
ABSTRACT
Macroautophagy/autophagy is an important cellular protein quality control process that clears intracellular aggregate-prone proteins. These proteins may cause neurodegenerative disorders such as Huntington disease (HD), which is mainly caused by the cytotoxicity of the mutant HTT/Hdh protein (mHTT). Thus, autophagy modulators may regulate mHTT levels and provide potential drug targets for HD and similar diseases. Meanwhile, autophagy function is also impaired in HD and other neurodegenerative disorders via unknown mechanisms. In a recent study, we identified a positive feedback mechanism that may contribute to mHTT accumulation and autophagy impairment in HD. Through genome-scale screening, we identified a kinase gene, HIPK3, as a negative modulator of autophagy and a positive regulator of mHTT levels in HD cells. Knocking down or knocking out HIPK3 reduces mHTT levels via enhancing autophagy in HD cells and in vivo in an HD knock-in mouse model. Interestingly, mHTT positively regulates HIPK3 mRNA levels in both HD cells and HD mouse brains, and this forms a positive feedback loop between mHTT and HIPK3. This loop potentially contributes to autophagy inhibition, mHTT accumulation, and disease progression in HD. The modulation of mHTT by HIPK3 is dependent on its kinase activity and its known substrate DAXX, providing potential HD drug targets. Collectively, our data reveal a novel kinase modulator of autophagy in HD cells, providing therapeutic entry points for HD and similar diseases.
KEYWORDS:
Autophagy is an important cellular protein quality control process that is highly relevant to neurodegenerative disorders. A common hallmark of most neurodegenerative disorders is the accumulation of misfolded and aggregation-prone proteins, which are degraded by autophagy. Conversely, autophagy function is impaired in most of these diseases. Thus, enhanced autophagy may provide a promising treatment strategy for these diseases by reducing the disease-causing proteins as well as correcting autophagy function.
Among different neurodegenerative disorders, Huntington disease (HD) is an appealing disease for translational research because of its monogenetic nature. HD is mainly caused by cytotoxicity of the mutant HTT protein (mHTT) encoded by the mutant HTT gene, and lowering the mHTT level is a promising treatment strategy for HD. In a recent effort to identify genetic modifiers of mHTT, we performed unbiased RNAi screening and identified the kinase HIPK3 as a novel modulator of the mHTT protein level. In several different HD models including human embryonic stem cell (ESC)-derived neurons, knock-in mouse striatal cells (STHdhQ7,111), HD patient fibroblasts, HD patient induced pluripotent stem cell (iPSC)-derived neurons, and in vivo striatal tissue from a knock-in mouse model (HdhQ7,111), knockdown or knockout of HIPK3 significantly reduces both the mHTT and the wild-type (WT) HTT protein level.
We then investigated the mechanism of HIPK3-mediated HTT regulation. Knockdown or knockout of HIPK3 shows no significant influence on the HTT mRNA level, suggesting that the regulation is post-transcriptional. Consistent with this, pulse-chase experiments confirmed that HIPK3 regulates HTT protein degradation. Autophagy inhibitors or knockdown of key autophagy genes abolishes the HIPK3-mediated HTT regulation, whereas proteasome inhibitors have no effect, suggesting that HIPK3 positively regulates HTT levels via inhibiting autophagy.
HIPK3 has not previously been shown to be an autophagy modulator. To confirm this, we tested a series of key autophagy markers in several different HD models. In knock-in mouse striatal cells (STHdhQ7,111), HD patient fibroblasts, HD patient iPSC-derived neurons, and in vivo striatal tissue from a knock-in mouse model (HdhQ7,111), knockdown or knockout of HIPK3 increases the LC3-II level, the LC3-puncta number and the BECN1 level, but reduces the SQSTM1/p62 level, consistent with an enhancement of autophagy flux. Thus, HIPK3 negatively regulates autophagy in HD cells. We further investigated the downstream mediator of HIPK3's effect on autophagy. The major substrate of HIPK3, DAXX, is a transcriptional suppressor of autophagy. HIPK3 directly interacts with DAXX and phosphorylates it. Knockdown or knockout of DAXX reduces autophagy flux and abolishes the effect of HIPK3 on autophagy, suggesting that HIPK3 regulates autophagy via DAXX. In summary, we identified HIPK3 as a novel negative regulator of autophagy in HD cells.
As a kinase, HIPK3 may serve as a potential drug target for HD. We confirmed that HIPK3's effect on mHTT is dependent on its kinase activity, because overexpression of HIPK3 increases mHTT levels whereas overexpression of 2 kinase-dead forms of HIPK3 (K226M and D322N) has the opposite effect. Treatment with a known HIPK3 inhibitor, AST-487, reduces mHTT in a dose-dependent manner in mouse and human HD cells. Knockdown of HIPK3 largely diminishes AST-487's effect, confirming that AST-487 reduces mHTT by inhibiting HIPK3. The results provide proof-of-concept evidence for targeting HD via HIPK3 inhibitors.
Interestingly, while HIPK3 regulates both mHTT and HTT levels in HD cells, its effect was not present in wild-type cells, whereas the effect was induced by expression of mHTT in wild-type cells. In the reverse direction, allele-specific knockdown of mHTT diminishes HIPK3's effect. Thus, the regulation of HTT by HIPK3 via autophagy is dependent on the presence of mHTT. We further revealed that the HIPK3 mRNA level is significantly higher in HD cells or in vivo striatal tissues compared to the WT controls. In addition, knockdown of HTT reduces HIPK3 mRNA levels in HD cells but not WT cells. Taken together, mHTT positively regulates HIPK3, and HIPK3 positively regulates mHTT via inhibiting autophagy (). This regulation forms a positive feedback loop that potentially contributes to mHTT accumulation, autophagy inhibition and disease progression in HD.
Figure 1. HIPK3 regulates the mHTT level via autophagy. mHTT upregulates HIPK3 expression and inhibits autophagy via DAXX. The decreased autophagy further increases the mHTT level and enhances HIPK3 expression, forming a positive feedback loop. This may contribute to mHTT accumulation and disease progression.
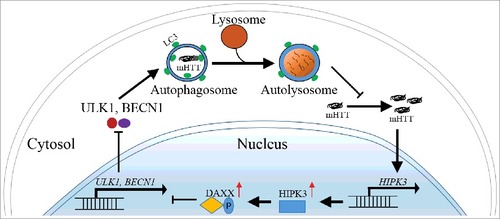
Collectively, our recent study identified HIPK3 as a novel kinase modulator of autophagy in HD cells. mHTT, HIPK3 and autophagy form a feedback loop that may contribute to disease protein accumulation and disease progression. Targeting HIPK3 by compound inhibitors may provide opportunities for drug discoveries for the treatment of HD and similar diseases.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are supported by the National Natural Science Foundation of China (91649105) and the National Key Research and Development Program of China (2016YFC0905100) for funding. Authors have no competing interests that might be perceived to influence the results and/or discussion reported in this paper.