the CXCL10 promoter by NF-kappaB and interferon regulatory factor 3 during hepatitis C virus infection. J Virol. 2014;88:1582-90. doi:10.1128/JVI.02007-13. PMID:24257594.
- Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4:a006049. doi:10.1101/cshperspect.a006049. PMID:22296764.
- Ohmori Y, Hamilton TA. Requirement for STAT1 in LPS-induced gene expression in macrophages. J Leukoc Biol. 2001;69:598-604. PMID:11310846.
- Doyle S, Vaidya S, O'Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, et al. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251-63. doi:10.1016/S1074-7613(02)00390-4. PMID:12354379.
- Fujita T, Reis LF, Watanabe N, Kimura Y, Taniguchi T, Vilcek J. Induction of the transcription factor IRF-1 and interferon-beta mRNAs by cytokines and activators of second-messenger pathways. Proc Natl Acad Sci U S A. 1989;86:9936-40. doi:10.1073/pnas.86.24.9936. PMID:2557635.
- Shultz DB, Rani MR, Fuller JD, Ransohoff RM, Stark GR. Roles of IKK-beta, IRF1, and p65 in the activation of chemokine genes by interferon-gamma. J Interferon Cytokine Res. 2009;29:817-24. doi:10.1089/jir.2009.0034.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722-37. doi:10.1038/nri3532. PMID:24064518.
- Newman AC, Scholefield CL, Kemp AJ, Newman M, McIver EG, Kamal A, Wilkinson S. TBK1 kinase addiction in lung cancer cells is mediated via autophagy of Tax1bp1/Ndp52 and non-canonical NF-kappaB signalling. PloS One. 2012;7:e50672. doi:10.1371/journal.pone.0050672. PMID:23209807.
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130-9. doi:10.1128/MCB.24.16.7130-7139.2004. PMID:15282312.
- Fan W, Tang Z, Chen D, Moughon D, Ding X, Chen S, Zhu M, Zhong Q. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy. 2010;6:614-21. doi:10.4161/auto.6.5.12189. PMID:20495340.
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275-85. doi:10.1128/MCB.00248-10. PMID:20421418.
- Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984-95. doi:10.1172/JCI25790. PMID:16585964.
- Kim J, Cha YN, Surh YJ. A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat Res. 2010;690:12-23. doi:10.1016/j.mrfmmm.2009.09.007. PMID:19799917.
- Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, Biswal S. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med. 2011;184:928-38. doi:10.1164/rccm.201102-0271OC. PMID:21799073.
- Kuhn AM, Tzieply N, Schmidt MV, von Knethen A, Namgaladze D, Yamamoto M, Brune B. Antioxidant signaling via Nrf2 counteracts lipopolysaccharide-mediated inflammatory responses in foam cell macrophages. Free Radic Biol Med. 2011;50:1382-91. doi:10.1016/j.freeradbiomed.2011.02.036.
- Awuh JA, Haug M, Mildenberger J, Marstad A, Do CP, Louet C, Stenvik J, Steigedal M, Damas JK, Halaas O, et al. Keap1 regulates inflammatory signaling in Mycobacterium avium-infected human macrophages. Proc Natl Acad Sci U S A. 2015;112:E4272-80. doi:10.1073/pnas.1423449112. PMID:26195781.
- Shembade N, Harhaj NS, Liebl DJ, Harhaj EW. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J. 2007;26:3910-22. doi:10.1038/sj.emboj.7601823. PMID:17703191.
- Saitoh T, Yamamoto M, Miyagishi M, Taira K, Nakanishi M, Fujita T, Akira S, Yamamoto N, Yamaoka S. A20 is a negative regulator of IFN regulatory factor 3 signaling. J Immunol. 2005;174:1507-12. doi:10.4049/jimmunol.174.3.1507.
- Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. The J Biol Chem. 2010;285:14999-5009. doi:10.1074/jbc.M110.109819. PMID:20304918.
- Husain S, Resende MR, Rajwans N, Zamel R, Pilewski JM, Crespo MM, Singer LG, McCurry KR, Kolls JK, Keshavjee S, et al. Elevated CXCL10 (IP-10) in bronchoalveolar lavage fluid is associated with acute cellular rejection after human lung transplantation. Transplantation. 2014;97:90-7. doi:10.1097/TP.0b013e3182a6ee0a. PMID:24025324.
- Zhuang J, Shan Z, Ma T, Li C, Qiu S, Zhou X, Lin L, Qi Z. CXCL9 and CXCL10 accelerate acute transplant rejection mediated by alloreactive memory T cells in a mouse retransplantation model. Exp Ther Med. 2014;8:237-42. PMID:24944628.
- Holm T, Andreassen AK, Aukrust P, Andersen K, Geiran OR, Kjekshus J, Simonsen S, Gullestad L. Omega-3 fatty acids improve blood pressure control and preserve renal function in hypertensive heart transplant recipients. Eur Heart J. 2001;22:428-36. doi:10.1053/euhj.2000.2369. PMID:11207085.
- Holm T, Berge RK, Andreassen AK, Ueland T, Kjekshus J, Simonsen S, Froland S, Gullestad L, Aukrust P. Omega-3 fatty acids enhance tumor necrosis factor-alpha levels in heart transplant recipients. Transplantation. 2001;72:706-11. doi:10.1097/00007890-200108270-00025. PMID:11544435.
- Kurinna S, Muzumdar S, Kohler UA, Kockmann T, Auf dem Keller U, Schafer M, Werner S. Autocrine and paracrine regulation of keratinocyte proliferation through a Novel Nrf2-IL-36 gamma pathway. J Immunol. 2016;196:4663-70. doi:10.4049/jimmunol.1501447.
- Lelouard H, Gatti E, Cappello F, Gresser O, Camosseto V, Pierre P. Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature. 2002;417:177-82. doi:10.1038/417177a. PMID:12000969.
- Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, Bazett-Jones DP, Brumell JH. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy. 2006;2:189-99. doi:10.4161/auto.2731. PMID:16874109.
- Sergin I, Bhattacharya S, Emanuel R, Esen E, Stokes CJ, Evans TD, Arif B, Curci JA, Razani B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Science Signaling. 2016;9:ra2. doi:10.1126/scisignal.aad5614. PMID:26732762.
- Lamark T, Perander M, Outzen H, Kristiansen K, Overvatn A, Michaelsen E, Bjorkoy G, Johansen T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278:34568-81. doi:10.1074/jbc.M303221200. PMID:12813044.
- Donohue E, Balgi AD, Komatsu M, Roberge M. Induction of covalently crosslinked p62 oligomers with reduced binding to polyubiquitinated proteins by the autophagy inhibitor verteporfin. PloS One. 2014;9:e114964. doi:10.1371/journal.pone.0114964. PMID:25494214.
- Decker EA, Akoh CC, Wilkes RS. Incorporation of (n-3) fatty acids in foods: Challenges and opportunities. J Nutr. 2012;142:610S-3S. doi:10.3945/jn.111.149328. PMID:22279141.
- Zhao Y, Zhang CF, Rossiter H, Eckhart L, Konig U, Karner S, Mildner M, Bochkov VN, Tschachler E, Gruber F. Autophagy is induced by UVA and promotes removal of oxidized phospholipids and protein aggregates in epidermal keratinocytes. J Invest Dermatol. 2013;133:1629-37. doi:10.1038/jid.2013.26. PMID:23340736.
- Awada M, Soulage CO, Meynier A, Debard C, Plaisancie P, Benoit B, Picard G, Loizon E, Chauvin MA, Estienne M, et al. Dietary oxidized n-3 PUFA induce oxidative stress and inflammation: role of intestinal absorption of 4-HHE and reactivity in intestinal cells. J Lipid Res. 2012;53:2069-80. doi:10.1194/jlr.M026179. PMID:22865918.
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213-23. PMID:20173742.
- Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576-91. doi:10.1074/jbc.M110.118976. PMID:20452972.
- Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochimica Et Biophysica Acta 2017;1863:585-97. doi:10.1016/j.bbadis.2016.11.005. PMID:27825853.
- Arisawa T, Tahara T, Shibata T, Nagasaka M, Nakamura M, Kamiya Y, Fujita H, Hasegawa S, Takagi T, Wang FY, et al. The relationship between Helicobacter pylori infection and promoter polymorphism of the Nrf2 gene in chronic gastritis. Int J Mol Med. 2007;19:143-8. PMID:17143558.
- Marzec JM, Christie JD, Reddy SP, Jedlicka AE, Vuong H, Lanken PN, Aplenc R, Yamamoto T, Yamamoto M, Cho HY, et al. Functional polymorphisms in the transcription factor NRF2 in humans increase the risk of acute lung injury. FASEB J. 2007;21:2237-46. doi:10.1096/fj.06-7759com. PMID:17384144.
- Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737-46. doi:10.1161/CIRCRESAHA.109.215715. PMID:20651288.
- Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys. 2014;564:83-8. doi:10.1016/j.abb.2014.09.005. PMID:25241054.
- Wang H, Khor TO, Saw CL, Lin W, Wu T, Huang Y, Kong AN. Role of Nrf2 in suppressing LPS-induced inflammation in mouse peritoneal macrophages by polyunsaturated fatty acids docosahexaenoic acid and eicosapentaenoic acid. Mol Pharm. 2010;7:2185-93. doi:10.1021/mp100199m. PMID:20831192.
- Ertel W, Kremer JP, Kenney J, Steckholzer U, Jarrar D, Trentz O, Schildberg FW. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood. 1995;85:1341-7. PMID:7858264.
- Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164:5564-74. doi:10.4049/jimmunol.164.11.5564.
- Vasconcellos LR, Dutra FF, Siqueira MS, Paula-Neto HA, Dahan J, Kiarely E, Carneiro LA, Bozza MT, Travassos LH. Protein aggregation as a cellular response to oxidative stress induced by heme and iron. Proc Natl Acad Sci U S A. 2016;113:E7474-82. doi:10.1073/pnas.1608928113. PMID:27821769.
- Ni C, Narzt MS, Nagelreiter IM, Zhang CF, Larue L, Rossiter H, Grillari J, Tschachler E, Gruber F. Autophagy deficient melanocytes display a senescence associated secretory phenotype that includes oxidized lipid mediators. Int J Biochem Cell Biol. 2016;81:375-82. doi:10.1016/j.biocel.2016.10.006.
- Li Z, Choi JH, Oh HJ, Park SH, Lee JB, Yoon KC. Effects of eye drops containing a mixture of omega-3 essential fatty acids and hyaluronic acid on the ocular surface in desiccating stress-induced murine dry eye. Curr Eye Res. 2014; 39:871-8. doi:10.3109/02713683.2014.884595. PMID:24559509.
- Sheridan DA, Bridge SH, Crossey MM, Felmlee DJ, Fenwick FI, Thomas HC, Neely RD, Taylor-Robinson SD, Bassendine MF. Omega-3 fatty acids and/or fluvastatin in hepatitis C prior non-responders to combination antiviral therapy - a pilot randomised clinical trial. Liver Int. 2014;34:737-47. doi:10.1111/liv.12316. PMID:24118830.
- Turowski JB, Pietrofesa RA, Lawson JA, Christofidou-Solomidou M, Hadjiliadis D. Flaxseed modulates inflammatory and oxidative stress biomarkers in cystic fibrosis: A pilot study. BMC Complement Altern Med. 2015;15:148. doi:10.1186/s12906-015-0651-2. PMID:25963404.
- Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev. 2015;26:311-27. doi:10.1016/j.cytogfr.2014.11.009.
- Bookout AL, Mangelsdorf DJ. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl Recept Signal. 2003;1:e012. doi:10.1621/nrs.01012. PMID:16604184.
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850-8. doi:10.1021/ac950914h. PMID:8779443.
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata JI, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149-63. doi:10.1016/j.cell.2007.10.035. PMID:18083104.