
ABSTRACT
Macroautophagy/autophagy is an essential, conserved self-eating process that cells perform to allow degradation of intracellular components, including soluble proteins, aggregated proteins, organelles, macromolecular complexes, and foreign bodies. The process requires formation of a double-membrane structure containing the sequestered cytoplasmic material, the autophagosome, that ultimately fuses with the lysosome. This review will define this process and the cellular pathways required, from the formation of the double membrane to the fusion with lysosomes in molecular terms, and in particular highlight the recent progress in our understanding of this complex process.
Introduction
The autophagy pathway starts at the birth of the phagophore and ends at death of the autophagosome. The cellular and molecular lifecycle of this pathway has occupied cell biologists since the late 1950s. Fundamental insights from yeast and other eukaryotes have provided details in some aspects of the pathway, in particular the identification of the machinery (the autophagy-related [ATG] proteins) while other aspects remain only partially understood. This review will cover the birth (formation) and death (maturation and fusion with lysosomes) of the autophagosome in mammalian cells. Much of the data has been obtained by studying the stress-induced birth of the autophagosome after amino acid starvation as this provides an acute change in the ATG proteins, either by alterations in subcellular localization and/or post-translational modifications. This review will cover formation (the birth) primarily by focusing on the ATG protein ATG9 and the membrane compartments involved in the early stage of formation, while the terminal stages (the death) of the autophagosome will be covered with more molecular detail as the machinery involved at the later stage is better understood. In particular, we will summarize the role of SNARE proteins, tethering factors and adaptors in autophagosome-lysosome fusion, the machinery of autophagic lysosome reformation and the membrane lipids involved in those steps.
Autophagosome formation
The ATG proteins
A dedicated cohort of ATG proteins assemble into functional complexes, which are activated and recruited to membranes to initiate autophagy (for a recent review seeCitation1 and ). In brief, these are the ULK complex, a serine-threonine kinase complex (ULK1, ULK2, ATG13, RBCC1/FIP200 [RB1-inducible coiled-coil protein 1], ATG101); the class III lipid kinase complex I producing phosphatidylinositol 3-phosphate (PIK3C3/VPS34, PIK3R4/p150, BECN1/Beclin 1, ATG14); the effector of phosphatidylinositol 3-phosphate (PtdIns3P), the WIPI proteins; 2 ubiqutin-like conjugation complexes, one which conjugates ATG12 to ATG5 in association with ATG16L1 (ATG7, ATG10) and one driving the lipidation of the Atg8 family members (ATG7, ATG3). The Atg8 family in mammals consists of at least 6 proteins (LC3A, B and C, GABARAP. GABARAPL1, and GABARAPL2/GATE-16). Finally, ATG9 is the only transmembrane protein in this cohort, and, while essential, its precise function remains to be determined. For additional information on ATG proteins not mentioned here as well as accessory regulatory proteins see a recent review.Citation2
Figure 1. Autophagy pathway in mammalian cells. The molecular pathway comprised of the core autophagy proteins is illustrated with their associated autophagy membrane compartments. The 4 major steps in the autophagic pathway are shown in red. PtdIns3P, phosphatidylinositol 3-phosphate; PE, phosphatidylethanolamine.
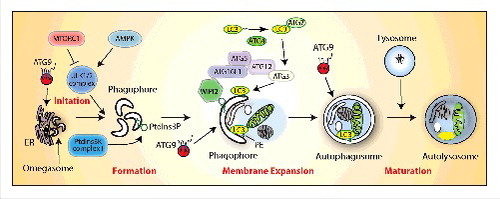
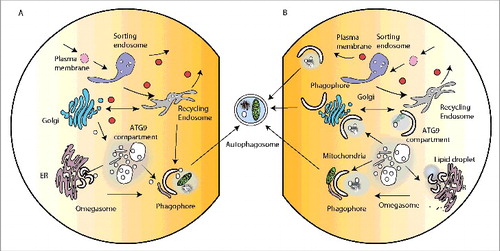
In mammalian cells, the initiation and formation of the autophagosome after a strong stimulus such as amino acid starvation, is now widely agreed to occur on the membrane of the endoplasmic reticulum through the formation of the omegasome.Citation3 However, it is clear that the formation of this intermediate, and growth of the phagophore, requires input from almost all the intracellular compartments in the cell. This view is supported by morphological data, which suggests the phagophore is in contact with many organelles.Citation4 Our current view is the requirement for these diverse organelles may be fulfilled by vesicular transport to and from the phagophore (), but could also occur through transient membrane contacts and exchange of proteins and lipids (). The latter could resolve puzzling properties of the phagophore that define it as distinct from all other compartments, and there is some evidence to support this hypothesis.
Figure 2. Intracellular organelles and membrane contacts facilitating autophagosome formation. (A) The major organelles required for the secretory pathway (ER, Golgi) and endocytosis (sorting endosome, recycling endosome) implicated in membrane contribution to phagophore formation. For simplicity not all ER-Golgi associated compartments (ERES, ERGIC, COP vesicles) are shown. Likewise not all endocytic-associated compartments (early endosome, late endosome, lysosome) are shown. (B) Membrane contact sites proposed or potentially implicated in phagophore expansion. The MAMs (mitochondria-associated membranes) are illustrated by contact of the phagophore with the mitochondria.
Role of ER, ER-associated compartments, and the ER-Golgi intermediate compartment (ERGIC)
Initiation of formation of the phagophore begins when the ULK kinase complex is activated, which corresponds to the translocation of the ULK complex at a discrete location on the ER that has been marked by ATG9.Citation5 Recruitment of the class III phosphatidylinositol 3-kinase (PtdIns3-kinase) follows, generating the ER domains or structures called the omegasome, which contain PtdIns3P. These domains are likely to be highly curved regions that favor recruitment of PtdIns3K complex I, thus driving further changes in the lipid composition to form the phagophore. This change in curvature may also facilitate exposure or clustering of PtdIns3P to allow recruitment of WIPI2B, which recruits the E3-like complex ATG12–ATG5-ATG16L1.Citation6 Evidence also supports the notion that ER-associated exit sites (ERES), or ERGIC can contribute membrane to the forming phagophore.Citation7,8 Further, there is good evidence in yeast that ERES, COPII vesicles and the ERGIC are contributing membranes or proteins to phagophores and this may be driven by SNARE-mediated fusion (for summary seeCitation9).
Role of the Golgi complex and endosomes
The Golgi complex is the distillery through which all secretory and post-Golgi proteins pass, become terminally glycosylated and undergo other post-translational modifications. The cytoplasmic surface of the Golgi complex contains receptors for incoming vesicles, and machinery that allows bidirectional vesicular transport through vesicles. Both in yeast and mammalian cells, the main connection between the Golgi complex and formation of autophagosomes is the multispanning membrane protein ATG9 (ATG9A in mammals). Genetic and biochemical evidence in yeast supports the role of the Golgi complexCitation10,11 and the Golgi-endosomal systemCitation12,13 as the source of the ATG9 vesicles and “Atg9 compartment.”
In mammalian cells at steady state, ATG9 resides in the Golgi complex, and it traffics through the endosomal system, controlled by the ULK complex, MAPK14/p38 MAP kinase,Citation14 RAB11 and TBC1D14,Citation15 TRAPPIII,Citation16 AP2 (adaptor related protein complex 2) containing clathrin coats,Citation17 and SH3GLB1/BIF1.Citation18 There is a role for the plasma membrane in this dynamic cycling of ATG9 vesicles originating from this source. This pathway involves AP2, the retromer, and TBC1D5 to sort ATG9 from the early sorting endosomeCitation19 and SNARES.Citation20 In addition, new evidence supports the need for metabolism of the lipid sphingomyelin in the biogenesis of the phagophore whereby an increase in sphingomyelin inhibits ATG9 recycling from endosomes.Citation21 Finally, phosphorylation of ATG9 may alter its function either directly, or indirectly through alterations in its association with cellular machinery. The best characterized of these regulatory pathways is through the ULK complex and AMPK.Citation22,23 Furthermore, recent data suggest a novel control of the endocytic pool of ATG9 trafficking through the Golgi by SRC kinase and ULK1 that also regulates the interaction with AP1 and AP2.Citation24 Recent data also point to a key role for the intracellular transport of ATG9 through endosomes, including early, sorting and recycling endosomes (). Several reports implicate the RAB11 (a small GTPase)-positive recycling endosomes, suggesting the recycling endosomes may play the most prominent role in this route: A direct transport route from recycling endosomes to the forming autophagosome has been identified, which is mediated by SNX18, a sorting nexin,Citation25 and evident after overexpression of TBC1D14, an effector of RAB11 and the TRAPPIII complex.Citation14 This pathway delivers to the forming phagophore ULK1, ATG16L1, and LC3 in vesicles that contain TF (transferrin).
These data point to the Golgi-endosomal system as being a key route for the production of ATG9 vesicles that could nucleate the domain forming the omegasome, directly fuse with the growing phagophore membrane or reside dynamically in a larger intermediate compartment called the ATG9 compartment () which appears as a collection of small vesicles, tubules and vacuoles.Citation11,25 The conservation of the ATG9 compartment in eukaryotes points to its essential role as a storage pool of vesicles that can be liberated and mobilized to aid the growth of the phagophore during induction by stress such as starvation. Further studies are needed to resolve the precise function of ATG9.
Role of membrane contacts and lipid droplets
Mitochondria-associated membranes (MAMs), or mitochondria-endoplasmic reticulum contact sites have been implicated in the formation of autophagosomes as potential sites that concentrate ATG formation machinery including ATG14 and STX17.Citation26 STX17, a Q-SNARE protein mainly resident on ER,Citation27 has also been implicated in the process of autophagosome closure.Citation28 In support of these data it had been previously shown that mitochondria are tethered to the ER, disruption of this tethering impairs starvation-induced autophagosome formation,Citation29 and the contacts with the ER allow transfer of both proteins and lipids from mitochondria.Citation30
Lipid droplets, formed at the ER, are important stores of neutral lipids. Lipid droplets have a unique surface covered with a single phospholipid bilayer and proteins, (for a recent review seeCitation31). Lipid droplets are in the proximity of, and likely in contact with, the forming autophagosome.Citation32 Mobilization of the neutral lipids stored in the lipid droplet, as well as a lipase PNPLA5 on the surface of the lipid droplet were recently shown to enhance autophagosome formation.Citation33 This pathway is likely to be conserved in yeast as the levels of enzymes required for synthesis of neutral lipids regulate autophagy.Citation34
Autophagosome-lysosome fusion
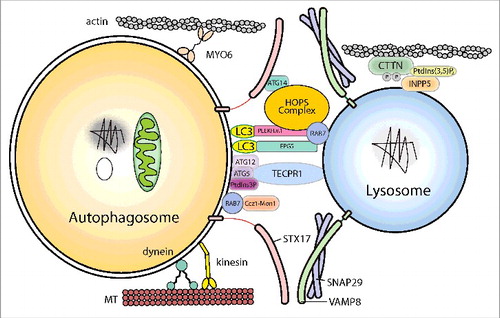
After closure of the phagophore, the double-membrane autophagosome matures and fuse with lysosomes to degrade its contents. The autophagosomes and lysosomes must first move closer together and then become tethered before SNARE-mediated fusion can occur. In the first step of autophagosome-lysosome fusion, the outer autophagosomal membrane fuses with the single lysosomal membrane. Full fusion is completed by degradation of the inner autophagosomal membrane by lysosomal hydrolases and exposure of the contents of the autophagosome to the lumen of the lysosome. So far, a large set of molecules, including cytoskeleton components and related motor proteins, tethering factors, phospholipids, and specific SNARE complexes, have been identified as important players in ensuring precise and efficient fusion ().
Figure 3. Schematic illustration of autophagosome-lysosome fusion. The cytoskeleton components and related motor proteins, the tethering factors, core machinery SNAREs and phospholipids involved in the fusion process are indicated. The detailed molecular mechanisms of each factor are discussed in the text. MT, microtubule.
Spatial positioning of autophagosomes and lysosomes
In order to achieve efficient fusion of autophagosomes and lysosomes, both organelles need to be physically close. Starvation causes perinuclear clustering of lysosomes, driven by changes in intracellular pH,Citation35 whereas autophagosomes are formed randomly in the peripheral region of the cell.Citation36 Once closed, autophagosomes are linked to and transported along microtubules,Citation37 and finally concentrate in the perinuclear region where lysosomes are located. Dynein, a minus end-directed microtubule motor, mediates the centripetal movement of autophagosomes. Dynein dysfunction results in decreased autophagosome-lysosome fusion.Citation36,38,39 In neurons, autophagosomes acquire retrograde mobility by fusing with dynein-loaded late endosomes to form amphisomes, which are then trafficked to the soma, the main location of mature acidic lysosomes.Citation40
Plus-end-directed microtubule motor kinesins can also affect autophagosome-lysosome fusion. Depletion of Klp98A causes reduced formation of starvation-induced autophagic vesicles and clustering of autophagosomes and lysosomes in the perinuclear region in flies. Despite the clustering of these vesicles, they do not fuse, which suggests that Klp98A may be involved in the formation of fusion-competent, mature autophagosomes.Citation41 It is worth noting that knockdown of FYCO1, an adaptor required for microtubule plus-end-directed transport of autophagic vesicles, also causes clustering of LysoTracker Red-negative autophagic vesicles in the perinuclear region.Citation42
Besides microtubule-based motors, actin-based motors, such as MYO6/myosin VI and MYO1/myosin I, also play a role in autophagosome-lysosome fusion by affecting autophagosome maturation and tethering.Citation43,44
Tethering factors required for fusion
Various tethering factors contribute to the fusion of autophagosomes with lysosomes. Broadly speaking, these factors fall into 3 categories: the HOPS (homotypic fusion and protein sorting) complex, RAB7, and adaptors that link autophagosomal or lysosomal components to the core tethering or fusion machinery.
The core tethering factor for autophagosome-lysosome fusion is the HOPS complex. HOPS is a conserved protein complex consisting of vacuolar protein sorting 11 (VPS11), Vps16, VPS18, Vps33A, VPS39, and Vps41.Citation45–47 The HOPS complex works as a specific tether for vacuolar/lysosome fusion events in both yeast and mammals.Citation48,49 All HOPS subunits are required for autophagosome-lysosome fusion.Citation50 The HOPS complex interacts with the Q-SNARE STX17 and facilitates assembly of the trans-SNARE complex to mediate autophagosome-lysosome fusion.Citation50,51 It is interesting to note that the HOPS complex specifically binds to STX17 on autophagosomes but not on mitochondria or the ER, which indicates that other factors are needed to ensure the specificity of autophagosome-lysosome fusion.
The fusion of vesicles with target organelles depends on consecutive RAB-mediated tethering steps. RABs are small GTPases that have GTP-loaded and GDP-loaded forms. GTP-loaded RABs can be recruited to membranes, where they bind effectors including tethering factors.Citation52 The role of RAB7 in membrane fusion has been extensively documented in yeast, fly and mammals. Ypt7 (the yeast homolog of RAB7) binds HOPS via Vps41 and Vps39.Citation47 Because RAB7/Ypt7 can bind membranes and membrane-anchored proteins, assembly of the RAB7/Ypt7-HOPS complex can bridge 2 opposing membranes, thus facilitating membrane fusion.Citation53 In the context of autophagosome-lysosome fusion, the requirement for RAB7 is well established.Citation54 RAB7 interacts with various tethering factors, and these interactions are likely to further enhance the specificity of autophagosome-lysosome fusion.Citation55-57 Not surprisingly, as well as RABs, various GTP/GDP exchange factors (GEFs) and RAB GTPase-activating proteins (GAPs) can also affect autophagosome-lysosome fusion.Citation58,59
The central question surrounding vesicle fusion is the mechanism of specificity. Although core SNAREs and core tethering proteins offering a degree of specificity, additional factors are required to ensure the exquisite precision of autophagosome-lysosome fusion. So far, various “adaptor” proteins have been demonstrated to fulfill this role. These adaptor proteins bind to components of autophagosomes or lysosomes, such as LC3/Atg8 and ATG12–ATG5, while also interacting with the SNARE complex and/or core tethering factors such as RAB7 and the HOPS complex, thus ensuring the specificity of fusion. For example, EPG5 can bind to RAB7 and the R-SNARE VAMP7-VAMP8 on late endosomes/lysosomes, and to both LC3/LGG-1 (the C. elegans homolog of LC3) and assembled STX17-SNAP29 on autophagosomes, stabilizing the assembly of STX17-SNAP29-VAMP7-VAMP8 complexes.Citation56 PLEKHM1 forms a bridge between LC3/GABARAP and the HOPS complex and tethers VPS41 to RAB7 on lysosomes.Citation55 TECPR1 represents another type of adaptor protein. Lysosome-localized TECPR1 facilitates the fusion between autophagosomes and lysosomes by binding to autophagosome-localized ATG12–ATG5 and PtdIns3P.Citation60 It is worth mentioning that the role of PLEKHM1 in autophagosome maturation appears to be cell-line specific.Citation61 Homologs of PLEKHM1 and TECPR1 are not involved in autophagosome maturation in C. elegans.Citation56
The core machinery for fusion: SNAREs
SNAREs are the core components of the fusion machinery. Assembly of the SNARE complex is sufficient to mediate membrane fusion in vitro. During membrane fusion, R-SNARE and Q-SNARE proteins on separate membranes assemble into trans-SNARE complexes to provide the force required for fusion. In mammalian systems, STX17 is the autophagosomal Q-SNARE. Upon autophagy induction, STX17 is recruited from the ER and mitochondria to completed autophagosomes. Autophagosomal STX17 then interacts with SNAP29 and the endosomal/lysosomal R-SNARE VAMP8 to form a trans-SNARE complex, which mediates autophagosome-lysosome fusion.Citation62
Various factors regulate the assembly of the autophagosomal SNARE complex. ATG14 binds to STX17 and stabilizes the STX17-SNAP29 binary Q-SNARE complex on autophagosomes, promoting membrane tethering and enhancing fusion events.Citation63 O-GlcNAcylation of the SNARE protein SNAP29 reduces its ability to from trans-SNARE complexes. It is worth noting that the level of O-GlcNAcylated SNAP29 depends on nutrient availability and is reduced by starvation.Citation64 This adds another level of regulation to autophagosome-lysosome fusion.
Lipids involved in fusion
Despite their low abundance, phosphoinositides (PIs) play key roles in various cellular processes including specification of membrane identity, regulation of cell signaling and control of membrane shaping.Citation65 PIs carry out their function by serving as membrane docking sites for a large array of effector proteins and as precursors of lipid second messengers. PIs are concentrated on the cytosolic face of cellular membranes and are able to diffuse quickly in membranes. Phosphorylation at the 3-, 4-, and 5-position of the inositol ring of phosphatidylinositol generates seven PI isoforms. The concerted action of PI and PtdIns kinases and phosphatases, which add or remove phosphate groups at various positions of the inositol ring, generate the unique PI signature of distinct membrane compartments.Citation65
So far, PtdIns3P, PtdIns4P and PtdIns(3,5)P2 have been shown to participate in autophagosome-lysosome fusion.Citation60,66–69 Changes in the PtdIns(3,5)P2 level on lysosomes, mediated by INPP5E (inositol polyphosphate-5-phosphatase E), contribute to the autophagosome-lysosome fusion process in neurons.Citation69 PtdIns(3,5)P2 is dephosphorylated by INPP5E, which resides on lysosomes. The decreased level of PtdIns(3,5)P2 enhances CTTN (cortactin)-dependent actin filament stabilization on lysosomes, facilitating autophagosome-lysosome fusion.Citation69
PtdIns3P is also implicated in autophagosome-lysosome fusion. As mentioned above, lysosomal TECPR1 binds to the ATG12–ATG5 conjugate on autophagosomes. TECPR1 only binds to PtdIns3P when it is in a complex with ATG12–ATG5. Deletion of the pleckstrin homology/PH domain from TECPR1 abolishes its ability to bind PtdIns3P and to mediate autophagosome-lysosome fusion, indicating that the TECPR1-PtdIns3P interaction is likely required for fusion.Citation60 In addition to PtdIns3P, PtdIns4P, which is generated on autophagosomes by PI4K2A/PI4KIIα, is also crucial for autophagosome-lysosome fusion.Citation68
Coordination of closure and fusion
The processes of autophagosome formation, closure, and fusion with lysosomes need to be tightly coordinated to guarantee successful delivery of the engulfed contents to lysosomes for degradation. Premature fusion of autophagosomes with lysosomes may cause insufficiency or failure of autophagy. This coordination is achieved by controlling the localization of STX17, the autophagosomal SNARE. STX17 is mainly localized on the ER and mitochondria under normal conditions; however, the completion or near-completion of autophagosomes triggers the relocation of STX17 to autophagosomes, thus making the completed autophagosomes fusion-competent.Citation28
Inhibitors of autophagosome-lysosome fusion
Bafilomycin A1 (BafA1) is widely used as an inhibitor of autophagosome-lysosome fusion. It has been generally assumed that BafA1 inhibits autophagosome-lysosome fusion by blocking vacuolar-type H+-translocating ATPase pump activity. However, Mauvezin et al. showed that BafA1 interferes with autophagosome-lysosome fusion and autolysosome acidification in 2 separate processes. BafA1 targets the vacuolar-type H+-translocating ATPase on the lysosome to prevent lumenal acidification, and independently inhibits Ca-P60A/SERCA to disrupt autophagosome-lysosome fusion.Citation70
Autophagic lysosome reformation
Upon fusion of lysosomes with the outer membrane of an autophagosome, the lysosomal contents enter the space between the 2 autophagosome membranes, and the degradation of the inner membrane of the autophagosome occurs in an LC3-dependent manner. Once the inner membrane breaks down, the process of autophagosomal cargo degradation begins. The degradation products, including amino acids and sugars, are transported out of the autolysosome through members of a family of lysosome efflux transporters, and the autolysosome volume is consequently reduced.
The limiting membrane of autolysosomes contain membranes from different sources including lysosomes, late endosomes and autophagosomes. Autolysosomes are not permanent structures and disintegrate once autophagy is terminated. The termination step of autophagy is a process dubbed as autophagic lysosome reformation (ALR).Citation71 During ALR, lysosomal membrane proteins are recycled from autolysosomes through tubular structures named reformation tubules. At the tips of these tubules, nascent lysosomes, called proto-lysosomes, are formed through a scission/budding process. Initially proto-lysosomes are not acidic and do not contain lysosome lumenal proteins, but eventually they mature into functional lysosomes in a protein synthesis-dependent manner.Citation72
ALR can be divided into a series of steps including triggering, initiation, cargo sorting, autolysosome tubulation, proto-lysosome scission, and proto-lysosome maturation (). In the past few years, an increasingly clear picture of the molecular regulation of ALR has started to emerge.
Figure 4. The overview of the autophagic lysosome reformation (ALR) process. mTOR reactivation trigger ALR. Clathrin-mediated buds driving initiation, microtubules and the motor protein KIF5B mediating tube elongation, and DNM2 (dynamin 2)-mediated proto-lysosome scission couple with the function of multiple phospholipids.
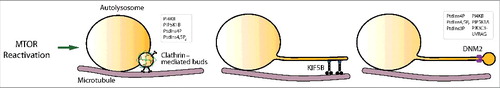
Upon starvation, autophagy is triggered by inhibition of mTOR (mechanistic target of rapamycin). Adding rapamycin after autolysosome formation blocks ALR, and adding serum to starved cells causes activation of mTOR and initiation of ALR.Citation71 In cells undergoing serum or glutamine starvation, which mimics in vivo starvation in a more physiologically relevant way, prolonged nutrient withdrawal causes elevated amino acid levels through the combined effect of autophagic degradation and increased amino acid uptake. Knockdown of SPNS/spinster, a lysosomal sugar efflux transporter, impairs the autolysosomal degradation capacity and attenuates mTOR reactivation, indicating that autophagic degradation contributes to reactivation of mTOR.Citation73 Prolonged starvation also causes activation of the general amino acid control pathway, which results in upregulation of amino acid transporters on the plasma membrane and contributes to mTOR reactivation.Citation74 At this point, the identity of the substrate of mTOR in the context of triggering ALR is still not clear.
Reformation tubules and proto-lysosomes do not contain autolysosomal lumenal proteins. Thus, a cargo retention mechanism must exist. PI4KB/PI4KIIIβ, a PtdIns4P kinase, appears to play key roles in autolysosomal cargo retention, as lysosomal lumenal components have uncontrolled access to reformation tubules in PI4KB− cells.Citation75 However, the mechanism by which PI4KB regulates cargo retention is unknown. Furthermore, it is still unclear how different membrane components are sorted out during ALR.
Autolysosome tubulation is initiated by formation of clathrin-coated, PtdIns(4,5)P2-enriched buds on autolysosomes. PtdIns(4,5)P2 generated by PIP5K1B recruits AP2 to the surface of autolysosomes, and the AP2 links in turn to clathrin. The formation of clathrin-coated buds creates PtdIns(4,5)P2-enriched microdomains, as one clathrin molecule can recruit 4 PtdIns(4,5)P2 molecules.Citation72 Through direct interaction with PtdIns(4,5)P2, KIF5B, a microtubule plus-end motor, is recruited in clusters to autolysosomes and generates a force that pulls reformation tubules out of the autolysosome. The autolysosome tubulation process has been successfully reconstituted in vitro using defined components. In this system, microtubules are polymerized and coated on a glass slide. Purified KIF5B is able to pull out tubules from PtdIns(4,5)P2-containing liposomes or purified autolysosomes in a clathrin-dependent manner.Citation76
Once tubular structures are formed, proto-lysosomes are generated from the tips of tubules through scission, which mimics the process of clathrin-mediated endocytosis. The core component of the scission machinery is the large GTPase DNM2 (dynamin 2), which functions by binding to PtdIns(4,5)P2.Citation77 The PtdIns(4,5)P2 involved in autolysosome tubulation and proto-lysosome scission is generated by different kinases. PIP5K1B regulates autolysosome tubulation whereas PIP5K1A mediates the formation of proto-lysosomes. A lack of PIP5K1A results in failure of proto-lysosome fission and elongation of reformation tubules.Citation72 In addition to PtdIns(4,5)P2, PtdIns3P and PtdIns4P are also involved in this process. Lysosomal PIK4B/PI4KIIIβ, which generates PtdIns4P, plays an important role in scission. Knockdown of PIK4B/PI4KIIIβ causes extensive autolysosome tubulation.Citation75 Lysosome-localized PtdIns3P produced by PIK3C3/VPS34-UVRAG is also important in the scission process.Citation78 At this moment, it is still not clear how different phospholipids and their kinases coordinate during the tubulation and scission processes.
Conclusions
The complexity of the life cycle of the autophagosome is just beginning to be unveiled. While the fundamental advance in our understanding was the identification of the Atg genes in yeast, the impact of this essential pathway on human health is now becoming apparent.Citation79 Integrating both the genetic data and the role of autophagy in the physiology of diseases requires an understanding of the molecular details driving the process, as well as the cellular pathways that control the membrane-mediated events. Here, we have provided a snapshot of the former and detailed the latter to highlight the recent progress the field has made. Understanding the molecular details of both the process itself, and the cellular pathways required for the process will be essential for development of therapeutics targeting autophagy in human diseases.
Abbreviations
| ALR | = | autophagic lysosome reformation |
| AP | = | adaptor protein |
| ATG | = | autophagy related |
| ERGIC | = | ER-Golgi intermediate compartment |
| HOPS | = | homotypic fusion and protein sorting |
| mTOR | = | mechanistic target of rapamycin |
| PI | = | phosphoinositides |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| PtdIns3P | = | phosphatidylinositol-3-phosphate |
| SNARE | = | Soluble NSF attachment protein receptor |
| ULK | = | unc-51 like autophagy activating kinase |
| VPS | = | vacuolar protein sorting |
Acknowledgments
The work in the laboratory of SAT is supported by the Francis Crick Institute which receives its core funding from Cancer Research UK (FC001187), the UK Medical Research Council (FC001187), and the Wellcome Trust (FC001187). Research in Li Yu's lab was supported by the Ministry of Science and Technology of the People's Republic of China 2016YFA0500202, 2015BAI08B01 and the National Natural Science Foundation of China 31671395, 31430053, 31321003.
Additional information
Funding
Related Research Data
References
- Hurley JH, Young LN. Mechanisms of Autophagy Initiation. Annu Rev Biochem. 2017 Jun 20;86:225–244. https://doi.org/10.1146/annurev-biochem-061516-044820. PMID:28301741.
- Galluzzi L. Molecular definitions of autophagy and related processes. EMBO J. 2017 Jul 3;36(13):1811–1836.
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. https://doi.org/10.1083/jcb.200803137. PMID:18725538.
- Biazik J, Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy. 2015;11:439–51. https://doi.org/10.1080/15548627.2015.1017178. PMID:25714487.
- Karanasios E, Walker SA, Okkenhaug H, Manifava M, Hummel E, Zimmermann H, Ahmed Q, Domart MC, Collinson L, Ktistakis NT. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun. 2016;7:12420. https://doi.org/10.1038/ncomms12420 PMID:27510922.
- Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Molecular cell. 2014;55:238–52. https://doi.org/10.1016/j.molcel.2014.05.021. PMID:24954904.
- Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife. 2013;2:e00947. https://doi.org/10.7554/eLife.00947. PMID:23930225.
- Davis S, Wang J, Ferro-Novick S. Crosstalk between the Secretory and Autophagy Pathways Regulates Autophagosome Formation. Dev Cell. 2017;41:23–32. https://doi.org/10.1016/j.devcel.2017.03.015. PMID:28399396.
- Lemus L, Goder V. A SNARE and specific COPII requirements define ER-derived vesicles for the biogenesis of autophagosomes. Autophagy. 2016;12:1049–50. https://doi.org/10.1080/15548627.2016.1164368. PMID:27124469.
- Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198:219–33. https://doi.org/10.1083/jcb.201202061. PMID:22826123.
- Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190:1005–22. https://doi.org/10.1083/jcb.200912089. PMID:20855505.
- Ohashi Y, Munro S. Membrane Delivery to the Yeast Autophagosome from the Golgi-Endosomal System. Mol Biol Cell. 2010;21:3998–4008. https://doi.org/10.1091/mbc.E10-05-0457. PMID:20861302.
- Shirahama-Noda K, Kira S, Yoshimori T, Noda T. TRAPPIII is responsible for vesicular transport from early endosomes to Golgi, facilitating Atg9 cycling in autophagy. Journal of Cell Science. 2013;126:4963–73. https://doi.org/10.1242/jcs.131318. PMID:23986483.
- Webber JL, Tooze SA. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 2009;29:27–40. https://doi.org/10.1038/emboj.2009.321. PMID:19893488.
- Longatti A, Lamb CA, Razi M, Yoshimura S-I, Barr FA, Tooze SA. TBC1D14 regulates autophagosome formation via Rab11 and recycling endosomes. J Cell Biol. 2012;197:659–75. https://doi.org/10.1083/jcb.201111079. PMID:22613832.
- Lamb CA, Nuhlen S, Judith D, Frith D, Snijders AP, Behrends C, Tooze SA. TBC1D14 regulates autophagy via the TRAPP complex and ATG9 traffic. EMBO J. 2016;35:281–301. https://doi.org/10.15252/embj.201592695. PMID:26711178.
- Imai K, Hao F, Fujita N, Tsuji Y, Oe Y, Araki Y, Hamasaki M, Noda T, Yoshimori T. Atg9A trafficking through the recycling endosomes is required for autophagosome formation. Journal of Cell Science. 2016;129:3781–91. https://doi.org/10.1242/jcs.196196. PMID:27587839.
- Takahashi Y, Tsotakos N, Liu Y, Young MM, Serfass J, Tang Z, Abraham T, Wang HG. The Bif-1-Dynamin 2 membrane fission machinery regulates Atg9-containing vesicle generation at the Rab11-positive reservoirs Oncotarget. 2016 Apr 12;7(15):20855–68.
- Popovic D, Dikic I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep. 2014;15:392–401. https://doi.org/10.1002/embr.201337995. PMID:24603492.
- Puri C, Renna M, Bento Carla F, Moreau K, Rubinsztein David C. Diverse Autophagosome Membrane Sources Coalesce in Recycling Endosomes. Cell. 2013;154:1285–99. https://doi.org/10.1016/j.cell.2013.08.044. PMID:24034251.
- Corcelle-Termeau E, Vindeløv SD, Hämälistö S, Mograbi B, Keldsbo A, Bräsen JH, Favaro E, Adam D, Szyniarowski P, Hofman P, et al. Excess sphingomyelin disturbs ATG9A trafficking and autophagosome closure. Autophagy. 2016;12:833–49. https://doi.org/10.1080/15548627.2016.1159378. PMID:27070082.
- Mack HID, Zheng B, Asara JM, Thomas SM. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy. 2012;8:1197–214. https://doi.org/10.4161/auto.20586. PMID:22932492.
- Weerasekara VK, Panek DJ, Broadbent DG, Mortenson JB, Mathis AD, Logan GN, Prince JT, Thomson DM, Thompson JW, Andersen JL, et al. Metabolic-Stress-Induced Rearrangement of the 14-3-3ζ Interactome Promotes Autophagy via a ULK1- and AMPK-Regulated 14-3-3ζ Interaction with Phosphorylated Atg9. Molecular and Cellular Biology. 2014;34:4379–88. https://doi.org/10.1128/MCB.00740-14. PMID:25266655.
- Zhou C, Ma K, Gao R, Mu C, Chen L, Liu Q, Luo Q, Feng D, Zhu Y, Chen Q, et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017;27:184–201. https://doi.org/10.1038/cr.2016.146. PMID:27934868.
- Orsi A, Razi M, Dooley H, Robinson D, Weston A, Collinson L, Tooze SA. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, is required for autophagy. Molecular biology of the cell. 2012;23:1860–73. https://doi.org/10.1091/mbc.E11-09-0746. PMID:22456507.
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. nature. 2013;495:389–93. https://doi.org/10.1038/nature11910. PMID:23455425.
- Steegmaier M, Yang B, Yoo J-S, Huang B, Shen M, Yu S, Luo Y, Scheller RH. Three Novel Proteins of the Syntaxin/SNAP-25 Family. Journal of Biological Chemistry. 1998;273:34171–9. https://doi.org/10.1074/jbc.273.51.34171. PMID:9852078.
- Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–41. https://doi.org/10.1126/science.aaf6136. PMID:27885029.
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–10. https://doi.org/10.1038/nature07534. PMID:19052620.
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–67. https://doi.org/10.1016/j.cell.2010.04.009. PMID:20478256.
- Kory N, Farese Jr RV, Walther TC. Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets. Trends in Cell Biology. 2016;26:535–46. https://doi.org/10.1016/j.tcb.2016.02.007. PMID:26995697.
- Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–5. https://doi.org/10.4161/auto.5.8.10274. PMID:19855179.
- Dupont N, Chauhan S, Arko-Mensah J, Castillo Eliseo F, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T, Deretic V. Neutral Lipid Stores and Lipase PNPLA5 Contribute to Autophagosome Biogenesis. Current Biology. 2014;24:609–20. https://doi.org/10.1016/j.cub.2014.02.008. PMID:24613307.
- Shpilka T, Welter E, Borovsky N, Amar N, Shimron F, Peleg Y, Elazar Z. Fatty acid synthase is preferentially degraded by autophagy upon nitrogen starvation in yeast. Proc Natl Acad Sci U S A. 2015;112:1434–9. https://doi.org/10.1073/pnas.1409476112. PMID:25605918.
- Heuser J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol. 1989;108:855–64. https://doi.org/10.1083/jcb.108.3.855. PMID:2921284.
- Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–87. https://doi.org/10.1111/j.1600-0854.2008.00701.x. PMID:18182013.
- Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem. 2006;281:36303–16. https://doi.org/10.1074/jbc.M607031200. PMID:16963441.
- Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33:109–22. https://doi.org/10.1247/csf.08005. PMID:18388399.
- Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, Brown SD, Rubinsztein DC. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–6. https://doi.org/10.1038/ng1591. PMID:15980862.
- Cheng XT, Zhou B, Lin MY, Cai Q, Sheng ZH. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol. 2015;209:377–86. https://doi.org/10.1083/jcb.201412046. PMID:25940348.
- Mauvezin C, Neisch AL, Ayala CI, Kim J, Beltrame A, Braden CR, Gardner MK, Hays TS, Neufeld TP. Coordination of autophagosome-lysosome fusion and transport by a Klp98A-Rab14 complex in Drosophila. J Cell Sci. 2016;129:971–82. https://doi.org/10.1242/jcs.175224. PMID:26763909.
- Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjørkøy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–69. https://doi.org/10.1083/jcb.200907015. PMID:20100911.
- Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol. 2012;14:1024–35. https://doi.org/10.1038/ncb2589. PMID:23023224.
- Brandstaetter H, Kishi-Itakura C, Tumbarello DA, Manstein DJ, Buss F. Loss of functional MYO1C/myosin 1c, a motor protein involved in lipid raft trafficking, disrupts autophagosome-lysosome fusion. Autophagy. 2014;10:2310–23. https://doi.org/10.4161/15548627.2014.984272. PMID:25551774.
- Rieder SE, Emr SD. A novel RING finger protein complex essential for a late step in protein transport to the yeast vacuole. Mol Biol Cell. 1997;8:2307–27. https://doi.org/10.1091/mbc.8.11.2307. PMID:9362071.
- Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci U S A. 2000;97:9402–7. https://doi.org/10.1073/pnas.97.17.9402. PMID:10944212.
- Wurmser AE, Sato TK, Emr SD. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J Cell Biol. 2000;151:551–62. https://doi.org/10.1083/jcb.151.3.551. PMID:11062257.
- Balderhaar HJ, Ungermann C. CORVET and HOPS tethering complexes – coordinators of endosome and lysosome fusion. J Cell Sci. 2013;126:1307–16. PMID:23645161 https://doi.org/10.1242/jcs.107805.
- Solinger JA, Spang A. Tethering complexes in the endocytic pathway: CORVET and HOPS. Febs J. 2013;280:2743–57. https://doi.org/10.1111/febs.12151. PMID:23351085.
- Takats S, Pircs K, Nagy P, Varga A, Karpati M, Hegedus K, Kramer H, Kovács AL, Sass M, Juhász G, et al. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell. 2014;25:1338–54. https://doi.org/10.1091/mbc.E13-08-0449. PMID:24554766.
- Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, Mizushima N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014;25:1327–37. https://doi.org/10.1091/mbc.E13-08-0447. PMID:24554770.
- Muller MP, Goody RS. Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases. 2017 Jan 5:1–17. https://doi.org/10.1080/21541248.2016.1276999.
- Lurick A, Gao J, Kuhlee A, Yavavli E, Langemeyer L, Perz A, Raunser S, Ungermann C. Multivalent Rab interactions determine tether-mediated membrane fusion. Mol Biol Cell. 2017;28:322–32. https://doi.org/10.1091/mbc.E16-11-0764. PMID:27852901.
- Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–97. https://doi.org/10.1242/jcs.01114. PMID:15138286.
- McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015;57:39–54. https://doi.org/10.1016/j.molcel.2014.11.006. PMID:25498145.
- Wang Z, Miao G, Xue X, Guo X, Yuan C, Zhang G, Chen Y4, Feng D5, Hu J, et al. The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Mol Cell. 2016;63:781–95. https://doi.org/10.1016/j.molcel.2016.08.021. PMID:27588602.
- Terawaki S, Camosseto V, Prete F, Wenger T, Papadopoulos A, Rondeau C, Combes A, Rodriguez Rodrigues C, Vu Manh TP, Fallet M, et al. RUN and FYVE domain-containing protein 4 enhances autophagy and lysosome tethering in response to Interleukin-4. J Cell Biol. 2015;210:1133–52. https://doi.org/10.1083/jcb.201501059. PMID:26416964.
- Hegedus K, Takats S, Boda A, Jipa A, Nagy P, Varga K, Kovács AL, Juhász G. The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Mol Biol Cell. 2016;27:3132–42. https://doi.org/10.1091/mbc.E16-03-0205. PMID:27559127.
- Itoh T, Kanno E, Uemura T, Waguri S, Fukuda M. OATL1, a novel autophagosome-resident Rab33B-GAP, regulates autophagosomal maturation. J Cell Biol. 2011;192:839–53. https://doi.org/10.1083/jcb.201008107. PMID:21383079.
- Chen D, Fan W, Lu Y, Ding X, Chen S, Zhong Q. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol Cell. 2012;45:629–41. https://doi.org/10.1016/j.molcel.2011.12.036. PMID:22342342.
- Tabata K, Matsunaga K, Sakane A, Sasaki T, Noda T, Yoshimori T. Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway via a novel Rab7-binding domain. Mol Biol Cell. 2010;21:4162–72. https://doi.org/10.1091/mbc.E10-06-0495. PMID:20943950.
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–69. https://doi.org/10.1016/j.cell.2012.11.001. PMID:23217709.
- Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520:563–6. https://doi.org/10.1038/nature14147. PMID:25686604.
- Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovács AL, Zhang Z, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014;16:1215–26. https://doi.org/10.1038/ncb3066. PMID:25419848.
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–7. https://doi.org/10.1038/nature05185. PMID:17035995.
- de Lartigue J, Polson H, Feldman M, Shokat K, Tooze SA, Urbe S, Clague MJ. PIKfyve regulation of endosome-linked pathways. Traffic. 2009;10:883–93. https://doi.org/10.1111/j.1600-0854.2009.00915.x. PMID:19582903.
- Cebollero E, van der Vaart A, Zhao M, Rieter E, Klionsky DJ, Helms JB, Reggiori F. Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr Biol. 2012;22:1545–53. https://doi.org/10.1016/j.cub.2012.06.029. PMID:22771041.
- Wang H, Sun HQ, Zhu X, Zhang L, Albanesi J, Levine B, Yin H. GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc Natl Acad Sci U S A. 2015;112:7015–20. https://doi.org/10.1073/pnas.1507263112. PMID:26038556.
- Nakamura S, Hasegawa J, Yoshimori T. Regulation of lysosomal phosphoinositide balance by INPP5E is essential for autophagosome-lysosome fusion. Autophagy. 2016;12:2500–1. https://doi.org/10.1080/15548627.2016.1234568. PMID:27715391.
- Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–8. https://doi.org/10.1080/15548627.2015.1066957. PMID:26156798.
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–6. https://doi.org/10.1038/nature09076. PMID:20526321.
- Rong Y, Liu M, Ma L, Du W, Zhang H, Tian Y, Cao Z, Li Y, Ren H, Zhang C, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012;14:924–34. https://doi.org/10.1038/ncb2557. PMID:22885770.
- Rong Y, McPhee CK, Deng S, Huang L, Chen L, Liu M, et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci U S A. 2011;108:7826–31. https://doi.org/10.1073/pnas.1013800108. PMID:21518918.
- Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, et al. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol. 2014;206:173–82. https://doi.org/10.1083/jcb.201403009. PMID:25049270.
- Sridhar S, Patel B, Aphkhazava D, Macian F, Santambrogio L, Shields D, et al. The lipid kinase PI4KIIIbeta preserves lysosomal identity. Embo J. 2013;32:324–39. https://doi.org/10.1038/emboj.2012.341. PMID:23258225.
- Du W, Su QP, Chen Y, Zhu Y, Jiang D, Rong Y, Zhang S, Zhang Y, Ren H, Zhang C, et al. Kinesin 1 Drives Autolysosome Tubulation. Dev Cell. 2016;37:326–36. https://doi.org/10.1016/j.devcel.2016.04.014. PMID:27219061.
- Schulze RJ, Weller SG, Schroeder B, Krueger EW, Chi S, Casey CA, McNiven MA. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J Cell Biol. 2013;203:315–26. https://doi.org/10.1083/jcb.201306140. PMID:24145164.
- Munson MJ, Allen GF, Toth R, Campbell DG, Lucocq JM, Ganley IG. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. Embo J. 2015;34:2272–90. https://doi.org/10.15252/embj.201590992. PMID:26139536.
- Levine B, Klionsky DJ. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker's yeast fuel advances in biomedical research. Proc Natl Acad Sci U S A. 2017;114:201–5. https://doi.org/10.1073/pnas.1619876114. PMID:28039434.