
ABSTRACT
Pathological developments leading to amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) are associated with misbehavior of several key proteins, such as SOD1 (superoxide dismutase 1), TARDBP/TDP-43, FUS, C9orf72, and dipeptide repeat proteins generated as a result of the translation of the intronic hexanucleotide expansions in the C9orf72 gene, PFN1 (profilin 1), GLE1 (GLE1, RNA export mediator), PURA (purine rich element binding protein A), FLCN (folliculin), RBM45 (RNA binding motif protein 45), SS18L1/CREST, HNRNPA1 (heterogeneous nuclear ribonucleoprotein A1), HNRNPA2B1 (heterogeneous nuclear ribonucleoprotein A2/B1), ATXN2 (ataxin 2), MAPT (microtubule associated protein tau), and TIA1 (TIA1 cytotoxic granule associated RNA binding protein). Although these proteins are structurally and functionally different and have rather different pathological functions, they all possess some levels of intrinsic disorder and are either directly engaged in or are at least related to the physiological liquid-liquid phase transitions (LLPTs) leading to the formation of various proteinaceous membrane-less organelles (PMLOs), both normal and pathological. This review describes the normal and pathological functions of these ALS- and FTLD-related proteins, describes their major structural properties, glances at their intrinsic disorder status, and analyzes the involvement of these proteins in the formation of normal and pathological PMLOs, with the ultimate goal of better understanding the roles of LLPTs and intrinsic disorder in the “Dr. Jekyll–Mr. Hyde” behavior of those proteins.
1. Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease, is a fatal form of the neurodegeration that represents a specific neuromuscular disease characterized by the degeneration of upper and lower motor neurons of the spinal cord leading to the progressive atrophy of abdominal, bulbar, limb, and thoracic muscles.Citation1,2 Respiratory failure is the most common cause of death of the afflicted patients, typically within 3–5 y after onset of the ALS symptoms.Citation3 Despite being much less abundant than other neurodegenerative maladies, such as Alzheimer disease (AD) or Parkinson disease (PD), ALS is considered as the most common neurodegenerative disorder that affects motor neurons in adults, and is particularly infamous for its devastating physiological effects and rapid lethality. As with many other neurodegenerative diseases, most ALS cases are sporadic, and only about 8–10% of ALS cases are inherited.Citation1-3 As it is a neurodegenerative disorder with the progressive degeneration of motor neurons in the motor cortex of the brain, the brainstem motor nuclei, and the anterior horns of the spinal cord, progression of ALS is characterized by the spatio-temporal spread of pathology, where early stages are manifested by local symptoms, such as subtle cramping or weakness in the bulbar or limb muscles, that rapidly culminate in the global paralysis of almost all skeletal muscles and invariably lead to death.Citation4 The characteristic symptom of ALS is progressive amyotrophy, or muscle wasting, caused by muscle denervation; i.e., the degeneration of spinal motor neurons results in atrophy of their target muscles.Citation4 Since degeneration of different sets of motor neurons would affect different regions of the body, ALS is clinically characterized by its considerable heterogeneity, and multiple subsets of the disease can be distinguished. For example, based on the clinical manifestations at symptom onset, patients with sporadic ALS can be grouped into limb- and bulbar-onset subtypes, where limb onset cases are associated with the limb motor symptoms, such as weakness or atrophy or fasciculation, and where bulbar onset cases are characterized by the presence of only bulbar signs, such as dysarthria, swallowing difficulty, and tongue fasciculation.Citation5,6 Importantly, patients with limb- and bulbar-onset ALS subtypes are characterized by the significant difference in brain atrophy patterns and in clinical progression, which is more rapid in bulbar-onset than in limb-onset ALS.Citation5,7 In addition to the classical (Charcot) ALS, this spectrum of disorders includes several other syndromes, such as ALS with multisystem involvement (e.g., ALS-dementia); flail arm or Vulpian-Bernhardt syndrome; flail leg syndrome, also known as pseudopolyneuritic form; progressive bulbar palsy, PBP; primary lateral sclerosis, PLS; and progressive muscular atrophy, PMA.Citation6 Among the pathological hallmarks of ALS are shrinking and degeneration of spinal motor neurons, and accumulation of rounded or thread-like deposits of aggregated proteins (collectively known as inclusions) in the cytoplasm of affected motor neurons.Citation4 These inclusions are often ubiquitinated, and, in most cases of ALS, contain ubiquitinated TARDBP/TDP-43 (TAR DNA binding protein) as the major component.Citation8
Because many ALS patients show cognitive impairment in addition to muscle atrophy, and develop frontotemporal lobar degeneration (FTLD, which is second to AD as a cause of dementia in patients under the age of 65, see ref. Citation9),Citation10 and because many FTLD patients show ALS symptoms,Citation11 it is thought now that ALS and FTLD constitute 2 parts of a clinical spectrum of a disease.Citation12 It was also pointed out that the overlapping pathogenesis of ALS and FTLD indicated that in these diseases, motor neuron degeneration and cognitive deficits might originate due to a common molecular basis.Citation2 In fact, both of these diseases are proteinopathies, because they are associated with the presence of deposits or inclusions containing various misfolded proteins, such as ATXN2 (ataxin 2); C9orf72; dipeptide repeat proteins, DPRs; FUS (FUS RNA binding protein); HNRNPA1 (heterogeneous nuclear ribonucleoprotein A1); HNRNPA2B1 (heterogeneous nuclear ribonucleoprotein A2/B1); NEK1 (NimA related kinase 1); OPTN (optineurin); SOD1 (superoxide dismutase 1); TARDBP (TAR DNA binding protein); UBQLN2 (ubiquilin 2); VCP (valosin containing protein); or members of the ubiquitin-proteasome system (UPS). Therefore, pathologically, ALS and FTLD can be grouped into multiple subtypes based on the proteins involved in the formation of corresponding inclusions. These corresponding subtypes are ALS–ATXN2, ALS–C9orf72 or ALS–DPR, ALS–FUS, ALS–HNRNPA1, ALS–HNRNPA2B1, ALS–NEK1, ALS–OPTN, ALS–SOD1, ALS–TARDBP, ALS–UBQLN2, and ALS–VCP, or FTLD–DPR, FTLD–FUS, FTLD–MAPT, FTLD–TARDBP, and FTLD–UPS.Citation2
Although all aforementioned subtypes are sporadic forms of ALS and FTLD, proteinaceous inclusions of many familial forms of these diseases contain defective and mutated forms of proteins typically seen in sporadic diseases. The corresponding examples of familial ALS cases include pathologies related to the mutated or otherwise altered forms of ATXN2 (polyQ expansion), C9orf72 (hexanucleotide repeat expansions of the intronic region of the C9orf72 gene between the noncoding exons 1a and 1b), FUS (mutations), OPTN (exon deletion, mutations), SOD1 (mutations), TARDBP (mutations), UBQLN2 (mutations), and VCP (mutations). In familial forms of FTLD, the genetic alterations include mutations in GRN/PGRN (granulin precursor) and MAPT, hexanucleotide repeat expansions near the C9orf72 gene,Citation13 and mutations in several rare FTLD-related genes (or FTLD/ALS-related genes), such as CHMP2B (charged multivesicular body protein 2B); CHCHD10 (coiled-coil-helix-coiled-coil-helix domain containing 10); FUS; HNRNPA1; HNRNPA2B1; OPTN; SQSTM1 (sequestosome 1); TBK1 (TANK binding kinase 1); TARDBP; and VCP.Citation14 It is important to note that sporadic ALS and FTLD often arise from spontaneous mutations that were not inherited.Citation6,15 For example, mutations in the TARDBP gene have been linked to both familial and sporadic ALS.Citation16,17 Furthermore, increased susceptibility to sporadic ALS is thought to be associated with mutations in APOE (apolipoprotein E),Citation18 or in the KSP repeat region of the NEFH (neurofilament heavy) gene,Citation19,20 as well as with the decreased expression of the glutamate transporter SLC1A2/EAAT2 (solute carrier family 1 member 2)Citation21,22 or alterations in the VEGF (vascular endothelial growth factor) gene.Citation23 Therefore, there is a reason to think that all ALS subtypes may have a genetic root cause, whether or not familial inherited.
There are many mechanisms by which different proteins implicated in ALS or FTLD can be related to the pathogenesis of these diseases. These could be grouped into 2 major categories: loss of physiological function (the protein does not do what it is supposed to do), and gain of pathological function (the protein does what it should not do). Various factors, such as mutations, posttranslational modifications (PTMs), processing, changes in the environmental conditions (pH, temperature, ionic strength, exposure to membrane or metal ions, or some other small molecules, etc.), toxic insults, and failure of the protective system can lead to misrecognition, mistrafficking, mislocalization, misfolding, and aggregation, eventually resulting in the loss of normal functionality or the gain of pathological functions. In addition to all these factors, many of which were targets of countless studies whose outputs are covered in numerous dedicated reviews, a new threat is coming into the light of modern research, namely, the ability of some of the proteins to be engaged in the physiological liquid-liquid phase transitions (LLPTs) leading to the formation of various proteinaceous membrane-less organelles (PMLOs, such as different cytoplasmic RNA granules, including stress granules [SGs]). The goal of this review is to look into this mechanism using several illustrative examples of ALS- and FTLD-related proteins. To this end, we will first introduce some of the ALS- and FTLD-related proteins that were shown to be engaged in the LLPTs or related to the PMLOs and describe normal and pathological functions of those proteins. Then, we will consider the roles of these proteins, such as SOD1, TARDBP, FUS, C9orf72, and dipeptide repeat proteins generated as a result of the translation of the intronic hexanucleotide expansions in the C9orf72 gene, PFN1, GLE1, PURA, FLCN, RBM45, SS18L1/CREST, HNRNPA1, HNRNPA2B1, ATXN2, MAPT, and TIA1 in the formation of PMLOs. We will also analyze a link between the normal and pathological LLPTs, and illuminate the role of liquid-liquid phase transitions and intrinsic disorder in the “Dr. Jekyll–Mr. Hyde” behavior of these proteins.
2. Intrinsic disorder in ALS- and FTLD-related proteins
2.1. A brief overview of the intrinsic disorder phenomenon: What are the intrinsically disordered proteins and what are they for?
The fact that numerous biologically active protein regions and even entire proteins do not possess stable tertiary and/or secondary structures is rapidly becoming the new reality in protein science. These naturally flexible proteins are known now as intrinsically disordered proteins (IDPs),Citation24 a term selected by the scientific community from several other names used in the early literature dealing with this topic (e.g., natively denatured,Citation25 natively unfolded,Citation26 intrinsically unstructured,Citation27 and natively disordered proteins,Citation28 among many other terms). In addition to proteins that are intrinsically disordered as a whole, a much larger cohort of proteins is described as hybrid proteins that contain both ordered domains and IDP regions (IDPRs).Citation29 These proteins/regions exist as highly dynamic structural ensembles possessing multiple rapidly interconverting conformations.Citation30,31 The IDPRs are characterized by remarkable spatiotemporal heterogeneity, and their structures can range from expanded, random coil-like ensembles, to pre-molten globule-like ensembles, which are the highly disordered entities containing some localized regions of transient secondary structure, to molten globule-like species with well-developed secondary structure and mobile side-chains.Citation32–34
There are multiple levels at which IDPs and IDPRs differ from structured globular proteins and domains. Such differences can be found in amino acid composition, charge, flexibility hydrophobicity, and sequence complexity. For example, IDPs/IDPRs are significantly depleted in so-called order-promoting residues (I, L, V, W, F, Y, C, and N), and contain more disorder-promoting residues, such as A, R, G, Q, S, P, E, and K.Citation24,35–37 Based on these and other remarkable differences between the amino acid sequences of ordered proteins/domains on the one side and IDPs/IDPRs on the other side, numerous disorder predictors were developed. A short list of commonly used predictors include members of the PONDR family,Citation35,38 CH-plot,Citation39 NORSp,Citation40 GlobPlot,Citation41,42 FoldIndex,Citation43 IUPred,Citation44 DisoPred,Citation45–47 DisCon,Citation48 MFDp2,Citation49 and many others. It was pointed out that, because different computational tools use different sequence attributes and different definitions of disorder, additional important information related to the peculiarities of predicted disorder can be retrieved via comparing and combining the results of several predictors on an individual protein of interest or on a protein dataset.Citation50-57 Furthermore, several methods were developed for the prediction of disordered protein binding regions including alpha-MoRF-Pred,Citation58 ANCHOR,Citation59 MoRFpred,Citation60 PepBindPred,Citation61 MFSPSSMpred,Citation62 DISOPRED3,Citation63 MoRFCHiBi,Citation64 fMoRFpred,Citation65 and DisoRDPbind,Citation66,67 indicating that functions of IDPRs and IDPs are predictable from protein sequences. Finally, because sites of the enzyme-catalyzed posttranslational modifications, such as phosphorylation,Citation68 acetylation, methylation, and ubiquitinationCitation69 are commonly located within the IDPRs, several computational tools utilizing this information have been developed, such as DisPhos (disorder-enhanced phosphorylation predictor), which can efficiently find IDPR-located phosphorylation sites with 76% accuracy for serine, 81% for threonine, and 83% for tyrosine.Citation68 More recently, another tool has been developed which is a unified sequence-based predictor of 23 types of posttranslational modification (PTM) sites.Citation69
The high abundance of intrinsic disorder in nature was illustrated in several computational studies of different proteomes where various disorder predictors were utilized. These same studies showed that the overall level of disorder of proteins increases with organism complexity, and that over half of eukaryotic proteins are predicted to have long IDPRs.Citation47,50,70,71
The conformational adaptability and structural plasticity of IDPs/IDPRs, their ability to quickly react in response to the changes in their environment and to fold under a variety of conditions,Citation24,27,39,72–81 combined with their unique capabilities to fold differently while interacting with different binding partners and to be able to engaged in promiscuous binding,Citation78,82 define a wide set of functions exerted by IDPs/IDPRs in different biological systems, and explain and determine the broad participation of IDPs/IDPRs in various biological processes,Citation34,83,84 where they can be involved in various signaling processes,Citation85,86 regulation of numerous pathways,Citation87–94 cell protection,Citation95 protein protection,Citation96,97 cellular homeostasis,Citation98,99 and controlled cell death.Citation100–104
These same structural features also define the ability of IDPs to modulate and control functions of their binding partners and to promote and coordinate the assembly of various complexes.Citation105 IDPs can be engaged in a wide spectrum of interactions, ranging from those leading to the formation of highly stable assemblies to weak but specific signaling interactions.Citation32 It is also important to remember that although binding of IDPs/IDPRs to their partners is often accompanied by the disorder-to-order transitions, many IDPs and IDPRs preserve significant amounts of disorder in their bound states,Citation32 leading to the formation of so-called disordered, dynamic, or fuzzy complexes, being bound to ordered proteins,Citation106–111 other disordered proteins,Citation112–114 membranes,Citation115,116 or nucleic acids. Overall, IDPs/IDPRs can form static, semi-static, and dynamic or fuzzy complexes.Citation33 Static and semi-static binding modes range from the interaction-induced folding of a whole molecule to binding-triggered gaining of local structure on the surface of a binding partner, and from penetrating deep inside the binding partner to wrapping around the binding partner.Citation117 IDPs can participate in one-to-many and many-to-one interactions, where one IDPR binds to multiple partners potentially gaining very different structures in the bound state, or where multiple unrelated IDPs/IDPRs bind to one partner.Citation32
Furthermore, there are several means by which biological activities of IDPs/IDPRs are tightly controlled and regulated in a precise and timely manner. These means include alternative splicing generating multiple protein isoforms with highly diverse regulatory elementsCitation118–120 and various PTMs, such as acetylation, glycosylation, phosphorylation, etc.Citation34,69,121,122 In fact, the multifunctionality of IDPs/IDPRs relies (at least in part) on the presence of multiple relatively short functional elements that are spread within their amino acid sequencesCitation32 and can be dramatically reshuffled by alternative splicing (AS),Citation119,120,123 which can generate a set of protein isoforms with a highly diverse set of regulatory elements.Citation118
2.2. Proteoforms: Intrinsic disorder and one sequence–many functions paradigm
The functional multifariousness of IDPs/IDPRs represents an important illustration for the paramount conclusion that the proteome size, not the genome size, defines the complexity of a biological system.Citation124 This is because the number of functionally divergent proteins is typically higher than the number of genes. For example, in a human cell, the number of protein-coding genes approaches 21,000,Citation125 while the number of functionally different proteins is substantially higher (in the range of a few hundred thousand, if not millions).Citation126–130 There are several mechanisms that contribute to this functional diversification of proteins and can affect a given gene product at the DNA, mRNA, or protein levels. Among such diversifiers are single or multiple point mutations, indels (i.e., insertions or deletions), and single nucleotide polymorphisms causing allelic variations contributing at the DNA level; AS, mRNA editing, the use of alternative promoters, alternative initiation of translation, and other pre-translational mechanisms affecting mRNA; and different PTMs that can change physico-chemical properties of a polypeptide chain. Therefore, instead of the “one gene–one protein–one function” view, the actual gene-protein relationship is much more complex, being better described by the “one gene–many proteins–many functions” model (see ).
Figure 1. Classic and modern protein structure-function relationships. Schematic representation of the classic “one gene–one protein–one function” paradigm (top part, blue) and its modification by alternative splicing and PTMs when affected genes encode ordered proteins (middle part, pink) or intrinsically disordered and hybrid proteins containing ordered and intrinsically disordered domains (bottom part, red). Reproduced with permission from ref. Citation132.
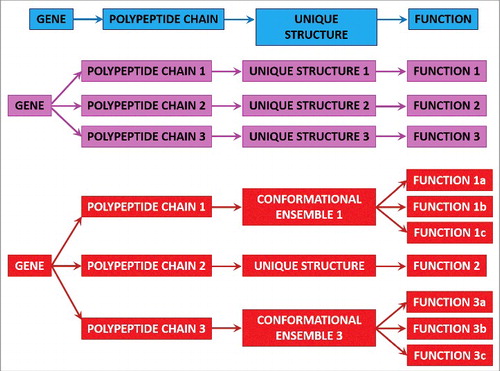
In other words, there are various means that ensure creation of a wide spectrum of rather different protein molecules from a single gene. This multilevel diversification of gene products is at the root of the proteoform concept,Citation131 where proteoforms “designate all of the different molecular forms in which the protein product of a single gene can be found, including changes due to genetic variations, alternatively spliced mRNA, and PTMs”.Citation131 Subsequently, it was pointed out that, in addition to the means for increasing the chemical variability of a polypeptide chain, the protein structural and functional diversity can be further extended via protein intrinsic disorder and function itself.Citation132–134 Here, any given protein can be considered as a basic (or intrinsic, or conformational) proteoform because it does exist as a dynamic conformational ensemble, members of which have different structures and can potentially have different functions. There are also inducible proteoforms generated by various allelic-, mRNA-, and PTM-based diversifications of the canonical protein sequence. Finally, because structural ensembles of both basic and induced proteoforms can be affected by protein function, interaction with specific partners, or placement inside the natural cellular environment (which is extremely crowded, characterized by the presence of high concentrations of various biological macromolecules,Citation135-137 has limited available volume,Citation138 and considerably restricted amounts of free waterCitation135,139–143), protein functionality can produce functioning proteoforms. Obviously, any member of the inducible (or modified) proteoform (i.e., any mutated, modified, or alternatively spliced form) or functioning proteoform is a conformational proteoform itself because it also represents a structural ensemble.Citation132–134 Curiously, alternative splicing generating multiple protein isoformsCitation118–120 and various enzymatically catalyzed PTMs, such as acetylation, glycosylation, phosphorylation, etc.,Citation34,69,121,122 are commonly located within the regions with intrinsic disorder, providing additional link between conformational/basic and inducible/modified proteoforms.
All these considerations, taken together, define the ability of a protein to have a multitude of structurally and functionally different states, which are constituents of the “protein structure-function continuum”, where a given protein exists as a dynamic conformational ensemble containing multiple proteoforms (conformational/basic, inducible/modified, and functioning) characterized by diverse structural and functional features.Citation132–134
2.3. Protein intrinsic disorder and human diseases
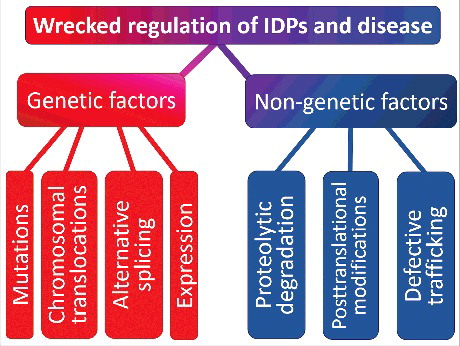
Since IDPs/IDPRs are common universal regulators and controllers, they are tightly controlled and regulatedCitation144 via multiple mechanisms, such as interaction with chaperonesCitation145–149 or nanny proteins,Citation150 binding to specific partners of proteinaceous and non-proteinaceous nature,Citation151–157 various PTMs,Citation87,158–161 and regulated degradation.Citation162–167 As the consequences of misbehavior of an important controller (reflected in the inability of such a protein to adopt and keep a functional state) or any distortion in the tightly controlled processes could be disastrous, it is not surprising that the development of particular pathological conditions is frequently associated with the dysfunction of IDPs manifested in protein misfolding and aggregation leading to the loss of normal function and gain of toxic function.Citation168–170 represents some of the factors related to this phenomenon and shows that the aberrant regulation at the genetic level or various posttranslational (nongenetic) mechanisms can cause pathogenic transformation in an IDP/IDPR.Citation171 The complexity of this picture, where numerous factors contribute to conformational diseases, is further increased by the ability of all these factors to not only work independently, but to also take action additively or synergistically.Citation171,172 Therefore, many IDPs and hybrid proteins contacting ordered domains and long IDPRs are related to the development of various diseases.Citation169,173,174 For example, in addition to the usual suspect, TP53/p53 (tumor protein p53),Citation175 some other well-known cancer-related proteins with experimentally confirmed IDPRs include APC (adenomatous polyposis coli),Citation176,177 TP53BPs/ASPPs (tumor protein p53 binding protein),Citation178,179 AXIN1 (axin 1),Citation176,180,181 BH3-only proteins,Citation182 BRCA1 (BRCA1, DNA repair associated),Citation183 CTAs/cancer-testis antigens,Citation184 CREBBP/CBP (CREB binding protein) and its paralog, EP300/p300 (E1A binding protein p300),Citation156 EWSR1 (EWS RNA binding protein 1),Citation185 proteins of human papillomavirus (HPV),Citation186 PTEN (phosphatase and tensin homolog),Citation187 CDKN1A/p21 (cyclin-dependent kinase inhibitor 1A) and CDKN1B/p27Kip1 (cyclin-dependent kinase inhibitor 1B),Citation92 SIRTs/NAD-dependent protein deacetylase sirtuins,Citation188,189 and many others. Many of the neurodegeneration-related proteins are also IDPs. Illustrative examples of highly disordered proteins engaged in various neurodegenerative diseases include a protein-chameleon, SNCA/α-synuclein,Citation190–195 associated with PD, dementia with Lewy bodies, AD, Down syndrome, and several other synucleinopathies, ataxin in spinocerebellar ataxia, prions in Creutzfeldt-Jakob disease and transmissible spongiform encephalopathies, amyloid β in AD, and MAPT in AD and many other tauopathies.Citation168,196 Among other human diseases for which involvement of IDPs was demonstrated are AIDS (HIV Rev protein),Citation197 cardiovascular disease (e.g., F2 [coagulation factor II, thrombin]),Citation198 cystic fibrosis (CFTR [cystic fibrosis transmembrane conductance regulator]),Citation199 and type II diabetes (IAPP/amylin [islet amyloid polypeptide]).Citation173
Figure 2. Factors causing pathogenic transformation in an IDP/IDPR. Schematic representation of different ways by which aberrant regulation at the genetic level or various posttranslational (non-genetic) mechanisms can cause pathogenic transformation in an IDP/IDPR. Reproduced with permission from ref. Citation171.
2.4. Intrinsic disorder in proteins related to ALS and FTLD
demonstrates that all ALS- and FTLD-related proteins considered in this review contain significant levels of intrinsic disorder. The analogous results for the dipeptide repeat proteins poly(GA), poly(GR), poly(GP), poly(PA), and poly(PR) synthesized as a result of the repeat-associated non-ATG (RAN) translation of the sense and antisense repeat RNAs derived from the intronic region of the C9orf72 gene are not shown in this figure. This is because of the well-known fact that the repeat-containing proteins are often intrinsically disordered, with the more perfect repeats being less structured.Citation200 Therefore, it is expected that these proteins should be mostly disordered, and in agreement with this expectation, all these proteins were predicted to be highly disordered.Citation201
Figure 3. Intrinsic disorder in proteins presented in this study. The intrinsic disorder predisposition of these proteins was evaluated by PONDR® VSL2. (A) SOD1; (B) TARDBP; (C) FUS; (D) C9orf72; (E) PFN1; (F) GLE1; (G) PURA; (H) FLCN; (I) RBM45; (J) SS18L1/CREST; (K) HNRNPA1; (L) HNRNPA2B1; (M) ATXN2; (N) TIA1; and (O) MAPT. In the corresponding plots, regions with disorder scores above the 0.5 threshold (shown as a thin black line) are predicted to be intrinsically disordered.
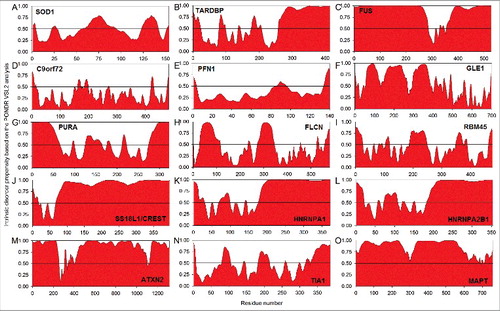
Results shown in are in agreement with the results reported in previous studies. In fact, prevalence of intrinsic disorder in proteins associated with various neurodegenerative diseases was the subject of several dedicated studies.Citation168,170,171,196,201–203 Also, some proteins associated with biological liquid-liquid phase transitions (LLPTs) and formation of various proteinaceous membrane-less organelles (PMLOs), including stress granules (SGs), are disordered.Citation204–206 As a result, in those studies, the intrinsic disorder status and biological roles of IDPRs were already systemized for several proteins considered in this review (SOD1, TARDBP, FUS, C9orf72 and DPRs, PFN1, TIA1, MAPT, HNRNPA1, HNRNPA2B1, and ATXN2).
Because interested readers can find details of those analyses in the corresponding publications,Citation168,196,205,206 there is no need to reproduce them here. Therefore, this section represents some data on the intrinsic disorder status of the remaining ALS-FTLD-related proteins that also play important roles in SG formation (GLE1, PURA, FLCN, RBM45, and SS18L1). It is important to keep in mind that the in-depth analysis of the intrinsic disorder predispositions of all these proteins and the detailed consideration of the potential roles of their IDPRs is outside the scope of this article.
Nucleoporin GLE1 (GLE1, RNA export mediator; UniProt ID: Q53GS7) is the nucleoporin responsible for the export of the poly(A)-tail containing mRNAs from the nucleus into the cytoplasmCitation207–210 and is also known as a multifunctional regulator of gene expression.Citation211 The N-terminal region (residues 1–30) of human GLE1 is required for interaction with the nuclear pore complex via the nucleoporin NUP155 (nucloporin 155),Citation212 whereas the coiled-coil domain containing 2 coiled-coil regions is required for GLE1 self-oligomerization and localization to the nuclear pore complex.Citation210 shows that the N-terminal half of this protein containing these functionally important regions is predicted to be mostly disordered.
A high level of disorder in this protein is also confirmed by the MobiDB database (http://mobidb.bio.unipd.it/)Citation213,214 which generates consensus disorder scores for a query protein by aggregating the output from 10 predictors. Such consensus MobiDB disorder content of human GLE1 is 25.50%. The conclusion on the presence of high levels of disorder in human GLE1 protein is further supported by , which shows the results of the analysis of this protein using the D2P2 database (http://d2p2.pro/)Citation215 that provides disorder evaluations by several computational tools and also shows some important disorder-related functional information, such as location of various curated PTMs and ANCHOR-predicted disorder-based protein-protein interaction sites,Citation59,216 known as molecular recognition features, MoRFs (see Citation58, 217–219). shows that human GLE1 is predicted to have 11 MoRFs, all located within the N-terminal half of the protein, and a multitude of PTMs, such as phosphorylation, ubiquitination, and acetylation. All this clearly indicates that abundant disorder serves several important functions for this protein.
Figure 4. Multifactorial analysis of intrinsic disorder in GLE1 (UniProt ID: Q53GS7) (A) and PURA (UniProt ID: Q00577) (B). Intrinsic disorder propensity and some important disorder-related functional information generated for the corresponding proteins by the D2P2 database are shown. Here, the green-and-white bar in the middle of the plot shows the predicted disorder agreement among 9 predictors, with green parts corresponding to disordered regions by consensus. The yellow bar shows the location of the predicted disorder-based binding sites (molecular recognition features, MoRFs), whereas colored circles at the bottom of the plot show the location of various PTMs.
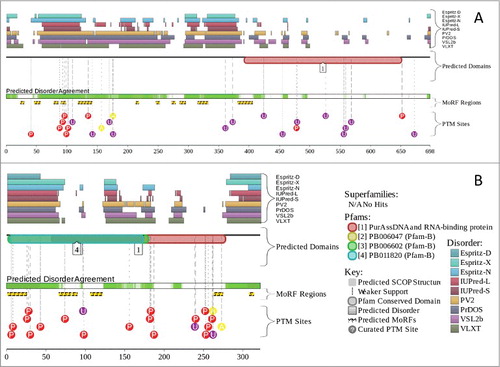
Transcriptional Activator PURA/Pur-α (UniProt ID: Q00577; MobiDB consensus disorder content is 41.93%) is a highly conserved multifunctional single-stranded DNA- and RNA-binding proteinCitation220 that serves as a transcriptional activator. and show that PURA is predicted to have intrinsically disordered N- and C-terminal tails and also has a disordered region in the middle of the sequence. It is predicted to have 6 MoRFs and several phosphorylation, ubiquitination, and acetylation sites spread throughout the sequence of this protein ().
FLCN (folliculin; UniProt ID: Q8NFG4; MobiDB consensus disorder content is 23.32%) is a tumor suppressor, that is predicted to contain 2 long IDPs and a disordered C-tail ( and ). The centrally located disordered region contains the coiled-coil domain (residues 287–310). FLCN is predicted to have 2 MoRFs located within the N-terminal IDPR, as well as several sites of phosphorylation and ubiquitination ().
Figure 5. Multifactorial analysis of intrinsic disorder in FLCN (UniProt ID: Q8NFG4) (A), SS18L1/CREST (UniProt ID: O75177) (B), and RBM45 (UniProt ID: Q8IUH3) (C). Plots (A) and (B) were generated using the D2P2 database. Here, keys are similar to those described in legends to . Because the D2P2 database does not have related information for human RBM45, plot (C) represents disorder profiles generated by PONDR® VLXT (black line), PONDR® FIT (red line), PONDR® VL3 (green line), PONDR® VSL2 (yellow line), IUPred_short (blue line) and IUPred_long (pink line). The cyan dashed line shows the mean disorder propensity calculated by averaging the disorder profiles of the individual predictors. The light pink shadow around the PONDR® FIT shows the error distribution. In these analyses, the predicted intrinsic disorder scores above 0.5 are considered to correspond to the disordered residues/regions, whereas regions with disorder scores between 0.2 and 0.5 are considered flexible.
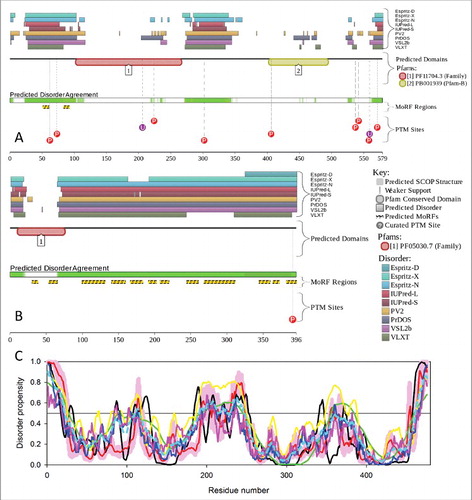
SS18L1/CREST (UniProt ID: O75177; MobiDB consensus disorder content is 86.16%) is a 396-residue-long, highly disordered transcription activator that has a disordered N-tail, 1 ordered domain that contains 2 MoRFs, and a long, highly disordered region covering the central and C-terminal parts of this protein ( and ). This disordered region serves as an excessive binding platform, because 11 of 13 MoRFs predicted in SS18L1 are located there. Overall, shows that 55.6% of human SS18L1 residues are expected to be engaged in disorder-based protein-protein interactions.
RBM45 (RNA binding motif protein 45; UniProt ID: Q8IUH3; MobiDB consensus disorder content is 18.07%) is an RNA-binding protein with binding specificity for poly(C). Although there is no information about this protein in D2P2, a multiparametric analysis of this protein by PONDR® VLXT, PONDR® FIT, PONDR® VL3, PONDR® VSL2, IUPred_short, and IUPred_long shown in suggests that this protein contains several disordered and flexible regions; that is, regions with disorder scores exceeding 0.5 and ranging between 0.2 and 0.5, respectively. Also, there are 3 MoRFs in human RBM45.
3. Stress granules, other proteinaceous membrane-less organelles, and biological liquid-liquid phase transitions
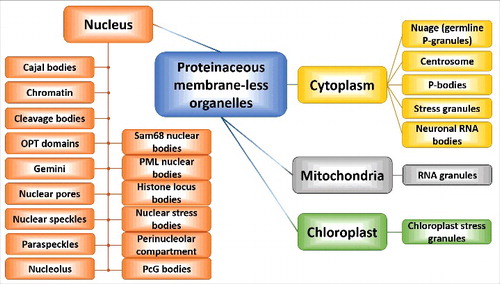
Colocalization of biological macromolecules (proteins and nucleic acids) at high concentrations within a small cellular micro-domain leads to the formation of various proteinaceous membrane-less organelles (PMLOs), including stress granules (SGs). These PMLOs are highly dynamic assemblages, which, due to the presence of various RNAs and proteins, are generally known as ribonucleoprotein (RNP) granules, RNP bodies, or RNP droplets.Citation221 Although such PMLOs can typically be found in the cytoplasm and the nucleoplasm of eukaryotic cells, they are also present in mitochondria and, in plants, in chloroplasts (). As a matter of fact, the known set of PMLOs exceeds 25 species. The incomplete list of cytoplasmic PMLOs includes centrosomes,Citation222 germline P-granules (germ cell granules or nuage),Citation223,224 neuronal RNA granules,Citation225 processing bodies or P-bodies,Citation226 and stress granules.Citation227 Most commonly known nuclear PMLOs are Cajal bodies,Citation228 chromatin,Citation229 cleavage bodies,Citation230 histone locus bodies,Citation231 nuclear gems (Gemini of Cajal bodies),Citation232,233 nuclear pores,Citation234 nuclear speckles or interchromatin granule clusters,Citation235 nuclear stress bodies,Citation236,237 nucleoli,Citation238 Oct1/PTF/ transcription (OPT) domains,Citation239 paraspeckles,Citation240 perinucleolar compartment,Citation241 PcG bodies (polycomb bodies, subnuclear organelles containing polycomb group proteins),Citation242 promyelocytic leukemia nuclear bodies or nuclear dots,Citation243 and the Sam68 nuclear body.Citation241 Mitochondria and chloroplasts have RNA granulesCitation244 and chloroplast SGs, respectively.Citation245 This realm of cytoplasmic, nuclear, mitochondrial and chloroplast PMLOs is depicted in . One should keep in mind that although in this review SGs are the most frequently discussed PMLOs, pathological cellular transformations may also engage other organelles (e.g., P-bodies, neuronal RNA granules, CREBBP-containing nuclear bodies, and paraspeckles among others, see below).
Figure 6. Diversity of PMLOs found in eukaryotic cells. Schematic representation of the multitude of cytoplasmic, nuclear, mitochondrial and chloroplast PMLOs.
The important feature of PMLOs that clearly differentiate them from other cellular organelles is the fact that, contrary to the “traditional” membrane-encapsulated organelles, the structural integrity of PMLOs is not supported by encapsulation in the membrane. Instead, their biogenesis is entirely controlled and mediated via a multitude of protein-protein, protein-RNA, and/or protein-DNA interactions.Citation246 Furthermore, this lack of membrane encapsulation defines the liquid-like behavior, such as dripping, relaxation to spherical structures upon fusion, and wetting, commonly described for various PMLOs.Citation223,227,247,248 Also, the lack of encapsulation defines the highly dynamic nature of PMLOs and ensures the direct contact of their components with the surrounding cytoplasm or nucleoplasm.Citation249,250 Based on all these considerations, as well as on the fact that PMLOs are just slightly denser than the rest of the cytoplasm or nucleoplasmCitation251,252 and possess high level of internal dynamics, these organelles are defined as liquid-droplet phases of the nucleoplasm/cytoplasm.Citation223,227,247,248,253,254 Curiously, although PMLOs are considered as a different liquid state of the cytoplasm or nucleoplasm, their major biophysical properties are rather similar to those of the rest of the intracellular fluid.Citation221 PMLOs have unique morphologies, specific distribution patterns, and, at the molecular level, their structures are determined by a specific set of resident proteins.
It is assumed that the formation of SGs and other PMLOs represents a result of the liquid-liquid phase transition (or fluid-fluid demixing).Citation221,223,247,255–258 Biophysically, besides the obvious requirement of the presence of high enough concentrations of the related constituents compartmentalized within a limited enough volume, the efficiency of such LLPTs depends on the flexibility and multivalency of the participating components and on the ability of these constituents to be engaged in multiple highly controllable low-affinity interactions.Citation253,259–270 It was emphasized that highly dynamic structures, a low complexity of amino acid sequences that can be considered as a string of repetitive amino acids or groups of amino acids, and the ability to be engaged in multiple low-affinity interactions with other proteins and nucleic acids of IDPs and IDPRs, make them ideal drivers of such LLPTs, and, in agreement with this hypothesis, many PMLOs abundantly contain IDPs/IDPRs.Citation205,206,271–273
Since SGs are the PMLOs which are most frequently considered in association with ALS, these RNP droplets are discussed below in greater detail. Readers interested in other PMLOs are can peruse recent reviews.Citation206,221,273,274 As with all other PMLOs, SGs are complex assemblages that include proteinaceous and RNA constituents and are typically formed as a specific cellular response to stressful conditions or to a variety of environmental and cellular cues. SGs include components of the translation-competent 48S complex (eukaryotic initiation factors EIF3 complex, EIF4G1/EIF4F, EIF4B, the small ribosomal subunits, PABPC1/PABP1 (poly[A] binding protein cytoplasmic 1), and stalled mRNA transcripts),Citation275 many other RNA binding proteins (RBPs; primary SG formers) that are involved and regulate mRNA stabilization, processing, translation, and transport,Citation276 and, being overproduced, are able to induce SG formation in the absence of stress (such as PABPC1, TIA1, TIA1L/TIAR [TIA1 cytotoxic granule associated RNA binding protein like 1], G3BP1 [G3BP stress granule assembly factor 1], CPEB [cytoplasmic polyadenylation element binding protein], FXR1 FMR1 autosomal homolog 1], FMR1/FMRP [fragile X mental retardation 1], and CASC3/MLN51 [cancer susceptibility 3]),Citation276–283 as well as a variety of other proteins that are recruited to the SGs via protein-protein interactions with primary SG formers.Citation275 Furthermore, these cytoplasmic foci contain various mRNA stalled at the pre-initiation stage and can therefore be considered as cytoplasmic messenger ribonucleoprotein particles that possess defined cytoprotective functions.Citation283 Among the biological functions ascribed to SGs are modification of the local patterns of translation within the cell, sequestration of various signaling molecules that regulate cell viability, and sequestration and silencing of mRNAs encoding house-keeping proteins.Citation275,284 Furthermore, under stress conditions SGs participate in shifting RNA translation towards cytoprotective proteins, such as chaperones and heat shock proteins.Citation285 The list of regulatory proteins sequestered by SGs includes several regulators of apoptosis, such as RACK1 (receptor for activated protein C kinase 1), TRAF2 (TNF receptor-associated factor 2),Citation281,286,287 and MTORC1 (mechanistic target of rapamycin complex 1) that serves as a regulator of cell growth and metabolism.Citation288,289
Of special importance is the fact that, unlike amyloid or amyloid-like fibrillation, the process of SG formation is completely reversible and is tightly controlled.Citation290 The formation of SGs, being an illustrative example of biological LLPT, is a very complex process. In brief, SG nucleation is initiated by several RNA-binding proteins, with the most commonly examined being TIA1, TIAL1, ZFP36/TTP (ZFP36 ring finger protein), and G3BP,Citation275 whereas SG maturation involves incorporation of many pro-apoptotic proteins (e.g., TRAF2, ROCK1 (Rho associated coiled-coil containing protein kinase 1), and RACK1 and other regulators of apoptosis (such as RPS6KA3/RSK2 [ribosomal protein S6 kinase A3] and FASTK [Fas activated serine/threonine kinase]), leading to the inhibition of the apoptotic response.Citation290 Among apoptosis-unrelated signaling and regulating proteins incorporated into the SGs are MAPK8/JNK1 (mitogen-activated protein kinase 8), MAP2K7/MKK7 (mitogen-activated protein kinase kinase 7), RHOA (ras homolog family member A), AKAP9/AKAP350 (A-kinase anchor protein 9), WDR62 (WD repeat domain 62), and HDAC6 (histone deacetylase 6).Citation290
An interesting recent development is the recognition of the importance of SGs for the pathology of several neurodegenerative diseases,Citation283,290–292 including ALS and FTLD.Citation283,293-299 Relation of SGs to neurodegeneration comes from the following observations:
| (i) | SG marker proteins, such as PABPC1 and G3BP, are found in cytosolic inclusion bodies of patients with AD, ALS, and FTLD, indicating that distorted SG dynamics might contribute to pathogenesis of these and other neurodegenerative diseasesCitation300; | ||||
| (ii) | in many neurodegenerative diseases, there is an obvious colocalization of SGs with insoluble protein aggregatesCitation290; | ||||
| (iii) | SGs frequently contain RNA-binding proteins related to the pathogenesis of various neurodegenerative diseases, such as TARDBP and FUS, related to the pathology of ALS and FTLD, SMN (survival motor neuron protein) related to the spinal muscular atrophy pathology, spinocerebellar ataxia 2-related ATXN2, OPTN related to primary open angle glaucoma and amyotrophic lateral sclerosis 12, and ANG (angiogenin) that is involved in amyotrophic lateral sclerosis 9 pathology; | ||||
| (iv) | Mutations in the synaptic functional regulator FMR1, which is a protein that plays a central role in neuronal development and synaptic plasticity through the regulation of alternative mRNA splicing, mRNA stability, mRNA dendritic transport, and postsynaptic local protein synthesis of a subset of mRNAsCitation301–305 and is associated with fragile X syndrome) or HNRNPA1 (an RNA-binding protein related to ALS-FTLD pathogenesis) cause severe impairment of SG dynamicsCitation300; | ||||
| (v) | also, MAPT,Citation290 PRNP/ PrP (prion protein),Citation306 HTT (huntingtin),Citation307 and some other Q/N-rich proteinsCitation308 are associated with SGs and modulate SG formation. | ||||
The roles of various proteins related to ALS and/or FTLD and involved in SG formation are considered below.
4. Normal and pathological functions of the ALS- and FTLD-related proteins and their roles in biogenesis of stress granules and other PMLOs
4.1. SOD1/Cu-Zn superoxide dismutase 1
4.1.1. Structural properties of SOD1
The SOD1 gene is located at position 22.11 within the long (q) arm of chromosome 21 (genetic location 21q22.11), and its coding region consists of 5 exons interrupted by 4 introns.Citation309 Struturally, human SOD1 (UniProt ID: P00441) is a homodimer whose protomers are held together by hydrophobic interactions.Citation310,311 The structure of this protein is characterized by the highly conserved topology found in all the intracellular eukaryotic SODs, where each protomer has 153 amino acids arranged in a Greek key β-barrel structure composed of 8 antiparallel β-strands.Citation312 Homodimeric SOD1 is characterized by a very high conformational stability, with the melting temperature (Tm) exceeding 90°C.Citation313 This high stability is determined by 3 factors associated with the posttranslational maturation of this protein, such as the coordination of 1 Zn2+ and 1 Cu2+ ion by each protomer of the homodimer, the formation of the intrachain disulfide bond Cys58-Cys147, and dimerization. In fact, disulfide-reduced apo-SOD1 (i.e, a protein with metal ions removed) has a Tm of ∼42°C,Citation314,315 which makes it highly succeptible for spontaneous partial unfolding followed by misfolding and aggregation under physiological conditions. Amyloid-like fibrils, which are a pathological hallmark in SOD1-related patients with familial ALS,Citation316 are formed only by the disulfide-reduced apo-SOD1.Citation317 This clearly indicates that intracellular deregulation of metal binding and/or disulfide formation might be among the key factors triggering the pathological misfolding of SOD1.Citation318 In agreement with this hypothesis, many ALS-related mutations of SOD1 affect the post-translational control of SOD1 maturation,Citation319,320 leading to an increase in the intracellular populations of the apo-,Citation321 and/or disulfide-reduced forms of this protein.Citation322
Reduction of the disulfide bond causes increased disorder in the apo-SOD1 generating a conformation significantly different from the structure of the wild-type SOD1 (WT-SOD1) protomer.Citation318 These observations are in strong agreement with a systematic computational analysis of the folding and structural dynamics of monomeric and dimeric forms of SOD1 in their holo- and apo-forms, both in the disulfide-intact and disulfide-reduced states by the all-atom discrete molecular dynamics (DMD) simulations, which revealed that although the disulfide bond formation has less pronounced stabilizing effects than the metal binding, the disulfide bond nevertheless has a significant contribution to protein dynamics.Citation323 It was also shown that the reduction of this disulfide bond is necessary for initiation of fibril formation.Citation324
4.1.2. Normal and pathological functions of SOD1
SOD1 is an antioxidant metalloenzyme that protects cells from oxidative stress (OS) by catalytic neutralization of the toxicity of superoxide radicals via a reaction that converts superoxide (O2−) to O2 ions and H2O2.Citation325–327
SOD1 is one of the genes that are most commonly involved in the ALS pathogenesis. SOD1 mutations have been associated with a great number of familial ALS (fALS) cases, and more than 170 mutations of SOD1 were found to cause ∼20% of the cases of fALS and ∼5% of the cases of sporadic ALS (sALS).Citation328–331 However, it is worth keeping in mind that the numbers reflecting the incidence of SOD1 mutations in ALS are changing over time, and it is currently accepted that ∼10% and ∼2% of fALS and sALS cases, respectively, are associated with mutations in the SOD1 gene.Citation332 Although many of the SOD1 mutations lead to a loss of function, at least some of the ALS-related mutations are thought to cause the enzyme to gain damaging properties,Citation333 such as ability to damage mitochondria, proteasomes, and protein chaperones.Citation334 Conformationally, many SOD1 mutants have similar properties, possessing solvent-exposed hydrophobic regions that increase the propensity of this protein to aggregate.Citation335
Interestingly, the “Dr. Jekyll–Mr. Hyde” behavior of SOD1 is not only triggered by mutations, because misfolded WT-SOD1 has also been implicated in sporadic ALS.Citation335 Curiously, although the discovery of the dominant mutations in the SOD1 gene made almost 25 years agoCitation336 marked the beginning of the molecular era of the analysis of ALS, no consensus on the main toxicity of mutant SOD1 has emerged as of yet.Citation4 Currently, there is a wide spectrum of potential toxic mechanisms proposed to explain how mutations in SOD1 might mediate the degeneration and death of motor neurons,Citation4 and such a lack of consensus serves as an indication of the overall complexity of this problem.
SOD1 protein maturation involves the appropriate coordination of Cu2+ (by His47, His49, His64, and His121) and Zn2+ ions (by His64, His72, His81, and Asp84) and the formation of a kinetically stable disulfide bond between Cys57 and Cys146.Citation337 A very unusual feature of SOD1 is its striking capability to maintain an intrasubunit disulfide bond even under the reducing conditions of the cytosol. In fact, in the vast majority of proteins the sulfhydryl groups of cysteine residues have a pKa >8.0. As a result, in the absence of oxidative stress and in the reducing environment of the cytoplasm at physiological pH, these sulfhydryl groups remain protonated, and cytoplasmic proteins, as a rule, do not contain disulfide bonds.Citation338,339 The CCS (copper chaperone for superoxide dismutase) is required for the formation of this disulfide bond in SOD1, because the spontaneous disulfide formation in SOD1 by O2 is a slow process, which is greatly accelerated by the Cu2+-bound form of CCS.Citation337 It was pointed out that both metal occupancy and disulfide status are crucial for the formation of a stable functional state of SOD1,Citation340 because without these factors the protein cannot gain its stable native conformation and cannot dimerize. Obviously, the formation of these functional homodimers with stable native conformation and intact disulfide bonds can also be altered by the pathological SOD1 mutations,Citation335 or by oxidation, demetallation, and other altered PTMs.Citation341–344 It was also pointed out that some aberrant PTMs induced by oxidative damage (e.g., appearance of oxidized carbonyl groups in the protein leading to its over-oxidation) force the WT-SOD1 to acquire binding and toxic properties of mutant forms of this protein.Citation341–344 This gives some support to the idea that SOD1 “is subject to misfolding following the loss of normal PTM and/or the induction of aberrant modification”.Citation335
There are several ways by which mutated and misfolded SOD1 and misfolded WT-SOD1 might act as a neurotoxin. First, pathogenic SOD1 may exert its neurotoxic effects via glutamate excitotoxicity, which causes an abnormally high release of calcium ions in the post-synaptic cleft that triggers subsequent neurotoxic events including mitochondrial dysfunction. Pathogenic SOD1 is theorized to cause this excitotoxicity due to the loss of SLC1A2/EAAT2/GLT1 (reducing reuptake of the neurotransmitter) and excessive glutamate efflux. Second, astrocytes expressing a SOD1 mutant lack the ability to effectively regulate their α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, whose subunit composition is in constant flux, which can cause different permeability. These mutant astrocytes lack the GRIA2/GluR2 subunit of these receptors that increases their permeability to Ca2+ ions. This sensitization, along with increased calcium production/decreased reuptake, act in tandem to cause excitotoxicity.Citation335 Third, OS by reactive oxygen species (ROS) is increased in cells with mutant SOD1. However, this stress is not due to loss of function by SOD1 but rather due to upregulation of the ROS. These ROS are formed via many different mechanisms including NAPDH oxidase and mitochondrial respiration.Citation335 Fourth, mutant SOD1 puts stress on endoplasmic reticulum (ER) proteins such as DERL1 (derlin 1), and the resulting ER stress has been linked to both familial and sporadic ALS. In particular, DERL1 is crucial for the elimination of misfolded proteins from the endoplasmic reticulum. Interactions between mutant SOD1 and DERL1 cause the inhibition of this process, resulting in ER stress and ultimately apoptosis via ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1).Citation335
The mitochondria is another target for mutant SOD1, which has been shown to accumulate exclusively on the cytosolic side of mitochondria in the spinal cord even before ALS symptoms are observed.Citation335 Binding of mutant SOD1 to several target proteins, including the integral membrane proteins BCL2 (BCL2, apoptosis regulator) and VDAC1 (voltage dependent anion channel 1), causes irregularities in mitochondrial homeostasis. Over-oxidized WT-SOD1 also forms a toxic complex with BCL2 in the lymphoblasts in some patients with sporadic ALS.Citation335
Vesicle transport is extremely important for long motor neurons to function normally, and distortion in this transport can potentially contribute to selective motor neuron death. Therefore, another pathological mechanism of mutant SOD1 has been linked to abnormal axonal synaptic vesicle transport in motor neurons during the early stages of ALS.Citation345 In fact, at an early asymptomatic ALS stage preceding loss of spinal motor neurons and peripheral axons, fast axonal transport is impaired in transgenic mice carrying G93A SOD1G93A or SOD1G86R mutationsCitation345,346 mutations equivalent to those found in subsets of human fALS. In line with the idea that the oxidized WT- SOD1 shares an aberrant conformation with the ALS-related mutants of this protein, a detailed analysis revealed that recombinant, oxidized WT- SOD1 and the wild-type protein immunopurified from sALS tissues were equally able to inhibit the kinesin-based fast axonal transport, acting similarly to the fALS-linked SOD1 mutants,Citation343 suggesting an overlapping pathogenic mechanism.Citation335
Finally, recent evidence suggests that SOD1 aggregation also exerts toxicity via prion-like propagation. This idea is supported by the fact that aggregating SOD1 is engaged in both cell-to-cell transmission and seeded aggregation. Both wild-type and mutant SOD1 form aggregates that are released from the injured cells as part of an exosome.Citation347 Also, mutant SOD1 can be released via a main secretory pathway mediated by CHGA (chromogranin A) and CHGB that serve as components of neurosecretory vessels.Citation348 Likely, this pathway can also be related to the release of the oxidized WT-SOD1 which was shown to interact with CHGB.Citation341,349 In addition, both wild-type and mutant SOD1 aggregates seed aggregation of WT- SOD1 in vivo, supporting the theory of prion-like propagation of SOD1-related pathology.Citation335
4.1.3. SOD1 and biogenesis of stress granules and other PMLOs
It has been established that both the WT-SOD1 and mutant SOD1 forms (mtSOD1 containing A4T, G41S, G85A, or G93A substitutions), as well as TARDBP, and YWHAB/14-3-3 protein α/β (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein beta) can be involved in interaction with NEFL (neurofilament light) mRNA and differentially modulate the stability of this mRNA, which is destabilized by mutant SOD1,Citation350 but stabilized by TARDBPCitation351 and YWHAB.Citation352 Furthermore, interaction of WT-SOD1 or mtSOD1 with YWHAB results in efficient disruption of the interaction of YWHAB with NEFL mRNA,Citation353 suggesting that SOD1 might have dual effects on the mRNA stability, via direct binding and by eliminating stabilizing YWHAB-NEFL mRNA interaction. Importantly, using RNA-IP-PCR in ALS spinal motor neurons, it has been also demonstrated that NEFL mRNA is sequestered in SGs and P-bodies, suggesting that NEFL mRNA processing is dramatically altered in ALS.Citation353 Although the colocalization with the NEFL mRNA-containing stress granules and P-bodies was unequivocally established for TARDBP, the ability of YWHAB, WT-SOD1 and mtSOD1 to interact with NEFL mRNA suggests that all these proteins can be related to alteration of the processing of this mRNA leading to its compartmentalization within both SGs and P-bodies.Citation353 It is important to note that the aforementioned NEFL, is not the only mRNA sequestered by SOD1 and TARDBP aggregates and SGs. As a matter of fact, it is now well-established that TARDBP and SGs in general bind up thousands of various RNAs, making NEFL mRNA, at best, a singular model RNA for the types of cellular mechanisms these promiscuous RNA-binding proteins and RNA granules might be involved with.Citation354–358
Direct involvement of SOD1 in the regulation of SG dynamics was recently established by showing the ability of mtSOD1 (L144F) to interact, in an RNA-dependent manner, with G3BP1 that plays a critical role in SG dynamics.Citation359 In fact, the aforementioned mtSOD1-G3BP1 interaction is rather specific (involving F380 and F382 residues of the RNA-binding RNA-recognition motif [RRM] domain of G3BP1), because almost no interaction is seen between the mtSOD1 and other RNA-binding proteins implicated in ALS.Citation359 It was also shown that the SG formation induced in N2A cells by hyperosmolar shock and arsenite treatment is delayed by the mtSOD1, indicating that one of the culprits of pathogenic SOD1 mutations in ALS could be related to the alterations of the RNA metabolism and the SG dynamics.Citation359
4.2. TARDBP/TDP-43 (TAR DNA binding protein)
4.2.1. Structural properties of TARDBP
TARDBP/TDP-43 is an RNA- and DNA-binding protein that is 414 amino acids long with a molecular mass of 44 kDa. TARDBP belongs to the family of the heterogeneous ribonucleoproteins (hnRNPs), members of which are RNA-binding proteins that bind to pre-mRNA and are involved in RNA splicing. The TARDBP gene is located on chromosome 1p36.2 and contains 6 transcribed exons.Citation360 Human TARDBP protein (UniProt ID: Q13148) has an N-terminal domain (NTD, residues 1–103), 2 RNA recognition motifs, RRM1 (residues 104–200) and RRM2 (residues 191–262), a nuclear export signal (NES), a nuclear localization signal (NLS), and a glycine-rich C-terminal domain (residues 274–413), which is known as a prion-like domain and is predicted to be highly disordered (). This RRM1-RRM2-C-tail domain organization is similar to that of hnRNP family proteins such as HNRNPA1 and HNRNPA2B1.Citation361 Although TARDBP is mainly a nuclear protein, it moves between the inside and outside of the nucleus, with this shuttling regulated by its NES and NLS motifs. The C-terminal domain is used by TARDBP for binding to single-stranded DNA, RNA, and proteins, and most of the ALS- and FTLD-related mutations found in TARDBP are located within this prion-like domain encoded by exon 6 of the TARDBP gene.Citation362 This prion-like domain has also been associated with the misfolding and self-aggregation of this protein.Citation363 According to comprehensive computational analysis, the C-terminal region of human TARDBP contains 4 disorder-based protein-protein interaction sitesCitation201 and, therefore, clearly serves as a major “docking port” for its binding partners.
Human TARDBP is present in 2 proteoforms generated by alternative splicing, with isoform #2 being different from the canonical form of the protein by having a changed N-terminal tail, where the 1MSEYIRVTEDENDEPIEICitation18 sequence is substituted to MPQMLAGEIWCMLSTIQK, and where the 19–134 region is missing.
4.2.2. Normal and pathological roles of TARDBP
TARDBP has many functions, most of which are related to RNA handling due to its ability to bind RNA through its RNA recognition motifs.Citation364 Some of these functions include regulation of transcription, pre-mRNA splicing, as well as processing, stability, and transport of mRNA.Citation365,366 In addition, TARDBP can participate in protein-protein interactions with itself and several other proteins. Examples of its binding partners include HNRNPA1, HNRNPA2B1 and FUS.Citation367 These proteins are required for TARDBP to perform some of its biological functions.Citation367 An important feature of TARDBP is its intrinsic ability to form aggregates, and this aggregation process is mediated by the C-terminal fragments of TARDBP.Citation368 TARDBP is a component of ubiquitin-positive MAPT-negative insoluble protein aggregates in both neurons and glial cells in sporadic ALS.Citation369 In addition, it was shown that the cleavage of TARDBP by CAPN (calpain) makes this protein more prone for aggregation than the full-length protein, and that both phosphorylated and nonphosphorylated TARDBP fragments resulting from this proteolytic cleavage remain intracellularly long enough to self-aggregate.Citation370 Furthermore, because the phosphorylated TARDBP becomes resistant to the CAPN cleavage it is likely that phosphorylation (and some other PTMs) may play a regulatory role in the TARDBP pathology of ALS and FTLD.Citation370 The pathological phosphorylation of TARDBP is controlled by the phosphatase PPP3/calcineurin that can bind to and catalyze the removal of pathological C-terminal phosphorylations of TARDBP.Citation371
In line with the hypothesis on the important roles of the TARDBP proteolytic cleavage for pathogenesis of neurodegeneration is the fact that various TARDBP fragments generated by the apoptotic CASP3 (caspase 3) and CASP7, as well as by CASP6 and CASP8 after activation by CASP3, accumulate in motor neurons and serve as a post mortem hallmark of different neurodegenerative diseases.Citation372 In fact, activation of the CASPs under some stress conditions might result in the fast appearance of the C-terminal fragments (CTFs) of TARDBP with the molecular mass of 35 kDa that accumulate in the cytoplasm, and the formation of this truncated form of TARDBP can be efficiently inhibited by the A90V mutation found in the FTLD/ALS patient with a family history of dementia,Citation373 whereas another mutant, D169G, is cleaved by CASP3 more efficiently than the wild-type TARDBP.Citation374 Also, the 35-kDa CTFs of TARDBP form ubiquitin-negative cytoplasmic inclusion bodies, which are mildly toxic, whereas the 25-kDa TARDBP CTFs are able to efficiently form toxic ubiquitin-positive inclusion bodies and sequester cellular RNA.Citation375 These observations are in line with the hypothesis that the CASP-mediated cleavage pattern of TARDBP determines the rate of clearance and cytotoxicity of this protein.Citation376 They also indicate that TARDBP possesses dual vulnerability to proteolytic attack by both CAPNs and CASPs.Citation377
4.2.3. TARDBP, SGs, and other PMLOs
Undoubtedly, among the ALS-FTLD-related RNA-binding proteins, the involvement of TARDBP in SG formation represents one of the best studied cases of the regulated protein aggregation phenomenon. One of the first reports indicating the existence of a potential link between TARDBP, SGs (which are liquid-like ribonucleoprotein complexes where protein synthesis is temporarily arrested), and ALS was published in 2009, when, based on axotomized motor neuron analysis, it was hypothesized that the upregulated TARDBP expression combined with the prominent localization of this protein to the cytosolic SGs might represent a physiological response to the motor neuron injury caused by degenerative processes, such as ALS.Citation378 In the same year, careful analysis of the response of a motoneuronal cell line to oxidative stress and environmental insults revealed that TARDBP is capable of efficient SG assembly, with the RRM1 (residues 104–200) and the 216–315 region of the TARDBP C-tail being crucial for the recruitment of this protein into SGs.Citation379 Also, analysis of the ALS spinal motor neurons revealed that TARDBP plays a fundamental role in compartmentalization of NEFL mRNA (processing of which is fundamentally altered in ALS) within SGs and P-bodies.Citation359 The SG recruitment of TARDBP was further evidenced by the large scale analysis of the TARDBP interacting proteins, many of which are known SG components.Citation364 Furthermore, this analysis revealed that the TARDBP interacting proteins can be functionally clustered into the cytoplasmic/translation- and nuclear/splicing-related PPI networks, suggesting that TARDBP can function in both the nucleus and the cytoplasm, playing multiple roles in RNA metabolism.Citation364
Many subsequent studies provided direct support to the idea that TARDBP, SGs, and ALS-FTLD are intimately linked. For example, an analysis of the cultured cells under the stress conditions revealed the existence of the TARDBP-SG association promoted by both the direct binding of TARDBP to specific SG proteins, such as TIA1, and via the direct interactions of TARDBP with RNA.Citation380 Furthermore, it was pointed out that the “regulated protein aggregation” leading to the SG formation might be associated with the ALS pathology, because the increased formation of TARDBP-containing SGs is associated with accumulation of TARDBP-based detergent insoluble complexes both in cell cultures and pathological brain tissues.Citation380 Also, treatment with the translational inhibitors that suppress or reverse SG formation inhibits the formation of inclusions mediated by the wild-type or mutant TARDBP,Citation380 suggesting that there is an interplay between TARDBP aggregation, SG formation, and the ALS-FTLD-associated TARDBP mutations, an understanding of which is vital to the clarification of the roles of TARDBP in neurodegeneration.Citation294 It was indicated that pathological TARDBP aggregates can be formed via at least 2 different mechanisms, either following the “independent model”, where the formation of such aggregates is independent of SG formation, or via the “precursor model,” where SG formation might seed the formation of TARDBP aggregates.Citation294 Therefore, TARDBP undergoes transitions between soluble, droplet, and aggregate phases, and these transitions might have important implications for the pathological aggregation and disease development.Citation381
There are also several studies emphasizing that the TARDBP-based formation of SGs, due to being regulated by various means, represents an illustrative example of “regulated protein aggregation”. For example, TARDBP involvement in SG formation in SH-SY5Y neuronal-like cells exposed to chronic oxidative stress induced by overnight treatment with the mitochondrial inhibitor paraquat is affected by inhibition of MAPK8/JNK1, MAPK3/ERK1 or MAPK14/p38.Citation382 In addition to oxidative stress, formation of TARDBP-positive SGs can be promoted by ER stress, which is induced in both familial and sporadic forms of ALS, and the extent of which is dependent on the overexpression of ALS-linked mutant TARDBP.Citation383 Intracellular levels of 4-hydroxynonenal (HNE, a marker of oxidative stress), are elevated in sporadic ALS patients. Treatment of cells with HNE induces noticeable aggregation and phosphorylation of TARDBP, affects cellular localization of this protein, and decreases its accumulation in SGs, suggesting that the aberrations in the SG dynamics induced by the enhanced HNE levels might represent an ALS risk factor.Citation384
The paraquat-induced formation of the TARDBP-positive SGs in SH-SY5Y cells is successfully suppressed by inhibiting the MAPK/ERK-induced phosphorylation of TARDBP by bioavailable bis(thiosemicarbazonato)copper(II) complexes.Citation385 Subsequent screening of various kinase inhibitors using an in vitro model of the formation of TARDBP-positve SGs revealed that the TARDBP accumulation is successfully decreased by the inhibitors of CDKs (cyclin-dependent kinases) and GSK3 (glycogen synthase kinase 3), and these same inhibitors are able to reverse the pre-formed TARDBP-positive SGs.Citation386 A more detailed analysis revealed that the SG integrity is dependent on CDK2 phosphorylation.Citation387 Here, only phosphorylated CDK2 is able to colocalize with TARDBP accumulated within SGs and to phosphorylate HNRNPK (heterogeneous nuclear ribonucleoprotein K) that interacts with TARDBP, whereas nonphosphorylated CDK2 fails to phosphorylate HNRNPK, preventing its incorporation into SGs and thereby preventing accumulation of TARDBP in SGs.Citation387
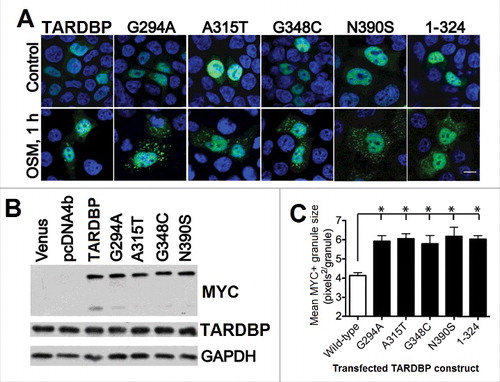
The fact that the ALS-linked mutations of TARDBP (A315T and Q343R) are able to increase the tendency of this protein not only to aggregate, but also to form SGs provides another important link between ALS pathology and aberrant SG dynamics.Citation388 provides an illustration for this idea by presenting effects of pathological TARDBP mutants, such as the fALS-associated mutation A315T, sALS-related mutations G294A and N390S, the fALS- and sALS-associated mutation G348C, and truncated TARDBP (residues 1–324) on SG formation and characteristics in HEK293T cells.Citation389 shows that although the wild-type and all mutant proteins are expressed at similar levels () and are predominantly localized to the nucleus in the unstressed HEK293T cells (see “Control” line in ), a multitude of SGs is formed in the cytoplasm of stressed HEK293T cells by each pathological mutant after one h of osmotic stress induced by 0.4 M sorbitol, and these “pathological” SGs are more numerous and noticeably larger than the SGs formed by the wild-type TARDBP under the same conditions (see for the quantification of the size of SGs formed by various TARDBP species).Citation389 However, not all ALS-related mutations in TARDBP are made equal, and a recent study showed that the A382T mutation causes a significant reduction in the number of SGs per cell, reduces the percentage of cells that form SGs, and leads to a significant decrease of viability, suggesting that the A382T mutation-induced loss of TARDBP function in SG nucleation reduces the ability of cells to respond to stress.Citation390
Figure 7. Effect of the ALS-related mutations within the C-terminal region of human TARDBP on the generation and morphology of SGs in response to osmotic stress. (A) Localization of the exogenous wild-type TARDBP, as well as transiently expressed pathological TARDBP mutants and TARDBP lacking the C-terminal tail in HEK293T cells. Exogenous TARDBP was stained with anti-MYC antibody; nuclei, with ToPro-3. Bar: 10 μm. (B) Levels of expression of proteins shown in panel (A). (C) Quantification of SG size (pixels2/granule) following 1 h of sorbitol stress. Shown are mean granule sizes ± SEM for wild-type (open bar) and mutant (filled bars) TARDBP (*, P < 0.05). Reproduced with permission from ref. Citation389.
Although many of the aforementioned findings seem to link TARDBP, SGs, and ALS pathogenesis, since alterations in SG dynamics have been suggested to play a key role in the TARDBP protein aggregation process,Citation283,292,294,299,391,392 this hypothesis remains hotly contested. This is because SGs are typically observed in studies that are usually conducted under gross overexpression of TARDBP or other ALS proteins, or in the presence of the nonpathologically relevant mutations to those proteins, or following harsh treatments with agents inducing cell stress, such as sodium arsenite or H2O2; SGs are not too often observed in ALS patients. To date, the primary criticism of the SG hypothesis is that it generated few “leads” that have withstood the test of time, proven highly reproducible in other laboratories, or proven more generally true in ALS patients. Despite these concerns, the important roles of TARDBP in regulation of SG biogenesis and dynamics are not under question in model studies when cells are stressed and undergo development of SG (and other PMLOs). Obviously, future studies are needed to shed more light on this important problem.
4.3. FUS/fused in sarcoma/translocated in liposarcoma (FUS RNA binding protein)
4.3.1. Structural properties of FUS
The second most common suspect linking aberrant SGs to ALS-FTLD is FUS (FUS RNA binding protein). In fact, FUS is an important player in ALS, mutations of which are responsible for a noticeable subset of both fALS and sALS,Citation393,394 where they account for ∼4% and 1% of total cases, respectively.Citation360,395 Furthermore, FUS mutations account for ∼35% of fALS in patients younger than 40 years old.Citation396 The FUS gene is positioned within chromosome 16 (its cytogenic location is 16p11.2, which is the short [p] arm of chromosome 16 at position 11.2) and has 15 exons. It encodes a 526 residue-long RBP, which belongs to the FET family of RPBs that includes FUS, EWSR1, and TAF15 (TATA-box binding protein associated factor 15).Citation397,398 Human FUS (UniProt ID: P35637) contains several distinct functional domains including a RNA-recognition motif and a highly-conserved C-terminal NLS.Citation395 This C-terminal domain is a hot-spot for the identified ALS-related mutations.Citation399
4.3.2. Biological and pathological roles of FUS
The FUS protein is expressed both in the nucleus and cytoplasm, and can shuttle between these 2 locations. Although the nucleus of neurons is a site of the predominant localization of FUS in the norm, characteristic FUS-immunoreactive inclusions are found in the cytoplasm of the ALS–FUS and FTLD–FUS patients, suggesting that the mislocalization of this protein to the cytoplasm might contribute to the neurodegeneration via the gain-of-toxicity mechanism.Citation399,400 This protein is involved in several biological functions, which include DNA repair,Citation401,402 regulation and control of transcription of target genes,Citation403 and RNA processing via regulation of pre-mRNA splicing.Citation354 Recent studies revealed that target gene expression can be significantly altered by FUS mutations due to the ability of mutant forms to bind to the target gene mRNA.Citation404 It was also indicated that the ALS-associated R521C mutation in human FUS is able to interfere with the normal function of the wild-type protein by interacting with it and by interfering with normal FUS binding to HDAC1 (histone deacetylase 1).Citation405 This mutant FUS also forms more stable complexes with the mRNA of BDNF (brain derived neurotrophic factor) than the wild-type protein and altered alternative splicing of this mRNA.Citation405 Furthermore, an interesting feed-forward regulatory loop has been established, where some mutant forms of FUS protein upregulate microRNAs MIR141 and MIR200A that, in their turn, affect FUS protein synthesis.Citation406 Although both TARDBP and FUS share the ability to form abnormal cytoplasmic aggregates in tissues of ALS and FTLD patients via interaction with mRNAs, these proteins bind different sets of cytoplasmic mRNAs that, however, converge on common cellular pathways, and differently affect the post-transcriptional fate of their partner mRNAs in the cytoplasm.Citation407 Similar to TARDBP, FUS can interact with RNA and with several members of the hnRNP family of RNA-binding proteins.Citation408 Although aggregation of FUS and TARDBP is observed in the cytoplasm of ALS patients, the related mechanisms of pathogenesis are assumed to be different.Citation370 This hypothesis is based on the observations that TARDBP aggregates are observed primarily in adult patients, whereas FUS aggregates are found in juvenile patients.Citation370
4.3.3. FUS and PMLOs
In ALS-FTLD, the cytoplasm-located SGs preferentially contain mutant FUS but not the wild-type protein,Citation409,410 suggesting that the pathogenic FUS mutations are related to the altered dynamics of SG assembly and reflect the presence of important differences between normal and disease physiology.Citation409–411 Furthermore, OS causes mutant FUS recruitment into SGs, leading to the sequestering of the wild-type FUS, disruption of RNA processing, and initiation of cell death.Citation412 The involvement of fALS-associated mutations in FUS cytoplasmic mislocalization is attributed to the fact that the majority of these mutations affect the NLS positioned within the C terminus of FUS, thereby impairing nuclear import of this protein.Citation400,413,414 Similar effects were described for the protein with a deleted C-terminal 32 amino acid residues containing an effective NLS.Citation415
All these observations emphasize the crucial roles of the ALS mutations or deletion of the NLS of FUS in the distortion of RNA metabolism via impairment of FUS nuclear localization and induction of the cytoplasmic inclusions and SGs.Citation415 The biological and pathological importance of the observed effects of the C-terminally truncated FUS was recently reemphasized by finding an ALS-related mutation in the FUS gene consisting of a 2-base pair deletion, c.1509_1510delAG, resulting in a truncated protein, p.G504Wfs*12, lacking the NLS.Citation416 Expression of this truncated FUS leads to the severe cytoplasmic mislocalization of mutant FUS and strong colocalization of this protein with SGs.Citation416
ALS-related mutations in FUS have a complex effect on SG dynamics. In fact, although SG assembly is delayed in cells expressing mutant FUS, once formed, the mutant-FUS-containing SGs are noticeably different from the SGs lacking FUS, being more dynamic, larger, and more abundant, and disassembling more rapidly once stress is removed.Citation417 ALS-linked C-terminal mutations have profound effects on cellular localization and functions of FUS protein, which is diffusely mislocalized in the cytoplasm in the form of SGs and spontaneously formed large aggregates. In addition to mutant FUS, these inclusions contain wild-type FUS and RNA-binding proteins HNRNPA1, HNRNPA2B1, and SMN1.Citation418 These large, spontaneously formed FUS mutant-derived aggregates in the cytoplasm sequester a variety of RNA binding proteins and mRNAs suggesting toxic gain of function leading to the disruption of the various aspects of RNA equilibrium and biogenesis.Citation418
In addition to ALS-related mutations, the methylation of FUS arginines by PRMT1 (protein arginine methyltransferase 1) plays an important role in regulation of the nuclear-cytoplasmic shuttling of wild-type FUS and its amyotrophic lateral sclerosis 6-associated mutants participating in the mutation-induced toxic gain of function.Citation419,420 Also, oxidative stress promotes the accumulation of mutant FUS in cytoplasmic SGs leading to the sequestration of the wild-type protein.Citation421
Curiously, the abilities of the endogenous wild-type FUS to translocate from the nucleus to the cytoplasm and to be engaged in SG formation are related to the physiological cellular response to the hyperosmolar stress induced by sorbitol, with the cell viability in response to sorbitol being mostly lost in cells with reduced expression of FUS.Citation422 It was pointed out that the recruitment of FUS in SGs might represent a unique cellular defense mechanism against irreversible aggregation of FUS protein mislocalized to the cytoplasm.Citation423
4.4. Protein C9orf72 and dipeptide repeat proteins Poly(GA), Poly(GR), Poly(GP), Poly(PA), and Poly(PR) generated due to the hexanucleotide repeat expansion of the intronic noncoding region of the C9orf72
4.4.1. Structural properties of C9orf72 and DPRs
The most common genetic change underlying both FTLD and ALS is related to the alterations of the C9orf72 (chromosome 9 open reading frame 72) gene. The cytogenic location of C9orf72 is 9p21.2, which is the short (p) arm of chromosome 9 at position 21.2. The C9orf72 gene consists of 12 exons, and multiple transcript variants encoding different isoforms can be generated by alternative splicing. The major pathological alteration of C9orf72 is the hexanucleotide repeat expansion (GGGGCC/CCCCGG) in the noncoding region located in an intronic region of the C9orf72 gene between its noncoding exons 1a and 1b.Citation424,425 This hexanucleotide repeat expansion can vary from 10 to thousands of repeats and is associated with 30 to 60% of fALS cases, 8% of sALS cases, 25% of FTLD cases, and up to 88% of cases in familial ALS-FTLD.Citation332,424–430 Said hexanucleotide repeat expansion is now considered as the most common cause of familial and sporadic TARDBP-positive FTLD and ALS (C9orf72-FTLD/ALS).Citation431,432
Although the hexanucleotide repeat expansion affects the noncoding region of the C9orf72 gene, the expanded repeats can be bi-directionally transcribed, generating numerous sense and antisense repeat RNAs.Citation433–436 Furthermore, the resulting sense and antisense repeat RNAs can be translated to generate a series of the dipeptide repeat proteins (DPRs or C9-related polypeptides synthesized via RAN-translation). When sense GGGGCC repeat RNAs are used as mRNAs in the RAN-translation, 3 different proteins, poly(GA), poly(GR), and poly(GP), are synthesized,Citation437,438 whereas RAN-translation of the antisense CCCCGG repeat RNAs generates poly(PA), poly(PR), and poly(GP) proteins.Citation434,436,439 Expanded repeat-containing RNAs are engaged in the formation of RNA foci that sequester RNA-binding proteins, whereas RAN-generated DPRs are found as aggregates throughout the CNS of C9orf72-ALS-FTLD patients.Citation434,436–440
Alternative splicing of human C9orf72 generates 2 proteoforms (UniProt ID: Q96LT7), a canonical or long isoform consisting of 481 residues, and a short isoform that has 222 residues due to lack of the C-terminal half of the sequence (residues 223–481).
As was already mentioned, due to their highly redundant sequences, all DPRs, including poly(GA), poly(GR), and poly(GP), poly(PA), and poly(PR),Citation433-436 synthesized by RAN-translation using the sense and antisense repeat RNAs generated as a result of the bidirectional transcription of the hexanucleotide repeat expansions of the C9orf72 gene are expected to be mostly disordered, and in agreement with this expectations, all these DPRs are also predicted to be highly disordered.Citation201
4.4.2. C9orf72 in the norm and pathology
Although the biological functions of the C9orf72 protein remained unknown for a long time, recent analysis revealed that this protein can be involved in interactions with CFL1 (cofilin 1) and other actin binding proteins, thereby regulating the axonal actin dynamics and also serving as a modulator of small GTPases.Citation441 Also, C9orf72 takes part in the regulation of endosomal trafficking.Citation442 Furthermore, in neuronal cell lines, primary cortical neurons and human spinal cord motor neurons, C9orf72 colocalizes with RAB/RAS-related proteins implicated in autophagy and endocytic transport—RAB1, RAB5, RAB7 and RAB11—indicating that C9orf72 might be related to autophagy regulation.Citation442
The repeat-positive patients are characterized by the reduced expression levels of both AS isoforms of human C9orf72.Citation443–445 However, the exact position of the C9orf72 loss-of-function mechanism in the C9-FTLD/ALS pathogenesis is not clear as of yet. In fact, although several studies ruled out a loss of function of the C9orf72 protein as a contributor to ALS pathologyCitation446 (because, e.g., cellular survival in vitroCitation447 or in vivoCitation448 of both cortical and motor neurons is not affected by the reduction of the C9orf72 levels), many laboratories continue to study the roles of C9orf72 in ALS pathology. Also, C9orf72-depleted motor neurons are characterized by reduced axonal actin dynamics, which is associated with the altered capacity of neurons to maintain axons and axon terminals in the cellular pathology underlying ALS and FTLD.Citation441 The reduction in the axonal actin dynamics is associated with a pathway involving phosphorylation of the actin binding protein CFL1 through the C9orf72-dependent modulation of the activity of the small GTPases ARF6 (ADP ribosylation factor 6) and RACK1 (receptor for activated C kinase 1) leading to the enhanced activity of LIMK1 LIM domain kinase 1) and LIMK2.Citation441 Furthermore, loss of C9orf72 in mouse brain neuroblastoma cells alters functions of a complex containing WDR41 (WD repeat domain 41) and SMCR8/Smith-Magenis syndrome chromosome region, candidate 8) that is involved in modulation of the GTPase functions of the members of RAS oncogene family, RAB8A and RAB39B, resulting in impaired autophagy in neurons.Citation449
It was pointed out that the mutations in C9orf72 not only cause the appearance of malformed RNA molecules, but also promote mislocalization of TARDBP to the cytoplasm.Citation450 Conversely, in various models, DPRs alter cellular functions and induce toxicity in different ways.Citation440 For example, the arginine-rich DPRs—poly(GR) and poly (PR)—display the highest toxicity in primary neurons and fly models, where they trigger nucleolar stress, nuclear transport defects, protein mislocalization, and RNA processing alterations.Citation447,451,452 In contrast, expression of poly(GA) proteins in primary neurons results in aggregation of the transport factor UNC119, proteasome impairment, and impairment of RAD23B/HR23B and nucleocytoplasmic transport proteins.Citation453–455 Finally, very little or almost no toxicity is reported for poly(GP) and poly(PA).Citation434,447
4.4.3. C9orf72, DPRs, and SGs
It has been established that the expanded repeats located within the noncoding region of the C9orf72 gene can be bidirectionally transcribed, and both sense and antisense repeat RNAs are engaged in the formation of RNA foci in the CNS of ALS and FTLD patients.Citation433–436 Besides RNA foci containing hexanucleotide sense and antisense repeat RNAs, the afflicted patients are characterized by the presence of diverse proteinaceous inclusions throughout the central nervous system (CNS).Citation456 These inclusions are composed of the DPRs (or C9-related polypeptides synthesized via RAN-translation) generated from the GGGGCC repeat RNA via repeat-associated non-ATG (RAN) translation. Therefore, the hexanucleotide repeat expansion of the C9orf72 gene produces a double-hit impact on the cells via sequestration of some of the RNA-binding proteins into RNA foci (e.g., as is observed when a GGGGCC31 repeat expression in NSC34 and HeLa cells causes sequestration of PURA and its binding partner, FMR1/FMRP, in the intracytosolic SGs)Citation457 and via formation of various intracellular inclusions (including SGs) containing RAN-generated DPRs.Citation458 Systematic analysis of the interactomes of all DPRs revealed that arginine-containing poly(GR) and poly(PR) are engaged in binding to RBPs and proteins with low complexity sequence domains, which are common components of several PMLOs, such as SGs, nucleoli, and the nuclear pore complex.Citation458 Furthermore, poly(GR) and poly(PR) alter dynamics and functions of multiple PMLOs.Citation458 For example, localization of these 2 DPRs to the nucleolus induces suppression of ribosomal RNA synthesis, impairs SG formation, and causes translocation of the key nucleolar component NPM1 (nucleophosmin 1), leading to nucleolar stress and cell death.Citation459 Finally, the C9orf72 protein itself is able to interact with several ALS-related proteins, such as UBQLN2, HNRNPA2B1, HNRNPA1, and ACTB (actin beta).Citation442 Furthermore, in cells overexpressing C9orf72, in addition to the formation of nuclear C9orf72 aggregates, prominent HNRNPA1- and HNRNPA2B1-positive SGs emerge, and the proportion of these HNRNPA1- and HNRNPA2B1-positive SGs noticeably increases when the cells overexpressing C9orf72 are treated with proteasome inhibitors.Citation442
4.5. PFN1 (profilin 1)
4.5.1. Structural properties of PFN1
Human PFN1 (profilin 1; UniProt ID: P07737) is a small, globular, actin-binding protein, mutations in which constitute a relatively rare cause of fALS,Citation460 mostly among patients with European ancestry.Citation461 The PFN1 gene is characterized by the cytogenetic location 17p13.2, which is the short (p) arm of chromosome 17 at position 13.2. It has 6 exons and can generate at least 6 alternatively spliced variants.
4.5.2. Normal and pathological functions of PFN1
Human PFN1 is a 140 residue-long ubiquitous eukaryotic protein that serves as a major regulator of filamentous actin growth via binding actin-ADP monomers, promoting the conversion of actin-ADP to actin-ATP, transporting the actin-ATP monomers, and interactions within FMNs/formins located at the growing end of actin filaments.Citation462,463 PFN1 also interacts with phosphoinositides, thereby serving as a link between the phosphatidylinositol cycle and actin polymerization.Citation464,465 Besides serving as an essential component of the cytoskeletal rearrangement signaling pathway, PFN1 can interact with a large cohort of proteins with poly-l-proline stretches.Citation466,467 Curiously, only mutant forms of PFN1 are engaged in the formation of insoluble aggregates, disrupt cytoskeletal structure, inhibit filamentous actin formation, and elevate UBC (ubiquitin C) and SQSTM1/p62 (sequestosome 1) levels in motor neurons, whereas the wild-type protein does not have such activities.Citation468
It was shown recently that the native conformation of PFN1 in vitro is severely destabilized by the ALS-linked mutations, and the turnover of the PFN1 mutant forms is dramatically accelerated in cells.Citation469 This mutation-induced structural destabilization of PFN1 defines the increased propensity of ALS-linked variants to aggregate and also their loss-of-function phenotypes.Citation469 In SH-SY5Y neuroblastoma cells, transient expression of ALS-linked PFN1 mutants (and not of the wild-type protein) causes the formation of cytoplasmic SQSTM1- and ubiquitin-positive aggregates that sequester endogenous TARDBP, induce accumulation of TARDBP, and promote conversion of normal TARDBP into its abnormal (detergent-insoluble and phosphorylated) form, suggesting the presence of the gain-of-toxic function scenario, where the ALS-related mutations equip PFN1 with a novel toxic function that causes TARDBP aggregation.Citation470
4.5.3. PFN1 and stress granules
Although mutations in the PFN1 gene are associated with fALS and provide a potential link between the actin cytoskeleton abnormalities and ALS pathogenesis, these ALS-linked PFN1 mutations are rare, and the actual role of mutations in this protein in neurodegeneration is poorly understood. Recently, some insights into the potential pathological mechanisms of the PFN1 mutations were gained using the budding yeast Saccharomyces cerevisiae that has a PFN1 ortholog (in fact, PFN1 is highly conserved through evolution) as a model and utilizing the power of yeast genetics in the form of an unbiased, genome-wide synthetic lethal screen with yeast cells lacking profilin (pfy1∆).Citation471 This analysis revealed that in the pfy1∆ background, deletion of some genes related to SGs and P-bodies is synthetically lethal, suggesting that PFN1 can be related to the SG and P-body dynamics.Citation471 This hypothesis is supported by the analysis of mouse primary cortical neurons and human cell lines that show association of PFN1 and the related protein PFN2 with the SGs, and revealed that SG dynamics is indeed altered by the ALS-linked mutations in PFN1.Citation471
4.6. Nucleoporin GLE1 (GLE1, RNA export mediator)
4.6.1. Structural properties of GLE1
The gene encoding the human nucleoporin GLE1 protein is located at position 34.11 of the long (q) arm of chromosome 9 (cytogenetic location: 9q34.11) and contains 22 exons, generating at least 11 transcript variants encoding different alternatively spliced isoforms. GLE1 serves as a shuttling mRNA export factor that is needed for the export of the poly(A) tail-containing mRNAs from the nucleus into the cytoplasm,Citation207-210 and also may play a role in the terminal step of mRNA transport through the nuclear pore complex (NPC) via interaction with nucleoporins NUPL2/CG1 (nucleoporin like 2) and NUP155.Citation472 Being an essential multifunctional modulator of DEAD-box RNA helicases in yeast, Gle1 is involved in translation initiation through regulation of a DEAD-box protein, the initiation factor Ded1,Citation473 and plays a role in termination of translation via interaction with another DEAD-box protein, Dbp5.Citation474 There are 2 alternative splicing-generated isoforms of GLE1 in human, with the canonical form (or GLE1B) consisting of 698 residues, and with the GLE1A form missing the C-terminal tail (residues 660–698) and harboring the change IEAI → YQAC at residues 656–659 constituting a new C terminus. Although GLE1B and GLE1A share 655 identical residues, these isoforms are characterized by different intracellular distribution. In fact, although the canonical GLE1B isoform is localized predominantly to the NPC, the short GLE1A isoform is spread diffusely throughout the nucleus and cytoplasm.Citation208 Localization of GLE1B to the NPC is ensured by the interaction of its N-terminal tail (residues 1–29) with NUP155,Citation212 as well as via binding of the C-terminal tail (residues 656–698) to NUPL2.Citation472 In addition to its NUP155- and NUPL2-binding motif located at the N- and C-terminal tails, respectively, GLE1 (UniProt ID: Q53GS7) possesses a region related to the mediation of protein shuttling between the nucleus and the cytoplasm (residues 444–483). This protein also has 2 coiled-coil regions (residues 151–277 and 306–356).
4.6.2. Functions and dysfunction of GLE1
Two rare deleterious ALS-associated mutations in GLE1 (a splice site 1965-2A>C and a nonsense 209C>A mutation) deplete GLE1 at the nuclear pore, thereby abrogating function of this protein in nuclear mRNA export.Citation475 The splice site mutation affects intron 14 of GLE1, where it destroys a splice acceptor site, resulting in the expression of a mutant protein, in which the C-terminal 44 residue-long region is replaced with a novel 88 amino acid C-terminal domain.Citation475 A nonsense c.209C>A mutation introduces a premature stop codon in exon 2 of GLE1 thereby encoding a truncated GLE1-S70X protein of only 69 residues.Citation475 The resulting c.209C>A mRNA is highly unstable and the truncated GLE1-S70X protein is not found.Citation475 In addition to these 2 deleterious mutations, some ALS patients possess a missense variation c.2089C>T encoding a mutant protein harboring the R697C substitution.Citation475 Functional implementations of this mutation have not been assessed as of yet.
Functional analysis revealed that the splice site mutant possesses functions ascribed to both GLE1A and GLE1B isoforms, suggesting that it is not subject to the normal mechanisms of GLE1 regulation.Citation476 Besides causing ALS, mutations in human GLE1 are associated with autosomal recessive conditions that lead to death of fetuses before the 32nd week of gestation or soon after birth, lethal congenital contracture syndrome 1 (LCCS1 caused by the T144 → TPFQ and R569H mutations) and lethal arthrogryposis with anterior horn cell disease (LAAHD caused by the V617M and I684T mutations).Citation477–479
4.6.3. GLE1 in PMLOs
One of the fALS forms associated with mutations in the GLE1 gene result in the production of a protein that is defective in SG regulation, and has some translation-related cytoplasmic functions.Citation480 As was already pointed out, GLE1 protein is present in cells as at least 2 isoforms generated by alternative splicing. These 2 forms are differently expressed (with the canonical form being more common), have different cellular localizations (with GLE1B being primarily localized to the nuclear envelope rim and with GLE1A being mostly found in the cytoplasm), and have different functions (with GLE1B interacting with NPC and playing a role in efficient and expedient mRNA export from the nucleus,Citation481 and with GLE1A being involved in essential cytoplasmic functions related to translational modulation and regulation of proper assembly and disassembly of SGsCitation480).Citation208,210,212,472 The effect of the ALS-related mutation on the GLE1 sequence can be considered from the GLE1A and GLE1B angles. From the GLE1B perspective, this mutation causes the replacement of the 44-amino acid NUPL2 binding site with a novel 88 amino acid C-terminal domain, whereas from the GLE1A perspective, the GLE1-c.1965-2A>C mutation in the GLE1 gene results in the replacement of a short C-tail with a long C-terminal domain. Because this new inserted domain cannot bind to the nucleoporin NUPL2,Citation475 it was expected that GLE1-IVS14-2A>C would be functionally similar to GLE1A.Citation476 In agreement with this hypothesis, GLE1-IVS14-2A>C is absent from the nuclear envelope rim, causes an increase in the cytoplasmic functions of GLE1, is recruited to SGs, and is able to regulate assembly and disassembly of SG2, resulting in the formation of larger SGs.Citation476 Furthermore, both the GLE1A and the GLE1-IVS14-2A>C form cytoplasmic “fibrous” aggregates that do not colocalize with the components of SGs but recruit TARDBP and the molecular chaperone HSP90.Citation476 However, in contrast to normal GLE1A that is not involved in mRNA export from the nucleus, GLE1-IVS14-2A>C is able to support some level of mRNA export in cells with endogenous GLE1 depletion.Citation476 Therefore, the ALS-related GLE1-IVS14-2A>C isoform is capable of acting as both GLE1A and GLE1B, and may contribute to ALS pathogenesis via distortion of the normal mechanisms of GLE1 regulation.Citation476
4.7. Transcriptional activator PURA/Pur-α (purine rich element binding protein A)
4.7.1. Structural properties of PURA
The gene encoding the transcriptional activator PURA/PUR1 (purine rich element binding protein A) has the cytogenetic location 5q31.3 (i.e., at position 31.3 of the long [q] arm of chromosome 5) and contains 5 exons. Originally, transactivator PURA was shown to bind to the purine-rich single strand of the PUR element located at a putative origin of replication site in the MYC gene.Citation482 Subsequent studies revealed that PURA is involved in (mostly negative) regulation of several genes, such as SPN/leukosialin/CD43 (sialophorin),Citation483 FAS (Fas cell surface death receptor),Citation484 APBB1/FE65 (amyloid beta precursor protein binding family B member 1),Citation485 MBP (myelin basic protein),Citation486 CHRNB4 (cholinergic receptor nicotinic beta 4 subunit),Citation487,488 and ACTA2 (actin, alpha 2, smooth muscle, aorta).Citation489
Human PURA (UniProt ID: Q00577) is a 322-residue long protein that possesses 2 regions with compositional biases, a Gly-rich domain (residues 11–53) and a Gln- and Glu-rich region (residues 293–322) that might serve as a part of the transcriptional activation domain, and a central DNA-binding domain containing 3 class I repeats and 2 class II repeats.Citation490
4.7.2. Biological and pathological roles of PURA
PURA is a highly conserved, ubiquitous and multifunctional protein engaged in a sequence-specific interaction with single-stranded DNA and RNA.Citation220 Among different functions ascribed to this protein are targeting mRNA to neuronal dendrites,Citation491–493 DNA replication, DNA repair, and gene transcription.Citation490,494,495 Being a single-stranded nucleic acid-binding protein, PURA possesses DNA helix-destabilizing helix unwinding activities.Citation496 The preferred DNA sequence recognized by PURA is composed of the GGN repeats, and binding of PURA to this recognition sequence is accompanied by the formation of multimeric complexes and is controlled and amended by interaction with other transcription factors.Citation490,497,498 In addition to interaction with single-stranded DNA and RNA, PURA can bind to a cohort of cellular regulatory proteins, such as the CCNA1 (cyclin A1)-CDK2 complex,Citation499,500 CCNT1 (cyclin T1),Citation501 CDK9,Citation501 E2F1 (E2F transcription factor 1),Citation502 RB1 (RB transcriptional corepressor 1),Citation503 SP1 (Sp1 transcription factor),Citation486 and YBX1/YB1 (Y-box binding protein 1).Citation504 Furthermore, this protein is considered to be a major player in the regulation of the cell cycle and can be involved in oncogenic transformation,Citation490 and its intracellular level varies during the cell cycle, peaking during mitosis and decreasing at the onset of S phase.Citation499
It was pointed out that PURA might be involved in the pathogenesis of the FUS mutation-associated form of ALS through the control of mRNA translation, indicating that altered protein synthesis may be implicated in the disease.Citation505
4.7.3. Roles of PURA in SG biogenesis
Engagement of PURA in ALS pathogenesis is related to the sequestration of this important RNA/DNA-binding protein in the inclusions containing the expanded GGGGCC repeat RNAs generated by the expansion of the hexanucleotide repeats in the first intron of C9orf72.Citation506-508 Also, in the presence of the GGGGCC31 repeats, PURA efficiently accumulates in the intracytosolic granules positively stained with SG markers.Citation457
Furthermore, PURA is able to specifically bind to the FUS C-terminal region, physically interact with the mutated FUS in an RNA-dependent manner, and colocalize with the mutated FUS to the SGs found in the motoneuronal cells derived from ALS patients.Citation505 In ALS patient cells carrying disease-causing FUS mutations and challenged with stress and in mammalian neuronal cells, PURA efficiently colocalizes with mutant FUS in constitutive SGs.Citation509 Because SG formation in mammalian cells is significantly reduced by the shRNA-mediated knockdown of endogenous PURA, it has been concluded that this protein is essential for SG assembly.Citation509
4.8. FLCN (folliculin)
4.8.1. Structural properties of FLCN
Human FLCN/BHD with cytogenetic location 17p11.2, which is the short (p) arm of chromosome 17 at position 11.2, contains 32 exons, and encodes a 579 residue-long protein (folliculin). FLCN serves as a tumor suppressor, mutations in which are associated with BHD syndrome, a rare inherited genodermatosis characterized by kidney tumors, lung cysts, and benign tumors of the hair follicle (hair follicle hamartomas).Citation510-513 Mutations in FLCN are also associated with primary spontaneous pneumothoraxCitation514-516 and familial renal cell carcinoma.Citation517 FLCN is highly conserved across species, with human protein showing 92% identity to the mouse FLCN, being 22–36% identical (44–56% similar) to the Drosophila melanogaster protein and 27–28% identical (44–52% similar) to the Caenorhabditis elegans ortholog.Citation511
Human FLCN (UniProt ID: Q8NFG4) has 1 coiled-coil region (residues 287–310) and might be present in 3 isoforms generated by alternative splicing, where isoforms #2 and #3 differ from the canonical form by missing long regions C-terminally located regions (residues 343–579 and 198–579) and possessing the DLEEESESWD…SGCGSWQPRK→GEAGVLL PGP…LREAHPPISV and CPGREGPIFF…FLLGKVRGII→SLVATEPVSV…ELREESCWTC changes at residues 291–342 and 134–197, respectively. Curiously, most cancer-related mutations in this protein are associated with the C-terminal truncations in FLCN suggesting that this domain might play a role in tumor suppression.Citation518 A crystal structure solved for the C-terminal domain of human FLCN (residues 341–566) shows that this domain is characterized by an αβ structure, where a core β-sheet and helices packed on the one side are followed by an all-helical region, and which is similar to the differentially expressed in normal cells and neoplasia (DENN) domain of DENND1B/connecdenn 2 (DENN domain containing protein 1B; PDB ID: 3V42, see ref. Citation518).
4.8.2. Functionality and dysfunctionality of FLCN
FLCN is related to energy and/or nutrient sensing and the MTOR (mechanistic target of rapamycin) pathway via the FLCN-interacting proteins FNIP1 and FNIP2 that also bind to AMP-activated protein kinase (AMPK), which is involved in energy sensing and negative regulation of MTOR activity.Citation519,520
FLCN is related to ALS via its direct interactions with TARDBP (where the residues 202–299 of FLCN interact with the RRM domains of TARDBP) leading to the modulation of TARDBP cytoplasmic translocation and aggregation.Citation521 FLCN is needed for TARDBP nuclear export, but not nuclear import.Citation521 The overexpression of FLCN is associated with enhanced TARDBP cytoplasmic mislocalization, whereas reduced FLCN levels lead to the preferential deposition of TARDBP to the nucleus.Citation521
4.8.3. FLCN and PMLOs
The FLCN-driven cytoplasmic mislocalization of TARDBP leads to SG formation and accumulation of TARDBP aggregates that are recognized by the UPS and autophagy-lysosome pathway.Citation521 Curiously, under stress conditions, FLCN is needed for the successful incorporation of TARDBP into the SGs, because in the arsenite-treated cells containing endogenous FLCN, TARDBP is preferentially found in the cytoplasm where it colocalizes with SGs, whereas after arsenite treatment of the FLCN-depleted cells, TARDBP dissociates from the SGs and shuttles back into the nucleus.Citation521
4.9. RBM45 (RNA binding motif protein 45)
4.9.1. Structural properties of RBM45
The gene encoding human RBM45/DRB1 (RNA binding motif protein 45) has the cytogenic location 2q31.2. The RBM45 gene has 8 exons and can generate 3 alternatively spliced isoforms.
The amino acid sequence of human RBM45 (UniProt ID: Q8IUH3) consists of 476 residues and possesses 3 RRMs (residues 26–106, 121–195, and 392–464) as well as a C-terminal nuclear localization sequence (within the C-tail residues 454–472).Citation522 There is also a homo-oligomer assembly (HOA) domain located within the linker region between RRM2 and RRM3 that promotes homo-oligomerization of RBM45 and is highly conserved across species.Citation522 In addition to the full-length canonical form, alternative splicing generates 2 proteoforms, where intron retention in isoform 2 generates a sequence missing the 225–226 dipeptide, missing a C-terminal domain (residues 331–476) and having a D330E substitution. Isoform 3 is minimally different for the canonical form, being characterized by the missing 225–226 dipeptide.
4.9.2. Biology and pathology of RBM45
RBM45 protein belongs to the family of neural RRM-type RNA-binding proteins that play a number of important roles in neural development.Citation523 It was also reported that human RBM45 preferentially interacts with the poly(C) RNA.Citation523 Although this protein is predominantly found in the nucleus, it contains a nuclear localization signal and can shuttle between the nucleus and cytoplasm.Citation524 Recent immunoprecipitation and mass spectrometry analysis revealed that 132 proteins can specifically interact with RBM45, and that this excessive protein-protein interaction (PPI) network includes many RBPs engaged with RBM45 mostly via RNA-dependent interactions in the nucleus.Citation525 Among the RBM45-interacting proteins discovered in this study were several ALS-linked RBPs, such as FUS, HNRNPA1, and TARDBP,Citation525 and among the biological processes and pathways ascribed to the RBM45-interacting proteins are RNA processing, RNA splicing, cytoplasmic RNA translation, and EIF2 (eukaryotic translation initiation factor 2) and EIF4 pathways.Citation525
Recently, it was reported that levels of RBM45 are statistically elevated in the cerebrospinal fluid of patients with sporadic and familial ALS, and cytoplasmic inclusion bodies containing RBM45, ubiquitin, and TARDBP are found in 91% of ALS, 100% of FTLD–TARDBP and 75% of Alzheimer disease cases.Citation526 Immunochemical analysis also revealed that RBM45 forms a punctate pattern within nuclei of neurons and glia in the brain and spinal cord that lack either TARDBP or ubiquitin. Patients with the C9orf72 hexanucleotide repeat expansion display the most extensive RBM45 pathology,Citation526 likely due to the aforementioned binding preference for poly(C) RNA. However, the generality of this conclusion is questionable, because only 3 C9orf72 patients were analyzed in this study. Subsequent studies also showed that RBM45 is able to bind and colocalize with the C-terminal fragment of TARDBP.Citation527 This protein also modulates the antioxidant response in ALS through interactions with KEAP1 (kelch like ECH associated protein 1), which is an inhibitor of the antioxidant response transcription factor NFE2L2 (nuclear factor, erythroid 2 like 2).Citation528 It was also shown that one of the pathological mechanisms linking RBM45 with ALS can be related to the mislocalization of this protein and perturbation of its nuclear-cytoplasmic trafficking that could be responsible for the induction of the formation and accumulation of toxic cytoplasmic aggregates.Citation524
4.9.3. RBM45 and SGs
The cytoplasmic inclusions containing RNA-binding protein RBM45, being present in neurons and glia in a majority of ALS and FTLD patients, incorporate TARDBP and are marked with ubiquitin.Citation526 It was pointed out that, while similar to TARDBP and FUS, RBM45 possesses RRMs, and does not have a characteristic glycine-rich prion-like domain (PrLD),Citation526 but contains a bipartite nuclear-localization sequence located at the C-terminal region.Citation522 Besides RNA binding, RBM45 is able to interact with the C-terminal domain of TARDBPCitation527 both in the nucleus and cytoplasm,Citation522 can bind to FUS in an RNA-dependent manner,Citation522 and is able to form homo-oligomers via the evolutionarily conserved motif located within the linker region between RRM2 and RRM3.Citation522
Although RBM45, TARDBP and FUS are predominantly localized to the nucleus in healthy cells, these proteins form cytoplasmic inclusions in cells of ALS-FTLD patients, with RBM45 homo-oligomerization being needed for the efficient association with other ALS-linked proteins.Citation522 Also, when cells are exposed to oxidative stress, all 3 proteins shuttle from the nucleus to the cytoplasm, where they are engaged in SG formation.Citation283,299,528,529 Altogether, currently available data suggest that RBM45 self-oligomerization in the cytoplasm plays a role in the incorporation of this protein into SGs, where it promotes RBM45 interaction with TARDBP.Citation522
4.10. SS18L1/CREST (SS18L1, nBAF chromatin remodeling complex subunit)
4.10.1. Structural properties of SS18L1
The SS18L1/CREST/KIAA0693 (SS18L1, nBAF chromatin remodeling complex subunit) gene is located on chromosome 20q13.33 and contains 11 exons, encoding a protein required for calcium-dependent dendritic growth and branching in cortical neurons.Citation530
Human SS18L1 (UniProt ID: O75177) is a 396-residue-long protein that has an N-terminal autoinhibitory-autoregulatory domain (residues 1–148) needed for SMARCA4/BRG1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4) binding,Citation530 followed by a methionine-rich domain (residues 149–232), a multifunctional domain (MFD, residues 246–317) related to the regulation of the subcellular localization and transactivation of SS18L1 and involved in the protein self-dimerization,Citation531 and a C-terminal transactivation domain (residues 325–396) related to the control of nuclear localization of SS18L1 (residues 334–396) containing a binding region crucial for interaction with CREBBP (residues 387–396).Citation530 Furthermore, there are 4 potential SH2-binding motifs in human SS18L1, residues 50–53, 353–356, 371–3789, and 391–394. The C-terminal half of SS18L1 (residues 190–396) is characterized by low sequence complexity (it possesses compositional bias, due to being enriched in Gln residues).Citation530 It was also pointed out that this C-terminal part satisfies the criteria for a PrLD,Citation530,532 serving as a primary cause of SS18L1 aggregation, a statement supported by the observation that protein aggregation is completely abolished by the deletion of this domain.Citation533
Human SS18L1is present as at least 5 isoforms generated by alternative splicing, where, in comparison with the full-length canonical form, isoforms 2, 3, and 4 are characterized by missing N-terminal regions 1–23, 1–86, and 1–131, respectively, and isoform 5 possesses the MSVAFA SARP…SKGKTAECTQ → MQSLSTEARY…ICPRSPPARR substitution at the 1–49 region.
4.10.2. Normal and pathological functions of SS18L1
SS18L1 is another protein whose mutations are associated with ALS.Citation532,534 SS18L1 is a nuclear calcium-regulated protein involved in transcriptional activation via its interaction with CREBBPCitation535 mediated by the last 9 residues of SS18L1, and in chromatin remodeling via binding to chromatin remodeling proteins ARID1A/BAF250 (AT-rich interaction domain 1A) and SMARCA4/BRG1.Citation530 In fact, SS18L1 serves as a constituent of the chromatin remodeling complex including SMARCD3/BAF (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily d, member 3) playing an important regulatory role at various stages of neural development by modulating transcription of specific sets of genes.Citation536 Because SS18L1-deficient mice display defective dendritic branching, early lethality, and motor disturbances, it has been hypothesized that the physiological function of this protein is related to the control of the normal development of neuronal dendritic trees.Citation530
Association of SS18L1 with ALS pathology is based on finding mutated forms of this protein (e.g., Q388stop and Ile123Met) in sporadic ALS,Citation532 and Ala264Thr and p.Gln222_Ser224del mutations in patients with the familial form of ALS.Citation534 Besides being involved in ALS, SS18L1 is associated with synovial sarcoma, which is an aggressive soft tissue tumor caused by the t(X;18)(p11;q11) translocation, where the SS18 gene on chromosome 18 is fused to one of the SSX genes on the X chromosome.Citation537-540 Among such fusions are SS18-SSX1, SS18-SSX2, and SS18-SSX4 that account for more than 95% of the synovial sarcomasCitation537-540 and a SS18L1-SSX1 fusion generated by the in-frame fusion of nucleotide 1216 (exon 10) of SS18L1 with nucleotide 422 (exon 6) of SSX1.Citation541 In the resulting chimeric SS18L1-SSX1 protein, the last 8 residues of SS18L1 are replaced by 78 C-terminal residues of the SSX1 protein, which is a transcriptional repressor domain.Citation541
4.10.3. SS18L1 and biogenesis of stress granules and other PMLOs
In the nucleus, SS18L1 is localized to the nuclear bodies of unknown nature that are positive for the histone acetyltransferase CREBBP/CBP, and the C-terminal 164 residues are needed for this nuclear body localization of SS18L1.Citation542 Furthermore, SS18L1 belongs to the set of paraspeckle proteins.Citation543 A recent study showed that SS18L1 is involved in interaction with FUS and possesses a strong tendency for aggregation.Citation533 In its aggregated form, SS18L1 affects morphology and integrity of paraspeckles via trapping other paraspeckle proteins,Citation533 thereby supporting a hypothesis that compromised paraspeckle formation can serve as a pathogenic factor in ALS-FTLD.Citation544,545 Finally, similar to many other ALS-FTLD-related proteins, SS18L1 is found in SGs.Citation533 Therefore, SS18L1 possesses an intricate ability to be recruited to at least 3 different types of PMLOs, such as CREBBP-containing nuclear bodies, paraspeckles, and SGs.
SS18L1 is one of the ALS-related proteins that contain a PrLD. Curiously, a recent comprehensive review indicated that almost 70 human RBPs possesses PrLDs, which are “low-complexity domains that possess a similar amino acid composition to prion domains in yeast, which enable several proteins, including yeast Sup35 and Rnq1, to form infectious conformers, termed prions.”Citation546 Human RBPs routinely utilize PrLDs in their numerous functions. Furthermore, the presence of PrLDs defines the ability of RBPs to undergo LLPTs leading to the formation of various PMLOs.Citation546
4.11. HNRNPA1 (heterogeneous nuclear ribonucleoprotein A1) and HNRNPA2B1
4.11.1. Structural properties of HNRNPA1 and HNRNPA2B1
Human genes HNRNPA1 (with cytogenetic location 12q13.13, which is the long [q] arm of chromosome 12 at position 13.13) and HNRNPA2B1 (located on chromosome 7p15.2, which is the short [p] arm of chromosome 7 at position 15.2) encode members of a family of ubiquitously expressed hnRNPs. The HNRNPA1 and HNRNPA2B1 genes have 13 and 15 exons, respectively, and are able to generate multiple spliced isoforms (at least 9 and 13, respectively).
Both HNRNPA1 and HNRNPA2B1 have similar domain organization. Human HNRNPA1 (UniProt ID: P09651) is 372-residue-long protein possessing 2 globular domains (A and B, residues 4–94 and 95–108) portions of which form RRMs (residues 14–97 and 105–184). Region 218–240 represents the RNA-binding RGG-box, whereas residues 320–357 constitute the nuclear targeting sequence (M9). The C-terminal part of HNRNPA1 (residues 195–372) is a Gly-rich PrLD. There are 2 alternatively spliced isoforms of this protein, where the HNRNPA1-B isoform is considered as a canonical full-length form of the protein, and the HNRNPA1-A isoform (which is 20 times more abundant than HNRNPA1-B) is considered different from the canonical form due to missing residues 252–303.Citation547-549 Analogously, human HNRNPA2B1 (UniProt ID: P22626) is a 353-residue-long protein that has 2 RRMs (residues 21–104 and 112–191), a low complexity, Gly-rich C-terminally located PrLD (residues 193–253), a nuclear targeting sequence (residues 308–347), and a nuclear localization signal (residues 9–15). Structural information is available for the N-terminal regions of both proteins (residues 2–196 of human HNRNPA1 containing 2 RNA recognition motifs, PDB ID: 2LYVCitation550; and residues 1–103 corresponding to the first RRM of the human HNRNPA2B1, PDB ID: 1X4B).
Both HNRNPA1 and HNRNPA2B1 proteins are present as 2 proteoforms generated by alternative splicing, with the HNRNPA2B1 proteoforms being different by a 12 amino acid insertion close to the N terminus,Citation547-549 but both containing C-terminally located PrLDs that encompass residues 197–353 in HNRNPA2B1 and residues 185–341 in hnRNPA2.Citation363,551 The HNRNPA1 proteoforms are different due to missing amino acids 252–303 in the shorter HNRNPA1-A isoform, but both harbor PrLDs located at their C-termini that span residues 186–320 in HNRNPA1-A and residues 186–372 in HNRNPA1-B.Citation363,551
4.11.2. Biological and pathological functions of HNRNPA1 and HNRNPA2B1
HNRNPA1 and HNRNPA2B1 are 2 ubiquitous RNA-binding proteins with a PrLD that are mutated in ALS.Citation552 HNRNPA1 is an RNP involved in several RNA metabolic processes, such as pre-mRNA splicing and transport of cellular RNAs, which it accomplishes by shuttling between the nucleus and the cytoplasm.Citation361,553-555 Curiously, different RNA-binding profiles were assigned to the cytoplasmic and nuclear HNRNPA1, with the cytoplasmic protein possessing a high affinity for the AU-rich elements,Citation556,557 and with the nuclear protein having a high affinity for a polypyrimidine stretch bordered by AG at the 3′ ends of introns.Citation558,559
HNRNPA2B1 is among the most abundant RBPs in eukaryotic cells and is highly expressed in brain and muscle. The major function of this RBP is to form the core of the RNP complex that associates with the nascent pre-mRNAs, packaging them into hnRNP particles and thereby playing important roles in various processes ranging from transcription, to pre-mRNA processing, to RNA nuclear export and subcellular location, to mRNA translation, and to modulating the stability of mature mRNAs.Citation560 Human HNRNPA2B1 is engaged in the formation of a multitude of HNRNP particles via interaction with several (at least 20) other HNRNPs and heterogeneous nuclear RNA.Citation560 The name to this protein is given due to the existence of 2 functional isoforms, HNRNPA2 and HNRNPB1, generated by alternative splicing, with the HNRNPB1 serving as a full-length canonical form and with the HNRNPA2 isoform missing residues 3–14 close to the N terminus.
Mutations in PrLD of human HNRNPA2B1 (Asp302Val) and HNRNPA1 (Asp314Val or Asp314Asn) are associated with ALS and multisystem proteinopathy (also known as inclusion body myopathy with early-onset Paget disease and FTLD). To gain some knowledge on the molecular mechanisms connecting mutations in HNRNPA1 and HNRNPA2B1 with fALS, a structure-based threading algorithm ZipperDB that is designed to predict the presence in a query protein of steric zippers, short (6-amino acid-long) segments able to form 2 self-complementary β-strands,Citation561 was recently used.Citation552 This analysis revealed that the potent steric zipper motifs were introduced by various ALS-associated mutations into the PrLDs of HNRNPA1 and HNRNPA2B1.Citation552 A subsequent study provided experimental support to this hypothesis and revealed that although the wild type versions of HNRNPA1 and HNRNPA2B1 have a noticeable intrinsic tendency to form self-seeding fibrils, this fibrillation propensity is dramatically enhanced by the disease-associated mutations that accelerate the formation of self-seeding fibrils that cross-seed polymerization of wild-type proteins.Citation551
4.11.3. HNRNPA1, HNRNPA2B1, and PMLOs
In addition to enhancing the formation of cytoplasmic inclusions, the ALS-related mutations foster the excess incorporation of HNRNPA2B1 and HNRNPA1 into SGs.Citation551 In fact, the constitutive SGs contain higher levels of mutant forms of the HNRNPA2B1 and HNRNPA1 proteins than the corresponding wild-type proteins. Furthermore, arsenite treatment of model cells results in more rapid incorporation of mutant proteins into SGs than the wild-type proteins, indicating that disease-related mutations alter the recruitment of HNRNPA2B1 and HNRNPA1 not only into the cytoplasmic inclusions but also into SGs.Citation551
4.12. ATXN2 (ataxin 2)
4.12.1. Structural properties of ATXN2
Human ATXN2 is a 1,313-residue-long protein involved in the trafficking of EGFR (epidermal growth factor receptor) via binding to SH3GL2 (SH3 domain containing GRB2 like 2, endophilin A1) and SH3GL3 (SH3 domain containing GRB2 like 3, endophilin A3), which are the brain-expressed members of the SH3GL/endophilin A family involved in synaptic vesicle endocytosis.Citation562 ATXN2 is a 1,313 residue-long protein most known for its involvement in the pathogenesis of spinocerebellar ataxia type 2 (SCA2) caused by the expansion of the polyglutamine (polyQ) region of this protein encoded by the expanded CAG trinucleotide repeat within the open reading frame of the ATXN2 gene located at position 24.12 on the long (q) arm of chromosome 12 (cytogenetic location: 12q24.12) and encoding multiple transcript variants due to alternative splicing and the presence of 26 exons. In fact, although CAG repeats are known to vary in length in normal alleles of ATXN2, the most common allele carries 22 CAG repeats and the maximal ATXN2 repeat size is 31, whereas expansion of the ATXN2 CAG repeat region to carry 34–59 repeats is associated with SCA2.Citation563-568
In addition to the canonical full-length form, human ATXN2 (UniProt ID: Q99700) has at least four alternatively spliced isoforms. Isoform 2 is missing the C-terminal tail (residues 996–1313) and harbors the PLYPIPMTPMPVNQAK → YQICPNSGKTSIIRVP substitution in the preceding region (residues 980–995). Isoform 3 is the shortest and the most diversified form of ATXN2, missing N-terminal (residues 1–981) and C-terminal tails (residues 1258–1313), missing the 1106–1123 region, and possessing YPIPMTPMPVNQAKTYR→MYYAVEILFNRQSAFFS and AHVQSGMVP→VIPALAN FL substitutions at residues 982–998 and 1249–1257, respectively, in addition to harboring the Ile1124Val substitution. In isoform 4, the C-tail (residues 1244–1313) is missing, whereas isoform 5 is characterized by missing regions 1–265 and 1106–1124 and possesses the ACPKLPYNKETSPSF YFAI → V substitution. The canonical form has several regions with compositional bias, such as three Pro-rich regions (residues 47–158, 551–734, and 929–1085), a poly-Pro region (residues 55–64), a poly-Gln region (residues 166–187), and a poly-Ser region (residues 213–223). It was indicated that several of these isoforms are present in mice and humans, might have different functions, and are expressed in different tissues, such as brain, spinal cord, cerebellum, heart, and placenta.Citation569
4.12.2. Normal and pathological roles of ATXN2
This cytoplasmic RNA-binding protein is considered as an ALS susceptibility factor, because intermediate-length polyglutamine expansions (Q27-Q33) in ATXN2 are associated with increased ALS risk.Citation570-572 Curiously, although SCA2 is associated with the polyQ expansions exceeding 34 repeats all encoded by pure CAG repeat expansions in the ATXN2 gene, the corresponding expansions associated with ALS are all interrupted, containing 1 to 3 CAA codons (also encoding glutamine) within the CAG repeats.Citation573 Despite the fact that CAG expansion in genes encoding several proteins such as ATXN1, ATXN2, ATXN3, ATXN7, TBP (TATA-box-binding protein), ATN1 (atrophin 1), and HTT (huntingtin) is associated with a set of polyQ diseases, expansions in polyQ disease genes other than ATXN2 are not associated with ALS, suggesting that the repeat-induced alterations in the biological functions of ATXN2 and not the mere presence of an expanded polyQ repeat represent the ALS risk factor.Citation574 Because no ATXN2-associated polyglutamine expansion was found in patients with sporadic and familial FTLD, it has been concluded that this polymorphism is ALS specific.Citation575 The pathological effect of the ATXN2 polyQ expansion in ALS is associated with the enhancement of the stress-induced activation of CASP3 and increase TARDBP pathological modifications, such as C-terminal cleavage and phosphorylation.Citation576 Furthermore, the presence of the ATXN2 intermediate-length expansion affects the morphology of the TARDBP inclusions, because the motor neurons of ALS patients harboring ATXN2 polyQ expansions contain primarily skein-like or filamentous TARDBP deposits and do not contain large round Lewy body-like inclusions abundantly found in ALS patients without ATXN2 polyQ expansions.Citation577
4.12.3. ATXN2 in the biogenesis of PMLOs
In addition to TARDBP and FUS, ALS-FTLD-related mutations, which are known contributing factors to SG formation due to the mislocalization of carrier proteins to the cytoplasm, ATXN2 is also known as a constituent protein of SGs in mammalian cells, suggesting the existence of the common pathological cascade formed by TARDBP, FUS, and ATXN2 and related to the aberrations in SG dynamics.Citation578 The importance of ATXN2 in the controlling dynamics of PMLOs is supported by the observations that this protein can interact with the DEAD/H-box RNA helicase DDX6 (DEAD-box protein 6), which is an important component of P-bodies and SGs, and that the alterations in the ATXN2 levels might interfere with the assembly of SGs and P-bodies.Citation579
Based on the analysis of cellular ALS models, it was also suggested that ATXN2 with an ALS-linked intermediate-length repeat (ATXN2 Q31) might serve as a potent modifier of the FUS-based pathology, promoting FUS translocation to the cytoplasm, inducing ER stress, and promoting Golgi fragmentation.Citation580 Furthermore, because ATXN2 colocalizes with FUS in motor neurons derived from sALS and FUS-linked fALS patients, colocalizes with FUS in the ER-Golgi compartments in neuronal cell lines and coprecipitates with FUS in ALS spinal cord lysates, it was hypothesized that the interplay between the mutated FUS and ATXN2 with intermediate-length polyQ expansions provides potential molecular and cellular mechanisms linked to ALS pathology.Citation580 Similarly, a connection between the TARDBP-based pathology and ATXN2 with intermediate-length polyQ expansions was noted based on the comparative analysis of the TARDBP-positive inclusions in the ALS cases with and without ATXN2 possessing intermediate-length polyQ expansions.Citation577 Here, fewer motor neurons in the ALS cases without extended ATXN2 contain abundant large round (likely SGs) and skein-like TARDBP inclusions, whereas only skein-like TARDBP inclusions are predominantly found in almost ALS cases harboring ATXN2 polyQ expansions.Citation577 It was also found that an increased ATXN2 expression might cause TARDBP and FUS mislocalization, leading to RNA dysregulation.Citation581,582
4.13. TIA1 (TIA1 cytotoxic granule associated RNA binding protein)
4.13.1. Structural properties of TIA1
In humans, the TIA1 gene is located on chromosome 2p13.3 (at position 13.3 of the short [p] arm of chromosome 2). It has 15 exons, generates at least 11 transcript variants and the gene product is an RNA-binding protein involved in alternative pre-RNA splicing and regulation of mRNA translation.Citation583 TIA1 is another RNP containing poly-glycine-rich PrLD that promotes reversible physiological aggregation of TIA1 into SGs,Citation584 but, under stress conditions, can initiate formation of pathological RNA granules.Citation585,586 TIA1 is characterized by a well-developed interactome, shown to co-immunoprecipitate with 163 brain proteins.Citation586 Therefore, based on the presence of a domain with profound intrinsic aggregation propensity and the overall high binding promiscuity of TIA1 it is not surprising to find that abnormal TIA1-containing deposits are found in the brain tissue of patients with AD, ALS, Creutzfeld-Jakob disease, frontotemporal dementia with Parkinsonism (FTDP-17), FTLD–TDP43, Huntington disease, and spinal muscular atrophy.Citation290,584,585
Human TIA1 (UniProt ID: P31483) is a 386-residue-long protein that contains 3 RRMs (residues 7–83, 106–184 and 214–286), which serve to bind RNA and a Gly-rich domain that mediates protein aggregation.Citation290 In addition to the canonical full-length proteoform, 2 isoforms are produced by alternative splicing. Here, isoform 2 has a SSTVVSTQRSQD → N substitution at the 93–104 region, whereas isoform 3 is missing the C-terminal tail (residues 215–386) and has a SNTKQLSYDEVVNQSSPSNC → CRCIGEEKEMW NFGEKYARF substitution in the preceding region (residues 195–214).
4.13.2. Biology and pathology of TIA1
TIA1 can serve as an apoptosis-promoting factor that is able to modulate alternative splicing of dedicated transcripts.Citation587 Careful analysis of the consequences of the Tia1 deletion in mouse spinal cord and cerebellum revealed several strong dysregulations, including noticeable effects on the cell cycle and apoptosis regulators, as well as on lipid storage and membrane trafficking factors.Citation588
4.13.3. TIA1 and SGs
TIA1 is thought to serve as one of the core RBPs responsible for the primary step (nucleation) of RNA granule formation followed by recruitment of secondary RBPs to form mature RNA granules, such as SGs. SGs act as key players in environmental stress- and viral infection-induced translational suppression due to their role in deciding the fate of sequestered mRNA by sorting it for re-initiation, storage, or degradation.Citation586 One of the facts supporting the involvement of TIA1 in SG biogenesis is formation of SGs in cells infected with poliovirus.Citation589 Such SGs, formed at early stages of infection, contain TIA1, translation initiation factors, RNA binding proteins, and mRNA.Citation589 TIA1 shuttling from the nucleus in response to environmental stress, and subsequent aggregation of a C-terminal proteolytic TIA1 fragment containing glutamine-rich PrLD play a crucial role in the formation of cytoplasmic SGs,Citation269,277,590,591 suggesting that the controlled prion-like aggregation of TIA1 might be related to the regulation of SG formation.Citation290,592 In fact, it was pointed out that TIA1 is one of a few key RBPs, such as TIAL1/TIAR, ZFP36, G3BP1, G3BP2, and FMR1, which play a crucial role in the initial stress-induced nucleation of SGs.Citation269,290,592 The central role of TIA1 and TIAL1 in SG biogenesis and related stress-induced inhibition of translation initiation is supported by the fact the stress-induced phosphorylation of translation initiation factor EIF2A is followed by the TIA1- and TIAL1-dependent recruitment of most cytoplasmic mRNAs to SGs.Citation277 In this way, TIA1 and TIAL1 act as translational silencers needed for controlling the duration of stress-induced translational arrest.Citation277
4.14. MAPT (microtubule associated protein tau)
4.14.1. Structural properties of MAPT
The MAPT gene located on chromosome 17q21.31 (at position 21.31 of the long [q] arm of chromosome 17) has 16 exons and the gene product is a cytosolic axonal protein that binds and stabilizes microtubules,Citation593,594 thereby serving as an important structural component of the microtubule-based transport system of the neurons.
Human MAPT (PMID: P10636) exists as a family of proteoforms migrating in SDS gel electrophoresis at close bands of 55–62 kDa. Alternative splicing of MAPT mRNA is (at least in part) responsible for this heterogeneity. In fact, although the MAPT gene has 16 exons, only 8 of them (1, 4, 5, 7, 9, 11, 12, and 13) are found in all isoforms of this protein and are therefore considered as constitutive, whereas the remaining exons (2, 3, 4A, 6, 8, 10, and 14) are subject to alternative splicing.Citation595 Because exons 1 and 14 are parts of the promoter and the 3′ untranslated region of the MAPT mRNA, respectively, they are not found in any isoform of MAPT. None of the brain-specific MAPT isoforms have exons 4A, 6, and 8, which are found only in the MAPT mRNAs of the peripheral tissue.Citation596,597 The variability of the brain-specific MAPT isoforms is caused by the alternative splicing of exons 2, 3, and 10 (see ref. Citation597) that generates 6 mRNA isoformsCitation598,599 containing 2+3+10+, 2+3+10−, 2+3−10+, 2+3−10−, 2−3−10+, and 2−3−10− combinations of these exons and encoding 6 protein isoforms. Because exons 2 and 3 each encode 29-residue-long sequences located in the N-terminal part of MAPT (designated N inserts), and because exon 10 encodes 1 of the 4 31-residue-long microtubule-binding repeats located within the C-terminal part of MAPT and encoded by exons 9, 10, 11, and 12 (designated as R regions), alternative splicing generates MAPT variants that differ from each other by having either 0, 1, or 2 N-terminal inserts, and 3 or 4 C-terminally located R-regions (designated, respectively, as 2N4R, 2N3R, 1N4R, 1N3R, 0N4R, and 0N3R). The longest isoform in the CNS (441 amino acids total) has 4 repeats (R1, R2, R3, and R4) and 2 inserts, whereas the shortest isoform (352 amino acids total) has 3 repeats (R1, R3, and R4) and no insert.Citation600,601 Although the normal cortex is characterized by equal expression levels of the 3- and 4-repeat forms, the ratio of isoforms is changed in tauopathies.Citation602,603 Furthermore, in the peripheral nervous system and almost in all CNS neurons that extend processes into the peripheral nervous system, the big MAPT (with a molecular mass of ∼110 kDa) is expressed that contains an additional 254-residue insert in the amino-terminal half encoded by the additional large exon (4a) included in the corresponding mRNA.Citation604-606
Phosphorylation and other PTMs serve as an additional source of MAPT microheterogeneity.Citation607 GSK3B, CDK5, and MAPK kinases are involved in phosphorylation of many residues of MAPT during brain developmentCitation608 and multiple sites of MAPT are phosphorylated by several kinases in vitro (for a review, see ref. Citation609). The primary targets of the in vitro phosphorylation are residues within the microtubule-interacting region (repeat-containing domain) and its flanking regions. Many of these sites are also phosphorylated in paired helical filaments (which are the major form of MAPT deposition in AD).Citation610,611 In fact, 10 major phosphorylation sites have been identified in MAPT isolated from paired helical filaments from patients with AD.Citation610,611
4.14.2. MAPT function and dysfunction
Among functions ascribed to MAPT in vitro are binding to microtubules, regulation of their assembly, and control of their dynamic instability.Citation612-616 In situ, the axons have high levels of MAPT,Citation617 and brain cells in general are estimated to have high proportion of this protein, which might account for 0.025–0.25% of total soluble brain protein.Citation618,619 Although at normal conditions, the levels of the microtubule-unbound MAPT are kept low, the increase of soluble MAPT caused by the impairment of the protein-degradation systemCitation620 or aberrant phosphorylationCitation621 might result in aggregation of this protein.Citation622
Aggregation of MAPT is associated with a broad spectrum of neurodegenerative diseases collectively known as tauopathies (e.g., AD, FTLD, ALS, progressive supranuclear palsy, cortico basal degeneration, Pick disease, etc.).Citation623 FTLD is a group of complex diseases characterized by high heterogeneity of their pathological features. In fact, pathologically, FTLD variants are diversified and classified based on the major specific protein found in their inclusions, with FTLD–MAPT (which is an FTLD variant where MAPT serves as the major specific protein of the inclusions) being one of the most common pathological subtypes of this disease.Citation624
Among the mechanisms connecting MAPT to ALS is cleavage of this protein and generation of the neurotoxic MAPT45–230 fragment that is highly abundant in lumbar and cervical spinal cord specimens obtained from ALS subjects, being present in the ALS upper motor neurons located in the precentral gyrus, and forming aggregates in the spinal cord of ALS patients.Citation625
4.14.3. MAPT stress granules, and other PMLOs
ALS and FTLD are tauopathies, which are a large group of neurodegenerative diseases associated with aggregation and dysfunction of MAPT. Curiously, the TIA1 protein, which is typically considered as a SG marker and participates in SG nucleation, was recently shown to be localized to the AD-related neurofibrillary tangles composed of the hyperphosphorylated and aggregated MAPT, with TIA1 being able to directly interact with MAPT.Citation585 Although normal cytosolic MAPT only weakly interacts with TIA1, the abnormal hyperphosphorylation promotes this interaction.Citation585 A dedicated study was recently conducted to check if, in its turn, MAPT can be associated with TIA1-containing SGs.Citation626 This study revealed that when MAPT-expressing cells are treated with arsenite, a prominent SG response is observed and some formed SGs contain MAPT.Citation626 Furthermore, when extracellular MAPT is internalized in the recipient cells (in line with the cell-to-cell propagation of the MAPT-related pathology modelCitation627), a prominent formation of SGs is observed, and MAPT is abundantly present in these cytoplasmic nonvesicular cellular bodies.Citation626 Because in these studies, a pseudohyperphosphorylated MAPT-E14 variant carrying 14 phosphomimetic (serine/threonine to glutamate) mutations is more efficient in induction of SG formation and is able to more strongly interact with TIA1, it has been hypothesized that the hyperphosphorylation-induced oligomerization may be an important factor promoting SG recruitment of the internalized MAPT species.Citation626 Based on these observations it was suggested that SGs and TIA1 might be important players in the cell-to-cell transmission of MAPT pathology.Citation626
5. ALS- and FTLD-related proteins, PMLOs, and autophagy
In order to appropriately balance sources of energy at critical times in development and in response to nutrient stress, a cell can utilize a self-degradative process known as autophagy (self-eating) that is responsible for the disposal of various intracellular species.Citation103,628-634 Macroautophagy (hereafter autophagy) provides a way for elimination of aberrant protein aggregates, superfluous or damaged organelles, and even entire bacteria.Citation635 Structures doomed for autophagy-based clearance are first sequestered by phagophores, the precursors to autophagosomes, which are large, double-membrane bound vesicles, and then delivered into the interior of the lysosome or vacuole, where they are digested by resident hydrolases.Citation635,636 When the autophagosome fuses with the lysosome, the autolysosome is formed, and the content of the autophagosome is catabolized.Citation637,638 It is important to remember that the formation of a new autophagosome does not cover all autophagy processes.Citation298 For example, in a chaperone-mediated autophagy, individual protein substrates are directly targeted to the lysosome by chaperones and then translocated across the lysosomal membrane, whereas in microautophagy, invagination of the lysosomal membrane directly engulfs the cytoplasmic content.Citation639 Because autophagy can sequester and degrade large protein complexes and entire organelles, it represents an important alternative to the ubiquitin-proteasome system.Citation298,638,640
In ALS, aberrant SGs cannot disassemble and therefore accumulate and are consequently degraded by autophagy.Citation641 There are several observations linking together ALS- and FTLD-related proteins, stress granules (and potentially other PMLOs), and autophagy. Furthermore, it is thought that there might be a special chaperone-mediated SG surveillance system, granulostasis, which might regulate composition and dynamics of SGs, thereby playing a role in ALS pathogenesis.Citation641,642 The first lines of evidence on the connection between autophagy and ALS have been derived from animal models.Citation298 For example, an autophagy pathway was shown to be enhanced in a transgenic mouse model of SOD1-ALS,Citation643 and also was observed in spinal cord tissues from SOD1-ALS patients.Citation644 In line with the granulostasis hypothesis,Citation642 recruitment of chaperones prevents formation of aberrant SGs in cells expressing ALS-linked variants of SOD1, and promotes disassembly of such SGs when the stress subsides.Citation645
TARDBP can also be involved in autophagy regulation, mostly by controlling biogenesis of autophagosomes and lysosomes.Citation646-649 For example, autophagosome-lysosome fusion is blocked by TARDBP loss of function.Citation648 Also, TARDBP targets a key component of the MTORC1 complex, RPTOR/raptor (regulatory associated protein of MTOR complex 1), thereby regulating the MTORC1-TFEB (transcription factor EB) signaling pathway.Citation649 Stability of RPTOR mRNA is decreased when TARDBP function is lost, leading to the enhancement of autophagosomal and lysosomal biogenesis in an MTORC1-dependent manner. Furthermore, loss of TARDBP is also associated with impaired autophagosome-lysosome fusion in an MTORC1-independent manner.Citation649
Another study in this area was a work by Farg et al., who established that besides regulating endosomal trafficking, C9orf72, via its interaction with RAB proteins related to autophagy and endocytic transport, such as RAB1, RAB5, RAB7 and RAB11, can be involved in regulation of autophagy in neurons.Citation442 This idea is further supported by the colocalization of C9orf72 with UBQLN2 and autophagosome marker MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta)-positive vesicles, and comigration of this protein with lysosome-stained vesicles in neuronal cells.Citation442 Finally, a possible dysregulation of trafficking in ALS patients bearing the C9orf72 repeat expansion was postulated based on the immunohistochemical analysis of motor neurons of ALS patients that revealed an increased colocalization between C9orf72 and RAB7 and RAB11 compared with controls.Citation442 Recently, it was shown that the loss of C9orf72 might cause deregulation of autophagy via reduction of the activity of the negative regulator of autophagy MTOR, and the increase of the levels and nuclear translocation of TFEB, indicating that C9orf72 might serve as a negative regulator of autophagy, playing an important role in coupling the cellular metabolic state with autophagy regulation.Citation650
An analysis of the role of autophagy in regulation of FUS-positive SGs was conducted in the same year by Ruy et al.Citation651 Here, it was shown that the FUS-positive SGs formed under oxidative stress in neurons bearing the ALS-linked FUSR521C mutation are colocalized to the zinc finger protein TRIM27 (tripartite motif containing 27) RFP-LC3-positive autophagosomes and are preferentially accumulated in autophagy-deficient neurons,Citation651 whereas rapamycin-enhanced autophagy is able to reduce the accumulation of FUS-positive SGs in an autophagy-dependent manner.Citation300,651 It was also indicated that autophagy activation not only reduces mutant FUS-positive SG accumulation, but also inhibits neurite fragmentation mediated by mutant FUS, and reduces cell death in response to oxidative stress in disease-associated cultured neurons.Citation300 Therefore, the enhancement of the autophagy activity leading to the reduction in the number of SGs containing mutant FUS can be neuroprotective,Citation651,652 whereas distortion of the autophagic process can contribute to the ALS-FTLD pathogenesis.Citation653
6. Conjointness of the LLPT-based pathogenicity: Collective responsibility of proteins involved in PMLO formation
Because any PMLO (physiological or aberrant) represents a proteinaceous assemblage containing multiple proteins, LLPT-based pathologies represent a collective responsibility of proteins involved in PMLO formation. In essence, such kind of pathogenicity driven by aberrant PMLOs represents an example of a misdeed conducted by good guys assembled with bad company in the wrong place at the wrong time. Several examples below show how ALS-related proteins might cooperate, while affecting dynamics of PMLOs.
Although in the norm, TARDBP and FUS are preferentially localized in the nucleus, being transported there via the nuclear import receptors in a RAN (RAN, member RAS oncogene family) GTPase-dependent manner,Citation413 they also play a role in regulation of the RNA life cycle, being involved in the normal biogenesis of the intracytoplasmic RNA-containing PMLOs (e.g., SGs) in addition to regulation of the transcription, processing, transport, and stability of RNA.Citation392 In ALS and FTLD, both of these proteins mislocalize to the cytoplasm, where they form cytoplasmic inclusions, affect SG dynamics, and impair the RNA quality control system.Citation293 For example, the rate of TARDBP incorporation into the SGs is accelerated by the ALS-related mutations in this protein, and the mutated TARDBP forms larger SGs than the wild-type protein.Citation389 Similarly, FUS with the ALS-associated mutations efficiently localizes to SGs causing their assembly into larger structures.Citation505 However, TARDBP and FUS do not act alone, being engaged in interaction with each other, and this interaction is enhanced by the ALS-related mutations in TARDBP.Citation654 Furthermore, ATXN2 with intermediate-length polyglutamine expansions that serves as an ALS susceptibility factorCitation570-572 and is an SG constituent RNA-binding protein, is associated with both FUS-Citation580 and TARDBP-based pathologies.Citation577 For example, longer polyglutamine expansions in ATXN2 enhance interaction of this protein with TARDBP and increase the efficiency of the TARDBP cytoplasmic mislocalization.Citation570 These observations suggest that the ALS-FTLD pathology can be at least in part driven by the mislocalization of these 3 RNA-binding proteins to the cytoplasm that results in triggering the aberrant LLPTs leading to the formation of abnormal cytoplasmic PMLOs and promoting impairment of the RNA quality control system.Citation293,578
TARDBP, FUS, and ATXN2 that interact with each other, potentially forming the core of the ALS-FTLD pathological cascadeCitation578 are not exceptions to the rule, and other proteins associated with ALS-FTLD pathogenicity may also contribute to the aforementioned PMLO-related collective responsibility. For example, among the known C9orf72 targets are some proteins related to ALS, such as actin, HNRNPA1, HNRNPA2B1, and UBQLN2, and overexpression of this protein is accompanied by the formation of both nuclear C9orf72 aggregates and prominent cytoplasmic SGs containing HNRNPA1 and HNRNPA2B1.Citation442 It was also indicated that HNRNPA1 and HNRNPA2B1 can interact with TARDBP via their PrLDs.Citation364,367,552 Mutations of these proteins trigger their mislocalization from the nucleus to the cytoplasm, promote their aggregation and formation of sarcoplasmic inclusions, and cause multisystem proteinopathy and ALS.Citation552 Curiously, pathological mutations in HNRNPA1 and HNRNPA2B1 also stimulate mislocalization of wild-type TARDBP from the nucleus to the cytoplasm and result in formation of TARDBP-positive cytoplasmic inclusions. Typically, the inclusions formed by TARDBP, HNRNPA2B1, and HNRNPA1 are physically separated and only occasionally overlap.Citation551 Similarly, pathological mutations in VCP promote formation of cytoplasmic inclusions by the wild type versions of TARDBP, HNRNPA2B1, and HNRNPA1,Citation551 suggesting that the same phenotypic changes leading to TARDBP pathology and disease can be promoted by perturbations of the VCP, HNRNPA2B1, and HNRNPA1 pathways.Citation552 This important phenomenon is illustrated by that represents the results of the analysis of the effect of mutant forms of VCP (A232E and R155H) on the formation of SGs and TARDBP recruitment to those SGs.Citation655 It is seen that the constitutive SGs containing TARDBP are formed only as a result of the overexpression of the mutated VCP, whereas no such effects were found when the wild-type VCP was overexpressed in HeLa cells.Citation655
Figure 8. Effect of mutated forms of VCP (A232E and R155H) on the formation of constitutive SGs and the recruitment of TARDBP to those SGs. HeLa cells were transfected with plasmid expressing wild-type or mutant VCP-GFP forms and stained for the SG marker EIF3B and TARDBP. Overexpression of mutant but not wild-type VCP resulted in the formation of TARDBP-containing SGs. Scale bar: 10 µm. Reproduced with permission from ref. Citation655.
PURA can interact with the C-terminal region of the mutated FUS in an RNA-dependent manner, and these 2 proteins are colocalized in the SGs present in ALS patients.Citation505 Furthermore, accumulation of PURA within the intra-cytosolic SGs is enhanced in the presence of the (GGGGCC)31 repeatsCitation457 due to the ability of this protein to interact with the GGGGCC expansions of the C9orf72 gene.Citation508 Interaction of FLCN with TARDBP can modulate shuttling of TARDBP between the nucleus and the cytoplasm,Citation521 thereby playing a role in regulation of SG dynamics. RBM45 shows some sequence similarity to TARDBP and FUS and forms both nuclear and cytoplasmic inclusions.Citation522 Furthermore, the homo-oligomeric form of this protein found in the cytoplasm can interact with both TARDBP and FUS, and accumulation of RBM45 in the cytoplasm leads to the formation of the TARDBP-positive SGs.Citation522 SS18L1 can both self-aggregate and interact with FUS.Citation533 Curiously, this protein serves as an illustration of the hypothesis that the ALS-FTLD pathology can be caused by misbehaving PMLOs other than SGs (e.g., compromised paraspeckle formationCitation544,545). In fact, the dynamics, morphology, and integrity of paraspeckles can all be affected by aggregated SS18L1 due to the ability of this species to entrap other paraspeckle proteins.Citation533
7. Concluding remarks: Linking PMLOs with neurodegeneration via protein intrinsic disorder
Observations presented in the previous sections clearly indicate that many proteins considered in this article and related to ALS and FTLD are either directly or indirectly involved in the LLPTs and formation of SGs and other PMLOs. Furthermore, being purified and analyzed in vitro, some of these proteins show high aggregation potential and the ability to undergo liquid-liquid demixing, leading to the formation of phase-separated dropletsCitation656 or polymer hydrogels.Citation657 It is assumed that these membrane-less cellular compartments represent liquid droplets that phase separately from the cytoplasm and stably coexist with their environments.Citation658 Although typically LLPTs leading to the formation and disintegration of PMLOs represent normal physiological processes related to the organization of macromolecules into compartments needed for the proper functioning of a cell, abnormal liquid-liquid phase transitions and aberrant PMLOs can be associated with age-related neurodegenerative diseases,Citation658 via impairment of the cytoplasmic RNA quality control systemCitation293 or some other related mechanisms.
It was also pointed out that LLPTs are often driven by IDPs containing low-complexity regions or domains.Citation205,206,226,269,271,657,659–666 In agreement with these earlier observations shows that the majority of the proteins considered in this article that are related to ALS and FTLD contain long IDPRs. Although C9orf72 is the most ordered protein in this set, the pathological involvement of this protein to neurodegeneration is related to the hexanucleotide repeat expansions of the intronic region of the C9orf72 gene leading to the bidirectional transcription of the expanded repeats associated with the generation of various sense and antisense repeat RNAsCitation433-436 that serve as mRNAs in RAN-translation to generate a series of DPRs.Citation434,436-439 All these DPRs including poly(GA), poly(GR), poly(GP), poly(PA), and poly(PR) proteins are highly disordered. Furthermore, many of the ALS- and FTLD-related proteins are able to interact with nucleic acids and many protein partners, and many of them are involved in SG formation, and are associated with the assembly of SG-like inclusions in ALS and FTLD. For example, in addition to being involved in the formation of SGs, TARDBP and FUS are associated with several other PMLOs, such as DNA damage repair sites, paraspeckles, other RNA granules, and transport granules.Citation299,402,664,667,668
In other words, intrinsic disorder might represent a feature unifying very different proteins in their relation to the pathogenesis of neurodegeneration via aberrant LLPTs and abnormal PMLOs. Therefore, it is likely that the “Dr. Jekyll–Mr. Hyde” behavior of proteins related to ALS and FTLD pathogenesis can at least in part be due to the ability of these proteins to be engaged in the intrinsic disorder-based LLPTs.
Abbreviations
| APBB1/FE65 | = | amyloid beta precursor protein binding family B member 1 |
| ACTA2 | = | actin, alpha 2, smooth muscle, aorta |
| ACTB | = | actin beta |
| AD | = | Alzheimer disease |
| AKAP9/AKAP350 | = | A-kinase anchor protein 9 |
| ALS | = | amyotrophic lateral sclerosis |
| AMPA | = | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AMPK | = | AMP-activated protein kinase |
| ANG | = | angiogenin |
| APC | = | adenomatous polyposis coli |
| APOE | = | apolipoprotein E |
| ARF | = | ADP ribosylation factor |
| ARID1A/BAF250 | = | AT-rich interaction domain 1A |
| AS | = | alternative splicing |
| ATN1 | = | atrophin 1 |
| ATXN | = | ataxin |
| ATXN1 | = | ataxin 1 |
| ATXN2 | = | ataxin 2 |
| ATXN3 | = | ataxin 3 |
| ATXN7 | = | ataxin 7 |
| AXIN1 | = | axin 1 |
| BCL2 | = | BCL2, apoptosis regulator |
| BDNF | = | brain derived neurotrophic factor |
| BHD | = | Birt-Hogg-Dubé syndrome |
| BRCA1 | = | BRCA1, DNA repair associated |
| C9orf72 | = | chromosome 9 open reading frame 72 |
| CASC3/MLN51 | = | cancer susceptibility 3 |
| CCNA1 | = | cyclin A1 |
| CCNT1 | = | cyclin T1 |
| CCS | = | copper chaperone for superoxide dismutase |
| CDK | = | cyclin dependent kinase |
| CDKN1A/p21 | = | cyclin dependent kinase inhibitor 1A |
| CDKN1B/p27Kip1 | = | cyclin dependent kinase inhibitor 1B |
| CHCHD10 | = | coiled-coil-helix-coiled-coil-helix domain containing 10 |
| CHMP2B | = | charged multivesicular body protein 2B |
| CHRNB4 | = | cholinergic receptor nicotinic beta 4 subunit |
| CFTR | = | cystic fibrosis transmembrane conductance regulator |
| CHGA | = | chromogranin A |
| CHGB | = | chromogranin B |
| CNS | = | central nervous system |
| CPEB | = | cytoplasmic polyadenylation element binding protein |
| CREB | = | cAMP response element binding |
| CREBBP/CBP | = | CREB binding protein |
| CTA | = | cancer/testis antigen |
| CTF | = | C-terminal fragment |
| DDX6 | = | DEAD-box protein 6 |
| DDX19B/DBP5 | = | DEAD-box helicase 19B |
| DEAD box | = | protein with Walker B motif with the amino acid sequence D-E-A-D |
| Ded1 | = | ATP-dependent RNA helicase ded1 |
| DENND1B/connecdenn 2 | = | DENN domain containing 1B |
| DERL1 | = | derlin 1 |
| DMD | = | discrete molecular dynamics |
| DPR | = | dipeptide repeat proteins |
| E2F1 | = | E2 transcription factor 1 |
| EIF2 | = | eukaryotic translation initiation factor 2 |
| EIF2A | = | eukaryotic translation initiation factor 2A |
| EIF3 | = | eukaryotic translation initiation factor 3 |
| EIF4B | = | eukaryotic translation initiation factor 4B |
| EIF4G1/EIF4F | = | eukaryotic translation initiation factor 4 gamma 1 |
| EP300 | = | E1A binding protein p300 |
| ER | = | endoplasmic reticulum |
| ERN1/IRE1 | = | endoplasmic reticulum to nucleus signaling 1 |
| EWSR1 | = | EWS RNA binding protein 1 |
| fALS | = | familial form of ALS |
| F2 | = | coagulation factor II, thrombin |
| FAS | = | Fas cell surface death receptor |
| FASTK | = | Fas activated serine/threonine kinase |
| FET | = | protein family including FUS, EWSR1, and TAF15 |
| FLCN | = | folliculin |
| FMR1/FMRP | = | fragile X mental retardation 1 |
| FNIP | = | folluculin interacting protein |
| FTLD | = | frontotemporal lobar degeneration |
| FUS | = | FUS RNA binding protein |
| FXR1 | = | FMR1 autosomal homolog 1 |
| G3BP1 | = | G3BP stress granule assembly factor 1 |
| G3BP2 | = | G3BP stress granule assembly factor 2 |
| GLE1 | = | GLE1, RNA export mediator |
| GLFG | = | G-L-F-G repeat region of nucleoporin |
| GRN/PGRN | = | granulin precursor |
| GSK3 | = | glycogen synthase kinase 3 |
| HDAC | = | histone deacetylase |
| HNE | = | 4-hydroxynonenal |
| HOA | = | homo-oligomer assembly domain |
| HNRNP | = | heterogeneous nuclear ribonucleoprotein |
| HNRNPA1 | = | heterogeneous nuclear ribonucleoprotein A1 |
| HNRNPA2B1 | = | heterogeneous nuclear ribonucleoprotein A2/B1 |
| HPV | = | human papillomavirus |
| HTT | = | huntingtin |
| IAPP/amylin | = | islet amyloid polypeptide |
| IDP | = | intrinsically disordered protein |
| IDPR | = | intrinsically disordered protein region |
| KEAP1 | = | kelch like ECH associated protein 1 |
| LCCS1 | = | lethal congenital contracture syndrome 1 |
| LAAHD | = | lethal arthrogryposis with anterior horn cell disease |
| LIMK | = | LIM domain kinase |
| LLPT | = | liquid-liquid phase transition |
| MAP1LC3B | = | microtubule associated protein 1 light chain 3 beta |
| MAP2K7/MKK7 | = | mitogen-activated protein kinase kinase 7 |
| MAPK3/ERK1 | = | mitogen-activated protein kinase 3 |
| MAPK8/JNK1 | = | mitogen-activated protein kinase 8 |
| MAPT | = | microtubule associated protein tau |
| MBP | = | myelin basic protein |
| MFD | = | multifunctional domain |
| MoRF | = | molecular recognition feature |
| mtSOD1 | = | mutant SOD1 |
| MTORC1 | = | mechanistic target of rapamycin complex 1 |
| NEFH | = | neurofilament heavy |
| NEFL/NFL | = | neurofilament light |
| NEK1 | = | NimA related kinase 1 |
| NES | = | nuclear export signal |
| NimA | = | never in mitosis A |
| NLS | = | nuclear localization signal |
| NPC | = | nuclear pore complex |
| NPM1 | = | nucleophosmin 1 |
| NFE2L2 | = | nuclear factor, erythroid 2 like 2 |
| NTD | = | N-terminal domain |
| NUP155 | = | nucleoporin 155 |
| NUPL2 | = | nucleoporin like 2 |
| OPTN | = | optineurin |
| OS | = | oxidative stress |
| PABPC1/PABP1 | = | poly(A) binding protein cytoplasmic 1 |
| P-body | = | processing body |
| PBP | = | progressive bulbar palsy |
| PD | = | Parkinson disease |
| PFN1 | = | profilin 1 |
| pfy1∆ | = | yeast cells lacking profilin |
| PLS | = | primary lateral sclerosis |
| PMA | = | progressive muscular atrophy |
| PMLO | = | proteinaceous membrane-less organelle |
| PPI | = | protein-protein interaction |
| PrLD | = | prion-like domain |
| PRNP/PrP | = | prion protein |
| PRMT1 | = | protein arginine methyltransferase 1 |
| PTEN | = | phosphatase and tensin homolog |
| PTM | = | posttranslational modification |
| PURA | = | purine rich element binding protein A |
| PV | = | poliovirus |
| RAB | = | RAB, member RAS oncogene family |
| RACK1 | = | receptor for activated C kinase 1 |
| RAD23B/HR23B | = | RAD23 homolog B, nucleotide excision repair protein |
| RAN | = | RAN, member RAS oncogene family |
| RAN-translation | = | repeat-associated non-ATG translation |
| RB1 | = | RB transcriptional corepressor 1 |
| RBM45 | = | RNA binding motif protein 45 |
| RBP | = | RNA binding protein |
| RHOA | = | ras homolog family member A |
| RNP | = | ribonucleoprotein |
| Rnq1 | = | [PIN+] prion protein RNQ1 |
| ROCK1 | = | Rho associated coiled-coil containing protein kinase 1 |
| ROS | = | reactive oxygen species |
| RPTOR/Raptor | = | regulatory associated protein of MTOR complex 1 |
| RRM | = | RNA-recognition motif |
| RPS6KA3/RSK2 | = | ribosomal protein S6 kinase A3 |
| sALS | = | sporadic form of ALS |
| SCA2 | = | spinocerebellar ataxia 2 |
| SG | = | stress granule |
| SIRT | = | sirtuin |
| SLC1A2/EAAT2 | = | solute carrier family 1 member 2 |
| SMARCA4/BRG1 | = | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4 |
| SMCR8 | = | Smith-Magenis syndrome chromosome region, candidate 8 |
| SMN | = | survival motor neuron protein |
| SOD1 | = | superoxide dismutase 1 |
| SP1 | = | Sp1 transcription factor |
| SPN/CD43 | = | sialophorin |
| SQSTM1/p62 | = | sequestosome 1 |
| SS18L1/CREST | = | SS18L1, nBAF chromatin remodeling complex subunit |
| Sup35 | = | eukaryotic peptide chain release factor GTP-binding subunit |
| TAF15 | = | TATA-box binding protein associated factor 15 |
| TANK | = | TRAF family member associated NFKB activator |
| TARDBP/TDP-43 | = | TAR DNA binding protein |
| TBK1 | = | TANK binding kinase 1 |
| TBP | = | TATA-box binding protein |
| TFEB | = | transcription factor EB |
| TIA1 | = | TIA1 cytotoxic granule associated RNA binding protein |
| TIAL1/TIAR | = | TIA1 cytotoxic granule associated RNA binding protein like 1 |
| TP53 | = | tumor protein p53 |
| TP53BP/ASPP | = | tumor protein p53 binding protein |
| TRAF2 | = | TNF receptor associated factor 2 |
| TRIM27 | = | tripartite motif containing 27 |
| UBC | = | ubiquitin C |
| UBQLN2 | = | ubiquilin 2 |
| UNC119/RG4 | = | unc-119 lipid binding chaperone |
| UPS | = | ubiquitin-proteasome system |
| VCP | = | valosin containing protein |
| VDAC1 | = | voltage dependent anion channel 1 |
| VEGF | = | vascular endothelial growth factor |
| WDR41 | = | WD repeat domain 41 |
| WDR62 | = | WD repeat domain 62 |
| WT-SOD1 | = | wild-type SOD1 |
| YBX1/YB1 | = | Y-box binding protein 1 |
| YWHAB | = | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein beta |
| ZFP36/TTP | = | ZFP36 ring finger protein |
Conflicts of interest
The author declares no conflict of interest.
Acknowledgments
I am thankful to Alexey Uversky and Prof. Daniel Klionsky for careful reading and editing the manuscript of this article.
Funding
This work was supported in part by a grant from the ALS Association to V.N.U. (grant number IIP-265).
References
- Leblond CS, Kaneb HM, Dion PA, Rouleau GA. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp Neurol. 2014;262(Pt B):91–101. doi:10.1016/j.expneurol.2014.04.013. PMID:24780888.
- Guerrero EN, Wang H, Mitra J, Hegde PM, Stowell SE, Liachko NF, Kraemer BC, Garruto RM, Rao KS, Hegde ML. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog Neurobiol. 2016;145–146:78–97. doi:10.1016/j.pneurobio.2016.09.004. PMID:27693252.
- Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012;124:339–52. doi:10.1007/s00401-012-1022-4. PMID:22903397.
- Taylor JP, Brown RH, Jr., Cleveland DW. Decoding ALS: From genes to mechanism. Nature. 2016;539:197–206. doi:10.1038/nature20413. PMID:27830784.
- Kim HJ, de Leon M, Wang X, Kim HY, Lee YJ, Kim YH, Kim SH. Relationship between clinical parameters and brain structure in sporadic amyotrophic lateral sclerosis patients according to onset type: A voxel-based morphometric study. PLoS One. 2017;12:e0168424. doi:10.1371/journal.pone.0168424. PMID:28095425.
- Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3. doi:10.1186/1750-1172-4-3. PMID:19192301.
- Norris F, Shepherd R, Denys E, U K, Mukai E, Elias L, Holden D, Norris H. Onset, natural history and outcome in idiopathic adult motor neuron disease. J Neurol Sci. 1993;118:48–55. doi:10.1016/0022-510X(93)90245-T. PMID:8229050.
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi:10.1126/science.1134108. PMID:17023659.
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–21. doi:10.1212/WNL.58.11.1615. PMID:12058088.
- Broustal O, Camuzat A, Guillot-Noel L, Guy N, Millecamps S, Deffond D, Lacomblez L, Golfier V, Hannequin D, Salachas F, et al. FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J Alzheimers Dis. 2010;22:765–9. PMID:21158017.
- Lipton AM, White CL, 3rd, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 2004;108:379–85. doi:10.1007/s00401-004-0900-9. PMID:15351890.
- Ferrari R, Kapogiannis D, Huey ED, Momeni P. FTD and ALS: A tale of two diseases. Curr Alzheimer Res. 2011;8:273–94. doi:10.2174/156720511795563700. PMID:21222600.
- Gasca-Salas C, Masellis M, Khoo E, Shah BB, Fisman D, Lang AE, Kleiner-Fisman G. Characterization of movement disorder phenomenology in genetically proven, familial frontotemporal lobar degeneration: A systematic review and meta-analysis. PLoS One. 2016;11:e0153852. doi:10.1371/journal.pone.0153852. PMID:27100392.
- Pottier C, Ravenscroft TA, Sanchez-Contreras M, Rademakers R. Genetics of FTLD: Overview and what else we can expect from genetic studies. J Neurochem. 2016;138(Suppl 1):32–53. doi:10.1111/jnc.13622. PMID:27009575.
- Gros-Louis F, Gaspar C, Rouleau GA. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:956–72. doi:10.1016/j.bbadis.2006.01.004. PMID:16503123.
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72. doi:10.1126/science.1154584. PMID:18309045.
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–4. doi:10.1038/ng.132. PMID:18372902.
- al-Chalabi A, Enayat ZE, Bakker MC, Sham PC, Ball DM, Shaw CE, Lloyd CM, Powell JF, Leigh PN. Association of apolipoprotein E epsilon 4 allele with bulbar-onset motor neuron disease. Lancet. 1996;347:159–60. doi:10.1016/S0140-6736(96)90343-8. PMID:8544551.
- Figlewicz DA, Krizus A, Martinoli MG, Meininger V, Dib M, Rouleau GA, Julien JP. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3:1757–61. doi:10.1093/hmg/3.10.1757. PMID:7849698.
- Tomkins J, Usher P, Slade JY, Ince PG, Curtis A, Bushby K, Shaw PJ. Novel insertion in the KSP region of the neurofilament heavy gene in amyotrophic lateral sclerosis (ALS). Neuroreport. 1998;9:3967–70. doi:10.1097/00001756-199812010-00036. PMID:9875737.
- Trotti D, Aoki M, Pasinelli P, Berger UV, Danbolt NC, Brown RH, Jr., Hediger MA. Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity. J Biol Chem. 2001;276:576–82. doi:10.1074/jbc.M003779200. PMID:11031254.
- Meyer T, Fromm A, Munch C, Schwalenstocker B, Fray AE, Ince PG, Stamm S, Grön G, Ludolph AC, Shaw PJ. The RNA of the glutamate transporter EAAT2 is variably spliced in amyotrophic lateral sclerosis and normal individuals. J Neurol Sci. 1999;170:45–50. doi:10.1016/S0022-510X(99)00196-3. PMID:10540035.
- Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383–94. doi:10.1038/ng1211. PMID:12847526.
- Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi:10.1016/S1093-3263(00)00138-8. PMID:11381529.
- Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–7. PMID:7929085.
- Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT, Jr. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–15. doi:10.1021/bi961799n. PMID:8901511.
- Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–31. doi:10.1006/jmbi.1999.3110. PMID:10550212.
- Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker AK. Natively disordered proteins. In: Buchner J, Kiefhaber T, editors. Handbook of protein folding. Weinheim (Germany): Wiley-VCH, Verlag GmbH & Co. KGaA; 2005. p. 271–353.
- Dunker AK, Babu MM, Barbar E, Blackledge M, Bondos SE, Dosztányi Z, Dyson HJ, Forman-Kay J, Fuxreiter M, Gsponer J, et al. What's in a name? Why these proteins are intrinsically disordered. Intrinsically Disord Proteins. 2013;1:e24157. doi:10.4161/idp.24157. PMID:28516007.
- van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, et al. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–631. doi:10.1021/cr400525m. PMID:24773235.
- Habchi J, Tompa P, Longhi S, Uversky VN. Introducing protein intrinsic disorder. Chem Rev. 2014;114:6561–88. doi:10.1021/cr400514h. PMID:24739139.
- Uversky VN. Intrinsic disorder-based protein interactions and their modulators. Curr Pharm Des. 2013;19:4191–213. doi:10.2174/1381612811319230005. PMID:23170892.
- Uversky VN. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta. 2013;1834:932–51. doi:10.1016/j.bbapap.2012.12.008. PMID:23269364.
- Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–64. doi:10.1016/j.bbapap.2010.01.017. PMID:20117254.
- Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi:10.1002/1097-0134(20010101)42:1%3c38::AID-PROT50%3e3.0.CO;2-3. PMID:11093259.
- Williams RM, Obradovic Z, Mathura V, Braun W, Garner EC, Young J, Takayama S, Brown CJ, Dunker AK. The protein non-folding problem: Amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput. 2001:89–100. PMID:11262981.
- Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic disorder and functional proteomics. Biophys J. 2007;92:1439–56. doi:10.1529/biophysj.106.094045. PMID:17158572.
- Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform Ser Workshop Genome Inform. 1999;10:30–40. PMID:11072340.
- Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–27. doi:10.1002/1097-0134(20001115)41:3%3c415::AID-PROT130%3e3.0.CO;2-7 10.1002/1097-0134(20001115)41:3%3c415::AID-PROT130%3e3.3.CO;2-Z. PMID:11025552.
- Liu J, Rost B. NORSp: Predictions of long regions without regular secondary structure. Nucleic Acids Res. 2003;31:3833–5. doi:10.1093/nar/gkg515. PMID:12824431.
- Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31:3701–8. doi:10.1093/nar/gkg519. PMID:12824398.
- Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: Implications for structural proteomics. Structure. 2003;11:1453–9. doi:10.1016/j.str.2003.10.002. PMID:14604535.
- Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–8. doi:10.1093/bioinformatics/bti537. PMID:15955783.
- Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–4. doi:10.1093/bioinformatics/bti541. PMID:15955779.
- Jones DT, Ward JJ. Prediction of disordered regions in proteins from position specific score matrices. Proteins. 2003;53(Suppl 6):573–8. doi:10.1002/prot.10528. PMID:14579348.
- Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–9. doi:10.1093/bioinformatics/bth195. PMID:15044227.
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–45. doi:10.1016/j.jmb.2004.02.002. PMID:15019783.
- Mizianty MJ, Zhang T, Xue B, Zhou Y, Dunker AK, Uversky VN, Kurgan L. In-silico prediction of disorder content using hybrid sequence representation. BMC Bioinformatics. 2011;12:245. doi:10.1186/1471-2105-12-245. PMID:21682902.
- Mizianty MJ, Uversky V, Kurgan L. Prediction of intrinsic disorder in proteins using MFDp2. Methods Mol Biol. 2014;1137:147–62. doi:10.1007/978-1-4939-0366-5_11. PMID:24573480.
- Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44:1989–2000. doi:10.1021/bi047993o. PMID:15697224.
- Ferron F, Longhi S, Canard B, Karlin D. A practical overview of protein disorder prediction methods. Proteins. 2006;65:1–14. doi:10.1002/prot.21075. PMID:16856179.
- Esnouf RM, Hamer R, Sussman JL, Silman I, Trudgian D, Yang ZR, Prilusky J. Honing the in silico toolkit for detecting protein disorder. Acta Crystallogr D Biol Crystallogr. 2006;62:1260–6. doi:10.1107/S0907444906033580. PMID:17001103.
- Bourhis JM, Canard B, Longhi S. Predicting protein disorder and induced folding: From theoretical principles to practical applications. Curr Protein Pept Sci. 2007;8:135–49. doi:10.2174/138920307780363451. PMID:17430195.
- Dosztanyi Z, Sandor M, Tompa P, Simon I. Prediction of protein disorder at the domain level. Curr Protein Pept Sci. 2007;8:161–71. doi:10.2174/138920307780363406. PMID:17430197.
- Dosztanyi Z, Tompa P. Prediction of protein disorder. Methods Mol Biol. 2008;426:103–15. doi:10.1007/978-1-60327-058-8_6. PMID:18542859.
- He B, Wang K, Liu Y, Xue B, Uversky VN, Dunker AK. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009;19:929–49. doi:10.1038/cr.2009.87. PMID:19597536.
- Dosztanyi Z, Meszaros B, Simon I. Bioinformatical approaches to characterize intrinsically disordered/unstructured proteins. Brief Bioinform. 2010;11:225–43. doi:10.1093/bib/bbp061. PMID:20007729.
- Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46:13468–77. doi:10.1021/bi7012273. PMID:17973494.
- Meszaros B, Simon I, Dosztanyi Z. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol. 2009;5:e1000376. doi:10.1371/journal.pcbi.1000376. PMID:19412530.
- Disfani FM, Hsu WL, Mizianty MJ, Oldfield CJ, Xue B, Dunker AK, Uversky VN, Kurgan L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics. 2012;28:i75–83. doi:10.1093/bioinformatics/bts209. PMID:22689782.
- Khan W, Duffy F, Pollastri G, Shields DC, Mooney C. Predicting binding within disordered protein regions to structurally characterised peptide-binding domains. PLoS One. 2013;8:e72838. doi:10.1371/journal.pone.0072838. PMID:24019881.
- Fang C, Noguchi T, Tominaga D, Yamana H. MFSPSSMpred: Identifying short disorder-to-order binding regions in disordered proteins based on contextual local evolutionary conservation. BMC Bioinformatics. 2013;14:300. doi:10.1186/1471-2105-14-300. PMID:24093637.
- Jones DT, Cozzetto D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics. 2015;31:857–63. doi:10.1093/bioinformatics/btu744. PMID:25391399.
- Malhis N, Gsponer J. Computational identification of MoRFs in protein sequences. Bioinformatics. 2015;31:1738–44. doi:10.1093/bioinformatics/btv060. PMID:25637562.
- Yan J, Dunker AK, Uversky VN, Kurgan L. Molecular recognition features (MoRFs) in three domains of life. Mol Biosyst. 2016;12:697–710. doi:10.1039/c5mb00640f. PMID:26651072.
- Peng Z, Kurgan L. High-throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res. 2015;43:e121. doi:10.1093/nar/gkv585. PMID:26109352.
- Peng Z, Wang C, Uversky VN, Kurgan L. Prediction of disordered RNA, DNA, and protein binding regions using DisoRDPbind. Methods Mol Biol. 2017;1484:187–203. doi:10.1007/978-1-4939-6406-2_14. PMID:27787828.
- Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–49. doi:10.1093/nar/gkh253. PMID:14960716.
- Pejaver V, Hsu WL, Xin F, Dunker AK, Uversky VN, Radivojac P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci. 2014;23:1077–93. doi:10.1002/pro.2494. PMID:24888500.
- Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–71. PMID:11700597.
- Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30:137–49. doi:10.1080/07391102.2012.675145. PMID:22702725.
- Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–84. doi:10.1016/S0022-2836(02)00969-5. PMID:12381310.
- Uversky VN. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002;11:739–56. doi:10.1110/ps.4210102. PMID:11910019.
- Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002;269:2–12. doi:10.1046/j.0014-2956.2001.02649.x. PMID:11784292.
- Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–82. doi:10.1021/bi012159+. PMID:12022860.
- Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–33. doi:10.1016/S0968-0004(02)02169-2. PMID:12368089.
- Dyson HJ, Wright PE. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv Protein Chem. 2002;62:311–40. doi:10.1016/S0065-3233(02)62012-1. PMID:12418108.
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi:10.1038/nrm1589. PMID:15738986.
- Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi:10.1016/j.sbi.2005.01.002. PMID:15718131.
- Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–84. doi:10.1002/jmr.747. PMID:16094605.
- Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. Febs J. 2005;272:5129–48. doi:10.1111/j.1742-4658.2005.04948.x. PMID:16218947.
- Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9(Suppl 1):S1. doi:10.1186/1471-2164-9-S1-S1.
- Ferreon AC, Ferreon JC, Wright PE, Deniz AA. Modulation of allostery by protein intrinsic disorder. Nature. 2013;498:390–4. doi:10.1038/nature12294. PMID:23783631.
- Cozzetto D, Jones DT. The contribution of intrinsic disorder prediction to the elucidation of protein function. Curr Opin Struct Biol. 2013;23:467–72. doi:10.1016/j.sbi.2013.02.001. PMID:23466039.
- Mitrea DM, Kriwacki RW. Regulated unfolding of proteins in signaling. FEBS Lett. 2013;587:1081–8. doi:10.1016/j.febslet.2013.02.024. PMID:23454209.
- Follis AV, Llambi F, Ou L, Baran K, Green DR, Kriwacki RW. The DNA-binding domain mediates both nuclear and cytosolic functions of p53. Nat Struct Mol Biol. 2014;21:535–43. doi:10.1038/nsmb.2829. PMID:24814347.
- Galea CA, Wang Y, Sivakolundu SG, Kriwacki RW. Regulation of cell division by intrinsically unstructured proteins: Intrinsic flexibility, modularity, and signaling conduits. Biochemistry. 2008;47:7598–609. doi:10.1021/bi8006803. PMID:18627125.
- Wang Y, Fisher JC, Mathew R, Ou L, Otieno S, Sublet J, Xiao L, Chen J, Roussel MF, Kriwacki RW. Intrinsic disorder mediates the diverse regulatory functions of the Cdk inhibitor p21. Nat Chem Biol. 2011;7:214–21. doi:10.1038/nchembio.536. PMID:21358637.
- Ou L, Waddell MB, Kriwacki RW. Mechanism of cell cycle entry mediated by the intrinsically disordered protein p27(Kip1). ACS Chem Biol. 2012;7:678–82. doi:10.1021/cb200487h. PMID:22276948.
- Follis AV, Galea CA, Kriwacki RW. Intrinsic protein flexibility in regulation of cell proliferation: Advantages for signaling and opportunities for novel therapeutics. Adv Exp Med Biol. 2012;725:27–49. doi:10.1007/978-1-4614-0659-4_3. PMID:22399317.
- Yoon MK, Mitrea DM, Ou L, Kriwacki RW. Cell cycle regulation by the intrinsically disordered proteins p21 and p27. Biochem Soc Trans. 2012;40:981–8. doi:10.1042/BST20120092. PMID:22988851.
- Mitrea DM, Yoon MK, Ou L, Kriwacki RW. Disorder-function relationships for the cell cycle regulatory proteins p21 and p27. Biol Chem. 2012;393:259–74. doi:10.1515/hsz-2011-0254. PMID:23029651.
- Moldoveanu T, Grace CR, Llambi F, Nourse A, Fitzgerald P, Gehring K, Kriwacki RW, Green DR. BID-induced structural changes in BAK promote apoptosis. Nat Struct Mol Biol. 2013;20:589–97. doi:10.1038/nsmb.2563. PMID:23604079.
- Frye JJ, Brown NG, Petzold G, Watson ER, Grace CR, Nourse A, Jarvis MA, Kriwacki RW, Peters JM, Stark H, et al. Electron microscopy structure of human APC/C(CDH1)-EMI1 reveals multimodal mechanism of E3 ligase shutdown. Nat Struct Mol Biol. 2013;20:827–35. doi:10.1038/nsmb.2593. PMID:23708605.
- Mei Y, Su M, Soni G, Salem S, Colbert CL, Sinha SC. Intrinsically disordered regions in autophagy proteins. Proteins. 2014;82:565–78. doi:10.1002/prot.24424. PMID:24115198.
- Chakrabortee S, Tripathi R, Watson M, Schierle GS, Kurniawan DP, Kaminski CF, Wise MJ, Tunnacliffe A. Intrinsically disordered proteins as molecular shields. Mol Biosyst. 2012;8:210–9. doi:10.1039/C1MB05263B. PMID:21909508.
- De Jonge N, Garcia-Pino A, Buts L, Haesaerts S, Charlier D, Zangger K, Wyns L, De Greve H, Loris R. Rejuvenation of CcdB-poisoned gyrase by an intrinsically disordered protein domain. Mol Cell. 2009;35:154–63. doi:10.1016/j.molcel.2009.05.025. PMID:19647513.
- Norholm AB, Hendus-Altenburger R, Bjerre G, Kjaergaard M, Pedersen SF, Kragelund BB. The intracellular distal tail of the Na+/H+ exchanger NHE1 is intrinsically disordered: Implications for NHE1 trafficking. Biochemistry. 2011;50:3469–80. doi:10.1021/bi1019989. PMID:21425832.
- Follis AV, Chipuk JE, Fisher JC, Yun MK, Grace CR, Nourse A, Baran K, Ou L, Min L, White SW, et al. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat Chem Biol. 2013;9:163–8. doi:10.1038/nchembio.1166. PMID:23340338.
- Peng Z, Xue B, Kurgan L, Uversky VN. Resilience of death: Intrinsic disorder in proteins involved in the programmed cell death. Cell Death Differ. 2013;20:1257–67. doi:10.1038/cdd.2013.65. PMID:23764774.
- Uversky AV, Xue B, Peng Z, Kurgan L, Uversky VN. On the intrinsic disorder status of the major players in programmed cell death pathways. F1000Res. 2013;2:190. doi:10.12688/f1000research.2-190.v1. PMID:24358900.
- Popelka H, Uversky VN, Klionsky DJ. Identification of Atg3 as an intrinsically disordered polypeptide yields insights into the molecular dynamics of autophagy-related proteins in yeast. Autophagy. 2014;10:1093–104. doi:10.4161/auto.28616. PMID:24879155.
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. doi:10.1080/15548627.2015.1100356. PMID:26799652.
- Na I, Meng F, Kurgan L, Uversky VN. Autophagy-related intrinsically disordered proteins in intra-nuclear compartments. Mol Biosyst. 2016;12:2798–817. doi:10.1039/C6MB00069J. PMID:27377881.
- Fuxreiter M, Toth-Petroczy A, Kraut DA, Matouschek A, Lim RY, Xue B, Kurgan L, Uversky VN. Disordered proteinaceous machines. Chem Rev. 2014;114:6806–43. doi:10.1021/cr4007329. PMID:24702702.
- Borg M, Mittag T, Pawson T, Tyers M, Forman-Kay JD, Chan HS. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc Natl Acad Sci U S A. 2007;104:9650–5. doi:10.1073/pnas.0702580104. PMID:17522259.
- Mittag T, Orlicky S, Choy WY, Tang X, Lin H, Sicheri F, Kay LE, Tyers M, Forman-Kay JD. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc Natl Acad Sci U S A. 2008;105:17772–7. doi:10.1073/pnas.0809222105. PMID:19008353.
- Mittag T, Kay LE, Forman-Kay JD. Protein dynamics and conformational disorder in molecular recognition. J Mol Recognit. 2010;23:105–16. doi:10.1002/jmr.961. PMID:19585546.
- Mittag T, Marsh J, Grishaev A, Orlicky S, Lin H, Sicheri F, Tyers M, Forman-Kay JD. Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase. Structure. 2010;18:494–506. doi:10.1016/j.str.2010.01.020. PMID:20399186.
- Sigalov AB, Kim WM, Saline M, Stern LJ. The intrinsically disordered cytoplasmic domain of the T cell receptor zeta chain binds to the nef protein of simian immunodeficiency virus without a disorder-to-order transition. Biochemistry. 2008;47:12942–4. doi:10.1021/bi801602p. PMID:19012413.
- Permyakov SE, Millett IS, Doniach S, Permyakov EA, Uversky VN. Natively unfolded C-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins. 2003;53:855–62. doi:10.1002/prot.10481. PMID:14635127.
- Sigalov A, Aivazian D, Stern L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry. 2004;43:2049–61. doi:10.1021/bi035900h. PMID:14967045.
- Sigalov AB, Zhuravleva AV, Orekhov VY. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie. 2007;89:419–21. doi:10.1016/j.biochi.2006.11.003. PMID:17174464.
- Pometun MS, Chekmenev EY, Wittebort RJ. Quantitative observation of backbone disorder in native elastin. J Biol Chem. 2004;279:7982–7. doi:10.1074/jbc.M310948200. PMID:14625282.
- Sigalov AB, Hendricks GM. Membrane binding mode of intrinsically disordered cytoplasmic domains of T cell receptor signaling subunits depends on lipid composition. Biochem Biophys Res Commun. 2009;389:388–93. doi:10.1016/j.bbrc.2009.09.002. PMID:19733547.
- Sigalov AB, Aivazian DA, Uversky VN, Stern LJ. Lipid-binding activity of intrinsically unstructured cytoplasmic domains of multichain immune recognition receptor signaling subunits. Biochemistry. 2006;45:15731–9. doi:10.1021/bi061108f. PMID:17176095.
- Uversky VN. Multitude of binding modes attainable by intrinsically disordered proteins: A portrait gallery of disorder-based complexes. Chem Soc Rev. 2011;40:1623–34. doi:10.1039/C0CS00057D. PMID:21049125.
- Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, Cortese MS, Sickmeier M, LeGall T, Obradovic Z, et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci U S A. 2006;103:8390–5. doi:10.1073/pnas.0507916103. PMID:16717195.
- Buljan M, Chalancon G, Dunker AK, Bateman A, Balaji S, Fuxreiter M, Babu MM. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr Opin Struct Biol. 2013;23:443–50. doi:10.1016/j.sbi.2013.03.006. PMID:23706950.
- Buljan M, Chalancon G, Eustermann S, Wagner GP, Fuxreiter M, Bateman A, Babu MM. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol Cell. 2012;46:871–83. doi:10.1016/j.molcel.2012.05.039. PMID:22749400.
- Collins MO, Yu L, Campuzano I, Grant SG, Choudhary JS. Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder. Mol Cell Proteomics. 2008;7:1331–48. doi:10.1074/mcp.M700564-MCP200. PMID:18388127.
- Kurotani A, Tokmakov AA, Kuroda Y, Fukami Y, Shinozaki K, Sakurai T. Correlations between predicted protein disorder and post-translational modifications in plants. Bioinformatics. 2014;30:1095–1103. doi:10.1093/bioinformatics/btt762. PMID:24403539.
- Colak R, Kim T, Michaut M, Sun M, Irimia M, Bellay J, Myers CL, Blencowe BJ, Kim PM. Distinct types of disorder in the human proteome: Functional implications for alternative splicing. PLoS Comput Biol. 2013;9:e1003030. doi:10.1371/journal.pcbi.1003030. PMID:23633940.
- Schluter H, Apweiler R, Holzhutter HG, Jungblut PR. Finding one's way in proteomics: A protein species nomenclature. Chem Cent J. 2009;3:11. doi:10.1186/1752-153X-3-11. PMID:19740416.
- Consortium TEP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi:10.1038/nature11247. PMID:22955616.
- Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson AC, Angelidou P, Asplund A, Asplund C, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4:1920–32. doi:10.1074/mcp.R500009-MCP200 10.1074/mcp.M500279-MCP200. PMID:16127175.
- Farrah T, Deutsch EW, Omenn GS, Sun Z, Watts JD, Yamamoto T, Shteynberg D, Harris MM, Moritz RL. State of the human proteome in 2013 as viewed through PeptideAtlas: Comparing the kidney, urine, and plasma proteomes for the biology- and disease-driven human proteome project. J Proteome Res. 2014;13:60–75. doi:10.1021/pr4010037. PMID:24261998.
- Farrah T, Deutsch EW, Hoopmann MR, Hallows JL, Sun Z, Huang CY, Moritz RL. The state of the human proteome in 2012 as viewed through PeptideAtlas. J Proteome Res. 2013;12:162–71. doi:10.1021/pr301012j. PMID:23215161.
- Reddy PJ, Ray S, Srivastava S. The quest of the human proteome and the missing proteins: Digging deeper. OMICS. 2015;19:276–82. doi:10.1089/omi.2015.0035. PMID:25933256.
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. A draft map of the human proteome. Nature. 2014;509:575-+. doi:10.1038/nature13302. PMID:24870542.
- Smith LM, Kelleher NL, Consortium for Top Down P. Proteoform: A single term describing protein complexity. Nat Methods. 2013;10:186–7. doi:10.1038/nmeth.2369. PMID:23443629.
- Uversky VN. p53 Proteoforms and intrinsic disorder: An illustration of the protein structure-function continuum concept. Int J Mol Sci. 2016;17:1874. doi:10.3390/ijms17111874.
- Uversky VN. Looking at the recent advances in understanding alpha-synuclein and its aggregation through the proteoform prism. F1000Res. 2017;6:525. doi:10.12688/f1000research.10536.1. PMID:28491292.
- Malaney P, Uversky VN, Dave V. PTEN proteoforms in biology and disease. Cell Mol Life Sci. 2017;74:2783–94. doi:10.1007/s00018-017-2500-6. PMID:28289760.
- Zimmerman SB, Trach SO. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J Mol Biol. 1991;222:599–620. doi:10.1016/0022-2836(91)90499-V. PMID:1748995.
- van den Berg B, Ellis RJ, Dobson CM. Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 1999;18:6927–33. doi:10.1093/emboj/18.24.6927. PMID:10601015.
- Rivas G, Ferrone F, Herzfeld J. Life in a crowded world. EMBO Rep. 2004;5:23–7. doi:10.1038/sj.embor.7400056. PMID:14710181.
- Ellis RJ, Minton AP. Cell biology: Join the crowd. Nature. 2003;425:27–8. doi:10.1038/425027a. PMID:12955122.
- Zimmerman SB, Minton AP. Macromolecular crowding: Biochemical, biophysical, and physiological consequences. Annu Rev Biophys Biomol Struct. 1993;22:27–65. doi:10.1146/annurev.bb.22.060193.000331. PMID:7688609.
- Fulton AB. How crowded is the cytoplasm? Cell. 1982;30:345–7. doi:10.1016/0092-8674(82)90231-8. PMID:6754085.
- Minton AP. Influence of excluded volume upon macromolecular structure and associations in ‘crowded’ media. Curr Opin Biotechnol. 1997;8:65–9. doi:10.1016/S0958-1669(97)80159-0. PMID:9013656.
- Ellis RJ. Macromolecular crowding: Obvious but underappreciated. Trends Biochem Sci. 2001;26:597–604. doi:10.1016/S0968-0004(01)01938-7. PMID:11590012.
- Minton AP. Protein folding: Thickening the broth. Curr Biol. 2000;10:R97–9. doi:10.1016/S0960-9822(00)00301-8. PMID:10679316.
- Gsponer J, Futschik ME, Teichmann SA, Babu MM. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science. 2008;322:1365–8. doi:10.1126/science.1163581. PMID:19039133.
- Rudiger S, Freund SM, Veprintsev DB, Fersht AR. CRINEPT-TROSY NMR reveals p53 core domain bound in an unfolded form to the chaperone Hsp90. Proc Natl Acad Sci U S A. 2002;99:11085–90. doi:10.1073/pnas.132393699. PMID:12163643.
- Martinez-Yamout MA, Venkitakrishnan RP, Preece NE, Kroon G, Wright PE, Dyson HJ. Localization of sites of interaction between p23 and Hsp90 in solution. J Biol Chem. 2006;281:14457–64. doi:10.1074/jbc.M601759200. PMID:16565516.
- Rodriguez F, Arsene-Ploetze F, Rist W, Rudiger S, Schneider-Mergener J, Mayer MP, Bukau B. Molecular basis for regulation of the heat shock transcription factor sigma32 by the DnaK and DnaJ chaperones. Mol Cell. 2008;32:347–58. doi:10.1016/j.molcel.2008.09.016. PMID:18995833.
- Didenko T, Duarte AM, Karagoz GE, Rudiger SG. Hsp90 structure and function studied by NMR spectroscopy. Biochim Biophys Acta. 2012;1823:636–47. doi:10.1016/j.bbamcr.2011.11.009. PMID:22155720.
- Karagoz GE, Duarte AM, Akoury E, Ippel H, Biernat J, Moran Luengo T, Radli M, Didenko T, Nordhues BA, Veprintsev DB, et al. Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell. 2014;156:963–74. doi:10.1016/j.cell.2014.01.037. PMID:24581495.
- Tsvetkov P, Reuven N, Shaul Y. The nanny model for IDPs. Nat Chem Biol. 2009;5:778–81. doi:10.1038/nchembio.233. PMID:19841623.
- Demarest SJ, Martinez-Yamout M, Chung J, Chen H, Xu W, Dyson HJ, Evans RM, Wright PE. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature. 2002;415:549–53. doi:10.1038/415549a. PMID:11823864.
- Ebert MO, Bae SH, Dyson HJ, Wright PE. NMR relaxation study of the complex formed between CBP and the activation domain of the nuclear hormone receptor coactivator ACTR. Biochemistry. 2008;47:1299–308. doi:10.1021/bi701767j. PMID:18177052.
- Chipuk JE, Fisher JC, Dillon CP, Kriwacki RW, Kuwana T, Green DR. Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proc Natl Acad Sci U S A. 2008;105:20327–32. doi:10.1073/pnas.0808036105. PMID:19074266.
- Ferreon JC, Lee CW, Arai M, Martinez-Yamout MA, Dyson HJ, Wright PE. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc Natl Acad Sci U S A. 2009;106:6591–6. doi:10.1073/pnas.0811023106. PMID:19357310.
- Ferreon JC, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc Natl Acad Sci U S A. 2009;106:13260–5. doi:10.1073/pnas.0906770106. PMID:19651603.
- Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J. 2009;28:948–58. doi:10.1038/emboj.2009.30. PMID:19214187.
- Lee CW, Martinez-Yamout MA, Dyson HJ, Wright PE. Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry. 2010;49:9964–71. doi:10.1021/bi1012996. PMID:20961098.
- Kostic M, Matt T, Martinez-Yamout MA, Dyson HJ, Wright PE. Solution structure of the Hdm2 C2H2C4 RING, a domain critical for ubiquitination of p53. J Mol Biol. 2006;363:433–50. doi:10.1016/j.jmb.2006.08.027. PMID:16965791.
- Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jäkel H, Kullmann M, Kriwacki RW, Hengst L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128:269–80. doi:10.1016/j.cell.2006.11.047. PMID:17254966.
- Zhan J, Easton JB, Huang S, Mishra A, Xiao L, Lacy ER, Kriwacki RW, Houghton PJ. Negative regulation of ASK1 by p21Cip1 involves a small domain that includes Serine 98 that is phosphorylated by ASK1 in vivo. Mol Cell Biol. 2007;27:3530–41. doi:10.1128/MCB.00086-06. PMID:17325029.
- Mitrea DM, Grace CR, Buljan M, Yun MK, Pytel NJ, Satumba J, Nourse A, Park CG, Madan Babu M, White SW, et al. Structural polymorphism in the N-terminal oligomerization domain of NPM1. Proc Natl Acad Sci U S A. 2014;111:4466–71. doi:10.1073/pnas.1321007111. PMID:24616519.
- Asher G, Tsvetkov P, Kahana C, Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005;19:316–21. doi:10.1101/gad.319905. PMID:15687255.
- Tsvetkov P, Asher G, Paz A, Reuven N, Sussman JL, Silman I, Shaul Y. Operational definition of intrinsically unstructured protein sequences based on susceptibility to the 20S proteasome. Proteins. 2008;70:1357–66. doi:10.1002/prot.21614. PMID:17879262.
- Tsvetkov P, Reuven N, Prives C, Shaul Y. Susceptibility of p53 unstructured N terminus to 20 S proteasomal degradation programs the stress response. J Biol Chem. 2009;284:26234–42. doi:10.1074/jbc.M109.040493. PMID:19617345.
- Wiggins CM, Tsvetkov P, Johnson M, Joyce CL, Lamb CA, Bryant NJ, Komander D, Shaul Y, Cook SJ. BIM(EL), an intrinsically disordered protein, is degraded by 20S proteasomes in the absence of poly-ubiquitylation. J Cell Sci. 2011;124:969–77. doi:10.1242/jcs.058438. PMID:21378313.
- Suskiewicz MJ, Sussman JL, Silman I, Shaul Y. Context-dependent resistance to proteolysis of intrinsically disordered proteins. Protein Sci. 2011;20:1285–97. doi:10.1002/pro.657. PMID:21574196.
- Tsvetkov P, Myers N, Moscovitz O, Sharon M, Prilusky J, Shaul Y. Thermo-resistant intrinsically disordered proteins are efficient 20S proteasome substrates. Mol Biosyst. 2012;8:368–73. doi:10.1039/C1MB05283G. PMID:22027891.
- Uversky VN. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front Biosci (Landmark Ed). 2009;14:5188–238. doi:10.2741/3594. PMID:19482612.
- Uversky VN, Oldfield CJ, Midic U, Xie H, Xue B, Vucetic S, Iakoucheva LM, Obradovic Z, Dunker AK. Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genomics. 2009;10(Suppl 1):S7. doi:10.1186/1471-2164-10-S1-S7. PMID:19594884.
- Uversky VN. Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: Another illustration of the D(2) concept. Expert Rev Proteomics. 2010;7:543–64. doi:10.1586/epr.10.36. PMID:20653509.
- Uversky VN. Wrecked regulation of intrinsically disordered proteins in diseases: Pathogenicity of deregulated regulators. Front Mol Biosci. 2014;1:6. doi:10.3389/fmolb.2014.00006. PMID:25988147.
- Uversky VN. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin Drug Discov. 2012;7:475–88. doi:10.1517/17460441.2012.686489. PMID:22559227.
- Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46. doi:10.1146/annurev.biophys.37.032807.125924. PMID:18573080.
- Uversky VN, Dave V, Iakoucheva LM, Malaney P, Metallo SJ, Pathak RR, Joerger AC. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem Rev. 2014;114:6844–79. doi:10.1021/cr400713r. PMID:24830552.
- Wells M, Tidow H, Rutherford TJ, Markwick P, Jensen MR, Mylonas E, Svergun DI, Blackledge M, Fersht AR. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci U S A. 2008;105:5762–7. doi:10.1073/pnas.0801353105. PMID:18391200.
- Xue B, Dunker AK, Uversky VN. The roles of intrinsic disorder in orchestrating the Wnt-pathway. J Biomol Struct Dyn. 2012;29:843–61. doi:10.1080/073911012010525024. PMID:22292947.
- Minde DP, Radli M, Forneris F, Maurice MM, Rudiger SG. Large extent of disorder in adenomatous polyposis coli offers a strategy to guard Wnt signalling against point mutations. PLoS One. 2013;8:e77257. doi:10.1371/journal.pone.0077257. PMID:24130866.
- Rotem S, Katz C, Friedler A. Insights into the structure and protein-protein interactions of the pro-apoptotic protein ASPP2. Biochem Soc Trans. 2007;35:966–9. doi:10.1042/BST0350966. PMID:17956256.
- Ahn J, Byeon IJ, Byeon CH, Gronenborn AM. Insight into the structural basis of pro- and antiapoptotic p53 modulation by ASPP proteins. J Biol Chem. 2009;284:13812–22. doi:10.1074/jbc.M808821200. PMID:19246451.
- Noutsou M, Duarte AM, Anvarian Z, Didenko T, Minde DP, Kuper I, de Ridder I, Oikonomou C, Friedler A, Boelens R, et al. Critical scaffolding regions of the tumor suppressor Axin1 are natively unfolded. J Mol Biol. 2011;405:773–86. doi:10.1016/j.jmb.2010.11.013. PMID:21087614.
- Xue B, Romero PR, Noutsou M, Maurice MM, Rudiger SG, William AM, Jr., Mizianty MJ, Kurgan L, Uversky VN, Dunker AK. Stochastic machines as a colocalization mechanism for scaffold protein function. FEBS Lett. 2013;587:1587–91. doi:10.1016/j.febslet.2013.04.006. PMID:23603389.
- Kvansakul M, Hinds MG. The structural biology of BH3-only proteins. Methods Enzymol. 2014;544:49–74. doi:10.1016/B978-0-12-417158-9.00003-0. PMID:24974286.
- Mark WY, Liao JC, Lu Y, Ayed A, Laister R, Szymczyna B, Chakrabartty A, Arrowsmith CH. Characterization of segments from the central region of BRCA1: An intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J Mol Biol. 2005;345:275–87. doi:10.1016/j.jmb.2004.10.045. PMID:15571721.
- Rajagopalan K, Mooney SM, Parekh N, Getzenberg RH, Kulkarni P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem. 2011;112:3256–67. doi:10.1002/jcb.23252. PMID:21748782.
- Ng KP, Potikyan G, Savene RO, Denny CT, Uversky VN, Lee KA. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proc Natl Acad Sci U S A. 2007;104:479–84. doi:10.1073/pnas.0607007104. PMID:17202261.
- Uversky VN, Roman A, Oldfield CJ, Dunker AK. Protein intrinsic disorder and human papillomaviruses: Increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J Proteome Res. 2006;5:1829–42. doi:10.1021/pr0602388. PMID:16889404.
- Malaney P, Pathak RR, Xue B, Uversky VN, Dave V. Intrinsic disorder in PTEN and its interactome confers structural plasticity and functional versatility. Sci Rep. 2013;3:2035. doi:10.1038/srep02035. PMID:23783762.
- McBurney MW, Clark-Knowles KV, Caron AZ, Gray DA. SIRT1 is a highly networked protein that mediates the adaptation to chronic physiological stress. Genes Cancer. 2013;4:125–34. doi:10.1177/1947601912474893. PMID:24020004.
- Sharma A, Costantini S, Colonna G. The protein-protein interaction network of the human Sirtuin family. Biochim Biophys Acta. 2013;1834:1998–2009. doi:10.1016/j.bbapap.2013.06.012. PMID:23811471.
- Uversky VN. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn. 2003;21:211–34. doi:10.1080/07391102.2003.10506918. PMID:12956606.
- Uversky VN. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J Neurochem. 2007;103:17–37. doi:10.1111/j.1471-4159.2007.04764.x. PMID:17623039.
- Uversky VN. Alpha-synuclein misfolding and neurodegenerative diseases. Curr Protein Pept Sci. 2008;9:507–40. doi:10.2174/138920308785915218. PMID:18855701.
- Uversky VN, Eliezer D. Biophysics of Parkinson's disease: Structure and aggregation of alpha-synuclein. Curr Protein Pept Sci. 2009;10:483–99. doi:10.2174/138920309789351921. PMID:19538146.
- Breydo L, Wu JW, Uversky VN. Alpha-synuclein misfolding and Parkinson's disease. Biochim Biophys Acta. 2012;1822:261–85. doi:10.1016/j.bbadis.2011.10.002. PMID:22024360.
- Alderson TR, Markley JL. Biophysical characterization of alpha-synuclein and its controversial structure. Intrinsically Disord Proteins. 2013;1:18–39. doi:10.4161/idp.26255. PMID:24634806.
- Uversky VN. The triple power of D(3): Protein intrinsic disorder in degenerative diseases. Frontiers in Bioscience. 2014;19:181–258. doi:10.2741/4204.
- Casu F, Duggan BM, Hennig M. The arginine-rich RNA-binding motif of HIV-1 Rev is intrinsically disordered and folds upon RRE binding. Biophys J. 2013;105:1004–17. doi:10.1016/j.bpj.2013.07.022. PMID:23972852.
- Cheng Y, LeGall T, Oldfield CJ, Dunker AK, Uversky VN. Abundance of intrinsic disorder in protein associated with cardiovascular disease. Biochemistry. 2006;45:10448–60. doi:10.1021/bi060981d. PMID:16939197.
- Baker JM, Hudson RP, Kanelis V, Choy WY, Thibodeau PH, Thomas PJ, Forman-Kay JD. CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat Struct Mol Biol. 2007;14:738–45. doi:10.1038/nsmb1278. PMID:17660831.
- Jorda J, Xue B, Uversky VN, Kajava AV. Protein tandem repeats - the more perfect, the less structured. FEBS J. 2010;277:2673–82. doi:10.1111/j.1742-4658.2010.07684.x. PMID:20553501.
- Santamaria N, Alhothali M, Harreguy Alfonso M, Breydo L, Uversky VN. Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cell Mol Life Sci. 2017;74:1297–1318. doi:10.1007/s00018-016-2416-6. PMID:27838743.
- Breydo L, Uversky VN. Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics. 2011;3:1163–80. doi:10.1039/c1mt00106j. PMID:21869995.
- Uversky VN. Flexible nets of malleable guardians: Intrinsically disordered chaperones in neurodegenerative diseases. Chem Rev. 2011;111:1134–66. doi:10.1021/cr100186d. PMID:21086986.
- Meng F, Na I, Kurgan L, Uversky VN. Compartmentalization and functionality of nuclear disorder: Intrinsic disorder and protein-protein interactions in intra-nuclear compartments. Int J Mol Sci. 2015;17:E24. doi:10.3390/ijms17010024. PMID:26712748.
- Uversky VN, Kuznetsova IM, Turoverov KK, Zaslavsky B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett. 2015;589:15–22. doi:10.1016/j.febslet.2014.11.028. PMID:25436423.
- Uversky VN. Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: Complex coacervates and membrane-less organelles. Adv Colloid Interface Sci. 2017;239:97–114. doi:10.1016/j.cis.2016.05.012. PMID:27291647.
- Watkins JL, Murphy R, Emtage JL, Wente SR. The human homologue of Saccharomyces cerevisiae Gle1p is required for poly(A)+ RNA export. Proc Natl Acad Sci U S A. 1998;95:6779–84. doi:10.1073/pnas.95.12.6779. PMID:9618489.
- Kendirgi F, Barry DM, Griffis ER, Powers MA, Wente SR. An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J Cell Biol. 2003;160:1029–40. doi:10.1083/jcb.200211081. PMID:12668658.
- Murphy R, Wente SR. An RNA-export mediator with an essential nuclear export signal. Nature. 1996;383:357–60. doi:10.1038/383357a0. PMID:8848052.
- Folkmann AW, Collier SE, Zhan X, Aditi, Ohi MD, Wente SR. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell. 2013;155:582–93. doi:10.1016/j.cell.2013.09.023. PMID:24243016.
- Folkmann AW, Dawson TR, Wente SR. Insights into mRNA export-linked molecular mechanisms of human disease through a Gle1 structure-function analysis. Adv Biol Regul. 2014;54:74–91. doi:10.1016/j.jbior.2013.10.002. PMID:24275432.
- Rayala HJ, Kendirgi F, Barry DM, Majerus PW, Wente SR. The mRNA export factor human Gle1 interacts with the nuclear pore complex protein Nup155. Mol Cell Proteomics. 2004;3:145–55. doi:10.1074/mcp.M300106-MCP200. PMID:14645504.
- Di Domenico T, Walsh I, Martin AJ, Tosatto SC. MobiDB: A comprehensive database of intrinsic protein disorder annotations. Bioinformatics. 2012;28:2080–1. doi:10.1093/bioinformatics/bts327. PMID:22661649.
- Potenza E, Domenico TD, Walsh I, Tosatto SC. MobiDB 2.0: An improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res. 2015;43:D315–D20. doi:10.1093/nar/gku982. PMID:25361972.
- Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, et al. D(2)P(2): Database of disordered protein predictions. Nucleic Acids Res. 2013;41:D508–16. doi:10.1093/nar/gks1226. PMID:23203878.
- Dosztanyi Z, Meszaros B, Simon I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–6. doi:10.1093/bioinformatics/btp518. PMID:19717576.
- Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–70. doi:10.1021/bi050736e. PMID:16156658.
- Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs). J Mol Biol. 2006;362:1043–59. doi:10.1016/j.jmb.2006.07.087. PMID:16935303.
- Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res. 2007;6:2351–66. doi:10.1021/pr0701411. PMID:17488107.
- Graebsch A, Roche S, Niessing D. X-ray structure of Pur-alpha reveals a Whirly-like fold and an unusual nucleic-acid binding surface. Proc Natl Acad Sci U S A. 2009;106:18521–6. doi:10.1073/pnas.0907990106. PMID:19846792.
- Brangwynne CP. Phase transitions and size scaling of membrane-less organelles. J Cell Biol. 2013;203:875–81. doi:10.1083/jcb.201308087. PMID:24368804.
- Decker M, Jaensch S, Pozniakovsky A, Zinke A, O'Connell KF, Zachariae W, Myers E, Hyman AA. Limiting amounts of centrosome material set centrosome size in C. elegans embryos. Curr Biol. 2011;21:1259–67. doi:10.1016/j.cub.2011.06.002. PMID:21802300.
- Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, Jülicher F, Hyman AA. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–32. doi:10.1126/science.1172046. PMID:19460965.
- Chuma S, Hosokawa M, Tanaka T, Nakatsuji N. Ultrastructural characterization of spermatogenesis and its evolutionary conservation in the germline: Germinal granules in mammals. Mol Cell Endocrinol. 2009;306:17–23. doi:10.1016/j.mce.2008.11.009. PMID:19063939.
- Kiebler MA, Bassell GJ. Neuronal RNA granules: Movers and makers. Neuron. 2006;51:685–90. doi:10.1016/j.neuron.2006.08.021. PMID:16982415.
- Decker CJ, Teixeira D, Parker R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol. 2007;179:437–49. doi:10.1083/jcb.200704147. PMID:17984320.
- Wippich F, Bodenmiller B, Trajkovska MG, Wanka S, Aebersold R, Pelkmans L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell. 2013;152:791–805. doi:10.1016/j.cell.2013.01.033. PMID:23415227.
- Strzelecka M, Trowitzsch S, Weber G, Luhrmann R, Oates AC, Neugebauer KM. Coilin-dependent snRNP assembly is essential for zebrafish embryogenesis. Nat Struct Mol Biol. 2010;17:403–9. doi:10.1038/nsmb.1783. PMID:20357773.
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi:10.1016/j.cell.2007.01.015. PMID:17320508.
- Li L, Roy K, Katyal S, Sun X, Bleoo S, Godbout R. Dynamic nature of cleavage bodies and their spatial relationship to DDX1 bodies, Cajal bodies, and gems. Mol Biol Cell. 2006;17:1126–40. doi:10.1091/mbc.E05-08-0768. PMID:16371507.
- Nizami Z, Deryusheva S, Gall JG. The Cajal body and histone locus body. Cold Spring Harb Perspect Biol. 2010;2:a000653. doi:10.1101/cshperspect.a000653. PMID:20504965.
- Matera AG, Frey MR. Coiled bodies and gems: Janus or gemini? Am J Hum Genet. 1998;63:317–21. doi:10.1086/301992. PMID:9683623.
- Gubitz AK, Feng W, Dreyfuss G. The SMN complex. Exp Cell Res. 2004;296:51–6. doi:10.1016/j.yexcr.2004.03.022. PMID:15120993.
- Grossman E, Medalia O, Zwerger M. Functional architecture of the nuclear pore complex. Annu Rev Biophys. 2012;41:557–84. doi:10.1146/annurev-biophys-050511-102328. PMID:22577827.
- Lamond AI, Spector DL. Nuclear speckles: A model for nuclear organelles. Nat Rev Mol Cell Biol. 2003;4:605–12. doi:10.1038/nrm1172. PMID:12923522.
- Biamonti G, Vourc'h C. Nuclear stress bodies. Cold Spring Harb Perspect Biol. 2010;2:a000695. doi:10.1101/cshperspect.a000695. PMID:20516127.
- Biamonti G. Nuclear stress bodies: A heterochromatin affair? Nat Rev Mol Cell Biol. 2004;5:493–8. doi:10.1038/nrm1405. PMID:15173828.
- Shav-Tal Y, Blechman J, Darzacq X, Montagna C, Dye BT, Patton JG, Singer RH, Zipori D. Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol Biol Cell. 2005;16:2395–413. doi:10.1091/mbc.E04-11-0992. PMID:15758027.
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol. 2011;193:97–108. doi:10.1083/jcb.201011083. PMID:21444690.
- Fox AH, Lam YW, Leung AK, Lyon CE, Andersen J, Mann M, Lamond AI. Paraspeckles: A novel nuclear domain. Curr Biol. 2002;12:13–25. doi:10.1016/S0960-9822(01)00632-7. PMID:11790299.
- Huang S. Review: Perinucleolar structures. J Struct Biol. 2000;129:233–40. doi:10.1006/jsbi.2000.4247. PMID:10806073.
- Pirrotta V, Li HB. A view of nuclear Polycomb bodies. Curr Opin Genet Dev. 2012;22:101–9. doi:10.1016/j.gde.2011.11.004. PMID:22178420.
- Maul GG, Negorev D, Bell P, Ishov AM. Review: Properties and assembly mechanisms of ND10, PML bodies, or PODs. J Struct Biol. 2000;129:278–87. doi:10.1006/jsbi.2000.4239. PMID:10806078.
- Antonicka H, Shoubridge EA. Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep. 2015;10:920–36. doi:10.1016/j.celrep.2015.01.030. PMID:25683715.
- Uniacke J, Zerges W. Stress induces the assembly of RNA granules in the chloroplast of Chlamydomonas reinhardtii. J Cell Biol. 2008;182:641–6. doi:10.1083/jcb.200805125. PMID:18710928.
- Dundr M, Misteli T. Biogenesis of nuclear bodies. Cold Spring Harb Perspect Biol. 2010;2:a000711. doi:10.1101/cshperspect.a000711. PMID:21068152.
- Brangwynne CP, Mitchison TJ, Hyman AA. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc Natl Acad Sci U S A. 2011;108:4334–9. doi:10.1073/pnas.1017150108. PMID:21368180.
- Feric M, Brangwynne CP. A nuclear F-actin scaffold stabilizes ribonucleoprotein droplets against gravity in large cells. Nat Cell Biol. 2013;15:1253–9. doi:10.1038/ncb2830. PMID:23995731.
- Phair RD, Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–9. doi:10.1038/35007077. PMID:10766243.
- Pederson T. Protein mobility within the nucleus–what are the right moves? Cell. 2001;104:635–8. doi:10.1016/S0092-8674(01)00258-6. PMID:11257216.
- Handwerger KE, Cordero JA, Gall JG. Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structure. Mol Biol Cell. 2005;16:202–11. doi:10.1091/mbc.E04-08-0742. PMID:15509651.
- Updike DL, Hachey SJ, Kreher J, Strome S. P granules extend the nuclear pore complex environment in the C. elegans germ line. J Cell Biol. 2011;192:939–48. doi:10.1083/jcb.201010104. PMID:21402789.
- Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–40. doi:10.1038/nature10879. PMID:22398450.
- Aggarwal S, Snaidero N, Pahler G, Frey S, Sanchez P, Zweckstetter M, Janshoff A, Schneider A, Weil MT, Schaap IA, et al. Myelin membrane assembly is driven by a phase transition of myelin basic proteins into a cohesive protein meshwork. PLoS Biol. 2013;11:e1001577. doi:10.1371/journal.pbio.1001577. PMID:23762018.
- Elbaum-Garfinkle S, Kim Y, Szczepaniak K, Chen CC, Eckmann CR, Myong S, Brangwynne CP. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci U S A. 2015;112:7189–94. doi:10.1073/pnas.1504822112. PMID:26015579.
- Hyman AA, Simons K. Cell biology. Beyond oil and water–phase transitions in cells. Science. 2012;337:1047–9. doi:10.1126/science.1223728. PMID:22936764.
- Hyman AA, Weber CA, Julicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30:39–58. doi:10.1146/annurev-cellbio-100913-013325. PMID:25288112.
- Wheeler JR, Matheny T, Jain S, Abrisch R, Parker R. Distinct stages in stress granule assembly and disassembly. Elife. 2016;5:e18413. doi:10.7554/eLife.18413. PMID:27602576.
- Mitrea DM, Cika JA, Guy CS, Ban D, Banerjee PR, Stanley CB, Nourse A, Deniz AA, Kriwacki RW. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. Elife. 2016;5:e13571. doi:10.7554/eLife.13571. PMID:26836305.
- Lee CF, Brangwynne CP, Gharakhani J, Hyman AA, Julicher F. Spatial organization of the cell cytoplasm by position-dependent phase separation. Phys Rev Lett. 2013;111:088101. doi:10.1103/PhysRevLett.111.088101.
- Lee C, Occhipinti P, Gladfelter AS. PolyQ-dependent RNA-protein assemblies control symmetry breaking. J Cell Biol. 2015;208:533–44. doi:10.1083/jcb.201407105. PMID:25713414.
- Rohatgi R, Nollau P, Ho HY, Kirschner MW, Mayer BJ. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J Biol Chem. 2001;276:26448–52. doi:10.1074/jbc.M103856200. PMID:11340081.
- Blasutig IM, New LA, Thanabalasuriar A, Dayarathna TK, Goudreault M, Quaggin SE, Li SS, Gruenheid S, Jones N, Pawson T. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol Cell Biol. 2008;28:2035–46. doi:10.1128/MCB.01770-07. PMID:18212058.
- Fromm SA, Kamenz J, Noldeke ER, Neu A, Zocher G, Sprangers R. In vitro reconstitution of a cellular phase-transition process that involves the mRNA decapping machinery. Angew Chem Int Ed Engl. 2014;53:7354–9. doi:10.1002/anie.201402885. PMID:24862735.
- Urry DW. Entropic elastic processes in protein mechanisms. I. Elastic structure due to an inverse temperature transition and elasticity due to internal chain dynamics. J Protein Chem. 1988;7:1–34. doi:10.1007/BF01025411. PMID:3076447.
- Urry DW. Free energy transduction in polypeptides and proteins based on inverse temperature transitions. Prog Biophys Mol Biol. 1992;57:23–57. doi:10.1016/0079-6107(92)90003-O. PMID:1549698.
- Urry DW. Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. J Phys Chem B. 1997;101:11007–28. doi:10.1021/jp972167t.
- Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally-responsive polypeptides. Nat Biotechnol. 1999;17:1112–5. doi:10.1038/15100. PMID:10545920.
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15:5383–98. doi:10.1091/mbc.E04-08-0715. PMID:15371533.
- Zhang H, Elbaum-Garfinkle S, Langdon EM, Taylor N, Occhipinti P, Bridges AA, Brangwynne CP, Gladfelter AS. RNA controls PolyQ protein phase transitions. Mol Cell. 2015;60:220–30. doi:10.1016/j.molcel.2015.09.017. PMID:26474065.
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–33. doi:10.1016/j.cell.2015.09.015. PMID:26406374.
- Lin Y, Protter DS, Rosen MK, Parker R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol Cell. 2015;60:208–19. doi:10.1016/j.molcel.2015.08.018. PMID:26412307.
- Mitrea DM, Kriwacki RW. Phase separation in biology; functional organization of a higher order. Cell Commun Signal. 2016;14:1. doi:10.1186/s12964-015-0125-7. PMID:26727894.
- Pak CW, Kosno M, Holehouse AS, Padrick SB, Mittal A, Ali R, Yunus AA, Liu DR, Pappu RV, Rosen MK. Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol Cell. 2016;63:72–85. doi:10.1016/j.molcel.2016.05.042. PMID:27392146.
- Anderson P, Kedersha N. Stress granules: The Tao of RNA triage. Trends Biochem Sci. 2008;33:141–50. doi:10.1016/j.tibs.2007.12.003. PMID:18291657.
- Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006;172:803–8. doi:10.1083/jcb.200512082. PMID:16520386.
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;147:1431–42. doi:10.1083/jcb.147.7.1431. PMID:10613902.
- Fan AC, Leung AK. RNA granules and diseases: A case study of stress granules in ALS and FTLD. Adv Exp Med Biol. 2016;907:263–96. doi:10.1007/978-3-319-29073-7_11. PMID:27256390.
- Kimball SR, Horetsky RL, Ron D, Jefferson LS, Harding HP. Mammalian stress granules represent sites of accumulation of stalled translation initiation complexes. Am J Physiol Cell Physiol. 2003;284:C273–84. doi:10.1152/ajpcell.00314.2002. PMID:12388085.
- Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol. 2005;169:871–84. doi:10.1083/jcb.200502088. PMID:15967811.
- Buchan JR, Parker R. Eukaryotic stress granules: The ins and outs of translation. Mol Cell. 2009;36:932–41. doi:10.1016/j.molcel.2009.11.020. PMID:20064460.
- Moser JJ, Fritzler MJ. Cytoplasmic ribonucleoprotein (RNP) bodies and their relationship to GW/P bodies. Int J Biochem Cell Biol. 2010;42:828–43. doi:10.1016/j.biocel.2009.11.018. PMID:19944184.
- Bentmann E, Haass C, Dormann D. Stress granules in neurodegeneration–lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J. 2013;280:4348–70. doi:10.1111/febs.12287. PMID:23587065.
- Liu-Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, Warren ST, Wolozin B. Local RNA translation at the synapse and in disease. J Neurosci. 2011;31:16086–93. doi:10.1523/JNEUROSCI.4105-11.2011. PMID:22072660.
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151:1257–68. doi:10.1083/jcb.151.6.1257. PMID:11121440.
- Kim WJ, Back SH, Kim V, Ryu I, Jang SK. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol Cell Biol. 2005;25:2450–62. doi:10.1128/MCB.25.6.2450-2462.2005. PMID:15743837.
- Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat Cell Biol. 2008;10:1324–32. doi:10.1038/ncb1791. PMID:18836437.
- Takahara T, Maeda T. Stress granules: The last refuge of TORC1? Cell Cycle. 2012;11:3707–8. doi:10.4161/cc.22044. PMID:22983004.
- Takahara T, Maeda T. Transient sequestration of TORC1 into stress granules during heat stress. Mol Cell. 2012;47:242–52. doi:10.1016/j.molcel.2012.05.019. PMID:22727621.
- Wolozin B. Regulated protein aggregation: Stress granules and neurodegeneration. Mol Neurodegener. 2012;7:56. doi:10.1186/1750-1326-7-56. PMID:23164372.
- Buchan JR. mRNP granules. Assembly, function, and connections with disease. RNA Biol. 2014;11:1019–30. doi:10.4161/15476286.2014.972208. PMID:25531407.
- Wolozin B. Physiological protein aggregation run amuck: Stress granules and the genesis of neurodegenerative disease. Discov Med. 2014;17:47–52. PMID:24411700.
- Ito D, Suzuki N. Conjoint pathologic cascades mediated by ALS/FTLD-U linked RNA-binding proteins TDP-43 and FUS. Neurology. 2011;77:1636–43. doi:10.1212/WNL.0b013e3182343365. PMID:21956718.
- Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G. TDP-43 aggregation in neurodegeneration: Are stress granules the key? Brain Res. 2012;1462:16–25. doi:10.1016/j.brainres.2012.02.032. PMID:22405725.
- Dormann D, Haass C. Fused in sarcoma (FUS): An oncogene goes awry in neurodegeneration. Mol Cell Neurosci. 2013;56:475–86. doi:10.1016/j.mcn.2013.03.006. PMID:23557964.
- Aulas A, Vande Velde C. Alterations in stress granule dynamics driven by TDP-43 and FUS: A link to pathological inclusions in ALS? Front Cell Neurosci. 2015;9:423. doi:10.3389/fncel.2015.00423. PMID:26557057.
- Bowden HA, Dormann D. Altered mRNP granule dynamics in FTLD pathogenesis. J Neurochem. 2016;138(Suppl 1):112–33. doi:10.1111/jnc.13601. PMID:26938019.
- Monahan Z, Shewmaker F, Pandey UB. Stress granules at the intersection of autophagy and ALS. Brain Res. 2016;1649:189–200. doi:10.1016/j.brainres.2016.05.022. PMID:27181519.
- Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201:361–72. doi:10.1083/jcb.201302044. PMID:23629963.
- Lee JA. Autophagy manages disease-associated stress granules. Oncotarget. 2015;6:30421–2. doi:10.18632/oncotarget.5902. PMID:26429862.
- Antar LN, Li C, Zhang H, Carroll RC, Bassell GJ. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol Cell Neurosci. 2006;32:37–48. doi:10.1016/j.mcn.2006.02.001. PMID:16631377.
- Didiot MC, Tian Z, Schaeffer C, Subramanian M, Mandel JL, Moine H. The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. 2008;36:4902–12. doi:10.1093/nar/gkn472. PMID:18653529.
- Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P, Castets M, Pognonec P, Khandjian EW, Moine H, Bardoni B, et al. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 2009;7:e16. doi:10.1371/journal.pbio.1000016. PMID:19166269.
- Ascano M, Jr., Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–6. doi:10.1038/nature11737. PMID:23235829.
- Kenny PJ, Zhou H, Kim M, Skariah G, Khetani RS, Drnevich J, Arcila ML, Kosik KS, Ceman S. MOV10 and FMRP regulate AGO2 association with microRNA recognition elements. Cell Rep. 2014;9:1729–41. doi:10.1016/j.celrep.2014.10.054. PMID:25464849.
- Goggin K, Beaudoin S, Grenier C, Brown AA, Roucou X. Prion protein aggresomes are poly(A)+ ribonucleoprotein complexes that induce a PKR-mediated deficient cell stress response. Biochim Biophys Acta. 2008;1783:479–91. doi:10.1016/j.bbamcr.2007.10.008. PMID:18023289.
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–407. doi:10.1091/mbc.12.5.1393. PMID:11359930.
- Furukawa Y, Kaneko K, Matsumoto G, Kurosawa M, Nukina N. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J Neurosci. 2009;29:5153–62. doi:10.1523/JNEUROSCI.0783-09.2009. PMID:19386911.
- Milani P, Gagliardi S, Cova E, Cereda C. SOD1 transcriptional and posttranscriptional regulation and its potential implications in ALS. Neurol Res Int. 2011;2011:458427. doi:10.1155/2011/458427. PMID:21603028.
- Tainer JA, Getzoff ED, Beem KM, Richardson JS, Richardson DC. Determination and analysis of the 2 A-structure of copper, zinc superoxide dismutase. J Mol Biol. 1982;160:181–217. doi:10.1016/0022-2836(82)90174-7. PMID:7175933.
- Parge HE, Getzoff ED, Scandella CS, Hallewell RA, Tainer JA. Crystallographic characterization of recombinant human CuZn superoxide dismutase. J Biol Chem. 1986;261:16215–8. PMID:3782115.
- Tainer JA, Getzoff ED, Richardson JS, Richardson DC. Structure and mechanism of copper, zinc superoxide dismutase. Nature. 1983;306:284–7. doi:10.1038/306284a0. PMID:6316150.
- Forman HJ, Fridovich I. On the stability of bovine superoxide dismutase. The effects of metals. J Biol Chem. 1973;248:2645–9. PMID:4697386.
- Furukawa Y, O'Halloran TV. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J Biol Chem. 2005;280:17266–74. doi:10.1074/jbc.M500482200. PMID:15691826.
- Lindberg MJ, Normark J, Holmgren A, Oliveberg M. Folding of human superoxide dismutase: Disulfide reduction prevents dimerization and produces marginally stable monomers. Proc Natl Acad Sci U S A. 2004;101:15893–8. doi:10.1073/pnas.0403979101. PMID:15522970.
- Kato S, Takikawa M, Nakashima K, Hirano A, Cleveland DW, Kusaka H, Shibata N, Kato M, Nakano I, Ohama E. New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: Inclusions containing SOD1 in neurons and astrocytes. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:163–84. doi:10.1080/14660820050515160. PMID:11464950.
- Furukawa Y, Kaneko K, Yamanaka K, O'Halloran TV, Nukina N. Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J Biol Chem. 2008;283:24167–76. doi:10.1074/jbc.M802083200. PMID:18552350.
- Furukawa Y, Anzai I, Akiyama S, Imai M, Cruz FJ, Saio T, Nagasawa K, Nomura T, Ishimori K. Conformational disorder of the most immature Cu, Zn-superoxide dismutase leading to amyotrophic lateral sclerosis. J Biol Chem. 2016;291:4144–55. doi:10.1074/jbc.M115.683763. PMID:26694608.
- Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown RH Jr. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2002;277:15923–31. doi:10.1074/jbc.M112087200. PMID:11854284.
- Tiwari A, Hayward LJ. Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J Biol Chem. 2003;278:5984–92. doi:10.1074/jbc.M210419200. PMID:12458194.
- Lelie HL, Liba A, Bourassa MW, Chattopadhyay M, Chan PK, Gralla EB, Miller LM, Borchelt DR, Valentine JS, Whitelegge JP. Copper and zinc metallation status of copper-zinc superoxide dismutase from amyotrophic lateral sclerosis transgenic mice. J Biol Chem. 2011;286:2795–806. doi:10.1074/jbc.M110.186999. PMID:21068388.
- Jonsson PA, Graffmo KS, Andersen PM, Brannstrom T, Lindberg M, Oliveberg M, Marklund SL. Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models. Brain. 2006;129:451–64. doi:10.1093/brain/awh704. PMID:16330499.
- Ding F, Dokholyan NV. Dynamical roles of metal ions and the disulfide bond in Cu, Zn superoxide dismutase folding and aggregation. Proc Natl Acad Sci U S A. 2008;105:19696–701. doi:10.1073/pnas.0803266105. PMID:19052230.
- Chattopadhyay M, Nwadibia E, Strong CD, Gralla EB, Valentine JS, Whitelegge JP. The disulfide bond, but not zinc or dimerization, controls initiation and seeded growth in amyotrophic lateral sclerosis-linked Cu,Zn superoxide dismutase (SOD1) fibrillation. J Biol Chem. 2015;290:30624–36. doi:10.1074/jbc.M115.666503. PMID:26511321.
- Seetharaman SV, Taylor AB, Holloway S, Hart PJ. Structures of mouse SOD1 and human/mouse SOD1 chimeras. Arch Biochem Biophys. 2010;503:183–90. doi:10.1016/j.abb.2010.08.014. PMID:20727846.
- Fridovich I. The biology of oxygen radicals. Science. 1978;201:875–80. doi:10.1126/science.210504. PMID:210504.
- McCord JM, Fridovich I. The biology and pathology of oxygen radicals. Ann Intern Med. 1978;89:122–7. doi:10.7326/0003-4819-89-1-122. PMID:208444.
- Acevedo-Arozena A, Kalmar B, Essa S, Ricketts T, Joyce P, Kent R, Rowe C, Parker A, Gray A, Hafezparast M, et al. A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis. Dis Model Mech. 2011;4:686–700. doi:10.1242/dmm.007237. PMID:21540242.
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat Rev Neurol. 2011;7:603–15. doi:10.1038/nrneurol.2011.150. PMID:21989245.
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat Rev Neurosci. 2006;7:710–23. doi:10.1038/nrn1971. PMID:16924260.
- Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain. 2013;136:2342–58. doi:10.1093/brain/awt097. PMID:23687121.
- Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi:10.1038/nn.3584. PMID:24369373.
- Kaur SJ, McKeown SR, Rashid S. Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene. 2016;577:109–18. doi:10.1016/j.gene.2015.11.049. PMID:26657039.
- Boillee S, Vande Velde C, Cleveland DW. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi:10.1016/j.neuron.2006.09.018. PMID:17015226.
- Hayashi Y, Homma K, Ichijo H. SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv Biol Regul. 2016;60:95–104. doi:10.1016/j.jbior.2015.10.006. PMID:26563614.
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi:10.1038/362059a0. PMID:8446170.
- Furukawa Y, Torres AS, O'Halloran TV. Oxygen-induced maturation of SOD1: A key role for disulfide formation by the copper chaperone CCS. EMBO J. 2004;23:2872–81. doi:10.1038/sj.emboj.7600276. PMID:15215895.
- Rietsch A, Beckwith J. The genetics of disulfide bond metabolism. Annu Rev Genet. 1998;32:163–84. doi:10.1146/annurev.genet.32.1.163. PMID:9928478.
- Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D. Protein disulfide bond formation in the cytoplasm during oxidative stress. J Biol Chem. 2004;279:21749–58. doi:10.1074/jbc.M312267200. PMID:15031298.
- Arnesano F, Banci L, Bertini I, Martinelli M, Furukawa Y, O'Halloran TV. The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem. 2004;279:47998–8003. doi:10.1074/jbc.M406021200. PMID:15326189.
- Ezzi SA, Urushitani M, Julien JP. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem. 2007;102:170–8. doi:10.1111/j.1471-4159.2007.04531.x. PMID:17394546.
- Rotunno MS, Bosco DA. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253. doi:10.3389/fncel.2013.00253. PMID:24379756.
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–403. doi:10.1038/nn.2660. PMID:20953194.
- Guareschi S, Cova E, Cereda C, Ceroni M, Donetti E, Bosco DA, Trotti D, Pasinelli P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc Natl Acad Sci U S A. 2012;109:5074–9. doi:10.1073/pnas.1115402109. PMID:22416121.
- Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999;819:120–31. doi:10.1016/S0006-8993(98)01351-1. PMID:10082867.
- Dupuis L, de Tapia M, Rene F, Lutz-Bucher B, Gordon JW, Mercken L, Pradier L, Loeffler JP. Differential screening of mutated SOD1 transgenic mice reveals early up-regulation of a fast axonal transport component in spinal cord motor neurons. Neurobiol Dis. 2000;7:274–85. doi:10.1006/nbdi.2000.0292. PMID:10964600.
- Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O'Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 2014;111:3620–5. doi:10.1073/pnas.1312245111. PMID:24550511.
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–18. doi:10.1038/nn1603. PMID:16369483.
- Ezzi SA, Lariviere R, Urushitani M, Julien JP. Neuronal over-expression of chromogranin A accelerates disease onset in a mouse model of ALS. J Neurochem. 2010;115:1102–11. doi:10.1111/j.1471-4159.2010.06979.x. PMID:20807312.
- Ge WW, Wen WY, Strong W, Leystra-Lantz C, Strong MJ. Mutant copper-zinc superoxide dismutase binds to and destabilizes human low molecular weight neurofilament mRNA. J Biol Chem. 2005;280:118–24. doi:10.1074/jbc.M405065200. PMID:15507437.
- Strong MJ, Volkening K, Hammond R, Yang WC, Strong W, Leystra-Lantz C, Shoesmith C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35:320–7. doi:10.1016/j.mcn.2007.03.007. PMID:17481916.
- Ge WW, Volkening K, Leystra-Lantz C, Jaffe H, Strong MJ. 14-3-3 protein binds to the low molecular weight neurofilament (NFL) mRNA 3 ′ UTR. Mol Cell Neurosci. 2007;34:80–7. doi:10.1016/j.mcn.2006.10.001. PMID:17098443.
- Volkening K, Leystra-Lantz C, Yang WC, Jaffee H, Strong MJ. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009;1305:168–82. doi:10.1016/j.brainres.2009.09.105. PMID:19815002.
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci. 2012;15:1488–97. doi:10.1038/nn.3230. PMID:23023293.
- Decker CJ, Parker R. P-bodies and stress granules: Possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol. 2012;4:a012286. doi:10.1101/cshperspect.a012286. PMID:22763747.
- Protter DS, Parker R. Principles and properties of stress granules. Trends Cell Biol. 2016;26:668–79. doi:10.1016/j.tcb.2016.05.004. PMID:27289443.
- Adjibade P, Mazroui R. Control of mRNA turnover: Implication of cytoplasmic RNA granules. Semin Cell Dev Biol. 2014;34:15–23. doi:10.1016/j.semcdb.2014.05.013. PMID:24946962.
- Sfakianos AP, Whitmarsh AJ, Ashe MP. Ribonucleoprotein bodies are phased in. Biochem Soc Trans. 2016;44:1411–6. doi:10.1042/BST20160117. PMID:27911723.
- Gal J, Kuang L, Barnett KR, Zhu BZ, Shissler SC, Korotkov KV, Hayward LJ, Kasarskis EJ, Zhu H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016;132:563–76. doi:10.1007/s00401-016-1601-x. PMID:27481264.
- Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi:10.1016/S1474-4422(10)70195-2. PMID:20864052.
- Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem. 1993;62:289–321. doi:10.1146/annurev.bi.62.070193.001445. PMID:8352591.
- Lim L, Wei Y, Lu Y, Song J. ALS-causing mutations significantly perturb the self-assembly and interaction with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol. 2016;14:e1002338. doi:10.1371/journal.pbio.1002338. PMID:26735904.
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi:10.1016/j.brainres.2012.01.016. PMID:22445064.
- Freibaum BD, Chitta RK, High AA, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9:1104–20. doi:10.1021/pr901076y. PMID:20020773.
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–68. doi:10.1038/nn.2779. PMID:21358643.
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T, Nishimura AL, Zupunski V, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–8. doi:10.1038/nn.2778. PMID:21358640.
- Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280:37572–84. doi:10.1074/jbc.M505557200. PMID:16157593.
- Baralle M, Buratti E, Baralle FE. The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochem Soc Trans. 2013;41:1536–40. doi:10.1042/BST20130186. PMID:24256250.
- Aizawa H, Sawada J, Hideyama T, Yamashita T, Katayama T, Hasebe N, Kimura T, Yahara O, Kwak S. TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 2010;120:75–84. doi:10.1007/s00401-010-0678-x. PMID:20372915.
- Yamashita T, Teramoto S, Kwak S. Phosphorylated TDP-43 becomes resistant to cleavage by calpain: A regulatory role for phosphorylation in TDP-43 pathology of ALS/FTLD. Neurosci Res. 2016;107:63–9. doi:10.1016/j.neures.2015.12.006. PMID:26723245.
- Liachko NF, Saxton AD, McMillan PJ, Strovas TJ, Currey HN, Taylor LM, Wheeler JM, Oblak AL, Ghetti B, Montine TJ, et al. The phosphatase calcineurin regulates pathological TDP-43 phosphorylation. Acta Neuropathol. 2016;132:545–61. doi:10.1007/s00401-016-1600-y. PMID:27473149.
- De Marco G, Lomartire A, Mandili G, Lupino E, Buccinna B, Ramondetti C, Moglia C, Novelli F, Piccinini M, Mostert M, et al. Reduced cellular Ca(2+) availability enhances TDP-43 cleavage by apoptotic caspases. Biochim Biophys Acta. 2014;1843:725–34. doi:10.1016/j.bbamcr.2014.01.010. PMID:24440855.
- Wobst HJ, Delsing L, Brandon NJ, Moss SJ. Truncation of the TAR DNA-binding protein 43 is not a prerequisite for cytoplasmic relocalization, and is suppressed by caspase inhibition and by introduction of the A90V sequence variant. PLoS One. 2017;12:e0177181. doi:10.1371/journal.pone.0177181. PMID:28510586.
- Chiang CH, Grauffel C, Wu LS, Kuo PH, Doudeva LG, Lim C, Shen CK, Yuan HS. Structural analysis of disease-related TDP-43 D169G mutation: Linking enhanced stability and caspase cleavage efficiency to protein accumulation. Sci Rep. 2016;6:21581. doi:10.1038/srep21581. PMID:26883171.
- Kitamura A, Nakayama Y, Shibasaki A, Taki A, Yuno S, Takeda K, Yahara M, Tanabe N, Kinjo M. Interaction of RNA with a C-terminal fragment of the amyotrophic lateral sclerosis-associated TDP43 reduces cytotoxicity. Sci Rep. 2016;6:19230. doi:10.1038/srep19230. PMID:26757674.
- Li Q, Yokoshi M, Okada H, Kawahara Y. The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat Commun. 2015;6:6183. doi:10.1038/ncomms7183. PMID:25630387.
- Yang Z, Lin F, Robertson CS, Wang KK. Dual vulnerability of TDP-43 to calpain and caspase-3 proteolysis after neurotoxic conditions and traumatic brain injury. J Cereb Blood Flow Metab. 2014;34:1444–52. doi:10.1038/jcbfm.2014.105. PMID:24917042.
- Moisse K, Volkening K, Leystra-Lantz C, Welch I, Hill T, Strong MJ. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: Implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009;1249:202–11. doi:10.1016/j.brainres.2008.10.021. PMID:19046946.
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V, Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111:1051–61. doi:10.1111/j.1471-4159.2009.06383.x. PMID:19765185.
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderwyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, et al. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5:e13250. doi:10.1371/journal.pone.0013250. PMID:20948999.
- Sun Y, Chakrabartty A. Phase to phase with TDP-43. Biochemistry. 2017;56:809–23. doi:10.1021/acs.biochem.6b01088. PMID:28112502.
- Meyerowitz J, Parker SJ, Vella LJ, Ng DCH, Price KA, Liddell JR, Caragounis A, Li QX, Masters CL, Nonaka T, et al. C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol Neurodegener. 2011;6:57. doi:10.1186/1750-1326-6-57. PMID:21819629.
- Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA, Wallace RH, Crouch PJ, Turner BJ, Horne MK, et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One. 2013;8:e81170. doi:10.1371/journal.pone.0081170. PMID:24312274.
- Kabuta C, Kono K, Wada K, Kabuta T. 4-Hydroxynonenal induces persistent insolubilization of TDP-43 and alters its intracellular localization. Biochem Biophys Res Commun. 2015;463:82–7. doi:10.1016/j.bbrc.2015.05.027. PMID:25998392.
- Parker SJ, Meyerowitz J, James JL, Liddell JR, Nonaka T, Hasegawa M, Kanninen KM, Lim S, Paterson BM, Donnelly PS, et al. Inhibition of TDP-43 accumulation by Bis(thiosemicarbazonato)-copper complexes. PLoS One. 2012;7:e42277. doi:10.1371/journal.pone.0042277. PMID:22879928.
- Moujalled D, James JL, Parker SJ, Lidgerwood GE, Duncan C, Meyerowitz J, Nonaka T, Hasegawa M, Kanninen KM, Grubman A, et al. Kinase inhibitor screening identifies cyclin-dependent kinases and glycogen synthase kinase 3 as potential modulators of TDP-43 cytosolic accumulation during cell stress. PLoS One. 2013;8:e67433. doi:10.1371/journal.pone.0067433. PMID:23840699.
- Moujalled D, James JL, Yang S, Zhang K, Duncan C, Moujalled DM, Parker SJ, Caragounis A, Lidgerwood G, Turner BJ, et al. Phosphorylation of hnRNP K by cyclin-dependent kinase 2 controls cytosolic accumulation of TDP-43. Hum Mol Genet. 2015;24:1655–69. doi:10.1093/hmg/ddu578. PMID:25410660.
- Liu-Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu YF, Kobrin K, Murphy GJ, Petrucelli L, Wolozin B, et al. ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J Neurosci. 2014;34:4167–74. doi:10.1523/JNEUROSCI.2350-13.2014. PMID:24647938.
- Dewey CM, Cenik B, Sephton CF, Dries DR, Mayer P, 3rd, Good SK, Johnson BA, Herz J, Yu G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol Cell Biol. 2011;31:1098–108. doi:10.1128/MCB.01279-10. PMID:21173160.
- Orru S, Coni P, Floris A, Littera R, Carcassi C, Sogos V, Brancia C. Reduced stress granule formation and cell death in fibroblasts with the A382T mutation of TARDBP gene: Evidence for loss of TDP-43 nuclear function. Hum Mol Genet. 2016;25:4473--83. doi:10.1093/hmg/ddw276. PMID:28172957.
- Vanderweyde T, Youmans K, Liu-Yesucevitz L, Wolozin B. Role of stress granules and RNA-binding proteins in neurodegeneration: A mini-review. Gerontology. 2013;59:524–33. doi:10.1159/000354170. PMID:24008580.
- Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. 2016;138(Suppl 1):95–111. doi:10.1111/jnc.13625. PMID:27015757.
- Kwiatkowski TJ, Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi:10.1126/science.1166066. PMID:19251627.
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi:10.1126/science.1165942. PMID:19251628.
- Deng H, Gao K, Jankovic J. The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol. 2014;10:337–48. doi:10.1038/nrneurol.2014.78. PMID:24840975.
- Millecamps S, Boillee S, Le Ber I, Seilhean D, Teyssou E, Giraudeau M, Moigneu C, Vandenberghe N, Danel-Brunaud V, Corcia P, et al. Phenotype difference between ALS patients with expanded repeats in C9orf72 and patients with mutations in other ALS-related genes. J Med Genet. 2012;49:258–63. doi:10.1136/jmedgenet-2011-100699. PMID:22499346.
- Svetoni F, Frisone P, Paronetto MP. Role of FET proteins in neurodegenerative disorders. RNA Biol. 2016;13:1089–1102. doi:10.1080/15476286.2016.1211225. PMID:27415968.
- Furukawa Y, Tokuda E. Aggregation of FET proteins as a pathological change in amyotrophic lateral sclerosis. Adv Exp Med Biol. 2017;925:1–12. doi:10.1007/5584_2016_32. PMID:27311318.
- Nolan M, Talbot K, Ansorge O. Pathogenesis of FUS-associated ALS and FTD: Insights from rodent models. Acta Neuropathol Commun. 2016;4:99. doi:10.1186/s40478-016-0358-8. PMID:27600654.
- Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–57. doi:10.1038/emboj.2010.143. PMID:20606625.
- Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci. 2013;16:1383–91. doi:10.1038/nn.3514. PMID:24036913.
- Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem. 2013;288:24731–41. doi:10.1074/jbc.M113.497974. PMID:23833192.
- Tan AY, Riley TR, Coady T, Bussemaker HJ, Manley JL. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc Natl Acad Sci U S A. 2012;109:6030–5. doi:10.1073/pnas.1203028109. PMID:22460799.
- Coady TH, Manley JL. ALS mutations in TLS/FUS disrupt target gene expression. Genes Dev. 2015;29:1696–706. doi:10.1101/gad.267286.115. PMID:26251528.
- Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest. 2014;124:981–99. doi:10.1172/JCI72723. PMID:24509083.
- Dini Modigliani S, Morlando M, Errichelli L, Sabatelli M, Bozzoni I. An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA-FUS regulatory circuitry. Nat Commun. 2014;5:4335. doi:10.1038/ncomms5335. PMID:25004804.
- Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, Ratti A. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem. 2012;287:15635–47. doi:10.1074/jbc.M111.333450. PMID:22427648.
- Shang Y, Huang EJ. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016;1647:65–78. doi:10.1016/j.brainres.2016.03.036. PMID:27033831.
- Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr., Sapp P, McKenna-Yasek D, Brown RH Jr, Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–75. doi:10.1093/hmg/ddq335. PMID:20699327.
- Daigle JG, Lanson NA, Jr., Smith RB, Casci I, Maltare A, Monaghan J, Nichols CD, Kryndushkin D, Shewmaker F, Pandey UB. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum Mol Genet. 2013;22:1193–205. doi:10.1093/hmg/dds526. PMID:23257289.
- Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D, Haass C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem. 2012;287:23079–94. doi:10.1074/jbc.M111.328757. PMID:22563080.
- Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H, Manser C, Miller CC, Hortobágyi T, et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet. 2013;22:2676–88. doi:10.1093/hmg/ddt117. PMID:23474818.
- Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol. 2011;69:152–62. doi:10.1002/ana.22246. PMID:21280085.
- Kino Y, Washizu C, Aquilanti E, Okuno M, Kurosawa M, Yamada M, Doi H, Nukina N. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2011;39:2781–98. doi:10.1093/nar/gkq1162. PMID:21109527.
- Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, Zhu H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging. 2011;32:2323 e27–40. doi:10.1016/j.neurobiolaging.2010.06.010. PMID:20674093.
- Kent L, Vizard TN, Smith BN, Topp SD, Vance C, Gkazi A, Miller J, Shaw CE, Talbot K. Autosomal dominant inheritance of rapidly progressive amyotrophic lateral sclerosis due to a truncation mutation in the fused in sarcoma (FUS) gene. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:557–62. doi:10.3109/21678421.2014.920033. PMID:24899262.
- Baron DM, Kaushansky LJ, Ward CL, Sama RR, Chian RJ, Boggio KJ, Quaresma AJ, Nickerson JA, Bosco DA. Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol Neurodegener. 2013;8:30. doi:10.1186/1750-1326-8-30. PMID:24090136.
- Takanashi K, Yamaguchi A. Aggregation of ALS-linked FUS mutant sequesters RNA binding proteins and impairs RNA granules formation. Biochem Biophys Res Commun. 2014;452:600–7. doi:10.1016/j.bbrc.2014.08.115. PMID:25173930.
- Yamaguchi A, Kitajo K. The effect of PRMT1-mediated arginine methylation on the subcellular localization, stress granules, and detergent-insoluble aggregates of FUS/TLS. PLoS One. 2012;7:e49267. doi:10.1371/journal.pone.0049267. PMID:23152885.
- Tradewell ML, Yu Z, Tibshirani M, Boulanger MC, Durham HD, Richard S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet. 2012;21:136–49. doi:10.1093/hmg/ddr448. PMID:21965298.
- Svetoni F, Caporossi D, Paronetto MP. Oxidative stress affects FET proteins localization and alternative pre-mRNA processing in cellular models of ALS. Free Radic Biol Med. 2014;75(Suppl 1):S51. doi:10.1016/j.freeradbiomed.2014.10.820. PMID:26461404.
- Sama RR, Ward CL, Kaushansky LJ, Lemay N, Ishigaki S, Urano F, Bosco DA. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J Cell Physiol. 2013;228:2222–31. doi:10.1002/jcp.24395. PMID:23625794.
- Shelkovnikova TA, Robinson HK, Connor-Robson N, Buchman VL. Recruitment into stress granules prevents irreversible aggregation of FUS protein mislocalized to the cytoplasm. Cell Cycle. 2013;12:3194–202. doi:10.4161/cc.26241. PMID:24013423.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9orf72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56. doi:10.1016/j.neuron.2011.09.011. PMID:21944778.
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. A hexanucleotide repeat expansion in C9orf72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68. doi:10.1016/j.neuron.2011.09.010. PMID:21944779.
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012;11:54–65. doi:10.1016/S1474-4422(11)70261-7. PMID:22154785.
- Daoud H, Suhail H, Sabbagh M, Belzil V, Szuto A, Dionne-Laporte A, Khoris J, Camu W, Salachas F, Meininger V, et al. C9orf72 hexanucleotide repeat expansions as the causative mutation for chromosome 9p21-linked amyotrophic lateral sclerosis and frontotemporal dementia. Arch Neurol. 2012;69:1159–63. doi:10.1001/archneurol.2012.377. PMID:22964911.
- Garcia-Redondo A, Dols-Icardo O, Rojas-Garcia R, Esteban-Perez J, Cordero-Vazquez P, Munoz-Blanco JL, Catalina I, González-Muñoz M, Varona L, Sarasola E, et al. Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in Spain and different populations worldwide. Hum Mutat. 2013;34:79–82. doi:10.1002/humu.22211. PMID:22936364.
- Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–9. doi:10.1212/WNL.59.7.1077. PMID:12370467.
- van der Zee J, Van Broeckhoven C. Dementia in 2013: Frontotemporal lobar degeneration-building on breakthroughs. Nat Rev Neurol. 2014;10:70–2. doi:10.1038/nrneurol.2013.270. PMID:24394289.
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012;11:323–30. doi:10.1016/S1474-4422(12)70043-1. PMID:22406228.
- Chio A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, Sendtner M, Brunetti M, Ossola I, Calvo A, et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9orf72. Brain. 2012;135:784–93. doi:10.1093/brain/awr366. PMID:22366794.
- Cooper-Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C, Highley JR, Hautbergue G, Rattray M, Kirby J, et al. C9orf72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PLoS One. 2015;10:e0127376. doi:10.1371/journal.pone.0127376. PMID:26016851.
- Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, et al. RAN proteins and RNA foci from antisense transcripts in C9orf72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A. 2013;110:E4968–77. doi:10.1073/pnas.1315438110. PMID:24248382.
- Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126:845–57. doi:10.1007/s00401-013-1200-z. PMID:24170096.
- Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, et al. Antisense transcripts of the expanded C9orf72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126:829–44. doi:10.1007/s00401-013-1192-8. PMID:24129584.
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R, et al. Unconventional translation of C9orf72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–46. doi:10.1016/j.neuron.2013.02.004. PMID:23415312.
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–8. doi:10.1126/science.1232927. PMID:23393093.
- Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126:881–93. doi:10.1007/s00401-013-1189-3. PMID:24132570.
- Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, Pasinelli P, Trotti D. Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep. 2016;17:645–52. doi:10.1016/j.celrep.2016.09.032. PMID:27732842.
- Sivadasan R, Hornburg D, Drepper C, Frank N, Jablonka S, Hansel A, Lojewski X, Sterneckert J, Hermann A, Shaw PJ, et al. C9orf72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci. 2016;19:1610–8. doi:10.1038/nn.4407. PMID:27723745.
- Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA4, Gleeson PA5, Blair IP2, Soo KY, et al. C9orf72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23:3579–95. doi:10.1093/hmg/ddu068. PMID:24549040.
- Waite AJ, Baumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9orf72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35:1779 e5–e13. doi:10.1016/j.neurobiolaging.2014.01.016. PMID:24559645.
- Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015;78:568–83. doi:10.1002/ana.24469. PMID:26174152.
- van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG2, Jiang J, et al. Novel clinical associations with specific C9orf72 transcripts in patients with repeat expansions in C9orf72. Acta Neuropathol. 2015;130:863–76. doi:10.1007/s00401-015-1480-6. PMID:26437865.
- Todd TW, Petrucelli L. Insights into the pathogenic mechanisms of Chromosome 9 open reading frame 72 (C9orf72) repeat expansions. J Neurochem. 2016;138(Suppl 1):145–62. doi:10.1111/jnc.13623. PMID:27016280.
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, et al. Antisense proline-arginine RAN dipeptides linked to C9orf72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84:1213–25. doi:10.1016/j.neuron.2014.12.010. PMID:25521377.
- Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9orf72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One. 2013;8:e83450. doi:10.1371/journal.pone.0083450. PMID:24349511.
- Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, Hu F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016;4:51. doi:10.1186/s40478-016-0324-5. PMID:27193190.
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi:10.1038/nature14973. PMID:26308891.
- Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–45. doi:10.1126/science.1254917. PMID:25081482.
- Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IOC, Pietrzyk J, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–4. doi:10.1126/science.1256800. PMID:25103406.
- May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grässer FA, Mori K, Kremmer E, Banzhaf-Strathmann J, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128:485–503. doi:10.1007/s00401-014-1329-4. PMID:25120191.
- Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M, et al. C9orf72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016;19:668–77. doi:10.1038/nn.4272. PMID:26998601.
- Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128:505–24. doi:10.1007/s00401-014-1336-5. PMID:25173361.
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobágyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. doi:10.1007/s00401-011-0911-2. PMID:22101323.
- Rossi S, Serrano A, Gerbino V, Giorgi A, Di Francesco L, Nencini M, Bozzo F, Schininà ME, Bagni C, Cestra G, et al. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J Cell Sci. 2015;128:1787–99. doi:10.1242/jcs.165332. PMID:25788698.
- Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell. 2016;167:774–88 e17. doi:10.1016/j.cell.2016.10.002. PMID:27768896.
- Tao Z, Wang H, Xia Q, Li K, Li K, Jiang X, Xu G, Wang G, Ying Z, . Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet. 2015;24:2426–41. doi:10.1093/hmg/ddv005. PMID:25575510.
- Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488:499–503. doi:10.1038/nature11280. PMID:22801503.
- Daoud H, Dobrzeniecka S, Camu W, Meininger V, Dupre N, Dion PA, Rouleau GA. Mutation analysis of PFN1 in familial amyotrophic lateral sclerosis patients. Neurobiol Aging. 2013;34:1311 e1–2. doi:10.1016/j.neurobiolaging.2012.09.001. PMID:23062600.
- Pollard TD, Cooper JA. Quantitative analysis of the effect of Acanthamoeba profilin on actin filament nucleation and elongation. Biochemistry. 1984;23:6631–41. doi:10.1021/bi00321a054. PMID:6543322.
- Carlsson L, Nystrom LE, Sundkvist I, Markey F, Lindberg U. Actin polymerizability is influenced by profilin, a low molecular weight protein in non-muscle cells. J Mol Biol. 1977;115:465–83. doi:10.1016/0022-2836(77)90166-8. PMID:563468.
- Lassing I, Lindberg U. Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature. 1985;314:472–4. doi:10.1038/314472a0. PMID:2984579.
- Metzler WJ, Farmer BT, 2nd, Constantine KL, Friedrichs MS, Lavoie T, Mueller L. Refined solution structure of human profilin I. Protein Sci. 1995;4:450–9. doi:10.1002/pro.5560040312. PMID:7795529.
- Metzler WJ, Bell AJ, Ernst E, Lavoie TB, Mueller L. Identification of the poly-L-proline-binding site on human profilin. J Biol Chem. 1994;269:4620–5. PMID:8308034.
- Kaiser DA, Goldschmidt-Clermont PJ, Levine BA, Pollard TD. Characterization of renatured profilin purified by urea elution from poly-L-proline agarose columns. Cell Motil Cytoskeleton. 1989;14:251–62. doi:10.1002/cm.970140211. PMID:2611892.
- Yang C, Danielson EW, Qiao T, Metterville J, Brown RH, Jr., Landers JE, Xu Z. Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc Natl Acad Sci U S A. 2016;113:E6209–E18. doi:10.1073/pnas.1605964113. PMID:27681617.
- Boopathy S, Silvas TV, Tischbein M, Jansen S, Shandilya SM, Zitzewitz JA, Landers JE, Goode BL, Schiffer CA, Bosco DA. Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc Natl Acad Sci U S A. 2015;112:7984–9. doi:10.1073/pnas.1424108112. PMID:26056300.
- Tanaka Y, Hasegawa M. Profilin 1 mutants form aggregates that induce accumulation of prion-like TDP-43. Prion. 2016;10:283–9. doi:10.1080/19336896.2016.1207033. PMID:27432186.
- Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J Neurosci. 2014;34:8083–97. doi:10.1523/JNEUROSCI.0543-14.2014. PMID:24920614.
- Kendirgi F, Rexer DJ, Alcazar-Roman AR, Onishko HM, Wente SR. Interaction between the shuttling mRNA export factor Gle1 and the nucleoporin hCG1: A conserved mechanism in the export of Hsp70 mRNA. Mol Biol Cell. 2005;16:4304–15. doi:10.1091/mbc.E04-11-0998. PMID:16000379.
- Bolger TA, Wente SR. Gle1 is a multifunctional DEAD-box protein regulator that modulates Ded1 in translation initiation. J Biol Chem. 2011;286:39750–9. doi:10.1074/jbc.M111.299321. PMID:21949122.
- Alcazar-Roman AR, Bolger TA, Wente SR. Control of mRNA export and translation termination by inositol hexakisphosphate requires specific interaction with Gle1. J Biol Chem. 2010;285:16683–92. doi:10.1074/jbc.M109.082370. PMID:20371601.
- Kaneb HM, Folkmann AW, Belzil VV, Jao LE, Leblond CS, Girard SL, Daoud H, Noreau A, Rochefort D, Hince P, et al. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24:1363–73. doi:10.1093/hmg/ddu545. PMID:25343993.
- Aditi Glass L, Dawson TR, Wente SR. An amyotrophic lateral sclerosis-linked mutation in GLE1 alters the cellular pool of human Gle1 functional isoforms. Adv Biol Regul. 2016;62:25–36. doi:10.1016/j.jbior.2015.11.001. PMID:26776475.
- Herva R, Leisti J, Kirkinen P, Seppanen U. A lethal autosomal recessive syndrome of multiple congenital contractures. Am J Med Genet. 1985;20:431–9. doi:10.1002/ajmg.1320200303. PMID:3993672.
- Nousiainen HO, Kestila M, Pakkasjarvi N, Honkala H, Kuure S, Tallila J, Vuopala K, Ignatius J, Herva R, Peltonen L. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40:155–7. doi:10.1038/ng.2007.65. PMID:18204449.
- Vuopala K, Ignatius J, Herva R. Lethal arthrogryposis with anterior horn cell disease. Hum Pathol. 1995;26:12–9. doi:10.1016/0046-8177(95)90109-4. PMID:7821908.
- Aditi, Folkmann AW, Wente SR. Cytoplasmic hGle1A regulates stress granules by modulation of translation. Mol Biol Cell. 2015;26:1476–90. doi:10.1091/mbc.E14-11-1523. PMID:25694449.
- Folkmann AW, Noble KN, Cole CN, Wente SR. Dbp5, Gle1-IP6 and Nup159 A working model for mRNP export. Nucleus. 2011;2:540–8. doi:10.4161/nucl.2.6.17881. PMID:22064466.
- Bergemann AD, Ma ZW, Johnson EM. Sequence of cDNA comprising the human pur gene and sequence-specific single-stranded-DNA-binding properties of the encoded protein. Mol Cell Biol. 1992;12:5673–82. doi:10.1128/MCB.12.12.5673. PMID:1448097.
- Da Silva N, Bharti A, Shelley CS. hnRNP-K and Pur(alpha) act together to repress the transcriptional activity of the CD43 gene promoter. Blood. 2002;100:3536–44. doi:10.1182/blood.V100.10.3536. PMID:12411317.
- Lasham A, Lindridge E, Rudert F, Onrust R, Watson J. Regulation of the human fas promoter by YB-1, Puralpha and AP-1 transcription factors. Gene. 2000;252:1–13. doi:10.1016/S0378-1119(00)00220-1. PMID:10903433.
- Zambrano N, De Renzis S, Minopoli G, Faraonio R, Donini V, Scaloni A, Cimino F, Russo T. DNA-binding protein Pur alpha and transcription factor YY1 function as transcription activators of the neuron-specific FE65 gene promoter. Biochem J. 1997;328(Pt 1):293–300. doi:10.1042/bj3280293. PMID:9359867.
- Tretiakova A, Steplewski A, Johnson EM, Khalili K, Amini S. Regulation of myelin basic protein gene transcription by Sp1 and Puralpha: Evidence for association of Sp1 and Puralpha in brain. J Cell Physiol. 1999;181:160–8. doi:10.1002/(SICI)1097-4652(199910)181:1%3c160::AID-JCP17%3e3.0.CO;2-H. PMID:10457364.
- Du Q, Tomkinson AE, Gardner PD. Transcriptional regulation of neuronal nicotinic acetylcholine receptor genes. A possible role for the DNA-binding protein Puralpha. J Biol Chem. 1997;272:14990–5. doi:10.1074/jbc.272.23.14990. PMID:9169473.
- Melnikova IN, Yang Y, Gardner PD. Interactions between regulatory proteins that bind to the nicotinic receptor beta4 subunit gene promoter. Eur J Pharmacol. 2000;393:75–83. doi:10.1016/S0014-2999(99)00884-5. PMID:10771000.
- Kelm RJ, Jr., Elder PK, Getz MJ. The single-stranded DNA-binding proteins, Puralpha, Purbeta, and MSY1 specifically interact with an exon 3-derived mouse vascular smooth muscle alpha-actin messenger RNA sequence. J Biol Chem. 1999;274:38268–75. doi:10.1074/jbc.274.20.14238 10.1074/jbc.274.53.38268. PMID:10608902.
- White MK, Johnson EM, Khalili K. Multiple roles for Puralpha in cellular and viral regulation. Cell Cycle. 2009;8:1–7. doi:10.4161/cc.8.3.7585. PMID:19182532.
- Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–25. doi:10.1016/j.neuron.2004.07.022. PMID:15312650.
- Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Mol Biol Cell. 2004;15:118a–a.
- Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Cell Struct Funct. 2004;29:108-.
- Darbinian N, Cui JQ, Basile A, Del Valle L, Otte J, Miklossy J, et al. Negative regulation of A beta PP gene expression by Pur-alpha. J Alzheimers Dis. 2008;15:71–82. doi:10.3233/JAD-2008-15106. PMID:18780968.
- Johnson EM, Kinoshita Y, Weinreb DB, Wortman MJ, Simon R, Khalili K, Winckler B, Gordon J. Role of pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res. 2006;83:929–43. doi:10.1002/jnr.20806. PMID:16511857.
- Darbinian N, Gallia GL, Khalili K. Helix-destabilizing properties of the human single-stranded DNA- and RNA-binding protein Puralpha. J Cell Biochem. 2001;80:589–95. doi:10.1002/1097-4644(20010315)80:4%3c589::AID-JCB1013%3e3.0.CO;2-0 10.1002/1097-4644(20010315)80:4%3c589::AID-JCB1013%3e3.3.CO;2-S. PMID:11169743.
- Gallia GL, Johnson EM, Khalili K. Puralpha: A multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res. 2000;28:3197–205. doi:10.1093/nar/28.17.3197. PMID:10954586.
- Johnson EM. The Pur protein family: Clues to function from recent studies on cancer and AIDS. Anticancer Res. 2003;23:2093–100. PMID:12894583.
- Itoh H, Wortman MJ, Kanovsky M, Uson RR, Gordon RE, Alfano N, Johnson EM. Alterations in Pur(alpha) levels and intracellular localization in the CV-1 cell cycle. Cell Growth Differ. 1998;9:651–65. PMID:9716182.
- Liu H, Barr SM, Chu C, Kohtz DS, Kinoshita Y, Johnson EM. Functional interaction of Puralpha with the Cdk2 moiety of cyclin A/Cdk2. Biochem Biophys Res Commun. 2005;328:851–7. doi:10.1016/j.bbrc.2005.01.038. PMID:15707957.
- Darbinian N, Sawaya BE, Khalili K, Jaffe N, Wortman B, Giordano A, Amini S. Functional interaction between cyclin T1/cdk9 and Puralpha determines the level of TNFalpha promoter activation by Tat in glial cells. J Neuroimmunol. 2001;121:3–11. doi:10.1016/S0165-5728(01)00372-1. PMID:11730934.
- Darbinian N, Gallia GL, Kundu M, Shcherbik N, Tretiakova A, Giordano A, Khalili K. Association of Pur alpha and E2F-1 suppresses transcriptional activity of E2F-1. Oncogene. 1999;18:6398–402. doi:10.1038/sj.onc.1203011. PMID:10597240.
- Johnson EM, Chen PL, Krachmarov CP, Barr SM, Kanovsky M, Ma ZW, Lee WH. Association of human Pur alpha with the retinoblastoma protein, Rb, regulates binding to the single-stranded DNA Pur alpha recognition element. J Biol Chem. 1995;270:24352–60. doi:10.1074/jbc.270.41.24352. PMID:7592647.
- Safak M, Gallia GL, Khalili K. Reciprocal interaction between two cellular proteins, Puralpha and YB-1, modulates transcriptional activity of JCVCY in glial cells. Mol Cell Biol. 1999;19:2712–23. doi:10.1128/MCB.19.4.2712. PMID:10082537.
- Di Salvio M, Piccinni V, Gerbino V, Mantoni F, Camerini S, Lenzi J, Rosa A, Chellini L, Loreni F, Carrì MT, et al. Pur-alpha functionally interacts with FUS carrying ALS-associated mutations. Cell Death Dis. 2015;6:e1943. doi:10.1038/cddis.2015.295. PMID:26492376.
- Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. 2013;110:7778–83. doi:10.1073/pnas.1219643110. PMID:23553836.
- Sareen D, O'Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9orf72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi:10.1126/scitranslmed.3007529. PMID:24154603.
- Sareen D, O'Rourke JG, Meera P, Muhammad AKMG, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, et al. Targeting RNA Foci in iPSC-derived motor neurons from ALS patients with a C9orf72 repeat expansion. Sci Transl Med. 2013;5:208ra149. doi:10.1126/scitranslmed.3007529. PMID:24154603.
- Daigle JG, Krishnamurthy K, Ramesh N, Casci I, Monaghan J, McAvoy K, Godfrey EW, Daniel DC, Johnson EM, Monahan Z, et al. Pur-alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathologica. 2016;131:605–20. doi:10.1007/s00401-015-1530-0. PMID:26728149.
- Schmidt LS, Nickerson ML, Warren MB, Glenn GM, Toro JR, Merino MJ, Turner ML, Choyke PL, Sharma N, Peterson J, et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dube syndrome. Am J Hum Genet. 2005;76:1023–33. doi:10.1086/430842. PMID:15852235.
- Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, Duray P, Merino M, Choyke P, Pavlovich CP, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2:157–64. doi:10.1016/S1535-6108(02)00104-6. PMID:12204536.
- Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke C, Turner M, Choyke P, Merino MJ, Pinto PA, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: A new series of 50 families and a review of published reports. J Med Genet. 2008;45:321–31. doi:10.1136/jmg.2007.054304. PMID:18234728.
- Kluger N, Giraud S, Coupier I, Avril MF, Dereure O, Guillot B, Richard S, Bessis D. Birt-Hogg-Dube syndrome: Clinical and genetic studies of 10 French families. Br J Dermatol. 2010;162:527–37. doi:10.1111/j.1365-2133.2009.09517.x. PMID:19785621.
- Painter JN, Tapanainen H, Somer M, Tukiainen P, Aittomaki K. A 4-bp deletion in the Birt-Hogg-Dube gene (FLCN) causes dominantly inherited spontaneous pneumothorax. Am J Hum Genet. 2005;76:522–7. doi:10.1086/428455. PMID:15657874.
- Frohlich BA, Zeitz C, Matyas G, Alkadhi H, Tuor C, Berger W, Russi EW. Novel mutations in the folliculin gene associated with spontaneous pneumothorax. Eur Respir J. 2008;32:1316–20. doi:10.1183/09031936.00132707. PMID:18579543.
- Ren HZ, Zhu CC, Yang C, Chen SL, Xie J, Hou YY, Xu ZF, Wang DJ, Mu DK, Ma DH, et al. Mutation analysis of the FLCN gene in Chinese patients with sporadic and familial isolated primary spontaneous pneumothorax. Clin Genet. 2008;74:178–83. doi:10.1111/j.1399-0004.2008.01030.x. PMID:18505456.
- Woodward ER, Ricketts C, Killick P, Gad S, Morris MR, Kavalier F, Hodgson SV, Giraud S, Bressac-de Paillerets B, Chapman C, et al. Familial non-VHL clear cell (conventional) renal cell carcinoma: Clinical features, segregation analysis, and mutation analysis of FLCN. Clin Cancer Res. 2008;14:5925–30. doi:10.1158/1078-0432.CCR-08-0608. PMID:18794106.
- Nookala RK, Langemeyer L, Pacitto A, Ochoa-Montano B, Donaldson JC, Blaszczyk BK, Chirgadze DY, Barr FA, Bazan JF, Blundell TL. Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. Open Biol. 2012;2:120071. doi:10.1098/rsob.120071. PMID:22977732.
- Baba M, Hong SB, Sharma N, Warren MB, Nickerson ML, Iwamatsu A, Esposito D, Gillette WK, Hopkins RF 3rd, Hartley JL, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. 2006;103:15552–7. doi:10.1073/pnas.0603781103. PMID:17028174.
- Takagi Y, Kobayashi T, Shiono M, Wang L, Piao X, Sun G, Zhang D, Abe M, Hagiwara Y, Takahashi K, et al. Interaction of folliculin (Birt-Hogg-Dube gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene. 2008;27:5339–47. doi:10.1038/onc.2008.261. PMID:18663353.
- Xia Q, Wang G, Wang H, Hu Q, Ying Z. Folliculin, a tumor suppressor associated with Birt-Hogg-Dube (BHD) syndrome, is a novel modifier of TDP-43 cytoplasmic translocation and aggregation. Hum Mol Genet. 2016;25:83–96. doi:10.1093/hmg/ddv450. PMID:26516189.
- Li Y, Collins M, Geiser R, Bakkar N, Riascos D, Bowser R. RBM45 homo-oligomerization mediates association with ALS-linked proteins and stress granules. Sci Rep. 2015;5:14262. doi:10.1038/srep14262. PMID:26391765.
- Tamada H, Sakashita E, Shimazaki K, Ueno E, Hamamoto T, Kagawa Y, Endo H. cDNA cloning and characterization of Drb1, a new member of RRM-type neural RNA-binding protein. Biochem Biophys Res Commun. 2002;297:96–104. doi:10.1016/S0006-291X(02)02132-0. PMID:12220514.
- Mashiko T, Sakashita E, Kasashima K, Tominaga K, Kuroiwa K, Nozaki Y, Matsuura T, Hamamoto T, Endo H. Developmentally regulated RNA-binding protein 1 (Drb1)/RNA-binding motif protein 45 (RBM45), a nuclear-cytoplasmic trafficking protein, forms TAR DNA-binding protein 43 (TDP-43)-mediated cytoplasmic aggregates. J Biol Chem. 2016;291:14996–5007. doi:10.1074/jbc.M115.712232. PMID:27226551.
- Li Y, Collins M, An J, Geiser R, Tegeler T, Tsantilas K, Garcia K, Pirrotte P, Bowser R. Immunoprecipitation and mass spectrometry defines an extensive RBM45 protein-protein interaction network. Brain Res. 2016;1647:79–93. doi:10.1016/j.brainres.2016.02.047. PMID:26979993.
- Collins M, Riascos D, Kovalik T, An J, Krupa K, Krupa K, Hood BL, Conrads TP, Renton AE, Traynor BJ, et al. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 2012;124:717–32. doi:10.1007/s00401-012-1045-x. PMID:22993125.
- Hans F, Fiesel FC, Strong JC, Jackel S, Rasse TM, Geisler S, Springer W, Schulz JB, Voigt A, Kahle PJ. UBE2E ubiquitin-conjugating enzymes and ubiquitin isopeptidase Y regulate TDP-43 protein ubiquitination. J Biol Chem. 2014;289:19164–79. doi:10.1074/jbc.M114.561704. PMID:24825905.
- Bakkar N, Kousari A, Kovalik T, Li Y, Bowser R. RBM45 modulates the antioxidant response in amyotrophic lateral sclerosis through interactions with KEAP1. Mol Cell Biol. 2015;35:2385–99. doi:10.1128/MCB.00087-15. PMID:25939382.
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–64. doi:10.1038/nrn3430. PMID:23463272.
- Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science. 2004;303:197–202. doi:10.1126/science.1089845. PMID:14716005.
- Pradhan A, Liu Y. A multifunctional domain of the calcium-responsive transactivator (CREST) that inhibits dendritic growth in cultured neurons. J Biol Chem. 2005;280:24738–43. doi:10.1074/jbc.M504018200. PMID:15866867.
- Chesi A, Staahl BT, Jovicic A, Couthouis J, Fasolino M, Raphael AR, Yamazaki T, Elias L, Polak M, Kelly C, et al. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci. 2013;16:851–5. doi:10.1038/nn.3412. PMID:23708140.
- Kukharsky MS, Quintiero A, Matsumoto T, Matsukawa K, An H, Hashimoto T, Iwatsubo T, Buchman VL, Shelkovnikova TA. Calcium-responsive transactivator (CREST) protein shares a set of structural and functional traits with other proteins associated with amyotrophic lateral sclerosis. Mol Neurodegener. 2015;10:20. doi:10.1186/s13024-015-0014-y. PMID:25888396.
- Teyssou E, Vandenberghe N, Moigneu C, Boillee S, Couratier P, Meininger V, Pradat PF, Salachas F, Leguern E, Millecamps S. Genetic analysis of SS18L1 in French amyotrophic lateral sclerosis. Neurobiol Aging. 2014;35:1213 e9- e12. doi:10.1016/j.neurobiolaging.2013.11.023. PMID:24360741.
- Walton MR, Dragunow I. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23:48–53. doi:10.1016/S0166-2236(99)01500-3. PMID:10652539.
- Staahl BT, Tang J, Wu W, Sun A, Gitler AD, Yoo AS, Crabtree GR. Kinetic analysis of npBAF to nBAF switching reveals exchange of SS18 with CREST and integration with neural developmental pathways. J Neurosci. 2013;33:10348–61. doi:10.1523/JNEUROSCI.1258-13.2013. PMID:23785148.
- Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, Chan AM, Gusterson BA, Cooper CS. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet. 1994;7:502–8. doi:10.1038/ng0894-502. PMID:7951320.
- Crew AJ, Clark J, Fisher C, Gill S, Grimer R, Chand A, Shipley J, Gusterson BA, Cooper CS. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. 1995;14:2333–40. PMID:7539744.
- de Leeuw B, Balemans M, Olde Weghuis D, Geurts van Kessel A. Identification of two alternative fusion genes, SYT-SSX1 and SYT-SSX2, in t(X;18)(p11.2;q11.2)-positive synovial sarcomas. Hum Mol Genet. 1995;4:1097–9. doi:10.1093/hmg/4.6.1097. PMID:7655467.
- Skytting B, Nilsson G, Brodin B, Xie Y, Lundeberg J, Uhlen M, Larsson O. A novel fusion gene, SYT-SSX4, in synovial sarcoma. J Natl Cancer Inst. 1999;91:974–5. doi:10.1093/jnci/91.11.974. PMID:10359553.
- Storlazzi CT, Mertens F, Mandahl N, Gisselsson D, Isaksson M, Gustafson P, Domanski HA, Panagopoulos I. A novel fusion gene, SS18L1/SSX1, in synovial sarcoma. Genes Chromosomes Cancer. 2003;37:195–200. doi:10.1002/gcc.10210. PMID:12696068.
- Pradhan A, Liu Y. The calcium-responsive transactivator recruits CREB binding protein to nuclear bodies. Neurosci Lett. 2004;370:191–5. doi:10.1016/j.neulet.2004.08.062. PMID:15488321.
- Naganuma T, Nakagawa S, Tanigawa A, Sasaki YF, Goshima N, Hirose T. Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 2012;31:4020–34. doi:10.1038/emboj.2012.251. PMID:22960638.
- Nishimoto Y, Nakagawa S, Hirose T, Okano HJ, Takao M, Shibata S, et al. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol Brain. 2013;6:31. doi:10.1186/1756-6606-6-31. PMID:23835137.
- Shelkovnikova TA, Robinson HK, Troakes C, Ninkina N, Buchman VL. Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum Mol Genet. 2014;23:2298–312. doi:10.1093/hmg/ddt622. PMID:24334610.
- Harrison AF, Shorter J. RNA-binding proteins with prion-like domains in health and disease. Biochem J. 2017;474:1417–38. doi:10.1042/BCJ20160499. PMID:28389532.
- Burd CG, Swanson MS, Gorlach M, Dreyfuss G. Primary structures of the heterogeneous nuclear ribonucleoprotein A2, B1, and C2 proteins: A diversity of RNA binding proteins is generated by small peptide inserts. Proc Natl Acad Sci U S A. 1989;86:9788–92. doi:10.1073/pnas.86.24.9788. PMID:2557628.
- Kamma H, Horiguchi H, Wan L, Matsui M, Fujiwara M, Fujimoto M, Yazawa T, Dreyfuss G. Molecular characterization of the hnRNP A2/B1 proteins: Tissue-specific expression and novel isoforms. Exp Cell Res. 1999;246:399–411. doi:10.1006/excr.1998.4323. PMID:9925756.
- Han SP, Friend LR, Carson JH, Korza G, Barbarese E, Maggipinto M, Hatfield JT, Rothnagel JA, Smith R. Differential subcellular distributions and trafficking functions of hnRNP A2/B1 spliceoforms. Traffic. 2010;11:886–98. doi:10.1111/j.1600-0854.2010.01072.x. PMID:20406423.
- Barraud P, Allain FH. Solution structure of the two RNA recognition motifs of hnRNP A1 using segmental isotope labeling: How the relative orientation between RRMs influences the nucleic acid binding topology. J Biomol NMR. 2013;55:119–38. doi:10.1007/s10858-012-9696-4. PMID:23247503.
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. Mutations in prion-like domains in hnRNPA2B1 and HNRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–73. doi:10.1038/nature11922. PMID:23455423.
- Shorter J, Taylor JP. Disease mutations in the prion-like domains of HNRNPA1 and HNRNPA2B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis. 2013;1:e25200. doi:10.4161/rdis.25200. PMID:25002999.
- Pinol-Roma S, Dreyfuss G. hnRNP proteins: Localization and transport between the nucleus and the cytoplasm. Trends Cell Biol. 1993;3:151–5. doi:10.1016/0962-8924(93)90135-N. PMID:14731609.
- Weighardt F, Biamonti G, Riva S. The roles of heterogeneous nuclear ribonucleoproteins (hnRNP) in RNA metabolism. Bioessays. 1996;18:747–56. doi:10.1002/bies.950180910. PMID:8831291.
- Geuens T, Bouhy D, Timmerman V. The hnRNP family: Insights into their role in health and disease. Hum Genet. 2016;135:851–67. doi:10.1007/s00439-016-1683-5. PMID:27215579.
- Hamilton BJ, Nagy E, Malter JS, Arrick BA, Rigby WF. Association of heterogeneous nuclear ribonucleoprotein A1 and C proteins with reiterated AUUUA sequences. J Biol Chem. 1993;268:8881–7. PMID:8473331.
- Henics T, Sanfridson A, Hamilton BJ, Nagy E, Rigby WF. Enhanced stability of interleukin-2 mRNA in MLA 144 cells. Possible role of cytoplasmic AU-rich sequence-binding proteins. J Biol Chem. 1994;269:5377–83. PMID:8106521.
- Swanson MS, Dreyfuss G. RNA binding specificity of hnRNP proteins: A subset bind to the 3′ end of introns. EMBO J. 1988;7:3519–29. PMID:3208740.
- Mayrand SH, Pederson T. Crosslinking of hnRNP proteins to pre-mRNA requires U1 and U2 snRNPs. Nucleic Acids Res. 1990;18:3307–18. doi:10.1093/nar/18.11.3307. PMID:2141400.
- He Y, Smith R. Nuclear functions of heterogeneous nuclear ribonucleoproteins A/B. Cell Mol Life Sci. 2009;66:1239–56. doi:10.1007/s00018-008-8532-1. PMID:19099192.
- Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc Natl Acad Sci U S A. 2010;107:3487–92. doi:10.1073/pnas.0915166107. PMID:20133726.
- Nonis D, Schmidt MH, van de Loo S, Eich F, Dikic I, Nowock J, Auburger G. Ataxin-2 associates with the endocytosis complex and affects EGF receptor trafficking. Cell Signal. 2008;20:1725–39. doi:10.1016/j.cellsig.2008.05.018. PMID:18602463.
- Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, Weber C, Mandel JL, Cancel G, Abbas N, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. 1996;14:285–91. doi:10.1038/ng1196-285. PMID:8896557.
- Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, Pearlman S, Starkman S, Orozco-Diaz G, Lunkes A, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14:269–76. doi:10.1038/ng1196-269. PMID:8896555.
- Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet. 1996;14:277–84. doi:10.1038/ng1196-277. PMID:8896556.
- Cancel G, Durr A, Didierjean O, Imbert G, Burk K, Lezin A, Belal S, Benomar A, Abada-Bendib M, Vial C, et al. Molecular and clinical correlations in spinocerebellar ataxia 2: A study of 32 families. Hum Mol Genet. 1997;6:709–15. doi:10.1093/hmg/6.5.709. PMID:9158145.
- Geschwind DH, Perlman S, Figueroa CP, Treiman LJ, Pulst SM. The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am J Hum Genet. 1997;60:842–50. PMID:9106530.
- Lorenzetti D, Bohlega S, Zoghbi HY. The expansion of the CAG repeat in ataxin-2 is a frequent cause of autosomal dominant spinocerebellar ataxia. Neurology. 1997;49:1009–13. doi:10.1212/WNL.49.4.1009. PMID:9339681.
- Affaitati A, de Cristofaro T, Feliciello A, Varrone S. Identification of alternative splicing of spinocerebellar ataxia type 2 gene. Gene. 2001;267:89–93. doi:10.1016/S0378-1119(01)00402-4. PMID:11311558.
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–75. doi:10.1038/nature09320. PMID:20740007.
- Lee T, Li YR, Ingre C, Weber M, Grehl T, Gredal O, de Carvalho M, Meyer T, Tysnes OB, Auburger G, et al. Ataxin-2 intermediate-length polyglutamine expansions in European ALS patients. Hum Mol Genet. 2011;20:1697–700. doi:10.1093/hmg/ddr045. PMID:21292779.
- Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V, Dubois B, Matthijs G, van den Berg LH, Robberecht W. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology. 2011;76:2066–72. doi:10.1212/WNL.0b013e31821f445b. PMID:21562247.
- Yu Z, Zhu Y, Chen-Plotkin AS, Clay-Falcone D, McCluskey L, Elman L, Kalb RG, Trojanowski JQ, Lee VM, Van Deerlin VM, et al. PolyQ repeat expansions in ATXN2 associated with ALS are CAA interrupted repeats. PLoS One. 2011;6:e17951. doi:10.1371/journal.pone.0017951. PMID:21479228.
- Lee T, Li YR, Chesi A, Hart MP, Ramos D, Jethava N, Hosangadi D, Epstein J, Hodges B, Bonini NM, et al. Evaluating the prevalence of polyglutamine repeat expansions in amyotrophic lateral sclerosis. Neurology. 2011;76:2062–5. doi:10.1212/WNL.0b013e31821f4447. PMID:21562248.
- Van Langenhove T, van der Zee J, Engelborghs S, Vandenberghe R, Santens P, Van den Broeck M, Mattheijssens M, Peeters K, Nuytten D, Cras P, et al. Ataxin-2 polyQ expansions in FTLD-ALS spectrum disorders in Flanders-Belgian cohorts. Neurobiol Aging. 2012;33:1004 e17–20. doi:10.1016/j.neurobiolaging.2011.09.025. PMID:22035589.
- Hart MP, Gitler AD. ALS-associated ataxin 2 polyQ expansions enhance stress-induced caspase 3 activation and increase TDP-43 pathological modifications. J Neurosci. 2012;32:9133–42. doi:10.1523/JNEUROSCI.0996-12.2012. PMID:22764223.
- Hart MP, Brettschneider J, Lee VM, Trojanowski JQ, Gitler AD. Distinct TDP-43 pathology in ALS patients with ataxin 2 intermediate-length polyQ expansions. Acta Neuropathol. 2012;124:221–30. doi:10.1007/s00401-012-0985-5. PMID:22526021.
- Ito D. [Conjoint pathological cascades mediated by RNA-binding proteins, TDP-43, FUS and ataxin-2]. Rinsho Shinkeigaku. 2012;52:1221–3. doi:10.5692/clinicalneurol.52.1221. PMID:23196570.
- Nonhoff U, Ralser M, Welzel F, Piccini I, Balzereit D, Yaspo ML, Lehrach H, Krobitsch S. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell. 2007;18:1385–96. doi:10.1091/mbc.E06-12-1120. PMID:17392519.
- Farg MA, Soo KY, Warraich ST, Sundaramoorthy V, Blair IP, Atkin JD. Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:717–28. doi:10.1093/hmg/dds479. PMID:23172909.
- Nihei Y, Ito D, Suzuki N. Roles of ataxin-2 in pathological cascades mediated by TAR DNA-binding protein 43 (TDP-43) and Fused in Sarcoma (FUS). J Biol Chem. 2012;287:41310–23. doi:10.1074/jbc.M112.398099. PMID:23048034.
- Toyoshima Y, Takahashi H. TDP-43 pathology in polyglutamine diseases: With reference to amyotrphic lateral sclerosis. Neuropathology. 2014;34:77–82. doi:10.1111/neup.12053. PMID:23889603.
- Forch P, Valcarcel J. Molecular mechanisms of gene expression regulation by the apoptosis-promoting protein TIA-1. Apoptosis. 2001;6:463–8. doi:10.1023/A:1012441824719. PMID:11595836.
- Thomas MG, Loschi M, Desbats MA, Boccaccio GL. RNA granules: The good, the bad and the ugly. Cell Signal. 2011;23:324–34. doi:10.1016/j.cellsig.2010.08.011. PMID:20813183.
- Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, Duff K, Wolozin B.. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. 2012;32:8270–83. doi:10.1523/JNEUROSCI.1592-12.2012. PMID:22699908.
- Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PE, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, et al. Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 2016;15:1455–66. doi:10.1016/j.celrep.2016.04.045. PMID:27160897.
- Wang I, Hennig J, Jagtap PK, Sonntag M, Valcarcel J, Sattler M. Structure, dynamics and RNA binding of the multi-domain splicing factor TIA-1. Nucleic Acids Res. 2014;42:5949–66. doi:10.1093/nar/gku193. PMID:24682828.
- Heck MV, Azizov M, Stehning T, Walter M, Kedersha N, Auburger G. Dysregulated expression of lipid storage and membrane dynamics factors in Tia1 knockout mouse nervous tissue. Neurogenetics. 2014;15:135–44. doi:10.1007/s10048-014-0397-x. PMID:24659297.
- White JP, Lloyd RE. Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation. J Virol. 2011;85:12442–54. doi:10.1128/JVI.05888-11. PMID:21957303.
- Anderson P, Nagler-Anderson C, O'Brien C, Levine H, Watkins S, Slayter HS, Blue ML, Schlossman SF. A monoclonal antibody reactive with a 15-kDa cytoplasmic granule-associated protein defines a subpopulation of CD8+ T lymphocytes. J Immunol. 1990;144:574–82. PMID:2104899.
- Zhang T, Delestienne N, Huez G, Kruys V, Gueydan C. Identification of the sequence determinants mediating the nucleo-cytoplasmic shuttling of TIAR and TIA-1 RNA-binding proteins. J Cell Sci. 2005;118:5453–63. doi:10.1242/jcs.02669. PMID:16278295.
- Ash PE, Vanderweyde TE, Youmans KL, Apicco DJ, Wolozin B. Pathological stress granules in Alzheimer's disease. Brain Res. 2014;1584:52–8. doi:10.1016/j.brainres.2014.05.052. PMID:25108040.
- Bryan JB, Nagle BW, Doenges KH. Inhibition of tubulin assembly by RNA and other polyanions: Evidence for a required protein. Proc Natl Acad Sci U S A. 1975;72:3570–4. doi:10.1073/pnas.72.9.3570. PMID:1059144.
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–62. doi:10.1073/pnas.72.5.1858. PMID:1057175.
- Andreadis A, Broderick JA, Kosik KS. Relative exon affinities and suboptimal splice site signals lead to non-equivalence of two cassette exons. Nucleic Acids Res. 1995;23:3585–93. doi:10.1093/nar/23.17.3585. PMID:7567473.
- Zhou J, Yu Q, Zou T. Alternative splicing of exon 10 in the tau gene as a target for treatment of tauopathies. BMC Neurosci. 2008;9(Suppl 2):S10. doi:10.1186/1471-2202-9-S2-S10. PMID:19090983.
- Liu F, Gong CX. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener. 2008;3:8. doi:10.1186/1750-1326-3-8. PMID:18616804.
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. Embo J. 1989;8:393–9. PMID:2498079.
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–26. doi:10.1016/0896-6273(89)90210-9. PMID:2484340.
- Himmler A, Drechsel D, Kirschner MW, Martin DW, Jr. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–8. doi:10.1128/MCB.9.4.1381. PMID:2498649.
- Himmler A. Structure of the bovine tau gene: Alternatively spliced transcripts generate a protein family. Mol Cell Biol. 1989;9:1389–96. doi:10.1128/MCB.9.4.1389. PMID:2498650.
- Goedert M. Introduction to the tauopathies. In: Dickson DW, editor. Neurodegeneration: The molecular pathology of dementia and movement disorders. Basel: ISN Neuropath Press; 2003. p. 82–5.
- Sergeant N, Delacourte A, Buee L. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta. 2005;1739:179–97. doi:10.1016/j.bbadis.2004.06.020. PMID:15615637.
- Goedert M, Spillantini MG, Crowther RA. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc Natl Acad Sci U S A. 1992;89:1983–7. doi:10.1073/pnas.89.5.1983. PMID:1542696.
- Georgieff IS, Liem RK, Couchie D, Mavilia C, Nunez J, Shelanski ML. Expression of high molecular weight tau in the central and peripheral nervous systems. J Cell Sci. 1993;105(Pt 3):729–37. PMID:8408300.
- Boyne LJ, Tessler A, Murray M, Fischer I. Distribution of Big tau in the central nervous system of the adult and developing rat. J Comp Neurol. 1995;358:279–93. doi:10.1002/cne.903580209. PMID:7560287.
- Kenessey A, Yen SH. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res. 1993;629:40–6. doi:10.1016/0006-8993(93)90478-6. PMID:8287279.
- Watanabe A, Hasegawa M, Suzuki M, Takio K, Morishima-Kawashima M, Titani K, Arai T, Kosik KS, Ihara Y. In vivo phosphorylation sites in fetal and adult rat tau. J Biol Chem. 1993;268:25712–7. PMID:8245007.
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: Effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323(Pt 3):577–91. doi:10.1042/bj3230577. PMID:9169588.
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, Titani K, Ihara Y. Hyperphosphorylation of tau in PHF. Neurobiol Aging. 1995;16:365–71; discussion 71–80. doi:10.1016/0197-4580(95)00027-C. PMID:7566346.
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, Ihara Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–9. doi:10.1074/jbc.270.2.823. PMID:7822317.
- Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116:207–25. doi:10.1016/0022-2836(77)90213-3. PMID:599557.
- Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116:227–47. doi:10.1016/0022-2836(77)90214-5. PMID:146092.
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–54. doi:10.1091/mbc.3.10.1141. PMID:1421571.
- Brandt R, Lee G. The balance between tau protein's microtubule growth and nucleation activities: Implications for the formation of axonal microtubules. J Neurochem. 1993;61:997–1005. doi:10.1111/j.1471-4159.1993.tb03613.x. PMID:8360696.
- Brandt R, Lee G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. J Biol Chem. 1993;268:3414–9. PMID:8429017.
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–8. doi:10.1083/jcb.101.4.1371. PMID:3930508.
- Drubin DG, Feinstein SC, Shooter EM, Kirschner MW. Nerve growth factor-induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J Cell Biol. 1985;101:1799–807. doi:10.1083/jcb.101.5.1799. PMID:2997236.
- Khatoon S, Grundke-Iqbal I, Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: A radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem. 1992;59:750–3. doi:10.1111/j.1471-4159.1992.tb09432.x. PMID:1629745.
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81. doi:10.1126/science.1113694. PMID:16020737.
- Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–7. doi:10.1038/nm0796-783. PMID:8673924.
- Gotzl JK, Lang CM, Haass C, Capell A. Impaired protein degradation in FTLD and related disorders. Ageing Res Rev. 2016;32:122–39. doi:10.1016/j.arr.2016.04.008. PMID:27166223.
- Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull. 2016;126:238–92. doi:10.1016/j.brainresbull.2016.08.018. PMID:27615390.
- van Swieten J, Spillantini MG. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17:63–73. doi:10.1111/j.1750-3639.2007.00052.x. PMID:17493040.
- Vintilescu CR, Afreen S, Rubino AE, Ferreira A. The neurotoxic TAU45-230 fragment accumulates in upper and lower motor neurons in amyotrophic lateral sclerosis subjects. Mol Med. 2016;22:477–86. doi:10.2119/molmed.2016.00095. PMID:27496042.
- Brunello CA, Yan X, Huttunen HJ. Internalized Tau sensitizes cells to stress by promoting formation and stability of stress granules. Sci Rep. 2016;6:30498. doi:10.1038/srep30498. PMID:27460788.
- Clavaguera F, Hench J, Goedert M, Tolnay M. Invited review: Prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol. 2015;41:47–58. doi:10.1111/nan.12197. PMID:25399729.
- Shintani T, Klionsky DJ. Autophagy in health and disease: A double-edged sword. Science. 2004;306:990–5. doi:10.1126/science.1099993. PMID:15528435.
- Klionsky DJ. Autophagy. Curr Biol. 2005;15:R282–3. doi:10.1016/j.cub.2005.04.013. PMID:15854889.
- Yorimitsu T, Klionsky DJ. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–52. doi:10.1038/sj.cdd.4401765. PMID:16247502.
- Jin M, Klionsky DJ. Regulation of autophagy: Modulation of the size and number of autophagosomes. FEBS Lett. 2014;588:2457–63. doi:10.1016/j.febslet.2014.06.015. PMID:24928445.
- Glick D, Barth S, Macleod KF. Autophagy: Cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi:10.1002/path.2697. PMID:20225336.
- Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015;25:354–63. doi:10.1016/j.tcb.2015.02.002. PMID:25759175.
- Ariosa AR, Klionsky DJ. Autophagy core machinery: Overcoming spatial barriers in neurons. J Mol Med (Berl). 2016;94:1217–27. doi:10.1007/s00109-016-1461-9. PMID:27544281.
- Reggiori F, Klionsky DJ. Autophagosomes: Biogenesis from scratch? Curr Opin Cell Biol. 2005;17:415–22. doi:10.1016/j.ceb.2005.06.007. PMID:15978794.
- Chan SN, Tang BL. Location and membrane sources for autophagosome formation - from ER-mitochondria contact sites to Golgi-endosome-derived carriers. Mol Membr Biol. 2013;30:394–402. doi:10.3109/09687688.2013.850178. PMID:24175710.
- Deter RL, Baudhuin P, De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J Cell Biol. 1967;35:C11–6. doi:10.1083/jcb.35.2.C11. PMID:6055998.
- Gomes LC, Scorrano L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta. 2013;1833:205–12. doi:10.1016/j.bbamcr.2012.02.012. PMID:22406072.
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi:10.1038/nature06639. PMID:18305538.
- Meijer WH, van der Klei IJ, Veenhuis M, Kiel JA. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy. 2007;3:106–16. doi:10.4161/auto.3595. PMID:17204848.
- Ganassi M, Mateju D, Bigi I, Mediani L, Poser I, Lee HO, Seguin SJ, Morelli FF, Vinet J, Leo G, et al. A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dynamism. Mol Cell. 2016;63:796–810. doi:10.1016/j.molcel.2016.07.021. PMID:27570075.
- Alberti S, Mateju D, Mediani L, Carra S. Granulostasis: Protein quality control of RNP granules. Front Mol Neurosci. 2017;10:84. doi:10.3389/fnmol.2017.00084. PMID:28396624.
- Morimoto N, Nagai M, Ohta Y, Miyazaki K, Kurata T, Morimoto M, Murakami T, Takehisa Y, Ikeda Y, Kamiya T, et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007;1167:112–7. doi:10.1016/j.brainres.2007.06.045. PMID:17689501.
- Sasaki S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2011;70:349–59. doi:10.1097/NEN.0b013e3182160690. PMID:21487309.
- Mateju D, Franzmann TM, Patel A, Kopach A, Boczek EE, Maharana S, Lee HO, Carra S, Hyman AA, Alberti S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017;36:1669–87. doi:10.15252/embj.201695957. PMID:28377462.
- Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179:485–500. doi:10.1083/jcb.200702115. PMID:17984323.
- Bose JK, Huang CC, Shen CK. Regulation of autophagy by neuropathological protein TDP-43. J Biol Chem. 2011;286:44441–8. doi:10.1074/jbc.M111.237115. PMID:22052911.
- Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Ren H, Chen D, Han J, Ying Z, et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016;35:121–42. doi:10.15252/embj.201591998. PMID:26702100.
- Ying Z, Xia Q, Hao Z, Xu D, Wang M, Wang H, Wang G. TARDBP/TDP-43 regulates autophagy in both MTORC1-dependent and MTORC1-independent manners. Autophagy. 2016;12:707–8. doi:10.1080/15548627.2016.1151596. PMID:27050460.
- Ji Y, Ugolino J, Brady NR, Hamacher-Brady A, Wang J. Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy. 2017;13:1254–55. doi:10.1080/15548627.2017.1299312. PMID:28319438.
- Ryu HH, Jun MH, Min KJ, Jang DJ, Lee YS, Kim HK, Lee JA. Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol Aging. 2014;35:2822–31. doi:10.1016/j.neurobiolaging.2014.07.026. PMID:25216585.
- Matus S, Bosco DA, Hetz C. Autophagy meets fused in sarcoma-positive stress granules. Neurobiol Aging. 2014;35:2832–5. doi:10.1016/j.neurobiolaging.2014.08.019. PMID:25444610.
- Majcher V, Goode A, James V, Layfield R. Autophagy receptor defects and ALS-FTLD. Mol Cell Neurosci. 2015;66:43–52. doi:10.1016/j.mcn.2015.01.002. PMID:25683489.
- Ling SC, Albuquerque CP, Han JS, Lagier-Tourenne C, Tokunaga S, Zhou H, Cleveland DW. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci U S A. 2010;107:13318–23. doi:10.1073/pnas.1008227107. PMID:20624952.
- Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153:1461–74. doi:10.1016/j.cell.2013.05.037. PMID:23791177.
- Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, Kremmer E, Ansorge O, Mackenzie IR, Neumann M, et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012;31:4258–75. doi:10.1038/emboj.2012.261. PMID:22968170.
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–67. doi:10.1016/j.cell.2012.04.017. PMID:22579281.
- Alberti S, Hyman AA. Are aberrant phase transitions a driver of cellular aging? Bioessays. 2016;38:959–68. doi:10.1002/bies.201600042. PMID:27554449.
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, et al. Cell-free formation of RNA granules: Bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768–79. doi:10.1016/j.cell.2012.04.016. PMID:22579282.
- Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155:1049–60. doi:10.1016/j.cell.2013.10.033. PMID:24267890.
- Malinovska L, Kroschwald S, Alberti S. Protein disorder, prion propensities, and self-organizing macromolecular collectives. Biochim Biophys Acta. 2013;1834:918–31. doi:10.1016/j.bbapap.2013.01.003. PMID:23328411.
- Toretsky JA, Wright PE. Assemblages: Functional units formed by cellular phase separation. J Cell Biol. 2014;206:579–88. doi:10.1083/jcb.201404124. PMID:25179628.
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell. 2015;57:936–47. doi:10.1016/j.molcel.2015.01.013. PMID:25747659.
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–77. doi:10.1016/j.cell.2015.07.047. PMID:26317470.
- Quiroz FG, Chilkoti A. Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat Mater. 2015;14:1164–71. doi:10.1038/nmat4418. PMID:26390327.
- Uversky VN. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol. 2016;44:18–30. doi:10.1016/j.sbi.2016.10.015. PMID:27838525.
- Hennig S, Kong G, Mannen T, Sadowska A, Kobelke S, Blythe A, Knott GJ, Iyer KS, Ho D, Newcombe EA, et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J Cell Biol. 2015;210:529–39. doi:10.1083/jcb.201504117. PMID:26283796.
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–43. doi:10.1016/j.neuron.2013.12.018. PMID:24507191.