
ABSTRACT
Myasthenia gravis is an autoimmune disorder of the neuromuscular junction manifested as fatigable muscle weakness, which is typically caused by pathogenic autoantibodies against postsynaptic CHRN/AChR (cholinergic receptor nicotinic) in the endplate of skeletal muscle. Our previous studies have identified CA3 (carbonic anhydrase 3) as a specific protein insufficient in skeletal muscle from myasthenia gravis patients. In this study, we investigated the underlying mechanism of how CA3 insufficiency might contribute to myasthenia gravis. Using an experimental autoimmune myasthenia gravis animal model and the skeletal muscle cell C2C12, we find that inhibition of CAR3 (the mouse homolog of CA3) promotes CHRN internalization via a lipid raft-mediated pathway, leading to accelerated degradation of postsynaptic CHRN. Activation of CAR3 reduces CHRN degradation by suppressing receptor endocytosis. CAR3 exerts this effect by suppressing chaperone-assisted selective autophagy via interaction with BAG3 (BCL2-associated athanogene 3) and by dampening endoplasmic reticulum stress. Collectively, our study illustrates that skeletal muscle cell CAR3 is critical for CHRN homeostasis in the neuromuscular junction, and its deficiency leads to accelerated degradation of CHRN and development of myasthenia gravis, potentially revealing a novel therapeutic approach for this disorder.
Introduction
Myasthenia gravis is an autoimmune disorder of the neuromuscular junction that is manifested as fatigable muscle weakness.Citation1,2 The disorder is typically caused by pathogenic autoantibodies against postsynaptic CHRN/AChR (cholinergic receptor nicotinic) in the endplate of skeletal muscle. In some cases, the targets of autoantibodies are non-CHRN components of the postsynaptic muscle endplate, such as MUSK (muscle, skeletal, receptor tyrosine kinase), and LRP4 (low density lipoprotein receptor-related protein 4).Citation3,4 Various T cell subsets have been reported to contribute to the pathogenesis of myasthenia gravis,Citation5-7 and thymectomy can relieve some clinical symptoms in CHRN antibody-positive patients, especially for those with thymoma.Citation8 Innate immune components, such as complement and toll-like receptors, are also involved in myasthenia gravis etiology, putatively through inflammation or other mechanisms.Citation9,10 In either case, these pathogenic immunological factors cause loss of functional CHRN in the postsynaptic endplate, leading to a decrease in endplate potential amplitudes in the neuromuscular junction that fall below the threshold required for muscle fiber activation, consequently resulting in neuromuscular transmission failure.Citation1
CAs (carbonic anhydrases), catalysts of a simple reaction that converts CO2 to bicarbonate ion and protons, are ubiquitously expressed metalloenzymes that participate in diverse physiological and pathological events, such as gluconeogenesis and tumorigencity.Citation11 There are 4 evolutionary gene families (α, β, γ, and δ), but only the α family exists in mammalian cells. Until now, 16 different isozymes of α-CA (referred to as CA hereafter) have been identified in mammals, with distinct tissue distribution, subcellular localization, and catalytic activity.Citation12 Five of the isozymes are cytosolic (CA1, CA2, CA3, CA7, and CA8) and five are membrane-bound (CA4, CA9, CA12, CA14, and CA15). CA5A and CA5B are found in mitochondria, while CA6 is a secretory form. CA3 is abundantly expressed in skeletal muscle and liver of both humans and rodents,Citation12 and has been linked to certain autoimmune diseases such as rheumatoid arthritis.Citation13 Our previous study demonstrated that CA3 was specifically decreased in skeletal muscle from patients afflicted with myasthenia gravis, suggesting the potential role of CA3 in the pathogenesis or manifestation of this disease.Citation14 However, the underlying mechanism of CA3 in the development of myasthenia gravis is poorly understood.
Eukaryotic cells utilize endocytosis for internalization of nutrients, regulation of signal transduction, elimination of pathogens, presentation of antigens and an array of other physiological processes.Citation15-18 Besides phagocytosis, endocytosis can be further categorized into 3 broad pathways: micropinocytosis, clathrin-mediated endocytosis (CME), and clathrin-independent endocytosis.Citation19,20 During pathogenesis of myasthenia gravis, autoantibody-induced endocytosis of CHRN is assumed to be a major mechanism of modulation at neuromuscular junctions.Citation1,21-24 Cytokines from T cells and complement also potentially promote postsynaptic CHRN endocytosis.Citation10,25 In addition, CHRN is internalized via a RAC-dependent, DNM (dynamin)-independent endocytic pathway to late endosomes, which usually represents a degradative pathway.Citation22 Indeed, a therapeutic strategy aimed to reduce the endocytosis of CHRN has been proposed, and could be a promising novel therapy for myasthenia gravis.Citation26,27
In the present study, we investigate the effects of CAR3 (the mouse homolog for human CA3) on CHRN endocytosis in muscle cells, and our results show that CAR3 activity reduces CHRN endocytosis and suppresses the downregulation of surface receptors, thereby alleviating myasthenia gravis symptoms. This novel mechanism highlights the essential role of CAR3 in regulating CHRN endocytosis and provides a potential therapeutic target for treatment of myasthenia gravis.
Results
Activation of CAR3 attenuates myasthenia in EAMG mice
Our previous studies have identified CA3 as a specific protein insufficient in skeletal muscle from myasthenia gravis patients,Citation14 but how CA3 insufficiency contributes to the disorder remains to be determined. In order to examine the underlying mechanism of CA3 insufficiency in myasthenia gravis, we established an experimental autoimmune myasthenia gravis (EAMG) animal model by immunizing mice with purified Torpedo californica (Pacific electric ray) chrn. EAMG mice displayed typical myasthenia gravis symptoms, such as reduced response to muscle electronic stimulus ( and ), elevated serum anti-CHRN antibodies (), and progressively decreased grip power (). Only a few activators for CA3 have been identified, including 1-(2-aminoethyl) piperazine (AP), serotonin, and morpholine, and only AP can efficiently activate CA3 with high specificity.Citation28 We added AP (2 μg/ml) to the drinking water of some EAMG mice starting on the 4th wk post immunization, the time point for a second immunization (boost) (). AP treatment considerably recovered the grip power () and alleviated experimental myasthenia gravis symptoms as compared to EAMG mice receiving just water (). Thus, our results suggest that CAR3 inhibition plays a central role in the pathogenesis of EAMG.
Figure 1. Activation of CAR3 attenuates myasthenia in EAMG mice. Female C57BL/6 mice (6- to 8-wk old) were immunized s.c. with Torpedo californica chrn in complete Freund's adjuvant, and were boosted 4 wk later. After 4 more wk, the mice were anesthesized with sodium pentobarbital, fixed and connected to the electrodes of an electromyography machine. Compound muscle action potential in response to repetitive stimulation of the nerve at 3-Hz was recorded (A), and evoked action potential decrements were displayed (B). (C) Blood was collected from mice tail veins and serum was prepared and subjected to ELISA assay for serum anti-CHRN. (D) Paw grip endurance of EAMG mice with or without treatment with AP as per the protocol in (E). (E) Schematic plan of treatment for the mice. EAMG mice were established as mentioned in (A), other than 1-(2-aminoethyl) piperazine (AP, 2 μg/ml) was given in the drinking water from the second immunization (boost). (F) Clinical score of EAMG mice with or without treatment with AP. The images shown are representative of 3 independent experiments. Data are mean ± SEM of 3 independent experiments (B, C, and E), *p < 0.05.
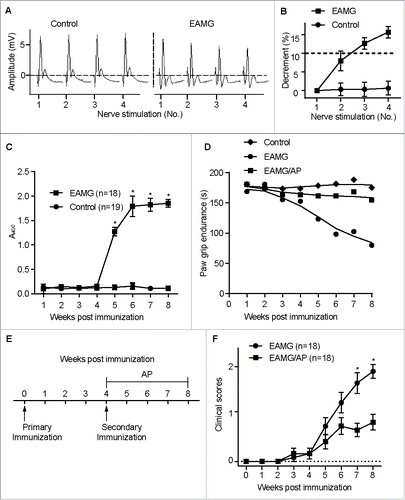
CAR3 regulates CHRN degradation in muscle cells
To further dissect how CAR3 works in the pathogenesis of myasthenia gravis, we examined the expression of CAR3 in EAMG mice given just water or water containing AP, the CAR3 agonist.Citation12 There were no detectable changes in protein expression of CAR3 in skeletal muscle whole cell lysates from control mice, EAMG mice, or EAMG mice with AP treatment ( and B). Because AP alleviates EAMG symptoms and the loss of CHRN is fundamental to the pathophysiology of myasthenia gravis, we investigated whether CAR3 regulates skeletal muscle CHRN degradation. Total CHRN protein expression was lower in skeletal muscle of EAMG mice than in control mice, whereas treatment of EAMG mice with AP prevented this loss in CHRN ( and D). However, there were no significant differences in mRNA for muscle-specific α1 subunit of Chrn (Chrna1) between these groups of mice (), implying the regulation of CAR3 on CHRN is post-transcriptional. Taken together, these data support the idea that CAR3 activation alleviates myasthenia gravis by reducing skeletal muscle CHRN protein degradation.
Figure 2. CAR3 regulates CHRN degradation in muscle cells. Gastrocnemius from control, EAMG and EAMG mice treated with AP was homogenized in lysis buffer containing 1% NP-40 and subject to SDS-PAGE and immunoblot analysis with anti-CAR3 (A) or anti-CHRN (C) antibodies. Densitometry quantification of total CAR3 (B) or CHRN (D) over ACTB/β-actin was performed using ImageJ software. (E) Total RNA was extracted from muscles, and quantitative reverse-transcribed PCR was performed using Chrna1-specific primer pairs. The results represent the mean ± SEM of 3 independent experiments (B, D, and E). *p < 0.05.
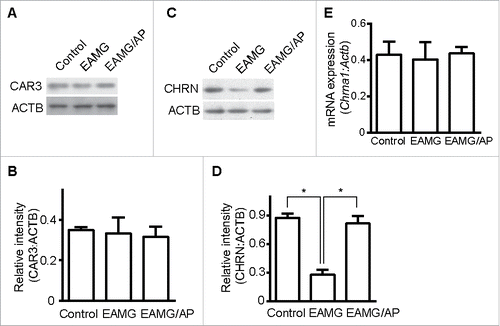
Endocytosis of CHRN via a lipid raft-mediated pathway
Endocytosis of membrane receptors is a dynamic process that regulates receptor expression, turnover and signaling.Citation15 Appropriate endocytosis of CHRN in skeletal muscle cells is essential for its homeostasis and function in neuromuscular transmission, whereas dysregulated endocytosis is involved in the pathogenesis of myasthenia gravis.Citation29 We used antibody-mediated cross-linking of CHRN to induce receptor endocytosis in C2C12 muscle cells. After incubating with Alexa Fluor 488-conjugated antibody, we used flow cytometry to quantify CHRN internalization. After 120 min, nearly 40% of surface CHRN was internalized following antibody treatment (). Previous investigators reported that CHRN in muscle cells is internalized via a clathrin-independent, RAC-dependent pathway.Citation22 Some small GTPases such as RHOA and RAC1 are involved in lipid raft-dependent endocytosis;Citation30,31 thus, we hypothesized that CHRN are internalized through a lipid raft-mediated pathway. We pretreated C2C12 muscle cells with methyl-beta-cyclodextrin (MβCD), which removes cholesterol from cell membranes and perturbs lipid rafts.Citation32 To exclude the effects of MβCD on apoptosis/necrosis of C2C12 cells, we stained the cells with ANXA5/annexin V and propidium iodide (PI), and found no significant change between MβCD-treated and control groups (Fig. S1). MβCD treatment largely eliminated CHRN endocytosis in C2C12 cells (). By contrast, treatment with Dynasore, a specific inhibitor of DNM GTPase that is required for CME, had no effect on CHRN internalization. Transfection of the dominant-negative RAC mutant, RAC1T17N/RAC N17, also prevented antibody-induced internalization of CHRN (, with representative FACS plots in Fig. S2A), consistent with a previous report.Citation22
Figure 3. Endocytosis of CHRN is mediated via a lipid raft-mediated pathway. (A) C2C12 cells were transfected with a plasmid encoding RAC1T17N, or pretreated with Dynasore (50 μM) or MβCD (5 μM) before incubation with Alexa Fluor 488-conjugated anti-CHRN antibody (mAb210) at 4°C for 1 h, then the cultures were switched to 37°C for different times to induce CHRN endocytosis. After acidic washes, the cells were fixed and analyzed using flow cytometry. (B) C2C12 cells were transfected with a plasmid encoding RAC1T17N, or pretreated with Dynasore (50 μM) or MβCD (5 μM) before being incubated with biotin-CHRN antibody (mAb210) at 4°C for 1 h, and then switched to 37°C for different times to induce CHRN endocytosis. After acidic washes, the cell lysate was prepared and subjected to SDS-PAGE and blotted with streptavidin-HRP. Shown is a representative image of 3 experiments (B), and the quantitative data are presented as the mean ± SEM of 3 experiments (A). *p < 0.05, between the MβCD group and the control group; # p < 0.05, between the RAC1T17N group and the control group.
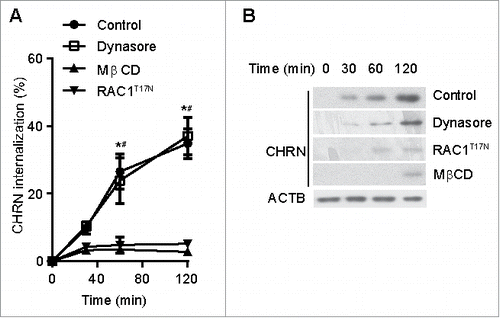
As an alternative approach, we monitored internalization of CHRN using immunoblotting after labeling C2C12 cells with biotin-conjugated CHRN antibody (biotin-mAb210). Endocytosis of CHRN was blocked by MβCD treatment and by transfection with a plasmid encoding dominant-negative RACT17N, whereas Dynasore treatment did not alter CHRN internalization (). These data are consistent with the idea that endocytosis of CHRN occurs via a lipid raft- and RAC-dependent, non-CME pathway in muscle cells.
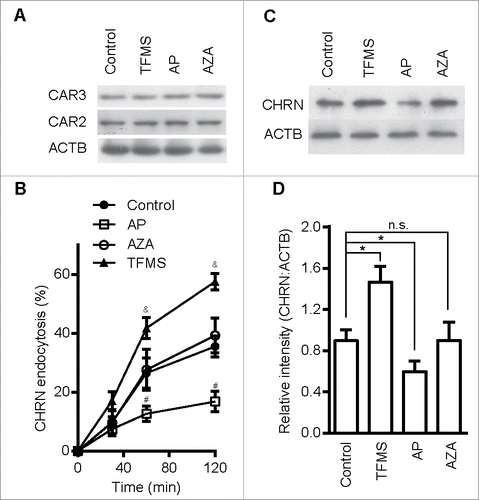
CAR3 specifically regulates CHRN endocytosis
Given that CAR3 insufficiency contributes to the pathogenesis of myasthenia gravis, and accelerated endocytosis of CHRN promotes receptor degradation, we hypothesized that CAR3 activation dampens CHRN endocytosis. To test our hypothesis, we pretreated C2C12 cells with the CAR3-specific agonist AP, the CAR3-specific antagonist trifluoromethanesulfonamide (TFMS), or acetazolamide (AZA), a carbonic anhydrase inhibitor that is less effective at inhibiting CAR3.Citation12 We observed no explicit effects of these compounds on apoptosis/necrosis of C2C12 cells based on ANXA5 and PI staining (Fig. S3). None of these compounds had an effect on CAR3 and CAR2 protein expression in C2C12 cells (). Treatment with TFMS accelerated the endocytosis of CHRN, whereas AP reduced CHRN endocytosis (, with representative FACS plots in Fig. S2B), consistent with the idea that CAR3 activation suppresses the endocytosis of surface CHRN. AZA, which has no observable effect on CAR3 enzyme activity, induced no detectable change in CHRN endocytosis (). In addition, endocytosis monitored with biotinylated CHRN antibody revealed that activation of CAR3 blocked CHRN endocytosis, whereas inhibition of CAR3 promoted CHRN endocytosis (). Collectively, our data indicate that CAR3 activity plays a role in mitigating endocytosis and subsequent degradation of CHRN in skeletal muscle cells.
Figure 4. CAR3 specifically regulates CHRN endocytosis. (A) C2C12 cells treated with AP (2 μg/ml), TFMS (2 mM) or AZA (1 mM) for 6 h, were lysed and subject to SDS-PAGE followed by immunoblot analysis using CAR3, CAR2 or ACTB antibodies. (B) C2C12 cells were treated with or without AP (2 μg/ml), TFMS (2 mM) or AZA (1 mM) for 6 h, followed by incubation with CHRN antibody (mAb210) at 4°C for 1 h, and then switched to 37°C for different times to induce CHRN endocytosis. After acidic washes, the cells were fixed and analyzed with flow cytometry. (C) C2C12 cells treated without or with AZA (1 mM), AP (2 μg/ml), TFMS (2 mM) for 6 h, and then labeled with biotin-CHRN antibody (mAb210). After further culture for 2 h, C2C12 cells were washed with acidic buffer, lysed and analyzed using SDS-PAGE and blotted with streptavidin-HRP. (D) The band densitometry was quantified using ImageJ software. Shown is a representative image of three experiments (A and C), and the quantitative data are presented as the mean ± SEM of 3 experiments (B). *p < 0.05, between the MβCD group and the control group; #p < 0.05, between the AP group and the control group; &p < 0.05, between the TFMS group and the control group.
CAR3 suppression leads to enhanced CHRN endocytosis
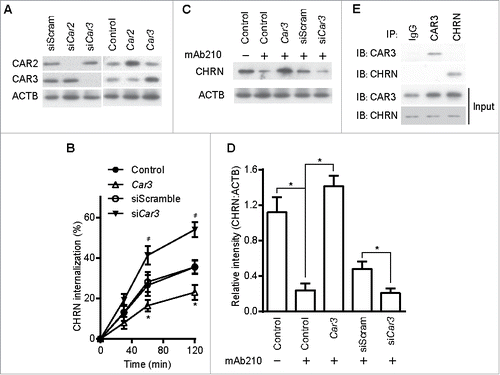
As an alternative approach to using pharmacological agents, we modified the expression of CAR3 using plasmid overexpression or siRNA-mediated knockdown. We transfected C2C12 cells with plasmids containing either Car3 or Car2 cDNA, or siRNA specific for each of these genes. The expression of CAR3 and CAR2 proteins was markedly increased following transfection with the respective cDNA plasmid (, right panel). C2C12 transfection with plasmids expressing siRNA for Car3 or Car2 resulted in decreased expression of the respective proteins (, left panel). Overexpression of CAR3 in C2C12 cells inhibited CHRN endocytosis, whereas siRNA-mediated CAR3 knockdown promoted it (), further confirming the central role of CAR3 in regulating the endocytosis of CHRN. Overexpression of CAR3 also attenuated CHRN degradation, whereas CAR3 knockdown enhanced receptor degradation ( and D). CAR3 is uniquely qualified for these roles, because neither overexpression nor knockdown of CAR2, another isoform of the CA family expressed in skeletal muscle cells, had an effect on CHRN endocytosis (Fig. S3). These data support the hypothesis that CAR3 has a specific role in regulation of CHRN endocytosis over other CA isoforms. However, we found no physical interaction between CAR3 and CHRN (), suggesting CAR3 renders its regulation of CHRN endocytosis through indirect mechanisms. Taken together, our results confirm that CAR3 is an important and specific regulator of CHRN endocytosis, which may serve as a fundamental mechanism in the pathogenesis of myasthenia gravis.
Figure 5. CAR3 suppression leads to enhanced CHRN endocytosis. C2C12 cells were transiently transfected with the indicated plasmids or specific siRNA using Lipofectamine 3000. 48 h later, the cells were either lysed and subjected to SDS-PAGE and immunoblotted with CAR2 or CAR3 antibody (A); or incubated with CHRN antibody (mAb210) at 4°C for 1 h, switched to 37°C for different times to induce CHRN endocytosis, subjected to acidic washes, then fixed and analyzed with flow cytometry (B); or were lysed and subjected to SDS-PAGE and immunoblotted with CHRN antibody (C). (D) The band densitometry was quantified using ImageJ software. (E) C2C12 cells were lysed and followed by immunoprecipitation with the indicated antibody, and then blotted with the specified antibodies. Data are mean ± SEM of 3 independent experiments, *p < 0.05, between the CAR3 group and the control group; #p < 0.05, between the siCar3 group and the siScram group.
CAR3 suppress endocytosis by repressing chaperone-assisted selective autophagy
Autophagy intersects with endocytosis at multiple steps and shares various molecular players, so it is reasonable to hypothesize that autophagy regulates endocytosis of CHRN.Citation33 To determine if autophagy is altered in myasthenia gravis, we measured the expression of autophagy regulators such as MAP1LC3A/B, BECN1, ATG5 and SQSTM1/p62 in skeletal muscle. Using immunoblot analysis, we observed significant changes in the expression of MAP1LC3A/B-II, but no visible changes in BECN1, ATG5 or SQSTM1 between control, EAMG, or EAMG mice treated with AP (). Staining of MAP1LC3A/B in skeletal muscle sections showed increased puncta in EAMG mice, but only sporadic puncta in that of control and the EAMG-AP group (), consistent with the immunoblot data. Additionally, knockdown of ATG7, an essential molecule for macroautophagy, in C2C12 cells using siRNA compromised the endocytosis of surface CHRN and the degradation of CHRN (). Using immunoprecipitation, we detected no interaction between CHRN and SQSTM1 in C2C12 cells (). CAR3 immunoprecipitated with BAG3 (BCL2-associated athanogene 3; ), which regulates autophagy.Citation34 As a co-chaperone, BAG3 interacts with HSPA8/HSC70 (heat shock protein 8), and contributes to chaperone-assisted selective autophagy (CASA).Citation35 CASA is essential for muscle maintainence.Citation36 To explore whether CASA is invovled in myasthenia gravis, we then detected the expression of HSPA8. Our data clearly demonstrated that the expression of HSPA8 was increased in muscle of myasthenic mice, whereas AP reversed the increase (), consistent with the development of myasthenia gravis symptoms in our mouse model (). These findings highlight the importance of CASA in the EAMG mouse model of myasthenia gravis. To investigate whether CHRN interacts with proteins from the CASA complex, such as BAG3 and HSPA8, we performed coimmunoprecipitation in C2C12 cells. The results show that CHRN interacted with BAG3 and HSPA8 (). We also observed that AP treatment inhibited, whereas TFMS treatment promoted, the degradation of FLNC (Fig. S4), a substrate of CASA-mediated degradation in muscle cells,Citation36 supporting the idea that CAR3 regulates CASA.
Figure 6. CAR3 suppresses endocytosis via a macroautophagy-mediated pathway. (A) Gastrocnemius from mice was homogenized in lysis buffer containing 1% NP-40, subject to SDS-PAGE and immunoblot analysis with the indicated antibody. Densitometric quantification of the indicated proteins over ACTB using ImageJ (right panel). (B) Gastrocnemius fixed, frozen sectioned, and permeabilized. After being blocked with 20% goat serum, the sections were incubated with anti-MAP1LC3A/B antibody and the appropriate Alexa Fluor 488-conjugated secondary antibody. After washing with PBS, the sections were examined with a confocal microscope and images were captured (left panel). Scale bar: 20 µm. MAP1LC3A/B-positive puncta were counted, and at least 50 cells were quantified. Puncta per cell are shown as mean ± SEM of 3 independent experiments (right panel). (C) C2C12 cells were transiently transfected with siScram or siAtg7 using Lipofectamine 3000. Forty-eight h later, the cells were either lysed and subjected to SDS-PAGE and immunoblot analysis with CHRN antibody (total CHRN, CHRN-t), or labeled with biotin-CHRN antibody (mAb210) and after 2 h the cells were washed with acidic buffer, lysed and analyzed by SDS-PAGE and immunblot analysis with streptavidin-HRP for endocytosed CHRN (CHRN-e). *p < 0.05, compared with the control group. Data are mean ± SEM of 3 independent experiments.
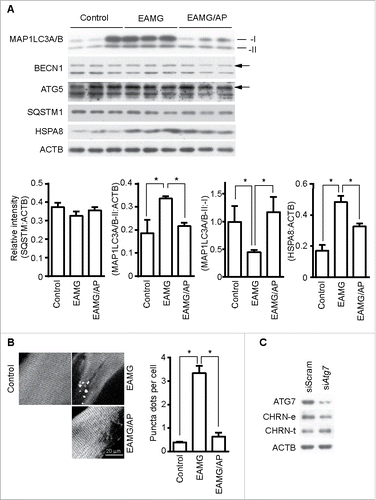
Figure 7. CAR3 suppresses endocytosis by repressing chaperone-assisted selective autophagy. (A) C2C12 cells were lysed, immunoprecipitated with the indicated antibody, then blotted with the specified antibodies. (B and C) C2C12 cells were lysed, followed by immunoprecipitation with the indicated antibody, and then blotted with the specified antibodies. (D) C2C12 cells were transiently transfected with specific siRNA using Lipofectamine 3000. Forty-eight h later, the cells were treated with TFMS (2 mM) for 6 h. Cell lysates from these cells were separated by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies. (E) C2C12 cells were transiently transfected with specific siRNA against Car3 using Lipofectamine 3000. Forty-eight h later, the cells with treated with TFMS (2 mM) for 6 h followed by incubation with CHRN antibody (mAb210) at 4°C for 1 h, and then switched to 37°C for different times to induce CHRN endocytosis. After acidic washes, the cells were fixed and analyzed with flow cytometry. (F) C2C12 cells were transiently transfected with siScram or siBag3 using Lipofectamine 3000. Forty-eight h later, the cells were lysed and subjected to SDS-PAGE and analyzed by immunoblotting with anti-BAG3 antibody. (G) C2C12 cells were transiently transfected with specific siRNA (siBag3) using Lipofectamine 3000. Forty-eight h later, either control C2C12 cells or siBag3-transfected cells were treated with vehicle or TFMS (2 mM) for 6 h followed by incubation with CHRN antibody (mAb210) at 4°C for 1 h, and then switched to 37°C for different times to induce CHRN endocytosis. After acidic washes, the cells were fixed and analyzed with flow cytometry. All immunoblotting and immunoprecipitation studies were performed 3 times. *p < 0.05, compared with the control group. Data are mean ± SEM of 3 independent experiments.
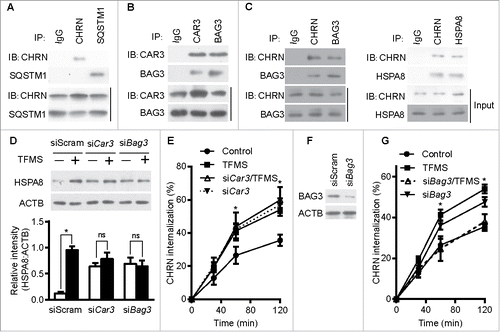
Based on our observations, we reasoned that CAR3 inhibits CASA in skeletal muscle. To determine if CASA is required for CAR3 to regulate CHRN endocytosis, we measured the expression of HSPA8 in C2C12 cells treated with or without TFMS, a specific CAR3 inhibitor. TFMS treatment increased the expression of HSPA8, and CAR3 knockdown maintained high expression of HSPA8 even in the absence of TFMS (based on the quantification of the amount of HSPA8 to that of ACTB; ). In addition, TFMS treatment promoted endocytosis of CHRN, and knockdown of CAR3 had a similar effect (). To gain further insight into whether regulation of CHRN endocytosis is dependent on BAG3, we took advantage of specific siRNA (siBag3) to knock down BAG3 in C2C12 cells (). Knockdown of BAG3 largely obviated the effect of TFMS, whereas knockdown of BAG3 without TFMS treatment led to increased CHRN endocytosis (), suggesting the essential role of CASA in CAR3-mediated regulation of CHRN endocytosis. Taken together, our results demonstrate that CAR3 suppresses endocytosis of CHRN by restraining CASA.
CAR3 regulates endocytosis of CHRN through ER stress
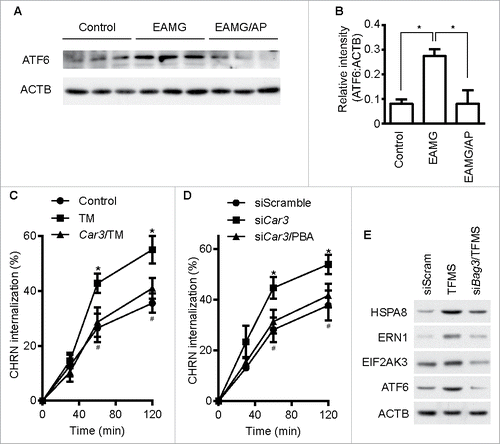
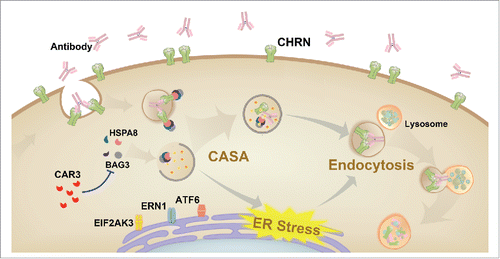
Endoplasmic reticulum (ER) stress is associated with myasthenia gravis because expression of the ER chaperone HSPA5/GRP78 is increased in skeletal muscle from patients with the disease.Citation37 We found that expression of the ER sensor ATF6 was increased in skeletal muscle from EAMG mice, and AP administration lowered its expression ( and B). These results suggest a potential role of ER stress in the pathogenesis of myasthenia gravis. Our previous study demonstrated that ER stress promotes CHRN degradation through accelerating endocytosis in muscle cells,Citation38 so we investigated whether CAR3 regulates ER stress in C2C12 cells. Overexpression of CAR3 largely reversed the tunicamycin-induced endocytosis of CHRN in C2C12 cells (). In addition, reduction of ER stress by treatment with 4-phenylbutyric acid (PBA), a chemical ER chaperone, diminished CHRN internalization in C2C12 cells with CAR3 knockdown (). To determine whether CASA regulates ER stress, we treated C2C12 cells with TFMS. TFMS promoted ER stress, whereas BAG3 knockdown largely obliterated this effect (). Therefore, our results indicate that CAR3 activity reduces endocytosis of CHRN through suppressing chaperone-assisted selective autophagy combined with dampening, at least partially, ER stress in skeletal muscle cells.
Figure 8. CAR3 regulates endocytosis of CHRN through ER stress. (A) Gastrocnemius from mice was homogenized in lysis buffer containing 1% NP-40, subject to SDS-PAGE and immunoblot analysis with the indicated antibody. (B) Densitometric quantification of the indicated proteins over ACTB was performed using ImageJ. (C) C2C12 cells were transiently transfected with the indicated plasmids using Lipofectamine 3000. Forty-eight h later, the cells were treated with tunicamycin (TM; 2 μM) for 12 h, followed by incubation with CHRN antibody (mAb210) at 4°C for 1 h, and then switched to 37°C for different times to induce CHRN endocytosis. After acidic washes, the cells were fixed and analyzed with flow cytometry. *p < 0.05, between the TM group and the control group; #p < 0.05, between the TM group and the Car3-TM group. (D) C2C12 cells were transiently transfected with the indicated specific siRNA using Lipofectamine 3000. Forty-eight h later, the cells with treated with PBA (2 μM) for 12 h followed by incubation with CHRN antibody (mAb210) at 4°C for 1 h, and then switched to 37°C for different times to induce CHRN endocytosis. After acidic washes, the cells were fixed and analyzed with flow cytometry. *p < 0.05, between siCar3 group and control group; #p < 0.05, between siCar3 group and siCar3-PBA group. (E) C2C12 cells were transiently transfected with siScramble or specific siRNA (siBag3) using Lipofectamine 3000. Forty-eight h later, the cells with treated with TFMS (2 mM) for 6 h followed by SDS-PAGE and immunoblot analysis with the indicated antibody. Data are mean ± SEM of 3 independent experiments (B, C, and D).
Discussion
In previous studies, we used 2-D gel electrophoresis combined with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry to identify a 25-kDa protein specifically insufficient in muscles from myasthenia gravis patients,Citation39-42 which was identified as CA3.Citation14 Several other isoforms of carbonic anhydrase are also expressed in skeletal muscle, such as CA2, CA4, CA9 and CA14.Citation43,44 Muscle contractile behaviors are usually normal when one specific carbonic anhydrase isoform is deficient,Citation44 suggesting the redundancy of CA isoforms in these tissues. However, we were the first to report that CA3 deficiency is associated with myasthenia gravis.Citation14
In order to further understand how CA3 insufficiency contributes to myasthenia gravis pathogenesis, we established an EAMG mouse model using Torpedo californica chrn. We also used the synthesized peptide α146-162 (LGIWT YDGTK VSISP ES), corresponding to region 146–162 of Torpedo chrn, as an antigen to establish the model. We found that injection of this synthetic peptide displayed similar efficiency to using the native Torpedo californica chrn, with more than 90% of the mice showing elevated anti-CHRN autoantibodies and considerable muscle weakness (Fig. S5). In contrast to that in human patients, we detected no considerable decrease for CAR3 expression in skeletal muscles from EAMG mice, putatively due to the short period of pathogenesis in this animal model. Because the activation profile of CA3 is distinct from all other CA isoforms investigated so far, only a few activators for CA3 have been identified, including 1-(2-aminoethyl) piperazine (AP), serotonin, and morpholine.Citation12 Among these, AP can efficiently activate CA3 with a KA of 0.32 μM, while inducing much less activation of other CA isoforms.Citation28 Administration of AP attenuated the development of myasthenia gravis-like symptoms, whereas administration of a CAR3 antagonist (TFMS) exacerbated such symptoms. A loss of CHRN is the fundamental pathophysiology for myasthenia gravis.Citation1,29 Accordingly, EAMG mice displayed increased CHRN degradation, and AP treatment reversed this loss of receptor. These finding support a central role of CAR3 in the pathogenesis of myasthenia gravis.
Among the patients with generalized myasthenia gravis, 80–90% of them are CHRN antibody positive; patients negative for anti- CHRN antibodies have serum autoantibodies against MUSK (approx. 40% in CHRN-negative patients), and others possess autoantibodies against other skeletal muscle antigens such as LRP4 and RYR1 (ryanodine receptor 1).Citation3,23,45 Obviously, autoantibodies are central to the pathogenesis of myasthenia gravis. Anti- CHRN antibodies, the most common in patients with the disease, contribute to myasthenia gravis through diverse mechanisms, including blockade of CHRN signaling, complement activation and accelerated CHRN degradation through endocytosis.Citation29 Usually, blockade of CHRN signaling and complement activation induced by antibody are regulated extracellularly, whereas CHRN endocytosis and degradation can be regulated both extra- and intracellularly. As CAR3 is a cytoplasmic molecule, we reason that CAR3 works through regulation of CHRN endocytosis.
Endocytosis of membrane proteins leads to either a recycling or degradation of products. For postsynaptic CHRN in mouse skeletal muscle cells, we found that CHRN is internalized through a clathrin-independent, lipid raft-dependent pathway that results in degradation, consistent with previous work.Citation22 However, other investigators report that endocytosis of CHRN is through a clathrin-dependent pathway in Xenopus muscle cells.Citation21 It is not clear whether the CHRN endocytosis pathway is species specific, but the significance underlying such differences might be of interest. Further understanding these endocytic pathways will shed more light on CHRN regulation and on the pathogenesis of myasthenia gravis. The present study reveals that CAR3 activation suppresses CHRN endocytosis, whereas AZA treatment or CAR3 overexpression had similar effects. These results emphasize the essential role of proper regulation of CHRN endocytosis and subsequent degradation in maintaining motor endplate function. CAR3 appears to play a specific role in these processes because overexpression or knockdown of another isoform of CAR expressed in skeletal muscle, CAR2,Citation43 lacks such effect.
Our study further reveals that CAR3-mediated regulation of CASA is a novel mechanism for CHRN endocytosis. Although autophagy has not been reported to contribute to the pathogenesis of myasthenia gravis, CASA has been shown to be essential for muscle maintenance,Citation36 whereas dysfunction of macroautophagy leads to severe reduction in muscle strength, degeneration of muscle fiber, and metabolic disorders.Citation46 In fact, a recent study showed that selective autophagy regulates CHRN turnover in a model of fasting-induced wasting of skeletal muscle through SQSTM1-involved degradation.Citation47 However, we observed no considerable changes in expression of SQSTM1 in EAMG mice, and this differential regulation of CHRN degradation by autophagy reflects a subtle function of CHRN in different diseases, an area worthy of future study. In the present study, we found that CAR3 physically interacts with BAG3, a co-chaperone of the HSPA8 system, and inhibits CASA. We observed an increase of HSPA8 expression in skeletal muscle from EAMG mice, and AP treatment attenuated this increase (). In addition, both BAG3 and HSPA8 interact with CHRN, suggesting that CASA may regulate CHRN endocytosis (). All of these findings point to the likelihood that CASA plays a role in the pathogenesis of myasthenia gravis. Increased MAP1LC3A/B expression by western blot, as well as the punctate structures observed by confocal microscopy of skeletal muscle from EAMG mice, support this assumption ( and B). Further study is needed to reveal the underlying mechanism of CASA in the pathogenesis of myasthenia gravis.
Autophagy may regulate CHRN endocytosis through inhibiting ER stress in skeletal muscle cells. ER stress affects various biological processes, such as immune responses,48 and contributes to physiological and pathological events such as metabolism,Citation49 senescence,Citation50 inflammationCitation51 and neurological disorders.Citation52 ER stress was increased in skeletal muscle from our experimental autoimmune myasthenia gravis mouse model, and treatment with the CAR3 agonist AP reversed the increase in ER stress. Moreover, AP diminished ER stress-induced endocytosis of CHRN, suggesting that the regulation of endocytosis by CAR3 is at least partially through reduction of ER stress. These findings unveil novel mechanisms of CAR3 in the pathogenesis of myasthenia gravis ().
Figure 9. Schematic diagram of the mechanisms of CAR3 in maintaining homeostasis of CHRN. CAR3 interacts with BAG3 to inhibit chaperone-assisted selective autophagy (CASA), which regulates endocytosis of CHRN through interaction between BAG3-HSPA8 and CHRN. CAR3 also reduces ER stress, preventing excessive degradation of CHRN in muscle cells.
Although no correlation has been confirmed between plasma CHRN antibody concentration and disease severity, changes in antibody concentration have been used to predict disease severity in patients given immunosuppressive drugs.Citation3 Functional activity of autoantibodies against CHRN is associated with the number of available receptors and is clinically relevant in the pathogenesis of myasthenia gravis.Citation53 In addition, because CHRN is also involved in other muscular disorders, such as congenital myasthenia syndromes and muscular atrophy,Citation54,55 further study of the regulation of CAR3 function in skeletal muscle is needed to identify a suitable therapeutic strategy for treating myasthenia gravis and other muscular disorders.
In summary, our study illustrates that CAR3 activity prevents CHRN endoycytosis via lipid rafts, a pathway typically leading to degradation of the receptor, and promotes its homeostasis in the cellular membrane. CAR3 deficiency allows CHRN endocytosis and consequent degradation of the receptor. Thus, insufficiency of CAR3 is pathogenic for myasthenia gravis. This novel mechanism highlights the essential role of CAR3 in regulation of CHRN endocytosis and may provide a novel therapeutic approach for treatment of myasthenia gravis and other neuromuscular disorders.
Materials and methods
Reagents and antibodies
Lysosome inhibitor E64d and proteasome inhibitor lactacystin were purchased from Sigma (E8640, and L6785). 1-(2-aminoethyl) piperazine (AP), trifluoromethanesulfonamide (TFMS), acetazolamide (AZA), 4-phenylbutyric acid (PBA), and methyl-β-cyclodextrin (MβCD) were obtained from Sigma (A55029, 638455, A6011, P21005, and C4555, respectively). CHRN antibody (mAb210, against α1/α3/α5 subunits) and SQSTM1 were obtained from Abcam (ab24719 [no longer available] and ab109012). Antibodies against CAR2 and CAR3 were purchased from Santa Cruz Biotechnology (sc-133111, and sc-50715). Antibodies for ACTB, MAP1LC3A/B, BECN1, ATG5, and ATG7 were obtained from Cell Signaling Technology (3700S, 12741, 3738, 12994, and 8558, respectively). Antibody for FLNC was obtained from Biorbyt (orb326498). HRP-conjugated anti-mouse and anti-rabbit secondary antibodies were purchased from Jackson ImmunoResearch (115–035-003, and 111–035-003). Biotinylation kit and Alexa Fluor 488 labeling kit for CHRN antibody were obtained from Thermo Fisher Scientific (90407, and A30006). Streptavidin-HRP and Tunicamycin were purchased from Sigma (18–152, and T7765). Dead Cell Apoptosis Kit with Annexin V Alexa Fluor™ 488 & Propidium Iodide was obtained from Invitrogen (V13245).
Mice
Female C57BL/6 mice (6- to 8-wk-old) were purchased from the Shanghai Laboratory Animal Center of the Chinese Academy of Sciences, and were kept under specific pathogen-free conditions in the animal center of Shanghai Jiao Tong University School of Medicine (Shanghai, China). All mouse experiments were approved by the Animal Welfare & Ethics Committee of the Shanghai Jiao Tong University School of Medicine. All efforts were made to minimize suffering.
Antigens and EAMG model
Torpedo californica chrn was provided by Dr. Baggi Fulvio (Neurological Institute “Carlo Besta”, Milan, Italy).Citation56 Peptide α146-162 (LGIWT YDGTK VSISP ES), corresponding to region 146–162 of Torpedo chrn, was synthesized and conjugated with poly-L-lysine by GL Biochem (Shanghai). The chrn (20 μg) in complete Freund's adjuvant (Sigma, F5881) in a total of volume of 200 μl per mouse was injected subcutaneously (s.c.) into 2 hind foot-pads and the shoulders, and mice were boosted 28 d later using the same protocol.Citation57 In some experiments, mice were immunized s.c. using the same protocol but with 50 μg of α146–162 synthesized peptide as antigen instead. The mice were checked weekly for signs of muscle weakness, and EAMG scores were assigned to each injected mouse based on the following scale:Citation58 grade 0, normal muscle strength and no muscle weakness, even after exercise (20 to 30 consecutive paw grips); grade 1, normal at rest but weak after exercise, with chin on the floor and inability to raise head, hunched back, and reduced mobility; grade 2, weakness at rest; and grade 3, moribund, dehydrated, and paralyzed.
Evaluation of EAMG using electromyography
Mice were anesthetized with sodium pentobarbital, fixed and connected to electrodes of an electromyography machine. A positive (stimulating) and a negative electrode were inserted into the sciatic notch and abdominal wall, respectively. A recoding electrode was inserted into the gastrocnemius muscle of the same leg. After stimulating the sciatic nerve with a set of 8 3-Hz supramaximal stimuli, the evoked muscle action responses were recorded using an EMG machine (Haishen NDI-200P+, Shanghai). The repetitive stimulation was performed 3 times for each mouse to determine the percentage of decrement. Responses to repeated stimulation were evaluated by comparing the first evoked response with the fourth one.
Paw grip endurance
Paw grip strength of mice was evaluated by paw grip endurance (PaGE) weekly from 1 wk post immunization, as previously described with modification.Citation59 The mouse was placed on the wire lid of a conventional housing cage, and was held approximately 50 cm over an open cage bottom. The mouse was prompted to fasten its grip by gently shaking, and then the lid was flipped upside down. The time was recorded until the mouse had fallen off the grid with at least both hind limbs, for an arbitrary maximum of 200 sec. Three attempts were allowed for each mouse, and the longest duration was recorded for data analysis.
Cell culture and transfection
The C2C12 cell line was purchased from American Type Culture Collection (ATCC, CRL-1772™) and cultured as described.Citation60 Briefly, C2C12 cells and HEK 293 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum. The cells were transfected using Lipofectamine 2000 or 3000 (Invitrogen, 11668019 and L3000015).
Overexpression of CAR3 in C2C12 cells was performed with lentivirus based on pLVX-IRES-zsGreen (Clontech Laboratories, 632187) containing the Car3 sequence amplified from mouse cDNA. siRNAs targeting Bag3, Car2, and Car3, and scrambled siRNA were purchased from Life Technologies (71816, 66069, and 60576), and knockdown assays were performed as previously described.Citation61
Immunoblotting
Immunoblotting was performed as previously described.Citation62 Briefly, muscle was homogenized in lysis buffer (Thermo Scientific, 78501) containing 1% NP-40 (Thermo Scientific Pierce, 28324) until no visible clumps were evident. Following brief vortexing, the lysates were centrifuged for 20 min at 15,000 g, and the supernatants were separated by SDS-PAGE and transferred to PVDF membranes. These membranes were blocked with 5% fat-free milk in phosphate-buffered saline (PBS, Gibco, 21600-044) with 0.1% Tween-20 (Sinopharm Chemicals, 30189328) and incubated with primary antibody followed by the proper HRP-conjugated secondary antibody. After subsequent washes, the immunoreactive bands were detected with ECL-Plus immunoblotting detection reagent (Amersham Pharmacia Biotech, RPN2132). ImageJ software (National Institutes of Health) was used to quantify band densitometry for immunoblot images.
RT-PCR
RT-PCR for Chrna1, Car2 and Car3 was performed using the primer pairs as follows: Chrna1 (forward: 5′ atg gaa tcc aga tga cta tg 3′, reverse: 5′ tgg ctg gcg gtg tcc agg tg 3′); Car2 (forward: 5′ gat aaa gct gcg tc aag ag 3′, reverse: 5′ agc ccc agt gaa agt gaa ac 3′); Car3 (forward: 5′ ata cgc tgc tga gct tca cc 3′, reverse: 5′ att ttg tcc agg gca tca ag 3′); Actb (forward: 5′ cat ggc att gtt acc aac tg 3′, reverse: 5′ cac ggt tgg cct tag ggt tc 3′). Total RNA was extracted from C2C12 cells using TRIzol® reagent (Invitrogen, 15596018), and the RNA was reverse transcribed using Superscript II Reverse Transcriptase (Invitrogen, 18064014) and random hexamer primers, followed by quantitative PCR using the FastStart Universal SYBR Green Master Kit (Roche, 04913914001) and an ABI PRISM 7900HT system (Applied Biosystems, Waltham, MA, USA). The reaction protocol used was 95°C 5 min, 35 cycles with 95°C 15 sec, 60°C 60 sec, and 72°C 5 min. The gene of interest expression was normalized to the reference gene, Actb, and was calculated with the 2−ΔΔCt method.
Endocytosis assay
For flow cytometry-based endocytosis assays, we used an antibody-probed endocytosis protocol, as previously described.Citation32 Cells were detached, incubated with Alexa Fluor 488-conjugated CHRN antibody (mAb210) at 4°C for 1 h, and then switched to 37°C for different periods of time to allow CHRN internalization. After acidic washes (0.1 M glycine, 0.1 M NaCl, pH 2.5) to remove uninternalized antibody, the cells were fixed with 3% paraformaldehyde and subsequently analyzed using a FACS Calibur flow cytometer (BD Bioscience, Mount View, CA, USA). The percentage of internalization was calculated using the formula [MFIT – MFItime0]/MFItotal × 100%.
We additionally utilized cell surface protein biotinylation to monitor endocytosis, as previously described with modification.Citation32 Briefly, cells at approximately 70–80% confluence were treated with biotin-conjugated CHRN antibody (mAb210) and were left 1 h at 4°C. Cells were then switched to 37°C for different periods of time to allow CHRN internalization. After acidic washes (0.1 M glycine, 0.1 M NaCl, pH 2.5) to remove uninternalized antibody, cellular extracts were prepared with 200 μl of lysis buffer (Thermo Scientific, 78501), and subjected to SDS-PAGE and immunoblot analysis using CHRN antibody. For some experiments, after internalization, cell lysate was directly subjected to SDS-PAGE and immunoblot analysis using CHRN antibody. We then quantitatively analyzed the band densitometry using ImageJ software.
ELISA for serum CHRN antibody analysis
Serum anti-CHRN antibodies were measured using an ELISA kit obtained from Mybiosource (MBS726941) according to the instructions of the manufacturer. After adding the stop solution, the intensity of end-reaction color was measured spectrophotometrically at 450 nm in a microplate reader Biotek Synergy 2 (Biotek, Winooski, VT, USA). Results were displayed as absorbance at 450 nm (A450).
Autophagy analyses
Autophagy was analyzed by immunoblotting as described previously.Citation62 Briefly, muscle was homogenized in lysis buffer containing 1% NP-40 until no visible clumps were evident, or C2C12 cells were treated with or without TFMS (2 mM) for 6 h and cell lysates were prepared with lysis buffer. Following brief vortexing and rotation, cell lysates were separated by SDS-PAGE and immunoblotted with anti-MAP1LC3A/B, BECN1, ATG5, or HSPA8 antibody to monitor expression during the formation of autophagosomes.
Immunofluorescence confocal microscopy
Gastrocnemius was removed from mouse and immediately fixed in freshly prepared 3% paraformaldehyde in PBS (Gibco, 21600–044). Muscles were embedded in OCT (Sakura Finetek, 4583) and 10-µm-thick frozen sections were prepared using a Cryostat Microm (Thermo, HM525). Sections were then permeabilized using 0.5% Triton X-100 (Sigma, X100) in PBS and blocked with 20% goat serum (Gibco, 16210064) in PBS. anti-MAP1LC3A/B antibody (Cell Signaling Technology, 12741) was incubated overnight at 4°C, followed by washing and incubation with goat anti-rabbit Alexa Fluor 488-IgG (Invitrogen, 31627) at room temperature for 1 h. After washing with PBS, the sections were mounted and confocal microscopy (TCS SP5, Leica Microsystems) was used for examining the slides and capturing the images.
Coimmunoprecipitation
Autophagy was analyzed by immunoblotting as described previously.Citation62 Briefly, after cell lysis with lysis buffer (Thermo Scientific, 78501), cell lysates were centrifuged for 20 min at 15,000 g, and the supernatants were incubated with the indicated antibody coated protein G-Sepharose (Amersham Biosciences, 17–0618-02) at 4°C overnight to form immunocomplexes, extensively washed, and then subjected to immnoblotting.
Statistical analysis
The 2-tailed Student t test or one-way ANOVA with a Turkey multiple comparison post hoc test were used for all statistical analyses in this study. A p value less than 0.05 was considered as statistically significant.
Abbreviations
| AP | = | 1-(2-aminoethyl) piperazine |
| AZA | = | acetazolamide |
| BAG3 | = | BCL2-associated athanogene 3 |
| CA(R) | = | carbonic anhydrase |
| CASA | = | chaperone-assisted selective autophagy |
| CHRN | = | cholinergic receptor nicotinic |
| CME | = | clathrin-mediated endocytosis |
| EAMG | = | experimental autoimmune myasthenia gravis |
| HSPA8 | = | heat shock protein 8 |
| LRP4 | = | low density lipoprotein receptor-related protein 4 |
| MβCD | = | methyl-beta-cyclodextrin |
| MFI | = | mean fluorescence intensity |
| MUSK | = | muscle, skeletal, receptor tyrosine kinase |
| PBA | = | 4-phenylbutyric acid |
| TMFS | = | trifluoromethanesulfonamide |
Disclosure of potential conflicts of interest
The authors declare that they have no conflict of interest.
1375633_Supplemental_Material.docx
Download MS Word (39 MB)Acknowledgments
We are grateful to Dr. Xiaoyun Huang from Fudan University Huashan Hospital for his help in monitoring electromyography of experimental autoimmune myasthenia gravis in mice.
Funding
This work was supported by National Natural Science Foundation of China 31570905, 81190133 (to C.X.), 81200967 (to A.D.), Shanghai Municipal Health and Family Planning Commission 201540050 (to A.D), Shanghai Municipal Education Commission 14YZ036 (to C.X.), Shanghai Early Career Fund for Outstanding Researchers 201465 (to C.X.).
Related Research Data
References
- Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8:475-90. doi:10.1016/S1474-4422(09)70063-8. PMID:19375665
- Spillane J, Higham E, Kullmann DM. Myasthenia gravis. BMJ. 2012;345:e8497. doi:10.1136/bmj.e8497. PMID:23261848
- Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14:1023-36. doi:10.1016/S1474-4422(15)00145-3. PMID:26376969
- Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7:365-8. doi:10.1038/85520. PMID:11231638
- Berrih-Aknin S, Le Panse R. Myasthenia gravis: a comprehensive review of immune dysregulation and etiological mechanisms. J Autoimmun. 2014;52:90-100. doi:10.1016/j.jaut.2013.12.011. PMID:24389034
- Gertel-Lapter S, Mizrachi K, Berrih-Aknin S, Fuchs S, Souroujon MC. Impairment of regulatory T cells in myasthenia gravis: Studies in an experimental model. Autoimmunity Rev. 2013;12:894-903. doi:10.1016/j.autrev.2013.03.009.
- Wang Z, Wang W, Chen Y, Wei D. T helper type 17 cells expand in patients with myasthenia-associated thymoma. Scand J Immunol. 2012;76:54-61. doi:10.1111/j.1365-3083.2012.02703.x. PMID:22486891
- Cavalcante P, Le Panse R, Berrih-Aknin S, Maggi L, Antozzi C, Baggi F, Bernasconi P, Mantegazza R. The thymus in myasthenia gravis: Site of "innate autoimmunity"? Muscle Nerve. 2011;44:467-84. doi:10.1002/mus.22103. PMID:21922466
- Wang YZ, Yan M, Tian FF, Zhang JM, Liu Q, Yang H, Zhou WB, Li J.. Possible involvement of toll-like receptors in the pathogenesis of myasthenia gravis. Inflammation. 2013;36:121-30. doi:10.1007/s10753-012-9526-6. PMID:22898741
- Tuzun E, Christadoss P. Complement associated pathogenic mechanisms in myasthenia gravis. Autoimmunity Rev. 2013;12:904-11. doi:10.1016/j.autrev.2013.03.003.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nature Rev Drug Discov. 2008;7:168-81. doi:10.1038/nrd2467.
- Harju AK, Bootorabi F, Kuuslahti M, Supuran CT, Parkkila S. Carbonic anhydrase III: a neglected isozyme is stepping into the limelight. J Enzyme Inhib Med Chem. 2013;28:231-9. doi:10.3109/14756366.2012.700640. PMID:22803676
- Robert-Pachot M, Desbos A, Moreira A, Becchi M, Tebib J, Bonnin M, Aitsiselmi T, Bienvenu J, Fabien N. Carbonic anhydrase III: a new target for autoantibodies in autoimmune diseases. Autoimmunity. 2007;40:380-9. doi:10.1080/08916930701417473. PMID:17612900
- Du AL, Ren HM, Lu CZ, Tu JL, Xu CF, Sun YA. Carbonic anhydrase III is insufficient in muscles of myasthenia gravis patients. Autoimmunity. 2009;42:209-15. doi:10.1080/08916930802668610. PMID:19301202
- Platta HW, Stenmark H. Endocytosis and signaling. Curr Opin Cell Biol. 2011;23:393-403. doi:10.1016/j.ceb.2011.03.008. PMID:21474295
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517-33. doi:10.1038/nrm3151. PMID:21779028
- Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol. 2009;10:609-22. doi:10.1038/nrm2748. PMID:19696798
- Aguilar RC, Wendland B. Endocytosis of membrane receptors: Two pathways are better than one. Proc Natl Acad Sci USA. 2005;102:2679-80. doi:10.1073/pnas.0500213102. PMID:15710869
- Doherty GJ, McMahon HT. Mechanisms of Endocytosis. Annu Rev Biochem. 2009;78:857-902. doi:10.1146/annurev.biochem.78.081307.110540. PMID:19317650
- Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597-608. doi:10.1038/nrm2755. PMID:19696797
- Lee CW, Zhang H, Geng L, Peng HB. Crosslinking-induced endocytosis of acetylcholine receptors by quantum dots. PLoS One. 2014;9:e90187. doi:10.1371/journal.pone.0090187. PMID:24587270
- Kumari S, Borroni V, Chaudhry A, Chanda B, Massol R, Mayor S, Barrantes FJ. Nicotinic acetylcholine receptor is internalized via a Rac-dependent, dynamin-independent endocytic pathway. J Cell Biol. 2008;181:1179-93. doi:10.1083/jcb.200709086. PMID:18591431
- Vincent A, Palace J, Hilton-Jones D. Myasthenia gravis. The Lancet. 2001;357:2122-8. doi:10.1016/S0140-6736(00)05186-2
- Drachman DB. Myasthenia Gravis. New Engl J Med 1994;330:1797-810. doi:10.1056/NEJM199406233302507. PMID:8190158
- Kaminski HJ, Li Z, Richmonds C, Lin F, Medof ME. Complement regulators in extraocular muscle and experimental autoimmune myasthenia gravis. Exp Neurol. 2004;189:333-42. doi:10.1016/j.expneurol.2004.06.005. PMID:15380483
- Kuncl RW, Wittstein I, Adams RN, Wiggins WW, Avila O, Pestronk A, McIntosh K, Lucas D, DeSilva S, Lehar M, et al. A novel therapy for myasthenia gravis by reducing the endocytosis of acetylcholine receptors. Ann N Y Acad Sci. 1993;681:298-302. doi:10.1111/j.1749-6632.1993.tb22900.x
- Kuncl RW, Drachman DB, Adams R, Lehar M. 3-Deazaadenosine: a therapeutic strategy for myasthenia gravis by decreasing the endocytosis of acetylcholine receptors. J Pharmacol Exp Ther. 1993;267:582-9.
- Vullo D, Nishimori I, Scozzafava A, Supuran CT. Carbonic anhydrase activators: Activation of the human cytosolic isozyme III and membrane-associated isoform IV with amino acids and amines. Bioorg Med Chem Lett. 2008;18:4303-7. doi:10.1016/j.bmcl.2008.06.075. PMID:18627905
- Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116:2843-54. doi:10.1172/JCI29894. PMID:17080188
- Hansen CG, Nichols BJ. Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci. 2009;122:1713-21. doi:10.1242/jcs.033951. PMID:19461071
- Grassart A, Dujeancourt A, Lazarow PB, Dautry‐Varsat A, Sauvonnet N. Clathrin‐independent endocytosis used by the IL‐2 receptor is regulated by Rac1, Pak1 and Pak2. EMBO Rep. 2008;9:356-62. doi:10.1038/embor.2008.28. PMID:18344974
- Xu C, Zhang YH, Thangavel M, Richardson MM, Liu L, Zhou B, Zheng Y, Ostrom RS, Zhang XA. CD82 endocytosis and cholesterol-dependent reorganization of tetraspanin webs and lipid rafts. FASEB J. 2009;23:3273-88. doi:10.1096/fj.08-123414. PMID:19497983
- Bento CF, Puri C, Moreau K, Rubinsztein DC. The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci. 2013;126:1059-69. doi:10.1242/jcs.123075. PMID:23620509
- Rodriguez AE, Lopez-Crisosto C, Pena-Oyarzun D, Salas D, Parra V, Quiroga C, Morawe T, Chiong M, Behl C, Lavandero S. BAG3 regulates total MAP1LC3B protein levels through a translational but not transcriptional mechanism. Autophagy. 2016;12:287-96. doi:10.1080/15548627.2015.1124225. PMID:26654586
- Kaushik S, Cuervo AM. Chaperones in autophagy. Pharmacol Res. 2012;66:484-93. doi:10.1016/j.phrs.2012.10.002. PMID:23059540
- Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Fürst DO, Saftig P, Saint R, Fleischmann BK, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20:143-8. doi:10.1016/j.cub.2009.11.022. PMID:20060297
- Iwasa K, Nambu Y, Motozaki Y, Furukawa Y, Yoshikawa H, Yamada M. Increased skeletal muscle expression of the endoplasmic reticulum chaperone GRP78 in patients with myasthenia gravis. J Neuroimmunol. 2014;273:72-6. doi:10.1016/j.jneuroim.2014.05.006. PMID:24882382
- Du A, Huang S, Zhao X, Zhang Y, Zhu L, Ding J, Xu C. Endoplasmic reticulum stress contributes to acetylcholine receptor degradation by promoting endocytosis in skeletal muscle cells. J Neuroimmunol. 2016;290:109-14. doi:10.1016/j.jneuroim.2015.11.024. PMID:26711579
- Du AL, Ren HM, Lu CZ, Huang J. [Expression and tissue and species specificity of P25 protein in patients with myasthenia gravis]. Chin Med J. 2004;84:103-6.
- Ren H, Lu C, Zhou Z, Chen X, Huang J. [Analysis of 25 kD protein content of skeletal muscles from the patients with myasthenia gravis]. Chin J Neurol. 2002;35:32-5.
- Ren H, Zhou Z, Chen X, Huang J, Lu C. [Analysis of protein components of skeletal muscle from the patients with myasthenia gravis]. Chin J Neurol. 2000;33:227-30.
- Ren H, Zhou Z, Chen X, Huang J, Lu C. [Comparison of protein components in normal and myasthenia gravis skeletal muscles]. Chin J Clin Neurosci. 2000;8:260-2.
- Beekley MD, Wetzel P, Kubis HP, Gros G. Contractile properties of skeletal muscle fibre bundles from mice deficient in carbonic anhydrase II. Pflugers Arch. 2006;452:453-63. doi:10.1007/s00424-006-0048-7. PMID:16601982
- Scheibe RJ, Mundhenk K, Becker T, Hallerdei J, Waheed A, Shah GN, Sly WS, Gros G, Wetzel P.. Carbonic anhydrases IV and IX: subcellular localization and functional role in mouse skeletal muscle. Am J Physiol Cell Physiol. 2008;294:C402-12. doi:10.1152/ajpcell.00228.2007. PMID:18003750
- Romi F, Gilhus NE, Aarli JA. Myasthenia gravis: clinical, immunological, and therapeutic advances. Acta Neurol Scand. 2005;111:134-41. doi:10.1111/j.1600-0404.2005.00374.x. PMID:15644074
- Dokladny K, Myers OB, Moseley PL. Heat shock response and autophagy–cooperation and control. Autophagy. 2015;11:200-13. doi:10.1080/15548627.2015.1009776. PMID:25714619
- Khan MM, Strack S, Wild F, Hanashima A, Gasch A, Brohm K, Reischl M, Carnio S, Labeit D, Sandri M, et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy. 2014;10:123-36. doi:10.4161/auto.26841. PMID:24220501
- Janssens S, Pulendran B, Lambrecht BN. Emerging functions of the unfolded protein response in immunity. Nat Immunol. 2014;15:910-9. doi:10.1038/ni.2991. PMID:25232821
- Mandl J, Mészáros T, Bánhegyi G, Csala M. Minireview: Endoplasmic Reticulum Stress: Control in Protein, Lipid, and Signal Homeostasis. Mol Endocrinol. 2013;27:384-93. doi:10.1210/me.2012-1317. PMID:23349523
- Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am J Physiol Cell Physiol. 2015;308:C415-25. doi:10.1152/ajpcell.00334.2014. PMID:25540175
- Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A Molecular Web: Endoplasmic Reticulum Stress, Inflammation, and Oxidative Stress. Front Cell Neurosci. 2014;8:213. doi:10.3389/fncel.2014.00213. PMID:25120434
- Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013;12:105-18. doi:10.1016/S1474-4422(12)70238-7. PMID:23237905
- Drachman DB, Adams RN, Josifek LF, Self SG. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med. 1982;307:769-75. doi:10.1056/NEJM198209233071301.
- Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K, Sideri A, Zouridakis M, Tzartos SJ. Muscle and neuronal nicotinic acetylcholine receptors. Structure, function and pathogenicity. FEBS J. 2007;274:3799-845. doi:10.1111/j.1742-4658.2007.05935.x. PMID:17651090
- Cisterna BA, Cardozo C, Saez JC. Neuronal involvement in muscular atrophy. Front Cell Neurosci. 2014;8:405. doi:10.3389/fncel.2014.00405. PMID:25540609
- Nessi V, Nava S, Ruocco C, Toscani C, Mantegazza R, Antozzi C, Baggi F. Naturally occurring CD4+CD25+ regulatory T cells prevent but do not improve experimental myasthenia gravis. J Immunol. 2010;185:5656-67. doi:10.4049/jimmunol.0903183. PMID:20881192
- Tuzun E, Berrih-Aknin S, Brenner T, Kusner LL, Le Panse R, Yang H, Tzartos S, Christadoss P. Guidelines for standard preclinical experiments in the mouse model of myasthenia gravis induced by acetylcholine receptor immunization. Exp Neurol. 2015;270:11-7. doi:10.1016/j.expneurol.2015.02.009. PMID:25697844
- Bo Wu, Elzbieta Goluszko a, Christadoss P. Experimental Autoimmune Myasthenia Gravis in the Mouse. In Current Protocols in Immunology; Coligan J, Bierer B, Margulies D, Shevach E, Strober W, editors; John Wiley & Sons: Hoboken, NJ, 1997; pp 15.8.1-15.8.19.
- Joo IS, Hwang DH, Seok JI, Shin SK, Kim SU. Oral administration of memantine prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Clin Neurol. 2007;3:181-6. doi:10.3988/jcn.2007.3.4.181. PMID:19513129
- Xu C, Liu J, Hsu L-C, Luo Y, Xiang R, Chuang T-H. Functional interaction of heat shock protein 90 and Beclin 1 modulates Toll-like receptor-mediated autophagy. FASEB J. 2011;25:2700-10. doi:10.1096/fj.10-167676. PMID:21543763
- Cai W, Du A, Feng K, Zhao X, Qian L, Ostrom RS, Xu C. Adenylyl Cyclase 6 Activation Negatively Regulates TLR4 Signaling through Lipid Raft-Mediated Endocytosis. J Immunol. 2013;191:6093-100. doi:10.4049/jimmunol.1301912.
- Xu C, Feng K, Zhao X, Huang S, Cheng Y, Qian L, Wang Y, Sun H, Jin M, Chuang TH, et al. Regulation of autophagy by E3 ubiquitin ligase RNF216 through BECN1 ubiquitination. Autophagy. 2014;10:2239-50. doi:10.4161/15548627.2014.981792. PMID:25484083