
ABSTRACT
Macroautophagy/autophagy is a conserved lysosomal degradation system that breaks down intracellular material through the formation of double-membrane autophagosomes in eukaryotic cells. Cargo receptors have been shown to play essential roles in capturing and delivering specific substrates into phagophores, the precursors to autophagosomes, for degradation. However, the detailed mechanism underlying selective recognition of the substrates for autophagic degradation remains poorly understood. Recently, we have revealed that IFN (interferon)-induced BST2 recruits the E3 ubiquitin ligase MARCH8 to catalyze the K27-linked ubiquitination of MAVS for CALCOCO2-directed autophagic degradation, hence inhibiting DDX58-mediated type I interferon signaling through a negative feedback loop.
BST2/tetherin (bone marrow stromal cell antigen 2) serves as a restriction factor for various viruses and plays pivotal roles in antiviral immune responses. To gain insights into whether BST2 affects type I IFN signaling, we generated a BST2-inducible cell line to examine the innate immune response to several kinds of viruses, and identified BST2 as a negative regulator of DDX58-mediated type I IFN signaling at the late stage of infection. Additionally, the inhibitory role of BST2 in type I IFN signaling is not dependent on its virus restriction activity. Further study revealed that BST2 specifically inhibits RNA virus-induced, but not DNA virus-induced type I IFN signaling by targeting via the signaling adaptor MAVS (). Notably, RNA virus infection can enhance the association between BST2 and MAVS. We next found that BST2 promotes the degradation of MAVS. In addition, our results showed that BST2-mediated MAVS degradation can be rescued by autophagy or autolysosome inhibitors, but not a proteasome inhibitor, suggesting that BST2 is involved in the autophagic degradation of MAVS. Moreover, BST2 accelerates the degradation of MAVS upon starvation-induced autophagy activation, and the BST2-triggered MAVS degradation is almost totally abolished in BECN1 and ATG5 knockout (KO) cells, in which autophagy flux is largely deficient.
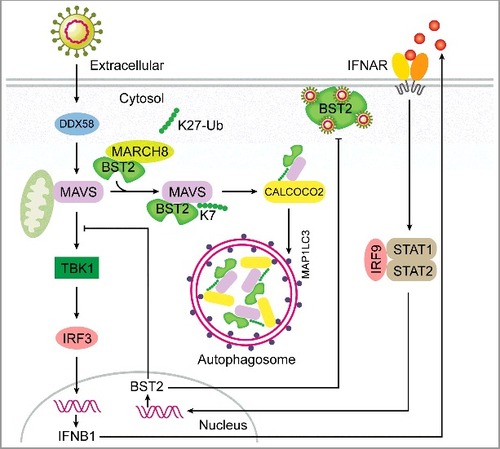
Figure 1. A working model to illustrate a negative feedback loop of DDX58-mediated type I IFN signaling generated by a BST2-MARCH8-MAVS-CALCOCO2 axis. The transcription of the BST2 gene is upregulated once type I IFN signaling is activated. BST2 works as a virus restriction factor by tethering the virus particles in the infected cells. Meanwhile, BST2 recruits the E3 ubiquitin ligase MARCH8 to catalyze the K27-linked ubiquitination of MAVS at K7. Through recognition of K27-linked polyubiquitin chains on MAVS, CALCOCO2 delivers MAVS to phagophores for subsequent degradation, which shuts down the sustained activation of TBK1-IRF3 signaling in a negative feedback loop.
As BST2 is not a cargo receptor, we hypothesized that BST2 might deliver MAVS to the cargo receptors for selective autophagic degradation. Although BST2 associates with most of the Atg8-family proteins, it only interacts with CALCOCO2/NDP52 rather than other cargo receptors. Furthermore, BST2 enhances the degradation of MAVS through CALCOCO2-mediated selective autophagy (). Next, we found that human CALCOCO2 interacts with MAVS through its LIM-L domain, which recognizes a ubiquitination signal and delivers ubiquitinated cargoes to phagophores. We hypothesized that ubiquitin chains on MAVS may work as a degradation signal for subsequent CALCOCO2 recognition. Intriguingly, CALCOCO2 is truncated in mice, lacking the LIM-L region. BST2 no longer regulates the autophagic degradation of MAVS in mouse cells as it cannot recognize MAVS.
To investigate the detailed mechanism underlying CALCOCO2-directed MAVS recognition, we detected the poly-ubiquitination of MAVS and observed that BST2 significantly increases this modification. Moreover, we found that BST2 markedly enhances the K27-linked ubiquitination of MAVS, but not the ubiquitination of MAVS with other linkages. Because BST2 is not an E3 ubiquitin ligase, it might function as a scaffold to link MAVS and potential E3 ubiquitin ligases. We next found that BST2 can interact with the E3 ligase MARCH8 and recruit it to MAVS (). MARCH8 deficiency significantly abrogates the inhibition of MAVS-induced type I IFN activation by BST2. Moreover, the K27-linked ubiquitination and degradation of MAVS mediated by BST2 also disappears in MARCH8 KO cells.
To identify the exact ubiquitination sites on MAVS, we generated a series of MAVS mutated constructs by substituting lysine (K) residues with arginines (R), and found that BST2 loses the ability to degrade the K7R MAVS mutant (MAVSK7R), and the MAVSK7R mutant displays an impaired K27-linked ubiquitination when compared with wild-type (WT) MAVS. We next investigated whether K27-linked polyubiquitin chains on K7 serve as a degradation signal for MAVS, and observed that the association between the MAVSK7R mutant and CALCOCO2 is decreased compared to that of WT MAVS. To test whether BST2-MARCH8 inhibits MAVS-activated type I IFN through K27-linked polyubiquitin chains on K7, we restored MAVS KO cells with WT or K7R MAVS, and found that the MAVSK7R mutant dramatically enhances the activation of type I IFN signaling.
Collectively, our study provides evidence that BST2 plays dual functions in antiviral immune responses and is indispensable for the deactivation of RNA virus-induced type I IFN signaling in human cells (). The BST2-MARCH8 axis is involved in the regulation of type I IFN signaling at the level of MAVS, which relies on CALCOCO2-mediated selective autophagy. Our work highlights the novel regulatory role of selective autophagy in type I IFN signaling. Because of the importance of BST2 in response to pathogen infection, our results provide new insights into the regulatory mechanisms of BST2 in type I IFN signaling and point out its potential risk as a pharmacological target.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.