
ABSTRACT
Post-translational modifications of autophagy-related (ATG) genes are necessary to modulate their functions. However, ATG protein methylation and its physiological role have not yet been elucidated. The methylation of non-histone proteins by SETD7, a SET domain-containing lysine methyltransferase, is a novel regulatory mechanism to control cell protein function in response to various cellular stresses. Here we present evidence that the precise activity of ATG16L1 protein in hypoxia/reoxygenation (H/R)-treated cardiomyocytes is regulated by a balanced methylation and phosphorylation switch. We first show that H/R promotes autophagy and decreases SETD7 expression, whereas autophagy inhibition by 3-MA increases SETD7 level in cardiomyocytes, implying a tight correlation between autophagy and SETD7. Then we demonstrate that SETD7 methylates ATG16L1 at lysine 151 while KDM1A/LSD1 (lysine demethylase 1A) removes this methyl mark. Furthermore, we validate that this methylation at lysine 151 impairs the binding of ATG16L1 to the ATG12–ATG5 conjugate, leading to inhibition of autophagy and increased apoptosis in H/R-treated cardiomyocytes. However, the cardiomyocytes with shRNA-knocked down SETD7 or inhibition of SETD7 activity by a small molecule chemical, display increased autophagy and decreased apoptosis following H/R treatment. Additionally, methylation at lysine 151 inhibits phosphorylation of ATG16L1 at S139 by CSNK2 which was previously shown to be critical for autophagy maintenance, and vice versa. Together, our findings define a novel modification of ATG16L1 and highlight the importance of an ATG16L1 phosphorylation-methylation switch in determining the fate of H/R-treated cardiomyocytes.
KEYWORDS:
Abbreviations
| ACTB | = | actin beta |
| ANXA5 | = | annexin A5 |
| ATG | = | autophagy related |
| BafA | = | bafilomycin A1 |
| BECN1beclin 1 | = | beclin 1autophagy related |
| CSNK2 | = | casein kinase 2 |
| coIP | = | coimmunoprecipitation |
| FL | = | full length |
| GFP | = | green fluorescent protein |
| GST | = | glutathione S-transferase |
| HBSS | = | Hanks balanced salts solution |
| H/R | = | hypoxia/reoxygenation |
| I/R | = | ischemia/reperfusion |
| KDM1A/LSD1 | = | lysine demethylase 1A |
| LAD | = | left anterior descending |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 beta |
| MEFs | = | mouse embryo fibroblasts |
| mRFP | = | monomeric red fluorescent protein |
| NRVCs | = | neonatal rat ventricular cardiomyocytes |
| PAS | = | phagophore assembly site |
| PE | = | phosphatidylethanolamine |
| PI | = | propidium iodide |
| PPP1 | = | protein phosphatase 1 |
| PTMs | = | post-translational modifications |
| PVDF | = | polyvinylidenedifluoride |
| SAM | = | S-adenosylmethionine |
| SQSTM1 | = | sequestosome 1 |
| SETD7 | = | SET domain containing lysine methyltransferase 7 |
| TUBA | = | tublin alpha |
| ULK1 | = | unc-51 like autophagy activating kinase 1. |
Introduction
Autophagy is an evolutionarily conserved degradation pathway and highly regulated membrane trafficking process, which is rapidly triggered by oxidative stress, pathogen infection, and nutrient starvation.Citation1 There are mainly 3 classes of autophagy: chaperone-mediated autophagy, microautophagy and macroautophagy.Citation2 During macroautophagy, from now on referred to as autophagy, a spherical structure called an autophagosome is formed via the expansion of double-membrane phagophores, by which intracellular components such as organelles or macromolecules are delivered to the lysosome/vacuole for degradation.Citation3 It has already been demonstrated that autophagy plays a critical role in diverse physiological processes, and is especially required for cell survival in several major pathologies such as neurodegenerative and cardiovascular syndromes.Citation4
So far, at least 19 core autophagy-related (ATG) proteins are required to govern the formation of autophagosomes.Citation5 These ATG proteins were first identified in yeast and are highly conserved in eukaryotes. They are recruited in a sequential manner and control the different steps of autophagy by comprising a set of functional units: the ULK1 (unc-51 like autophagy activating kinase 1/Atg1 protein kinase complex, the phosphatidylinositol 3-kinase complex, the phosphatidylinositol 3-phosphate–binding proteins, ATG9, and the LC3 and ATG12–ATG5-ATG16L1 ubiquitin-like conjugation systems.Citation6 Before LC3 lipidation, LC3 and ATG12–ATG5-ATG16L1 are the final ATG proteins recruited to the phagophore.Citation7 In the ATG12–ATG5-ATG16L1 complex, ATG16L1 associates with the ATG12–ATG5 conjugate to form a dimeric complex, which functions as an E3-like factor in the LC3 system to recruit and activate the E2-like protein ATG3, thereby promoting conjugation of the LC3 to phosphatidylethanolamine (PE), and subsequently converting LC3B-I to LC3B-II.Citation8 Thus, the site of ATG12–ATG5-ATG16L1 conjugate recruitment plays a pivotal role in autophagosome formation because it determines the location of LC3-II formation. However, the molecular mechanism by which ATG16L1 binds the ATG12–ATG5 complex in mammalian autophagy remains largely unknown.
ATG proteins are mainly regulated via post-translational modifications (PTMs).Citation9 PTMs include ubiquitination, sumoylation, phosphorylation, and acetylation, which are essential for modulating the biological functions of ATG proteins.Citation10 ATG protein phosphorylation is the most well studied PTM and our previous study also demonstrated that the phosphorylation and dephosphorylation of ATG16L1 by CSNK2 and PPP1, respectively, regulates autophagosome formation.Citation11 Recently, lysine methylation of non-histone proteins has been shown to act as a new regulatory mechanism to control protein functions and be involved in stress response.Citation12 However, very little was known about lysine methylation of ATG proteins.
For the first time, here we address the regulation of the core ATG protein ATG16L1 by lysine methylation through identifying ATG16L1-interacting proteins and its effects on autophagy and apoptosis in hypoxia/reoxygenation (H/R)-treated cardiomyocytes. Our comparative analysis reveals that ATG16L1 directly interacts with SETD7 and KDM1A. We furthermore demonstrate that ATG16L1 is a bona fide substrate of SETD7 and methylated at K151, which impairs the formation of the ATG12–ATG5-ATG16L1 complex, thereby resulting in inhibition of autophagy and increase of apoptosis in H/R-treated cardiomyocytes. We also provided evidence for the negative crosstalk between methylation and phosphorylation of ATG16L1 by in vitro and in vivo assays. CSNK2 was less efficient at phosphorylating ATG16L1 that had been pre-methylated at K151 by SETD7. Conversely, pre-phosphorylated ATG16L1 at S139 by CSNK2 was a poor substrate of SETD7, suggesting that agents and drugs modulating KDM1A, SETD7 or CSNK2 activity may have protective effects on apoptosis of the cardiomyocytes, mainly depending on the ATG16L1 status.
Results
H/R induces autophagic activity but decreases ATG16L1 methylation in rat NRVC and H9c2 cell lines
Given that hypoxia regulates lysine methylation of HIF1A/HIF-1α which mediates hypoxic responses and regulates gene expression involved in autophagy,Citation13 we wondered whether hypoxia could change lysine methylation of ATG16L1 in cardiomyocytes and whether lysine methylation of ATG16L1 might be an important regulatory mechanism underlying ATG16L1-mediated autophagy. To test these ideas, we first carried out LC3B conversion and GFP-LC3 puncta formation experiments to investigate the autophagic activity in H9c2 cells and primary neonatal rat ventricular cardiomyocytes (NRVCs) that were subjected to 4 h hypoxia and then followed by reoxygenation (H/R) for 3 h. The conversion of LC3B-I to LC3B-II indicates the formation of autophagosomes, and the ratio of LC3B-II to TUBA or LC3B-I acts as the primary indicator of autophagy induction.Citation14 In agreement with a previous report,Citation11 the H/R treatment indicates more autophagy than the control group as shown in the immunoblotting of LC3B-II (A). To elucidate whether LC3B-II accumulation results from autophagy induction or is due to a block in downstream steps, we examined autophagic flux. Because SQSTM1 (sequestosome 1), a polyubiquitin-binding protein, can be selectively sequestered within autophagosomes through direct binding to LC3B and subsequently degraded in lysosomes during autophagy, the total cellular levels of SQSTM1 can be used as a marker to reflect autophagic flux.Citation15 We observed that the expression of SQSTM1 in NRVCs and H9c2 cells was markedly decreased under H/R (A). However, the late-stage autophagy inhibitor bafilomycin A1 (BafA) significantly blocked H/R-induced SQSTM1 degradation in NRVCs and H9c2 cells, whereas it markedly increased LC3B-II levels under H/R (A).
Figure 1. H/R induces autophagic activity but decreases ATG16L1 methylation in rat NRVC and H9c2 cell lines. (A) H9c2 and NRVC cells were treated with or without 4-h hypoxia (3% O2, 5% CO2, and 92% N2) and then followed by 3-h reoxygenation (5% CO2 and 95% O2) with or without BafA (20 nM). The protein levels of LC3B-I, LC3B-II, SQSTM1, and TUBA were analyzed using western blot. (B) NRVCs expressing GFP-LC3 were exposed to H/R, and then GFP-LC3 localization was examined by fluorescence microscopy. Representative images are shown (left panel). Quantitative analysis of LC3 dots is shown (middle panel). Before establishing stable NRVCs-GFP-LC3 cells, NRVC percentages of transfection using GFP-LC3 plasmid were calculated (right panel). (C) NRVC cells were immunoprecipitated with anti-ATG16L1 and western blots were performed with the indicated antibodies. (D) NRVCs were incubated with Hanks balanced salts solution (HBSS) for 4 h. Then cell extracts were collected and ATG16L1 methylation assay was determined using western blots with the indicated antibodies. (E) Doxorubicin induces autophagic flux in NRVCs.
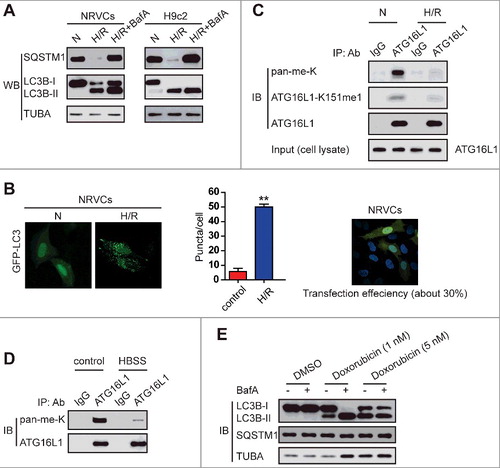
GFP-LC3 puncta appear in the cytoplasm and represent the recruitment of LC3B proteins to phagophores and their presence on autophagosomes.Citation16 Thus, we transfected NRVCs with a GFP-LC3 plasmid using BioT Transfection Reagent and transfection efficiency was approximately 30% (B, right panel). To independently assess autophagosome number, NRVCs were transfected with a GFP-LC3 construct and subsequently exposed to H/R. As shown in B, H/R substantially upregulated the numbers of GFP-LC3 puncta (left panel), suggesting that H/R induced autophagosome accumulation in NRVCs. Thus, both assays indicate that H/R promotes the autophagic activity of cultured cardiomyocytes.
The regulation of ATG16L1 by various PTMs has been uncovered.Citation11 However, lysine methylation of ATG16L1 and its effects on ATG16L1-dependent autophagy have not been examined. Next, we evaluated whether H/R stimulation of NRVCs changes ATG16L1 methylation at a lysine residue(s). Through immunoprecipitation of ATG16L1 in cardiomyocytes and using an anti-methyl lysine (anti-pan-me-K) antibody or site-specific methylation antibody (anti-ATG16L1-K151me1), ATG16L1 in NRVCs was observed to be methylated at a lysine residue, and the levels of ATG16L1 methylation declined during H/R, whereas total levels of ATG16L1 did not change (C). Additionally, nutrient deprivation, a common stimulus of autophagy, also repressed ATG16L1 methylation (D). Given that doxorubicin treatment was reported to induce autophagy in cardiomyocytes (E),Citation17 we examined whether doxorubicin treatment affected the levels of lysine methyl-ATG16L1. We observed that the methyl-ATG16L1 level in NRVC cells decreased upon doxorubicin treatment (data not shown). Thus, alterations in the levels of ATG16L1 lysine methylation due to environmental change suggest that lysine methylation maybe act as a novel regulatory mechanism of ATG16L1 and possibly affect autophagy in H/R-treated cardiomyocytes.
ATG16L1 interacts with SETD7 in vitro and in vivo
To identify the methyltransferase responsible for ATG16L1 methylation, we performed a mass spectrometry analysis of Flag-tagged ATG16L1 from the lysate of HEK293T cells. Intriguingly, SETD7 was identified as an ATG16L1-interacting protein based on LC-MS/MS analysis (Table S1), suggesting the possible involvement of SETD7 in methylation of ATG16L1. To test this hypothesis, we first examined the interaction between ATG16L1 and SETD7. We transfected MYC-tagged SETD7 with HA-tagged ATG16L1 or HA empty vector (Ai), as well as HA-ATG16L1 with MYC-SETD7 or MYC empty vector (Aii), into HEK293T cells. Then co-immunoprecipitation was carried out. It was observed that HA-ATG16L1 or MYC-SETD7 could efficiently pull down MYC-SETD7 (Ai) or HA-ATG16L1 (Aii), respectively, indicating that ATG16L1 interacts with SETD7. To determine whether these proteins interact endogenously, we performed co-immunoprecipitation assays in NRVC cells by using a monoclonal antibody against ATG16L1. The results show that endogenous ATG16L1 interacts with endogenous SETD7 (B).
Figure 2. ATG16L1 interacts with SETD7 in vitro and in vivo. (A) Coimmunoprecipitation of MYC-tagged SETD7 with HA-tagged ATG16L1, and vice versa. HEK293T cells were transfected with a plasmid expressing MYC-tagged SETD7, the HA empty vector, or a plasmid expressing HA-tagged ATG16L1. The anti-HA antibody-conjugated agarose beads were used for immunoprecipitation (A); a plasmid expressing HA-tagged ATG16L1 was transfected with the MYC empty vector, or a plasmid expressing MYC-tagged SETD7 into HEK293T cells and anti-MYC antibody-conjugated agarose beads were used for immunoprecipitation (A). (B) Endogenous interaction between ATG16L1 and SETD7. Western blot analysis of SETD7 after coimmunoprecipitation with endogenous ATG16L1 or mouse IgG control. (C) ATG16L1 directly interacted with SETD7. Bacterially produced GST-ATG16L1 was used to pull down bacterially produced His-SETD7 in vitro. (D) Schematic diagram of SETD7 (FL) and its deletion constructs. The interaction between SETD7 and ATG16L1 domains is indicated by the plus signs (+). The FL or fragments of GST-SETD7 were incubated with His-ATG16L1.The immunoblotting was carried out to detect the interaction with an anti-His antibody. (E) Schematic diagram of ATG16L1 (FL) and its deletion constructs. The interaction between SETD7 and ATG16L1 domains is indicated by the plus signs (+). GST affinity-isolation assays for GST-tagged ATG16L1 domains and His-tagged SETD7. The association of the GST-ATG16L1 domains with His-SETD7 was detected by immunoblotting with an anti-His antibody. (F) Proximity ligation assay (PLA) of the interaction between ATG16L1 and SETD7 in NRVCs with or without H/R treatment. Representative images are shown.
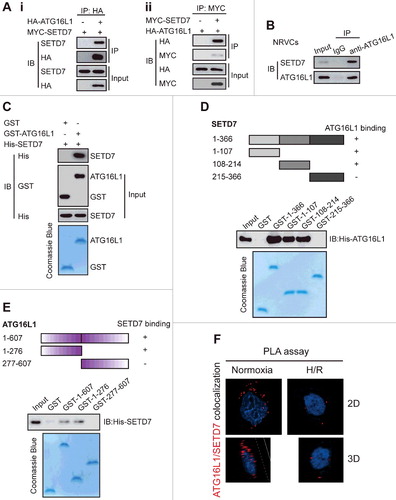
To examine the interaction more directly, we performed a GST affinity isolation experiment. When GST-ATG16L1 and His-SETD7 were mixed together, GST-ATG16L1 especially pulled down His-SETD7 (C), indicating that SETD7 directly binds ATG16L1.
To further map the domains of SETD7 for ATG16L1 binding, we constructed and purified the full-length (FL) and several fragments of GST-SETD7, and then performed a GST affinity-isolation assay. It was observed that both the N-terminal (amino acids 1–107) and middle (amino acids 108–214) fragments of SETD7 bound to ATG16L1. Nevertheless, the association of GST-SETD7 (amino acids 215–366) with GST-ATG16L1 failed to be detected (D). To determine whether SETD7 can directly bind to, and to which domain of, ATG16L1, the regions of ATG16L1 for SETD7 binding were mapped by incubating the FL or fragments of GST-ATG16L1 with His-SETD7. His-SETD7 was found to mainly interact with the FL and the 1–276 aa fragment of GST-ATG16L1, but not with the C-terminal fragment of ATG16L1 and GST alone (E). Proximity ligation assay further confirmed the interaction between SETD7 and ATG16L1 occurred in the cytoplasm, not the nucleus (F). Therefore, we conclude that SETD7 directly interacts with ATG16L1.
SETD7 monomethylates ATG16L1 at lysine 151 in vitro and in vivo
Given that SETD7 has recently been shown to methylate several nonhistone proteins,Citation18 we hypothesized that ATG16L1 was methylated by SETD7. To test this idea, we first examined whether SETD7 interacted with methylated ATG16L1 in setd7−/- and Setd7+/+ mouse embryonic fibroblasts (MEFs). Consistent with findings described above, co-IP of native ATG16L1 from Setd7+/+ MEFs confirmed that SETD7 was in a complex with ATG16L1 (A). When ATG16L1 was immunoprecipitated and probed with 2 different anti-methyl lysine antibodies (ab23366 antibody recognizing both mono- and dimethylated lysine, and ab7315 antibody primarily recognizing dimethylated lysine), we found that ATG16L1 was monomethylated in Setd7+/+ MEFs but not in setd7−/− MEFs (A). These results suggest that SETD7 may directly monomethylate ATG16L1.
Figure 3. SETD7 monomethylates ATG16L1 in vitro and in vivo. (A) Anti-ATG16L1 immunoprecipitates from high-density Setd7+/+ and setd7−/− MEFs were immunoblotted with ATG16L1, SETD7, methyl-lysine (mono-, dimethylated) (ab23366), and dimethyl-lysine (ab7315) antibodies, respectively. (B) ATG16L1 was methylated by SETD7 in vivo. Stably expressed control, SETD7WT, or SETD7H297A HEK293T cells were generated. Then the samples were precipitated using an anti-ATG16L1 antibody and analyzed using an anti-pan-me-K antibody to detect methylation of ATG16L1. (C) Stably expressed shcontrol or shSETD7 HEK293T cells were generated. The cell lysates were then precipitated with an anti-ATG16L1 antibody and probed with an anti-pan-me-K antibody to detect methylation of ATG16L1. (D) Using H3-SAM, recombinant SETD7, and ATG16L1, an in vitro methylation assay was carried out. Coomassie Brilliant Blue R-250 staining and autoradiography were used to show methylation and protein levels, respectively. (E) The indicated proteins were used to perform in vitro methylation analysis. To detect the methylation of ATG16L1, an anti-pan-methyl-lysine antibody (pan-me-K) was used. Coomassie Brilliant Blue staining was used to indicate the protein levels.
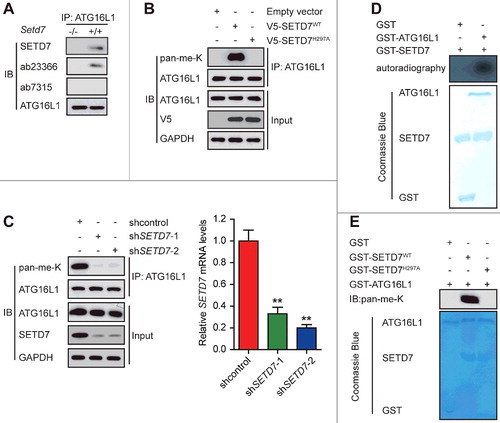
To further investigate whether the methylation of ATG16L1 is mediated by SETD7, HEK293T cells were transfected with pcDNA, wild-type SETD7 (SETD7WT), or methylase-deficient mutant SETD7 (SETD7H297A), and endogenous ATG16L1 was immunoprecipitated and subjected to immunoblotting analysis with an anti-pan-methyl-lysine antibody. The levels of the methylated ATG16L1 were significantly increased in the HEK293T-SETD7WT cell line, but the same phenomenon was not observed in the HEK293T-SETD7H297A cells (B). Conversely, the methylation of ATG16L1 was almost abolished in the HEK293T-shSETD7 cells compared with the HEK293T-shcontrol cells (C). As shown in Fig. S1, SETD7 knockdown and overexpression also modulated ATG16L1 methylation in NRVCs. Together, these experiments demonstrate that SETD7 methylates ATG16L1 in vivo.
To investigate whether ATG16L1 is methylated directly by SETD7, an in vitro methylation assay was performed by incubating SETD7 with 3H-SAM. As shown in D, GST-ATG16L1 was methylated by SETD7, but not by a catalytic mutant, SETD7H297A (E), confirming that SETD7 methylates ATG16L1 in vitro.
Next, we sought to identify the SETD7-methylated residue(s) in ATG16L1. The amino acid sequence of ATG16L1 was compared with the sequences surrounding the SETD7 methylation sites of several known substrates (A). It was found that the conserved lysine residues at position 151 and 440 and their adjacent residues of ATG16L1 strongly conform to the consensus SETD7 recognition sequence designated by a K/R-S/T/A-K motif (in which the methylated lysine site is underlined). When we aligned the full amino acid sequences of ATG16L1 proteins of various species, it was observed that the identified ATG16L1 methylation sequences were evolutionarily conserved (A). Moreover, K151R mutation almost completely abolished ATG16L1 methylation by SETD7 in vitro, whereas K440R mutation had no effect (B). Together, our data suggest that K151 is most likely the major, if not the sole, site within ATG16L1 that is methylated by SETD7.
Figure 4. SETD7 monomethylates ATG16L1 on lysine 151 in vitro and in vivo. (A) Alignment of the consensus amino acid residues adjacent to lysine targeted by SETD7, and identification of a putative SETD7 methylation site in ATG16L1. The consensus SETD7 recognition sequence, the lysines targeted for methylation by SETD7 in known substrates, and the methylated lysine are shown in red in each case. (B) In vitro methylation of various purified ATG16L1 mutants by SETD7. (C) An anti-ATG16L1-K151me1-specific antibody is characterized. ATG16L1 peptide substrates: 137–153, TISDLETECLDLRTKLC; K151me, TISDLETECLDLRTKmeLC; S139P, TIpSDLETECLDLRTKLC, (me, methylation; p, phosphorylation). (D) GST-ATG16L1 (1 μg) was methylated in vitro with or without 0.5 μg recombinant SETD7. The mixture sample was analyzed with the anti-ATG16L1-K151me1 antibody, in the presence of 10 μg/ml competing wild-type or mono-methylated K151 peptide. (E) The effect of ectopically expressed SETD7 and SETD7H297A on K151me1 of endogenous ATG16L1 in HEK293T cells was analyzed. (F) Methylation analysis of Flag-ATG16L1 and mutants by SETD7 was performed in transfected HEK293T cells. (G) In vitro methylation was carried out with GST-ATG16L1, H3-SAM recombinant His-SETD7, and His-SETD7H297A. Coomassie Brilliant Blue staining and immunoblotting were used to display methylation and protein levels, respectively. (H) Western blotting was subsequently performed to analyze the methylation status of ATG16L1 after mouse hearts were subjected to I/R.
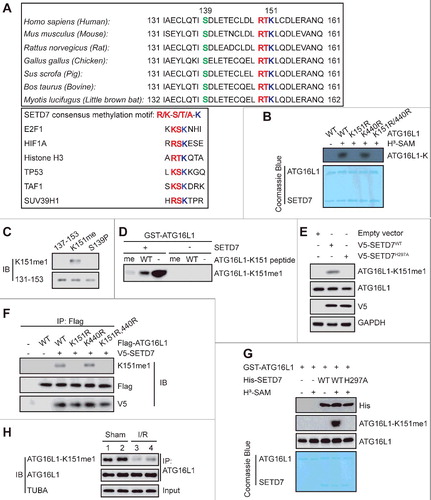
To quickly find whether methylation of ATG16L1 occurred in cells, we prepared an antibody, anti-ATG16L1-K151me1, which specifically recognizes monomethylated ATG16L1-K151 but not the unmethylated peptide (C). This antibody indeed recognized methylated GST-ATG16L1 after treatment with SETD7, with minimal reactivity against untreated GST-ATG16L1 (D). Further, the unmodified ATG16L1 peptide poorly prevented the binding of anti-ATG16L1-K151me1 antibody to methylated GST-ATG16L1, whereas the methylated peptide prevented the binding of the anti-ATG16L1-K151me1 antibody (D).
Using this ATG16L1-K151me1-specific antibody, we observed that overexpression of V5 epitope-tagged SETD7 in the HEK293T cells led to substantially increased endogenous ATG16L1-K151me1, while the enzymatic deficient SETD7H297A mutant reduced the level of ATG16L1-K151me1 (E). Furthermore, V5-SETD7 modified K151me1 in cells expressing Flag-ATG16L1WT and the Flag-ATG16L1K440R mutant, but not with the K151R mutant (F). The specificity of anti-ATG16L1-K151me1 toward ATG16L1 was further confirmed using an in vitro cell-free methylation reaction with purified recombinant GST-ATG16L1 and His-SETD7 in the presence of S-adenosylmethionine (G). To further evaluate whether methylation of ATG16L at K151 occurs in any pathophysiologically relevant settings, we established a mouse model with myocardial ischemia/reperfusion (I/R). As expected, myocardial I/R significantly repressed methylation of ATG16L1 at K151 compared with the sham group (H). Collectively, these results demonstrate that SETD7 methylates ATG16L1 at K151 both in vitro and in vivo.
ATG16L1 is demethylated by KDM1A
Our mass spectrometric analysis of the ATG16L1 immunocomplex from HEK293T cells uncovered that KDM1A, one component of a BRAF-HDAC complex,Citation19 was present in the immunocomplex (Table S1). Given that KDM1A can demethylate several non-histone protein substrates of SETD7, such as TP53 and DNMT1,Citation20 we hypothesized that KDM1A might reverse SETD7-induced ATG16L1 methylation. To confirm this idea, we first used co-immunoprecipitation to investigate whether KDM1A associates with ATG16L1. We found that transfected KDM1A and ATG16L1 interacted with each other in HEK293 cells (A). HDAC2 which is in a stable compound with KDM1A,Citation19 also co-immunoprecipitated with ATG16L1 (A). Additionally, we confirmed the interaction between endogenous KDM1A and ATG16L1 in NRVC cells with or without treatment by H/R (B). We also observed similar results in H9c2 cells (data not shown). Then we examined whether KDM1A interacted directly with ATG16L1. Affinity-isolation assays indicate that GST-ATG16L1, but not GST alone, associated with Flag-KDM1A, suggesting a direct interaction between ATG16L1 and KDM1A (C).
Figure 5. ATG16L1 is demethylated by KDM1A. The interaction between ectopic. (A) and endogenous (B) KDM1A and ATG16L1 in HEK293 (A) and NRVC cells (B) was detected using reciprocal co-immunoprecipitation assays. (C) GST affinity-isolation experiments were performed. (D) Schematic diagram of KDM1A (full length) and its deletion constructs. The interaction between ATG16L1 and KDM1A domains is indicated by plus signs (+). GST affinity-isolation assays for GST-tagged KDM1A domains and His-tagged ATG16L1 was detected by immunoblotting with the indicated antibodies. SWIRM, a small alpha-helical domain of approximately 85 amino acid residues found in eukaryotic chromosomal proteins; AOL, C-terminal amine oxidase-like (AOL) domain. (E) Proximity ligation assay (PLA) of the interaction between ATG16L1 and KDM1A in NRVCs with or without H/R treatment. (F) In vitro demethylation assay of monomethylated ATG16L1 by KDM1A was carried out. (G) ATG16L1 methylation in NRVC cells infected with shcontrol or shKdm1a was performed by immunoprecipitation. (H) Pargyline at the indicated concentrations was used to treat NRVC cells. ATG16L1 and its methylation were performed using immunoprecipitation.
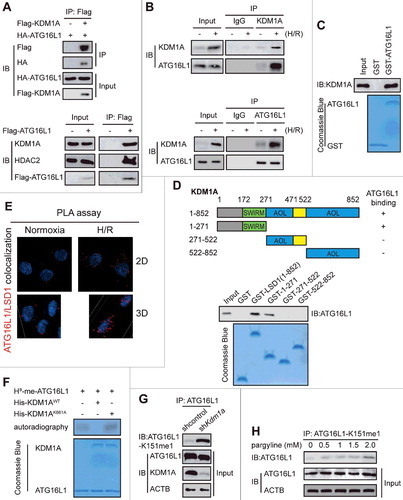
To identify which domain of KDM1A directly binds to ATG16L1, we purified the FL and several fragments of GST-KDM1A and then performed a GST affinity-isolation assay. It was observed that the N-terminal fragment (amino acids 1–271) of KDM1A bound to ATG16L1 (D). Proximity ligation assay further confirmed the interaction between KDM1A and ATG16L1 occurs in the cytoplasm, but not the nucleus (E). All of the above data indicate that KDM1A directly interacts with ATG16L1.
To demonstrate whether KDM1A demethylates ATG16L1-K151 and counteracts the function of SETD7, we used a chemically modified K151me-containing ATG16L1 to carry out an in vitro demethylase assay. It was observed that KDM1A demethylated K151, whereas KDM1AK661A could not (F), which was further confirmed by the increase in K151 methylation in KDM1A-knockdown NRVC cells (G). Moreover, upon treatment with a KDM1A inhibitor, pargyline, ATG16L1 methylation increased in a concentration-dependent manner (H). This demethylase activity against ATG16L1 suggests that KDM1A may regulate ATG16L1 activity by directly modulating lysine methylation of ATG16L1.
Methylation of ATG16L1 on K151 selectively blocked its interaction with the ATG12–ATG5 conjugate
Next, we set out to examine how methylation of ATG16L1 affects autophagy. Given that ATG16L1 is required for LC3–PE (LC3-II) biogenesis in vivo through recruiting the ATG12–ATG5 conjugate to the phagophore assembly site (PAS), we hypothesized that methylation of ATG16L1 might regulate the interaction between ATG16L1 and the ATG12–ATG5 conjugate. It was found that, compared to the control, SETD7WT, but not the methyltransferase activity-dead mutant SETD7H297A, significantly impaired the association of ATG16L1 with the ATG12–ATG5 conjugate (A).
Figure 6. Methylation of ATG16L1 at K151 selectively decreased its interaction with the ATG12–ATG5 conjugate. (A) H9c2 cells were transfected with plasmids containing empty vector, HA-SETD7WT, or its inactive mutant HA-SETD7H297A, and then exposed to H/R. Immunoprecipitation assay indicated that SETD7-dependent methylation of ATG16L1-V5 purified from H9c2 cells inhibited its binding to the ATG12–ATG5 conjugate. (B) In vitro binding assay showed that upon SETD7-mediated methylation, ATG16L1 purified from E. coli presented reduced binding, which was reversed by demethylation following (R)-RFI-2 treatment. Protein input and H3 uptake into ATG16L1 are also shown. (C) Indicated E. coli-generated ATG16L1 and GST-ATG12–ATG5 fusion protein were used to perform the affinity isolation assay. Compared to untreated wild-type ATG16L1, SETD7 methylation or untreated ATG16L1K151Q and K151R mutant showed opposite ATG12–ATG5 conjugate binding. Immunoblotting was carried out using the indicated antibodies. (D) NRVCs were or were not subjected to H/R treatment. Endogenous interaction between ATG16L1 and SETD7 or KDM1A was analyzed using co-immunoprecipitation with anti-ATG16L1. Immunoblotting was carried out using the indicated antibodies.
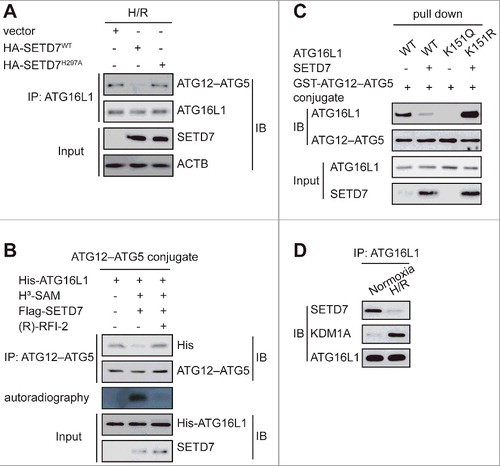
Because the E. coli bacterial system lacks SETD7, we used this system to further validate the effect of SETD7-mediated methylation of ATG16L1 on the formation of the ATG12–ATG5-ATG16L1 complex. Our data confirmed that methylation of ATG16L1 by SETD7 abolished its binding to the ATG12–ATG5 conjugate, which was reversed by demethylation of ATG16L1 by (R)-RFI-2, a potent and specific inhibitor of SETD7Citation21 (B). These data suggest that ATG16L1 demethylation by KDM1A is essential for its binding to the ATG12–ATG5 conjugate.
Then we further confirmed the role of ATG16L1 methylation on K151 in its binding to the ATG12–ATG5 conjugate. To this end, we generated a methylation mimetic mutant of ATG16L1 in which the K151 residue was substituted with glutamate (Q) as a mimic of methyl-lysine (K151Q mutant), and another mutant with arginine (R) as a mimic of the nonmethylated state of the ATG12–ATG5-ATG16L1 complex. The results indicated that only the methylation-deficient mutation, ATG16L1K151R significantly promoted its binding to the ATG12–ATG5 conjugate (C).
We also evaluated the interaction of SETD7 and KDM1A with ATG16L1 in NRVCs at baseline and in response to H/R. As shown in D, the interaction of SETD7 and KDM1A with ATG16L1 was observed at baseline and in response to H/R. However, H/R significantly promoted the interaction between KDM1A and ATG16L1 while decreasing the binding of SETD7 to ATG16L1, further supporting the above findings.
Collectively, these results demonstrate that methylation of ATG16L1 on K151 negatively regulates autophagy through decreasing ATG16L1 binding to the ATG12–ATG5 conjugate. Notably, this decrease is unique to the ATG12–ATG5 conjugate and not RB1CC1 (data not shown).
SETD7 overexpression reduces H/R-induced autophagy and increases apoptosis in cardiomyocytes
Next, we determined the functional importance of methylation of ATG16L1 induced by SETD7 for the inhibition of autophagy. As expected, overexpression of SETD7WT, but not the inactive mutant SETD7H297A, markedly inhibited H/R-induced autophagy (A and B), and enhanced the ratio of dead H9c2 cells (Cii and Ai). However, the K151R mutant, but not WT ATG16L1 or the ATG16L1 methylation mimic K151Q, reversed the enhanced ratio of dead H9c2 cells and reduced autophagy (B, C and A), which further validated that the effect of SETD7 on autophagy was mainly due to the methylation of ATG16L1 at K151. Using SETD7-deficient and -silenced MEF cells and HEK293T cells, we also provided strong evidence that the endogenous SETD7 significantly inhibited autophagy (Fig. S2A and B). We then wanted to clarify whether the methyltransferase activity of SETD7 is essential to affect autophagy. As shown in B, the treatment of (R)-PFI-2 not only greatly enhanced the levels of LC3B-II in H9c2 cells but also blocked SETD7-mediated methylation of ATG16L1 (C), whereas SETD7 knockdown in H9c2 cells ultimately rescued the increased LC3B-II induced by (R)-PFI-2 (data not shown). Additionally, as shown in Fig. S3, ATG16L1 methylation by SETD7 also regulated cardiomyocyte autophagy in response to starvation. These results suggest that methyltransferase activity of SETD7 is required to inhibit the protein function of ATG16L1. Together, we conclude that SETD7 specifically affects autophagy via the methylation of ATG16L1 at K151, which is consistent with our hypothesis that the reversible methylation of ATG16L1 at K151 by SETD7 and KDM1A decides the association of ATG16L1 with the ATG12–ATG5 conjugate.
Figure 7. SETD7 overexpression significantly decreased H/R-induced autophagy in cardiomyocytes. (A) H9c2 cells were transfected with plasmids including empty vector, HA-SETD7WT, or its inactive mutant HA-SETD7H297A, and then exposed to H/R. The lysates were analyzed using LC3B antibody. (B and C) The indicated plasmids were transfected into H9c2 cells with mRFP-GFP-LC3, and then exposed to H/R. Representative images of LC3 puncta are displayed (Bii and Cii), and the number of puncta was quantified (Biii and Ciii). Western blots were performed with the indicated antibodies (Bi and Ci).
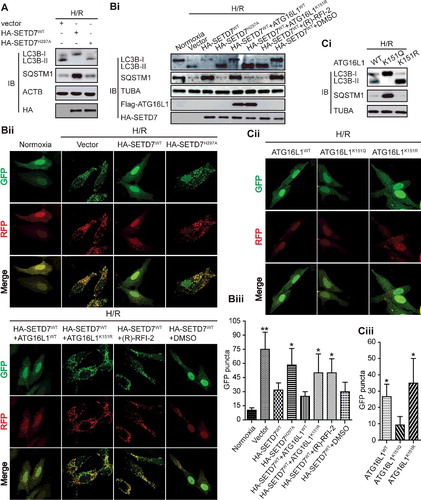
Figure 8. SETD7 overexpression significantly increased H/R-induced apoptosis in cardiomyocytes. (Ai and ii) The indicated plasmids were transfected into H9c2 cells and then exposed to H/R. Apoptosis was measured by ANXA5-PI staining. (Aiii) Apoptotic enzymes CASP3 and CASP7 activity was measured for the indicated H9c2 cells. The data from 3 independent experiments are presented as the mean ± SD. (B and C) Western blotting and immunoprecipitation assays were respectively used to indicate that a potent and selective inhibitor of SETD7, (R)-PFI-2 (1 μM, 18 h), inhibited LC3B-II expression and ATG16L1 methylation at K151.
H/R and autophagy regulate the expression of SETD7 and KDM1A
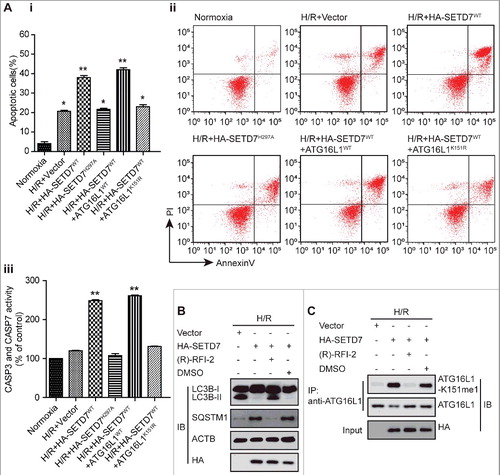
To further elucidate the relationship among SETD7, KDM1A, autophagy, and H/R, we investigated whether H/R and autophagy in turn affected the levels of SETD7 and KDM1A expression. Under H/R, the levels of SETD7 protein and mRNA expression were significantly suppressed in the 2 kinds of examined cardiomyocytes, whereas the expression of KDM1A had the opposite change, as revealed by western blots and semi-quantitative RT-PCR assays (A and B). The similar phenotype was also observed in HEK293T cells (data not shown).
Figure 9. SETD7 expression was considerably suppressed whereas KDM1A expression was increased under H/R conditions. (A and B) As indicated by western blots and semi-quantitative real-time PCR assays, under H/R conditions, SETD7 expression was significantly decreased, and the levels of KDM1A were increased in NRVC and H9c2 cells compared with that under normoxic conditions. (C) The protein levels of SETD7 and KDM1A were examined in NRVC and H9c2 cells with or without 5 mM 3-MA treatment. (D) NRVC and H9c2 cells were incubated with 10 nM BafA for 2 h after being treated with 5 mM 3-MA for 12 h. The cell lysates were analyzed with anti-LC3B antibody. ACTB was used as a loading control. (E) NRVC and H9c2 cells were transiently transfected with scrambled or siRNA of Atg7 for 48 h. Total cell extracts were analyzed by western blotting using the indicated antibodies. (F) H9c2 cells were transfected with a plasmid encoding SETD7 (Fi) or siKdm1a (Fii) and treated them with H/R, or H9c2 cells were without any treatment. The cell lysates were analyzed with the indicated antibodies.
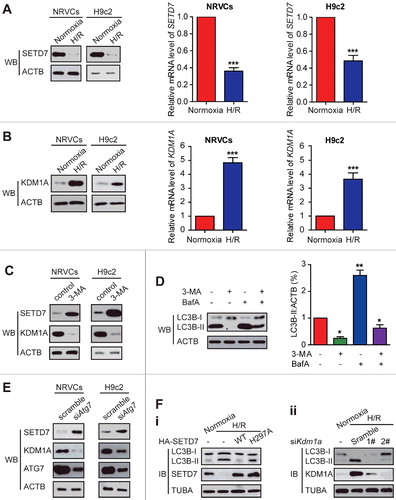
We then examined whether autophagy was involved in SETD7 and KDM1A production. To this end, we first used the autophagy inhibitor 3-MA in NRVC and H9c2 cells. After treatment with 5 mM 3-MA, the levels of SETD7 were enhanced whereas the KDM1A expression was blocked compared to the control (C). As expected, LC3-II levels were decreased by treatment with 3-MA, whereas co-incubation with 3-MA and BafA decreased the accumulation of LC3-II (D), suggesting 3-MA inhibited autophagic flux in NRVC and H9c2 cells. Supporting these results, when NRVC and H9c2 cells were transfected with Atg7 siRNA, the levels of SETD7 were substantially increased, whereas the levels of KDM1A were reduced compared to the controls (E). The above data clearly showed that autophagy regulates the expression of SETD7 and KDM1A. To determine whether these changes contribute to H/R-induced autophagy, we transfected H9c2 cells with a Setd7-encoding plasmid or siKdm1a and treated them with H/R. It was observed that WT SETD7, but not the H297A mutant, overexpression reverted the H/R-induced autophagy, whereas KDM1A knockdown also inhibited the H/R-induced autophagy (F). These findings further strengthen the role of methylation in regulation of autophagy. Collectively, our findings uncover that H/R treatment and autophagosome formation contribute to KDM1A and SETD7 production.
Crosstalk between ATG16L1 methylation and phosphorylation
In response to H/R, ATG16L1 protein is activated through distinct mechanisms, including direct phosphorylation by CSNK2 in particular at serine 139, which is close to the K151 site.Citation11 In agreement with previous reports, we could detect enhanced levels of phosphorylated ATG16L1 in H/R-treated H9c2 cells (A and B). Importantly, ATG16L1 phosphorylation was significantly increased in SETD7-depleted cells, but almost abolished in SETD7 overexpressing cells (Ai, Aii and B). Consistent with these data, KDM1A knockdown significantly decreased ATG16L1 phosphorylation (Aiii). These data suggest that SETD7-mediated methylation at lysine 151 may interfere with H/R-induced activating phosphorylation modification of ATG16L1. Confirming this notion, we found that doxorubicin-mediated phosphorylation of the wild-type ATG16L1 or methylated K151Q mutant, but not the K151R mutant form of ATG16L1, was diminished by SETD7 overexpression, whereas overexpression of methylase-deficient SETD7 had no effect (B and C). In turn, S139 phosphorylation of ATG16L1 inhibited the K151 methylation of ATG16L1 (D). These findings point to a ‘‘two-way’’ crosstalk working model (E), in which methylation and phosphorylation at 2 residues of ATG16L1 influence the generation of each other in a mutually exclusive manner.
Figure 10. Crosstalk between methylation at K151 and phosphorylation at S139 of ATG16L1. (A and B) Immunoprecipitation was performed using anti-ATG16L1 antibody with extracts from untreated or H/R-treated NRVC cells transfected with a plasmid encoding WT SETD7, or shSetd7, or shKdm1a, WT ATG16L1, or ATG16L1K151R expression vectors. Western blot analysis was performed with anti-ATG16L1-K151 or anti-phospho-Ser/Thr antibodies. (C) ATG16L1 protein was mixed with SETD7 with or without 1 μM cold SAM and then the mixture was subjected to in vitro phosphorylation with CSNK2 and γ32P-ATP. (D) ATG16L1 protein was preincubated with CSNK2 with or without 0.5 mM cold ATP and then the mixture was subjected to in vitro methylation reactions with SETD7 and H3-SAM. (E) Working model: (i) H/R decreases SETD7 expression and lysine methylation of ATG16L1, while it increases the KDM1A level and CSNK2 activity in cardiomyocytes; (ii) SETD7 methylates ATG16L1 at K151 in vivo and in vitro, whereas pre-phosphorylated ATG16L1 at S139 by CSNK2 blocks ATG16L1 methylation; and ATG16L1 methyl mark at K151 is removed by KDM1A; (iii) this methylation at K151 impairs the binding of ATG16L1 to the ATG12–ATG5 conjugate, leading to inhibition of autophagy and increased apoptosis in H/R-treated cardiomyocytes.
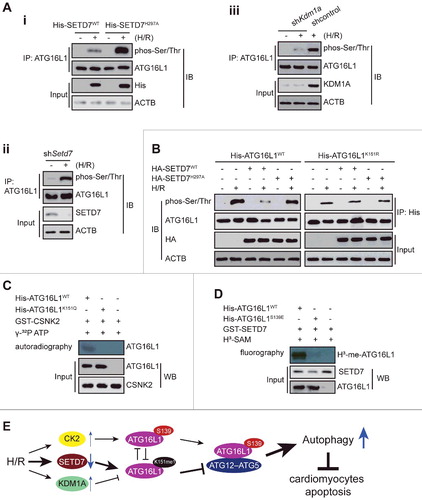
Discussion
In this study, we investigate the involvement of SETD7 and KDM1A in the autophagy process by modifying the ATG16L1 protein. We have demonstrated that SETD7-mediated lysine methylation of ATG16L1 is a determinant for the inhibition of autophagy, which contributes to H/R-associated cardiomyocyte apoptosis. Also, we have shown that a switch of methylation to phosphorylation of ATG16L1 triggers an adaptive pathway to mitigate cardiomyocyte damage. The above conclusions mainly resulted from the following findings: (i) H/R decreases SETD7 expression and lysine methylation of ATG16L1, while increasing the KDM1A level in cardiomyocytes; (ii) SETD7 methylates ATG16L1 at K151 in vivo and in vitro, whereas an enzymatically dead mutant cannot methylate ATG16L1, and a specific inhibitor of SETD7 blocks ATG16L1 methylation; (iii) ATG16L1 methyl mark at K151 is removed by KDM1A; (iv) upon being subjected to H/R, the cardiomyocytes with shRNA-knocked down SETD7 or inhibition of SETD7 by a chemical inhibitor display an increased autophagy and decreased apoptosis; and (v) pre-methylated ATG16L1 at K151 by SETD7 was less efficiently phosphorylated by CSNK2. Conversely, pre-phosphorylated ATG16L1 at S139 by CSNK2 was also a poor substrate of SETD7, suggesting a negative crosstalk between lysine methylation and phosphorylation of ATG16L1. To our knowledge, this is the first report establishing a direct connection between lysine methylation of ATG16L1 involved in autophagy initiation in H/R-treated cardiomyocytes. Overall, these data support the view that the PTMs of ATG16L1 potently influence cardiomyocyte apoptosis during H/R.
One very crucial finding in the present study is that ATG16L1 is a novel substrate of SETD7. In response to stress including hypoxia, as the central regulators of the autophagy signaling pathway, ATG protein can be modified many times by the same PTM at different residues, or by various PTMs on a single protein site in a sequential and separate manner.Citation22,Citation23 Through fine-tuning the activity of ATG proteins, distinct PTMs might display various downstream effects in autophagy. For example, during autophagy, different protein kinases regulate the assembly and activity of the ULK1/Atg1 complex by phosphorylating Atg1/ULK1 at distinct amino acid sites in yeast and mammalian cells.Citation24 During LPS-induced autophagy, E3 ligases such as USP14 or TRAF6 regulates the interaction between BECN1 and other ATG proteins through controlling lysine 63-linked ubiquitination of BECN1;Citation25,Citation26 it is well-known that EP300-dependent acetylation of ATG7 can inhibit autophagy.Citation27 Although these PTMs in the different stage of autophagy play important roles in regulating the functions of ATG proteins, whether ATG proteins at lysine residues can directly be methylated or demethylated and the underlying biological consequences remain poorly described, and involved factors remain unknown.
In the present study, we provided substantial evidence that ATG16L1 is methylated by SETD7, leading to the downregulation of autophagy during H/R. We also demonstrated that H/R and autophagy suppressed the levels of SETD7 expression. Thus, our data uncovered a novel crosstalk. These data also suggest that by regulating ATG16L1 lysine methylation, fine-tuning SETD7 and KDM1A may protect cells in response to hypoxia from exaggerating reactions by affecting cell apoptosis and autophagy, indicating a new role of ATG protein lysine methylation in response to hypoxia stress. Therefore, it is possible that the destruction of this PTM under some pathogenic conditions may result in the loss of control of ATG16L1 activity. For this reason, animal models should be established to further elucidate the role of SETD7-mediated ATG16L1 methylation and the physiological relevance of SETD7 in the autophagy signaling pathway. Supporting our data, one critical autophagy-related transcription factor, FOXO3, has recently been reported as being a substrate of SETD7.Citation28 Methylation of FOXO3 by SETD7 decreases FOXO3 protein stability.Citation28 Additionally, 2 recent reportsCitation29,Citation30 suggest a possible role of KDM1A in regulation of autophagy and clearance of misfolded protein, which is consistent with the working model of the current study. So far, 41 autophagy-related (Atg) genes have been identified.Citation31 However, the roles of SETD7-mediated methylation on ATG proteins and corresponding mechanisms have not been elucidated. Given the ability of SETD7 to positively or negatively regulate various substrates,Citation32 a complete description of the process, mechanism, and function of these PTMs in the regulation of autophagy will be needed, which maybe provide new therapeutic targets for the treatment of autophagy-associated diseases.
Notably, in this study, based on the ‘RSK’ sequence localized in ATG16L1, namely, K/R-S/T-K, on which several putative substrates (E2F1, TAF10 and TP53) of SETD7 have been identified,Citation32 we reasoned that ATG16L1 might be a substrate of SETD7. Although subsequent experiments confirmed this hypothesis, we still cannot rule out the possibility that SETD7 may also methylate other proteins without harboring this consensus motif; for example, SETD7 monomethylated CTNNB1/β-catenin and RB1 at lysine residues 180 and 810, respectively.Citation33,Citation34 Moreover, we also cannot exclude the possibility that in addition to the K151 in ATG16L1, SETD7 can also methylate other sites. In fact, besides the domain of ATG16L1 that harbors K151, SETD7 also binds to amino acids 1–276. Thus, other sites in ATG16L1 also become possible target sites of SETD7. However, because the antibody used in this study is specific to K151 methylation in ATG16L1, we could not detect sites other than K151 in the assays. Nonetheless, it is evident that regardless of the unrecognized other methylated sites, ATG16L1 activity is significantly inhibited by SETD7-mediated methylation, because (R)-PFI-2, a particular SETD7 methyltransferase activity inhibitor, releases the suppressive role of SETD7 on ATG16L1 activity. We need more studies to identify unknown methylated sites by SETD7 to fully elucidate the role of SETD7 on ATG16L1 methylation. Besides, our finding that SETD7 recognizes a methyl-degron in ATG16L1 also raises an intriguing question as to whether SETD7 can methylate other ATG proteins.
It is also noteworthy that our unpublished data also found that SMYD3 co-immunoprecipitated with ATG16L1; SOX2 can be methylated by SETD7 and SMYD3 at the same K119 site.Citation35 Thus, additional methyltransferases may also methylate ATG16L1 at K151. In other words, it is possible that multiple methyltransferases may methylate ATG16L1 and redundantly control its activity.
Another exciting finding in this report is that the crosstalk of phosphorylation and lysine methylation dictates the action of ATG16L1. Our results clearly indicate that the delicate activity of ATG16L1 essential for maintenance of autophagy is controlled not only at the level of transcription but also at the post-translational level by the balanced activities of CSNK2 and SETD7 or KDM1A, pointing to a dynamic interplay between methylation and other posttranslational modifications during H/R. ATG16L1 was shown previously to be phosphorylated at S139 by CSNK2, and this change is essential for H/R-induced autophagy and inactivation of the proapoptotic pathway.Citation11 The current study extends these findings and provides compelling evidence that phosphorylation of ATG16L1 at S139 is mutually exclusive with methylation at K151; vice vera, methylation also precludes phosphorylation of ATG16L1 at a nearby amino acid. The above data suggest the following working model: SETD7 and KDM1A are important to maintain a substantial pool of methylated ATG16L1 in the cells, which, upon H/R, can be subjected to CSNK2-mediated phosphorylation. Phosphorylated ATG16L1 by CSNK2 cannot be methylated by SETD7, which promotes autophagy and inhibits apoptosis in the cardiomyocytes. The above data also suggest the complexity of crosstalk and cellular signaling pathways. Additionally, like most proteins, ATG16L1 contains many modifiable lysines, cysteines, serines, and threonines, which favor modifications on these residues in response to stress response. Thus, one key remaining question is how the proteome of different post-translational modifications on ATG16L1 affects general and selective autophagic flux. For example, it is still unknown whether ATG16L1 can be modified by acetylation, and whether acetylation can affect its methylation, phosphorylation, and its function. Therefore, more work is needed for a complete description of the PTMs of ATG16L1 in the regulation of autophagy, which could provide key therapeutic targets in mitigating serious pathologies associated with diseases.
Taken together, we propose a novel working model elucidating how a methyl-phospho switch controls ATG16L1 protein activity and function in H/R-treated cardiomyocytes. Increased KDM1A expression in H/R-treated cardiomyocytes permits appropriate autophagy and thus correlates with apoptosis downregulation. Thus, we provide a novel role for the SETD7-KDM1A-CSNK2 axis among their many diverse functions. This discovery opens a potential new paradigm of reversible ATG16L1 methylation in affecting autophagy.
Materials and Methods
Cell line and culture conditions
H9c2 cardiomyocytes derived from rat myocardium and HEK293T were obtained (ATCC, CRL-1446, CRL-3216) and cultured in DMEM with 10% fetal bovine serum without penicillin/streptomycin. Setd7-null and Setd7 wild-type mouse embryonic fibroblasts (MEFs) were incubated in DMEM supplemented with 1 × nonessential amino acids (Sigma-Aldrich, D6046), sodium pyruvate (110 mg/l), 10% fetal bovine serum, and 1% penicillin–streptomycin. All cells were cultured in a humidified atmosphere of 5% CO2 at 37°C, and the medium was changed every 2 d. H9c2 cells were treated with 4 h of hypoxia and then followed by 3 h of reoxygenation. An incubator with O2 control, 5% CO2, and balanced with N2 (NBS Galaxy 48R) was used to culture the cells under hypoxic condition (1% O2).
Primary neonatal rat ventricular cardiomyocytes (NRVCs) were isolated and cultured as described previously.Citation36 Briefly, whole hearts from 2- to 5-d-old rats were isolated and rinsed in a balanced salt solution (137 mM NaCl, 0.338 mM NaH2PO4, 1.26 mM CaCl2, 0.6 U/mL of pancreatin [Sigma-Aldrich, P7545], 5.5 mM MgSO4, 5.3 mM KCl, 4.17 mM NaHCO3, 0.8 mM MgSO4, 0.44 mM KH2PO4, pH 7.3-7.4), with collagenase (95 U/mL; Sigma-Aldrich, C0130) 5 times. Then the pellet was resuspended in buffer A (horse serum [Sigma-Aldrich, H1270]:M199 [Sigma-Aldrich, M-4530] mixed 4:1, streptomycin [100 µg/mL], fetal calf serum [5%; Sigma-Aldrich, F7524], and penicillin [100 U/mL]). After allowing nonmyocyte cells to be removed on noncoated culture dishes for 1.5 h, neonatal cardiomyocytes contained in the remaining cell suspension was collected and plated in culture dishes coated with collagen I (Sigma-Aldrich, S3315) in buffer A. The cells were cultured in 5% CO2 at 37°C. After 72 h, the culture medium was replaced, and cardiomyocytes were allowed to reach confluence before use. Additionally, NRVCs were transfected with BioT Transfection Reagent (Morganville Scientific, SKU: BT0100) according to the manufacturer's instruction. HBSS (Gibco, 14025)
Recombinant protein purification and GST affinity-isolation assays
The ATG12–ATG5 conjugate and GST-ATG16L1 were produced in E. coli Rosetta pLysS (Novagen, 70956-3) by co-expression of ATG12, and hexahistidine-tagged ATG5.Citation11,Citation37 Briefly, E. coli Rosetta pLySS was grown and induced with 1 mM IPTG (Sigma-Aldrich, I6758) to an OD600 = 0.8 for 4 h at 37°C. Then bacteria were resuspended in resuspension buffer (10 mM imidazole [Sigma-Aldrich, CI2399], 2.5 mM Pefablock [Roth, 11429868001], 300 mM NaCl, 50 mM HEPES [Sigma-Aldrich, H3375], pH 7.5, 1 mM MgCl2, 2 mM 2-mercaptoethanol, 5 µg/ml DNase [Sigma-Aldrich, D5025]), and disrupted through sonication. Then a HisTrap column (GE Healthcare, 17-5247-01) and a step-wise (0-250 mM) imidazole gradient were used to elute proteins. Finally, Amicon Ultra centrifugal filter (MW cut-off 30 kDa; Sigma-Aldrich, Z717185) and a 16/60 S200 size exclusion column (GE Healthcare, 17-1069-01) was used to concentrate and purify the proteins. GST-tagged ATG16L1 was expressed in E. coli Rosetta pLysS from pOPTG-ATG16L1 (provided by MH Lin from Zhejiang University) and grown at 37°C to an OD600 = 0.8 using 1 mM IPTG for 4 h. TEV protease (Sigma-Aldrich, T4455) was used to cleave off protein from the beads and the protein was eluted using a buffer containing 1 mM DTT, 150 mM NaCl, 50 mM HEPES, pH 7.5.
Using glutathione-sepharose beads (GenScript, L00206) based on a standard procedure, GST-ATG12–ATG5 complex, and GST-ATG16L1 proteins were purified from E. coli BL21. Briefly, the beads binding purified GST-ATG16L1 or GST control were present in HSE buffer (150 mM NaCl, 20 mM HEPES, pH 7.4, 1% Triton X-100 [Fisher, BP151-100], 5 mM EDTA). Then these beads were mixed with purified SETD7, KDM1A, ATG12–ATG5, or cell lysates for 1 h at 4°C, and washed with 0.1% Triton X-100 in phosphate-buffered Ca2+- and Mg2+-free saline (Sigma-Aldrich, D5773), followed by SDS-PAGE and immunoblotting. HEK293T cells were lysed in buffer (1% Triton X-100, 50 mM NaF, 1 × Complete protease inhibitor [Roche, 11836153001]) on ice. Then the supernatant fraction was added to glutathione beads pre-coated with wild-type and mutant GST-ATG16L1 after centrifugation (16,000 × g) for 20 min.
Plasmid construction and siRNA
The human wild-type SETD7 gene, the enzymatically dead H297A mutant, and the corresponding fragments were PCR amplified and subcloned into the pCMV-MYC plasmid (provided by XW Wang from Rutgers University) and the lentivirus (LV) vector pHAGE-CMV-MCS-ZsGreen (provided by XW Wang from Rutgers University). Human enzymatically inactive KDM1AK661A and WT KDM1A were amplified by PCR and inserted into pSUPERpLV-TH (provided by Dr. BB Wang from Rutgers University) or the vector pTP6 (provided by H Zhang from China Three Gorges University). Tandem-tagged LC3 (mRFP-GFP-LC3) and GFP-LC3 expression vector were kindly provided by Dr. BB Wang (Rutgers University). Other plasmids used were as follows: ATG5 (Addgene, 22948; deposited by Noboru Mizushima), FLAG-ATG16L1 constructs (Addgene, 24302; deposited by Noboru Mizushima), and ATG12 (Addgene, 27049; deposited by Jayanta Debnath). A kit (Stratagene, 200523) for site-directed PCR mutagenesis was used to generate all point mutations and verified by sequencing. The siRNA sequences against different DNA targets are: KDM1A/LSD1-1 (Santa Cruz Biotechnology, sc-60970): 5’-CGGACAAGCTGTTCCTAAA-3’; KDM1A/LSD1-2 5’-GAACTCCATCAGCAATACA-3’; SETD7 (Santa Cruz Biotechnology, sc-45883-SH): Scramble, 5′-GTTGGCACCAGCAGCGCAC-3′, SET7/9-1, 5′-GGGAGTTTACACTTACGAA-3′, SET7/9-2, 5′-GCCTTGTAGGAGAAGTAAA-3′.
Chemical reagents and antibodies
Cell culture reagents were purchased from Invitrogen. Reagents used were as follows: (R)-PFI-2 (Tocris, CAS 1627607-87-7), Pargyline Hydrochloride (Sigma-Aldrich, P8013), doxorubicin (Sigma-Aldrich, D1515) and bafilomycin A1 (Sigma-Aldrich, SML1661). Transfection was done with Oligofectamine (Invitrogen Life Technologies, 12252-011) as recommended by the manufacturer. The following antibodies were used: anti-KDM1A/LSD1 (Diagenode, pAb-067-050), anti-KDM1A/LSD1 (Abcam, Ab17721), anti-MYC (Santa Cruz Biotechnology, SC-90), anti-ACTB (Sigma-Aldrich, A1978), anti-GAPDH (Bosterbio, 0411), anti-V5 antibody (Invitrogen, R960-25), anti-SETD7 (Abcam, ab124708), anti-FLAG (Sigma-Aldrich, F3165), anti-HA (Sigma-Aldrich, 9658-100), anti-pan-me-K antibody (Abcam, ab7315), anti-SQSTM1 (Sigma-Aldrich, P0068), rabbit polyclonal anti-LC3B antibody (Sigma-Aldrich, L7543), anti-SETD7 antibody (Merck Millipore, 04–0805), glutathione S-transferase (GST) antibodies (Sigma-Aldrich, A7340-5 mL), anti-His (Bethyl Laboratories, A190-112A), anti-ATG16L1 (Abcam, ab47946), rabbit anti-ATG12–ATG5 conjugate antibody (Biocompare, CPBT-66438RA), rabbit polyclonal antibodies to phosphoserine (Abcam, ab9332), phosphothreonine (Abcam, ab9337), phosphoserine/threonine (Abcam, ab17464), anti-TUBA (Abcam, ab6046). ATG16L1-K151me1, p-S139 and 137–153 antibodies were generated by immunizing rabbits with K151-monomethylated, S139-phosphorylated and unmodified 137–153 peptides conjugated with keyhole limpet hemacyanin, respectively. Briefly, rabbits were immunized with keyhole limpet hemocyanin-conjugated peptides. The tested sera were harvested after 4 more antigen boosts. To rule out nonspecific recognition of the nonmethylated antigen, the collected sera were precleared through incubating with nonmethylated peptide and being screened using affinity chromatography. Finally, via CNBr-activated Sepharose 4 Fast Flow (GE Healthcare, 17-0981-01), the specific polyclonal antibodies were purified by methylated peptide affinity chromatography.
Flow cytometry
H9c2 cells were transfected with BioT Transfection Reagent according to the manufacturer's instructions, then collected. The assay was performed according to the manufacturer's instructions. Cells were treated as indicated, harvested, washed with phosphate-buffered saline (Sigma-Aldrich, P4417). Analysis of cell death was performed using ANXA5 (BD, 556419). Briefly, cells were suspended in ANXA5 binding buffer (10 mM HEPES, pH 7.4, 2.5 mM CaCl2, 140 mM NaCl) and stained with ANXA5. Samples were analyzed on the FACSCanto™ II Flow Cytometer (BD Biosciences). FlowJo V7 software (Tree Star Inc., USA) was used to calculate the percentage of cells positive for ANXA5 and propidium iodide (PI). Both ANXA5 and PI-negative staining represents viable cells, early apoptotic cells were positive for ANXA5 staining, both ANXA5 and PI-positive staining means late apoptosis, but necrotic cells were positive for PI staining.
According to the manufacturer's instructions, the Caspase-Glo 3/7 reagent (Promega, G8091) was used to measure CASP3 and CASP7 activities in apoptotic cells. The luminescence emitted from the cleaved substrates was measured using a VICTOR X3 Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA).
In vitro methylation analysis
In a reaction buffer containing 5 mM MgCl2, 4 mM DTT, 50 mM Tris-HCl, pH 8.5, active SETD7 protein and 1 μM H3-SAM (PerkinElmer, NET155V250UC), the mixture was kept for 60 min at 30°C. ATG16L1 K151 peptide or GST-ATG16L1 proteins (1 μg) as substrates were added. The final volume of the reaction mixture was adjusted to 10 μl. Finally, the reaction mixture was stopped and subjected to western blot. The analysis was conducted to detect methylation with an anti-ATG16L1-K151me1 antibody.
Demethylase assay
As described previously,Citation38 the demethylation assay was performed. At 37°C, ATG16L1 methylated at K151 or its peptide was bound to IgG-sepharose (GE Healthcare, 17-0969-01), and then the samples were incubated in buffer 1 (50 mM Tris, pH 8.5, 5% glycerol [EMD Millipore, 4750], 5 mM MgCl, 0.5% BSA [RPI, A30075], 50 mM KCl) plus 10 mM ATP [NEB, P0756S] and KDM1A with or without 1 × 10–3 M pargyline and 1 µg of nucleosomes purified from the indicated cells. Six h later, the reaction mixture was analyzed by immunoblotting with the indicated antibodies.
Co-immunoprecipitation and immunoblotting
The indicated cells were lysed using a low-stringency lysis buffer (50 mM Tris-HCl, pH 7.5, 120 mM NaCl, 0.5 mM EDTA, 0.5% Nonidet P-40 [Sigma-Aldrich, 74385], 10% glycerol) and approximately 0.5 mg of whole-cell extract was used for each immunoprecipitation. Antibodies against KDM1A, ATG16L1, SETD7, ATG12–ATG5, and control rabbit IgGs (Cell Signaling, 2729) were used to carry out the immunoprecipitations overnight. Immunocomplexes were collected with a mixture of protein A and protein G-Sepharose (Santa Cruz, sc-2003) and washed 3 times in washing buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 0.1% Triton X-100, 5% glycerol and 0.5% dithiothreitol). Immunocomplexes were eluted in loading buffer and loaded onto a 7.5% SDS-PAGE gel and blotted onto nitrocellulose membrane. TBST (0.1% Tween 20 [Sigma-Aldrich, P7949] in TBS [50 mM Tris-HCl, pH 7.5, 150 mM NaCl]) buffer including 1% non-fat milk was used to block the membranes and then was incubated overnight at 4°C using specifically indicated antibodies against KDM1A, Flag, His, HA, ATG16L1, SETD7, and ATG12–ATG5. After washing 3 times with TBST, membranes were incubated with the indicated concentration of HRP-conjugated secondary antibodies (GE Healthcare, NA934) for 1 h in the blocking solution at room temperature. Then the membranes were washed with TBST 5 times. Using a chemiluminescence detector (LAS-3000, Fujifilm) and ECL plus (GE Healthcare), immunoreactive bands were then detected, and the intensity of the western blot bands was quantified using NIH ImageJ software.
Myocardial ischemia/reperfusion (I/R) mouse model
C57BL6/J mouse hearts were subjected to ischemia/reperfusion (I/R) in vivo as described previously.Citation11 This animal study was approved by the Institutional Animal Care Committee. Briefly, mice were anesthetized with pentobarbital sodium (50 mg/kg; Sigma-Aldrich, P3761) through intraperitoneal injection. Then the left anterior descending (LAD) coronary artery was ligated after performing a left thoracotomy using a 6-0 silk suture with a slip knot. To cause I/R injury, mice were subjected to 30 min of LAD ischemia followed by 60 min of reperfusion. The sham group was treated with a suture passed under the LAD or not. Finally, mice were euthanized with pentobarbital sodium (200 mg/kg) by an intraperitoneal injection, and the whole heart tissue was collected for further assays. The Langendorff-perfused isolated mouse heart model of I/R was performed as described previously.Citation39 In brief, hearts were isolated from C57Bl/6J male mice and cannulated via the aorta and perfused in a retrograde fashion with Krebs-Henseleit buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.25 mM CaCl2, 1.2 mM KH2PO4, 25 mM NaHCO3, 11 mM glucose). The hearts were perfused for 20 min to achieve stabilization and then paced at 420 beats/min for baseline functional measurements. Global ischemia was achieved by closing an in-line stopcock to stop perfusion for 20 min followed by 45 min of reperfusion by re-opening the stopcock. The whole heart tissue was collected for further assays.
Proximity ligation assay
Proximity ligation assayCitation40 was carried out using Duolink® In Situ Red Starter Kit Mouse/Rabbit (Sigma-Aldrich, DUO92101) according to the manufacturer's instructions. Confocal microscopy image capture and analysis was performed on a Nikon A1 and the Nikon elements software suite.
Analysis of co-binding proteins to ATG16L1 by mass spectrometry
HEK293T cells (2.5 × 106 cells per 55-cmCitation2 dish) were plated and cotransfected with 3 µg of plasmids encoding Flag-tagged-ATG16L1 or ATG12, ATG5, and ATG16L1. The cells were harvested after 48 h and lysed in 700 µl lysis buffer (50 mM Tris, pH 7.4, protease inhibitor cocktail, 150 mM NaCl, 1% Triton X-100). Lysates were then incubated with Strep-Tactin Sepharose (30 µl slurry, 2 h, 4°C; IBA GmbH, 2-1201-002), washed in wash buffer (0.1% Triton X-100, 20 mM Tris, pH 7.4, 150 mM NaCl) 4 times, and eluted with 2.5 mM desthiobiotin (IBA GmbH, 2-1000-001). The co-purified proteins were digested with trypsin (Promega, V5280) after SDS-PAGE and band excision. Ano-flow liquid chromatography with online tandem mass spectrometry (LC-MS/MS) was used to identify the peptide mixtures separated. Tandem mass spectra assays were performed and then the Mascot Server (Matrix Science) were used to search against a nonredundant human database from the NCBI database.
Statistics
The SPSS 16.0 statistical software package (Abbott Laboratories, USA) was used for all statistical analyses. The data were analyzed using the unpaired Student t test, one-way ANOVAs, or a 2-tailed to determine statistical significance. All values are shown with standard deviation in the figures. A value of *P < 0.05 was considered to be statistically significant plus **P < 0.01 and ***P < 0.001.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
supp_data_KAUP_A_1389357.zip
Download Zip (1.1 MB)Additional information
Funding
Related Research Data
References
- Schiattarella GG, Hill JA. Therapeutic targeting of autophagy in cardiovascular disease. J Molecular and Cellular Cardiology. 2016;95:86–93. doi:10.1016/j.yjmcc.2015.11.019. PMID:26602750
- Nakayama H, Nishida K, Otsu K. Macromolecular degradation systems and cardiovascular aging. Circ Res. 2016;118(10):1577–1592. doi:10.1161/CIRCRESAHA.115.307495. PMID:27174951
- Leon LJ, Gustafsson ÅB. Staying young at heart: autophagy and adaptation to cardiac aging. J Mol Cell Cardiol. 2016;95:78–85. doi:10.1016/j.yjmcc.2015.11.006. PMID:26549356
- Vásquez‐Trincado C, García‐Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, Lavandero S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. 2016;594(3):509–525. doi:10.1113/JP271301. PMID:26537557
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy K151chanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10(7):458–467. doi:10.1038/nrm2708. PMID:19491929
- Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S, et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol. 2013;203(1):115–128. doi:10.1083/jcb.201304188. PMID:24100292
- Kaufmann A, Beier V, Franquelim HG, Wollert T. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell. 2014;156(3):469–481. doi:10.1016/j.cell.2013.12.022. PMID:24485455
- Reggiori F, Klionsky DJ. Autophagosomes: biogenesis from scratch? Curr Opin Cell Biol. 2005;17(4):415–422. doi:10.1016/j.ceb.2005.06.007. PMID:15978794
- Cebollero E, Reggiori F. Regulation of autophagy in yeast Saccharomyces cerevisiae. Biochim Biophys Acta (BBA)-Mol Cell Res. 2009;1793(9):1413–1421. doi:10.1016/j.bbamcr.2009.01.008.
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(7):713–720. doi:10.1038/ncb2788. PMID:23817233
- Song H, Pu J, Wang L, Xiao J, Liu Q, Chen J, Zhang M, Liu Y, Ni M, et al. ATG16L1 phosphorylation is oppositely regulated by CSNK2/casein kinase 2 and PPP1/protein phosphatase 1 which determines the fate of cardiomyocytes during hypoxia/reoxygenation. Autophagy. 2015;11(8):1308–1325. doi:10.1080/15548627.2015.1060386. PMID:26083323
- Biggar KK, Li SS-C. Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol. 2015;16(1):5–17. doi:10.1038/nrm3915. PMID:25491103
- Pierre CC, Longo J, Bassey-Archibong BI, Hallett RM, Milosavljevic S, Beatty L, Hassell JA, Daniel JM. Methylation-dependent regulation of hypoxia inducible factor-1 alpha gene expression by the transcription factor Kaiso. Biochim Biophys Acta (BBA)-Gene Reg Mech. 2015;1849(12):1432–1441. doi:10.1016/j.bbagrm.2015.10.018.
- Guedes-Dias P, de Proença J, Soares TR, Leitão-Rocha A, Pinho BR, Duchen MR, Oliveira JM. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochim Biophys Acta (BBA)-Mol Basis Dis. 2015;1852(11):2484–2493. doi:10.1016/j.bbadis.2015.08.012.
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12(1):1–222. doi:10.1080/15548627.2015.1100356. PMID:26799652
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi:10.1016/j.cell.2010.01.028. PMID:20144757
- Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J Biol Chem. 2010;285(1):793–804. doi:10.1074/jbc.M109.070037. PMID:19901028
- Kassner I, Andersson A, Fey M, Tomas M, Ferrando-May E, Hottiger MO. SET7/9-dependent methylation of ARTD1 at K508 stimulates poly-ADP-ribose formation after oxidative stress. Open Biology. 2013;3(10):120173. doi:10.1098/rsob.120173. PMID:24088713
- Ceballos-Chávez M, Rivero S, García-Gutiérrez P, Rodríguez-Paredes M, García-Domínguez M, Bhattacharya S, Reyes JC. Control of neuronal differentiation by sumoylation of BRAF35, a subunit of the LSD1-CoREST histone demethylase complex. Proc Natl Acad Sci. 2012;109(21):8085–8090. doi:10.1073/pnas.1121522109.
- Nicholson TB, Chen T. LSD1 demethylates histone and non-histone proteins. Epigenetics. 2009;4(3):129–132. doi:10.4161/epi.4.3.8443. PMID:19395867
- Barsyte-Lovejoy D, Li F, Oudhoff MJ, Dong A, Zeng H, Wu H, Freeman SA, Schapira M, Senisterra GA, et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc Natl Acad Sci. 2014;111(35):12853–12858. doi:10.1073/pnas.1407358111.
- Popelka H, Klionsky DJ. Post‐translationally‐modified structures in the autophagy machinery: an integrative perspective. FEBS J. 2015;282(18):3474–3488. doi:10.1111/febs.13356. PMID:26108642
- Wani WY, Boyer-Guittaut M, Dodson M, Chatham J, Darley-Usmar V, Zhang J. Regulation of autophagy by protein post-translational modification. Lab Invest. 2015;95(1):14–25. doi:10.1038/labinvest.2014.131. PMID:25365205
- Mao K, Klionsky DJ. AMPK activates autophagy by phosphorylating ULK1. Circ Res. 2011;108(7):787–788. doi:10.1161/RES.0b013e3182194c29. PMID:21454792
- Xu D, Shan B, Sun H, Xiao J, Zhu K, Xie X, Li X, Liang W, Lu X, Qian L, et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes & Development. 2016;30(15):1718–1730. doi:10.1101/gad.285122.116.
- Shi C-S, Kehrl JH. Traf6 and A20 differentially regulate TLR4-induced autophagy by affecting the ubiquitination of Beclin 1. Autophagy. 2010;6(7):986–987. doi:10.4161/auto.6.7.13288. PMID:20798608
- Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009;284(10):6322–6328. doi:10.1074/jbc.M807135200. PMID:19124466
- Xie Q, Hao Y, Tao L, Peng S, Rao C, Chen H, You H, Dong M-q, Yuan Z. Lysine methylation of FOXO3 regulates oxidative stress‐induced neuronal cell death. EMBO Rep. 2012;13(4):371–377. doi:10.1038/embor.2012.25. PMID:22402663
- Feng S, Jin Y, Cui M, Zheng J. Lysine-Specific Demethylase 1 (LSD1) Inhibitor S2101 Induces Autophagy via the AKT/mTOR Pathway in SKOV3 Ovarian Cancer Cells. Med Sci Monit: Int Med J Exp Clinical Res. 2016;22:4742. doi:10.12659/MSM.898825. PMID:27914215
- Periz G, Lu J, Zhang T, Jablonski AM, Kalb R, McCampbell A, Wang J. Regulation of protein quality control by UBE4B and LSD1 through p53-mediated transcription. PLoS Biol. 2015;13(4):e1002114. doi:10.1371/journal.pbio.1002114. PMID:25837623
- Xie Y, Kang R, Sun X, Zhong M, Huang J, Klionsky DJ, Tang D. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11(1):28–45. doi:10.4161/15548627.2014.984267. PMID:25484070
- Lezina L, Aksenova V, Ivanova T, Purmessur N, Antonov A, Tentler D, Fedorova O, Garabadgiu AV, Talianidis I, Melino G, et al. KMTase Set7/9 is a critical regulator of E2F1 activity upon genotoxic stress. Cell Death & Differentiation. 2014;21(12):1889–1899. doi:10.1038/cdd.2014.108.
- Shen C, Wang D, Liu X, Gu B, Du Y, Wei F-Z, Cao L-L, Song B., Lu X, Yang Q, et al. SET7/9 regulates cancer cell proliferation by influencing β-catenin stability. FASEB J. 2015;29(10):4313–4323. doi:10.1096/fj.15-273540. PMID:26116705
- Zhao L, Zhang Y, Gao Y, Geng P, Lu Y, Liu X, Yao. R, Hou P, Liu D, Lu J, et al. JMJD3 promotes SAHF formation in senescent WI38 cells by triggering an interplay between demethylation and phosphorylation of RB protein. Cell Death & Differentiation. 2015;22(10):1630–1640. doi:10.1038/cdd.2015.6.
- Suzuki S, Nozawa Y, Tsukamoto S, Kaneko T, Imai H, Minami N. Histone methyltransferase Smyd3 regulates early embryonic lineage commitment in mice. Reproduction. 2015;150(1):21–30. doi:10.1530/REP-15-0019. PMID:25918436
- Hadad I, Veithen A, Springael JY, Sotiropoulou PA, Da Costa AM, Miot F, Naeije R, De Deken X, Mc Entee K. Stroma cell-derived factor-1α signaling enhances calcium transients and beating frequency in rat neonatal cardiomyocytes. PLoS One. 2013;8(2):e56007. doi:10.1371/journal.pone.0056007. PMID:23460790
- Romanov J, Walczak M, Ibiricu I, Schüchner S, Ogris E, Kraft C, Martens S. Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012;31(22):4304–4317. doi:10.1038/emboj.2012.278. PMID:23064152
- Shi Y, Lan F, Matson C, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi:10.1016/j.cell.2004.12.012. PMID:15620353
- Wan TC, Ge Z-D, Tampo A, Mio Y, Bienengraeber MW, Tracey WR, Gross GJ, Kwok WM, Auchampach JA. The A3 adenosine receptor agonist CP-532,903 [N6-(2, 5-dichlorobenzyl)-3′-aminoadenosine-5′-N-methylcarboxamide] protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel. J Pharmacol Exp Ther. 2008;324(1):234–243. doi:10.1124/jpet.107.127480. PMID:17906066
- Gauthier T, Claude-Taupin A, Delage-Mourroux R, Boyer-Guittaut M, Hervouet E. Proximity Ligation In situ Assay is a Powerful Tool to Monitor Specific ATG Protein Interactions following Autophagy Induction. PLoS One. 2015;10(6):e0128701. doi:10.1371/journal.pone.0128701. PMID:26034986