
ABSTRACT
In yeast, Tom22, the central component of the TOMM (translocase of outer mitochondrial membrane) receptor complex, is responsible for the recognition and translocation of synthesized mitochondrial precursor proteins, and its protein kinase CK2-dependent phosphorylation is mandatory for TOMM complex biogenesis and proper mitochondrial protein import. In mammals, the biological function of protein kinase CSNK2/CK2 remains vastly elusive and it is unknown whether CSNK2-dependent phosphorylation of TOMM protein subunits has a similar role as that in yeast. To address this issue, we used a skeletal muscle-specific Csnk2b/Ck2β-conditional knockout (cKO) mouse model. Phenotypically, these skeletal muscle Csnk2b cKO mice showed reduced muscle strength and abnormal metabolic activity of mainly oxidative muscle fibers, which point towards mitochondrial dysfunction. Enzymatically, active muscle lysates from skeletal muscle Csnk2b cKO mice phosphorylate murine TOMM22, the mammalian ortholog of yeast Tom22, to a lower extent than lysates prepared from controls. Mechanistically, CSNK2-mediated phosphorylation of TOMM22 changes its binding affinity for mitochondrial precursor proteins. However, in contrast to yeast, mitochondrial protein import seems not to be affected in vitro using mitochondria isolated from muscles of skeletal muscle Csnk2b cKO mice. PINK1, a mitochondrial health sensor that undergoes constitutive import under physiological conditions, accumulates within skeletal muscle Csnk2b cKO fibers and labels abnormal mitochondria for removal by mitophagy as demonstrated by the appearance of mitochondria-containing autophagosomes through electron microscopy. Mitophagy can be normalized by either introduction of a phosphomimetic TOMM22 mutant in cultured myotubes, or by in vivo electroporation of phosphomimetic Tomm22 into muscles of mice. Importantly, transfection of the phosphomimetic Tomm22 mutant in muscle cells with ablated Csnk2b restored their oxygen consumption rate comparable to wild-type levels. In sum, our data show that mammalian CSNK2-dependent phosphorylation of TOMM22 is a critical switch for mitophagy and reveal CSNK2-dependent physiological implications on metabolism, muscle integrity and behavior.
List of abbreviations
| ACTB | = | actin beta |
| ACTN2 | = | actinin alpha 2 |
| Atg32 | = | Yeast mitochondrial outer membrane protein required to initiate mitophagy |
| Atp2 | = | yeast beta subunit of the F1 sector of mitochondrial F1Fo ATP synthase |
| CMAP | = | compound muscle action potential |
| Cox4 | = | subunit IV of yeast cytochrome c oxidase |
| BCL2L13 | = | BCL2 like 13 |
| BECN1 | = | Beclin 1 |
| BN-PAGE | = | blue native polyacrylamide gel electrophoresis |
| BNIP3 | = | BCL2 interacting protein 3 |
| BNIP3L | = | BCL2 interacting protein 3 like |
| CSNK1 | = | casein kinase 1 |
| CSNK2 | = | casein kinase 2 |
| CSNK2A1 | = | casein kinase 2 alpha 1 |
| CSNK2A2 | = | casein kinase 2 alpha 2 |
| CSNK2B | = | casein kinase 2 beta |
| FACS | = | fluorescence-activated cell sorting |
| FUNDC1 | = | FUN14 domain containing 1 |
| GFP | = | green fluorescent protein |
| GST | = | glutathione S-transferase |
| FCCP | = | carbonyl cyanide p-trifluoromethoxyphenylhydrazone |
| FDB | = | flexor digitorum brevis |
| GSK3B | = | glycogen synthase kinase 3 beta |
| HSA-Cre | = | reflects reporter mice which use the human ACTA1 (actin, alpha 1, skeletal muscle) promoter 5’ in front of the bacterial cre recombinase gene |
| IMM | = | inner membrane of mitochondria |
| LAMP2 | = | lysosomal associated membrane protein 2 |
| MAP1LC3B | = | microtubule associated protein 1 light chain 3 beta |
| Mdh1 | = | yeast mitochondrial malate dehydrogenase 1 |
| MDH2 | = | malate dehydrogenase 2 |
| mEPC | = | miniature end plate current |
| MFN2 | = | mitofusin 2 |
| NDUFA10 | = | NADH:ubiquinone oxidoreductase subunit A10 |
| NLS | = | nuclear localization signal |
| NMJ | = | neuromuscular junction |
| OCR | = | oxygen consumption rate |
| OMM | = | outer membrane of mitochondria |
| OPTN | = | optineurin |
| PARL | = | presenilin associated rhomboid like |
| PD | = | Parkinson disease |
| PECAM1 | = | platelet and endothelial cell adhesion molecule 1 |
| PINK1 | = | PTEN induced putative kinase 1 |
| PKA | = | cAMP-dependent protein kinase A |
| PRKN/PARK2 | = | parkin RBR E3 ubiquitin protein ligase |
| qPCR | = | quantitative PCR |
| RFP | = | red fluorescent protein |
| RT | = | reverse transcriptase |
| SDHA | = | succinate dehydrogenase complex flavoprotein subunit A |
| SDS-PAGE | = | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| shRNA | = | short hairpin RNA |
| SLC25A4/5/31 | = | solute carrier family 25 member 4/5/31 |
| SQSTM1 | = | sequestosome 1 |
| TMRM | = | tetramethyl rhodamine methyl ester |
| TOMM | = | translocase of outer mitochondrial membrane |
| TOMM20 | = | translocase of outer mitochondrial membrane 20 |
| TOMM22 | = | translocase of outer mitochondrial membrane 22 |
| TOMM40 | = | translocase of outer mitochondrial membrane 40 |
| TOMM70 | = | translocase of outer mitochondrial membrane 70 |
| TUNEL | = | TdT-mediated dUTP-biotin nick end labeling |
| VDAC1/2/3 | = | voltage dependent anion channel 1/2/3. |
Introduction
Mitochondria are specialized organelles that supply the cells with energy, play important roles in different metabolic pathways, contribute to the maintenance of ion concentrations and regulate apoptosis.Citation1 The vast majority of mitochondrial proteins is encoded by nuclear DNA and has to be translocated from the cytosol into the mitochondria. The import of nearly all mitochondrial proteins requires a preprotein translocase of the outer mitochondrial membrane (TOMM complex).Citation2 Upon interaction of typical precursor proteins with the TOMM receptors, precursor proteins are transferred to the central receptor TOMM22, which acts in cooperation with TOMM20, and subsequently to the import channel TOMM40.Citation3,Citation4 Adjacent to TOMM20 and TOMM22, there is a third receptor, TOMM70, which is mainly required for the import of noncleavable hydrophobic precursors, like the metabolite carriers of the inner membrane.Citation5–7 After passing through the TOMM complex, the precursor proteins use different machineries to reach their functional destination in the 4 mitochondrial subcompartments: outer membrane of mitochondria (OMM), intermembrane space, inner membrane of mitochondria (IMM), and mitochondrial matrix.Citation3,Citation4,Citation8 In yeast, it has recently been reported that protein kinase CK2/casein kinase 2 (in mammals CSNK2) constitutively phosphorylates the receptor Tom22 at S44 and S46, an event which is critical for the biogenesis of further TOMM subunits and promotes mitochondrial protein import.Citation9,Citation10 Up to now, nothing is known about CSNK2-dependent TOMM22 phosphorylation in mammals. Unfortunately, the finding in yeast does not help to understand whether CSNK2-dependent TOMM22 phosphorylation occurs in mice because mouse and yeast TOMM22/Tom22 primary structures show only approx. 25% identity, and mouse TOMM22 does not contain the CSNK2-target sites S44 and S46, as in yeast.
Dysfunction of mitochondria causes fragmentation of the mitochondrial network and might induce mitophagy, a specific type of autophagy.Citation2,Citation11 Aberrant mitochondrial quality control has been linked to cellular abnormalities and cell death. Genetic findings support causal contribution of mitochondrial dysfunction to Parkinson disease (PD).Citation12–17 Among the mutated genes in familial PD are PRKN/Parkin (Park2 in mice) and PINK1 (PTEN induced putative kinase 1).Citation15–19 PINK1, which is partially imported into mitochondria, and PARK2, an E3 ubiquitin ligase that localizes to the cytosol, both are involved in selective clearance of damaged mitochondria.Citation20,Citation21 In healthy cells, full-length PINK1 (65 kDa) inserts into the IMM with its presequence in a membrane potential-dependent manner. Thereafter, PINK1 is processed by the protease PARL, that cleaves within the transmembrane segment and generates a destabilizing amino terminus, followed by degradation of cleaved PINK1 (53 kDa) by the ubiquitin-proteasome system.Citation22–25 Hence, under physiological conditions the protein level of PINK1 in cells is extremely low. Dissipation of the inner membrane potential of damaged mitochondria leads to an accumulation of PINK1 (65 kDa) at the OMM. PINK1 is then neither sequestered into the IMM, nor processed, but it remains on the OMM where it is stabilized by TOMM7,Citation26 and recruits PRKN/PARK2, which induces mitophagy.Citation27 Recently, it has been reported that PINK1 activates PRKN/PARK2 by phosphorylation of ubiquitin.Citation28,Citation29 Then PRKN/PARK2 ubiquitinates outer mitochondrial membrane proteins and thereby labels damaged mitochondria for removal.Citation2 PINK1, like almost all mitochondrial proteins, is synthesized in the cytosol as a preprotein, targeted to the surface of the organelle, and then translocated across the OMM utilizing the TOMM complex.Citation30–32 Previously, it has been found that for the membrane-potential dependent import of PINK1 into the mitochondria the import receptor TOMM70, but not TOMM40, is used.Citation23 Others reported that PINK1 can be crosslinked with TOMM20.Citation33 Altogether, it is still a matter of debate what protein PINK1 binds to on OMMs.Citation26,Citation33
The protein kinase CSNK2/CK2 is a tetramer composed of 2 catalytically active α- and 2 β-subunits, and is important for cell proliferation, differentiation, and survival.Citation34 Previously, we have shown that CSNK2 binds, and in some cases also phosphorylates, several protein members at neuromuscular junctions (NMJs) of mice and thereby stabilizes the postsynaptic apparatus.Citation35,Citation36 In the absence of the β-subunit of CSNK2, CSNK2B, in skeletal muscle fibers, mice develop an age-dependent muscle weakness and a decrease of grip strength.Citation35
Here, we asked whether CSNK2 phosphorylates TOMM22 in skeletal muscle fibers of mice and whether this influences mitochondrial physiology. We show that (1) CSNK2 phosphorylates TOMM22 at serine 15 and threonine 43, (2) CSNK2-dependent TOMM22 phosphorylation is not involved in the regulation of mitochondrial protein import in vitro, (3) skeletal muscles from skeletal muscle Csnk2b cKO mice contain dysfunctional mitochondria, and (4) PINK1, PRKN/PARK2, MFN2 (mitofusin 2), OPTN (optineurin) and SQSTM1/p62 are involved in the removal of impaired mitochondria. Ablation of Csnk2b in skeletal muscle fibers leads to a significant increase of cytosolic SQSTM1-associated aggregates which returned to normal values after introduction of phosphomimetic TOMM22 mutants into muscle fibers in vivo. Importantly, (1) transfection of the inactive TOMM22 mutant, TOMM22S15A,T43A, in muscle cells correlates with the appearance of SQSTM1-associated aggregates in comparison with transfection of wild-type TOMM22, and (2) transfection of a phosphomimetic TOMM22 mutant into cultured muscle cells from skeletal muscle Csnk2b cKO mice restored their oxygen consumption rate comparable to wild-type levels. Altogether, CSNK2-dependent TOMM22 phosphorylation in mice is not required for mitochondrial protein import, or for TOMM complex biogenesis. CSNK2-dependent phosphorylation of TOMM22 in mice rather appears to regulate mitochondrial homeostasis through mitophagy.
Results
The ablation of Csnk2b in skeletal muscle fibers of mice results in impaired muscle histology, increased number of central nuclei and apoptotic cells
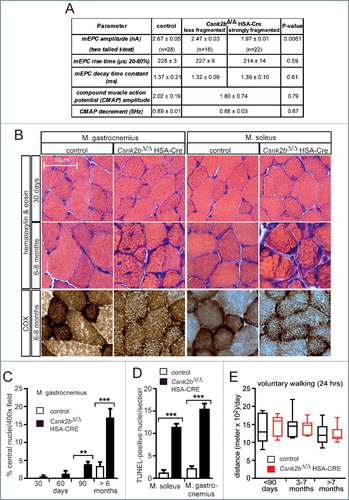
Previously, we have shown that protein kinase CSNK2 interacts with, and in some cases phosphorylates, several proteins of neuromuscular junctions (NMJ).Citation35,Citation36 Thereby, CSNK2 appears to ensure proper maintenance of CHRN (cholinergic receptor nicotinic) aggregates at the postsynaptic apparatus of NMJs.Citation35 In our in vivo model conditional deletion of Csnk2b in striated skeletal muscle fibers was achieved by use of a Cre driver mouse using the human ACTA1 (actin, alpha 1, skeletal muscle) promoter (previously denominated HSA-Cre mouse).Citation37,Citation38 Muscle-specific homozygous conditional knockout (cKO) csnk2b mice (csnk2b∆/∆, HSA-Cre), in comparison with control mice of the same litter (Csnk2b+/∆, HSA-Cre), have less grip strength and their postsynaptic endplates cover the full spectrum from almost nonfragmented to heavily fragmented patterns.Citation35 No difference was detected between wild-type (Csnk2b+/+) and heterozygous mice with one deleted Csnk2b allele (Csnk2b+/∆, HSA-Cre) or mice with floxed Csnk2b alleles (Csnk2b+/loxP, Csnk2bloxP/loxP), arguing against any haploinsufficiency.Citation35 In this manuscript, control mice (Csnk2b+/loxP, Csnk2bloxP/loxP or Csnk2b+/∆, HSA-Cre), were compared with corresponding muscles of homozygous skeletal muscle cKO Csnk2b mice (csnk2b∆/∆, HSA-Cre) of the same litter. Since CSNK2 is known to be ubiquitously expressed and pleiotropic,Citation39 we asked whether the extent of muscle weakness in the csnk2b∆/∆, HSA-Cre mice only depends on fragmented NMJs. First, we recorded neural transmission at the NMJs of control and csnk2b∆/∆, HSA-Cre mice (A). Previously, a reduction of the amplitude of miniature end plate currents (mEPCs) has been observed in the csnk2b∆/∆, HSA-Cre diaphragms.Citation35 We questioned whether this change explains the full extent of muscle weakness in csnk2b∆/∆, HSA-Cre mice. We decided to compare neural transmission in the csnk2b∆/∆, HSA-Cre and control diaphragms in more detail by differentiating between heavily fragmented or almost not fragmented NMJs.Citation35 Neural transmission was only affected in muscle fibers containing strongly fragmented NMJs (∼ 25% of total), reflected by ∼ 20% lower amplitudes of mEPC (A).Citation35 No significant decrease of mEPC amplitudes was observed in the csnk2b∆/∆, HSA-Cre muscle fibers that contain less fragmented NMJs (A). Nerve-dependent extracellular recordings demonstrated neither a change of compound muscle action potential (CMAP) amplitudes, nor a different CMAP decrement at 5 Hz between control and the csnk2b∆/∆, HSA-Cre diaphragms (A). Hence, the extent of muscle weakness of the csnk2b∆/∆, HSA-Cre mice cannot solely be explained by impaired neural transmission. Considering that CSNK2 accumulates at NMJs, but is still expressed along the whole muscle fiber,Citation35 we speculated that CSNK2 might also have extrasynaptic targets in muscle fibers.Citation35,Citation36 Towards identification of such targets, we performed different histological staining to screen for changes in the csnk2b∆/∆, HSA-Cre skeletal muscle fibers in comparison with controls. We decided to analyze both, glycolytic (gastrocnemius, tibialis anterior), and oxidative (soleus, diaphragm) muscles. A comparative look at hematoxylin and eosin stained cross-sections of gastrocnemius and soleus muscles of control and the csnk2b∆/∆, HSA-Cre mice (6 to 8 months old) revealed more of reduced fiber diameters in csnk2b∆/∆, HSA-Cre muscles (B). Cross-sections of the csnk2b∆/∆, HSA-Cre muscles had a more granular appearance, interstitial fibrosis was visible between fibers, and some fibers looked even split (B). Cytochrome oxidase (COX) histochemical staining of muscle cross sections typically labels fibers with high mitochondrial content, like slow fiber types (type I) or fast oxidative type fibers (type IIa), with a dark color. Adult csnk2b∆/∆, HSA-Cre skeletal muscle type I and IIa fibers showed a less intense staining; especially in soleus muscle that is mainly composed of type I and IIa muscle fiber types (B), indicating lower mitochondrial content in the csnk2b∆/∆, HSA-Cre muscle fibers. Other histological staining methods, like the Gomori trichrome, succinate dehydrogenase and nicotinamide adenine dinucleotide dehydrogenase staining, of adult control and csnk2b∆/∆, HSA-Cre muscle cross-sections also pointed to a potential impairment of oxidative metabolism in csnk2b∆/∆, HSA-Cre muscle fibers.Citation40 In comparison with adult, in young csnk2b∆/∆, HSA-Cre mice (30 d), histochemical staining of muscle cross-sections did not show any abnormalities, like changes in fiber diameter (B). Moreover, skeletal muscles from adult csnk2b∆/∆, HSA-Cre mice contained a high number of central nuclei, which indicated regenerative events (C). To find out whether the increase of central nuclei in the csnk2b∆/∆, HSA-Cre skeletal muscles pointed to degenerative changes, the number of TUNEL-positive nuclei per hind limb muscle cross-section was quantified and turned out being significantly increased in the csnk2b∆/∆, HSA-Cre compared to control muscle (D). To understand whether the lower mitochondrial content in the csnk2b∆/∆, HSA-Cre muscle fibers which was indicated by COX staining might be indirectly influenced by muscle contractile activity, voluntary walking distances of mice were recorded but the measurements indicated no differences between control and csnk2b∆/∆, HSA-Cre mice at young age and during adulthood (E).
Figure 1. csnk2b∆/∆, HSA-Cre muscle fibers exhibit impairment of neural transmission and morphological abnormalities. (A) The table summarizes electrophysiological recordings of intracellular miniature endplate current (mEPC) amplitude, rise, and decay time constants at NMJs of control and csnk2b∆/∆, HSA-Cre mice. A statistically significant difference was only found for mEPC amplitude of strongly fragmented, in comparison with less fragmented, NMJs of csnk2b∆/∆, HSA-Cre mice. No difference was detected between less fragmented NMJs of the csnk2b∆/∆, HSA-Cre in comparison with NMJs of control mice. Extracellular recordings of compound muscle action potential (CMAP) and CMAP decrement (5Hz) did not reveal any changes between control and csnk2b∆/∆, HSA-Cre diaphragm muscles. Three mice per genotype were recorded. N = number of NMJs. (B) Representative images of hematoxylin and eosin or COX stained hind limb muscle cross sections are shown. Note, dark colored mitochondrial rich fibers (type I and IIa) are less colored in the adult 6- to 8-month-old csnk2b∆/∆, HSA-Cre muscles. Moreover, the histological stainings of cross-sections of the csnk2b∆/∆, HSA-Cre are of more granular appearance in comparison with controls. (C) Number of central nuclei were counted in control or csnk2b∆/∆, HSA-Cre gastrocnemius muscle cross-sections of adult 6- to 8-mo-old mice and presented as graph. N = 5 mice per genotype. (D) Quantification of the total number of TUNEL-positive nuclei of muscle fibers of 6- to 8-mo-old mice of control or csnk2b∆/∆, HSA-Cre muscles soleus and gastrocnemius per muscle cross-section. N = 5 mice per genotype. (E) Voluntary walking distance of control and csnk2b∆/∆, HSA-Cre mice is shown in relation to the age of the mice. N = 5 mice per genotype. Note, differences of walking distances between control and csnk2b∆/∆, HSA-Cre mice are not significant (P values >0.05).
The amount and functionality of mitochondria are affected in csnk2b∆/∆, HSA-Cre skeletal muscle fibers
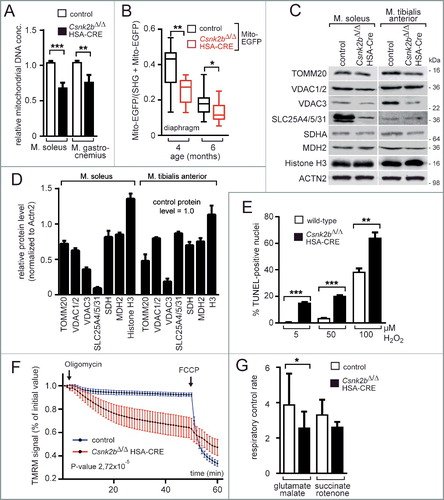
We speculated that the diminished oxidative capacity of csnk2b∆/∆, HSA-Cre muscle fibers is caused by mitochondrial impairments. We employed different strategies to analyze whether mitochondrial number or physiology is affected in csnk2b∆/∆, HSA-Cre muscle fibers from adult 6- to 8-mo-old mice. First, lower mitochondrial genome copy numbers were quantified in the snk2b∆/∆, HSA-Cre muscles soleus and gastrocnemius in comparison with corresponding control muscles (A). Second, a lower mitochondrial amount was detected in the snk2b∆/∆, HSA-Cre, mito-EGFP compared to control muscle with GFP labeled mitochondria (Csnk2b+/∆, HSA-Cre, mito-EGFP) (B). For this, a conditional reporter mouse line, which contains a mitochondrially located EGFP fluorescent protein,Citation41 was bred with csnk2b∆/∆, HSA-Cre mice. Offspring that inherited the EGFP allele, with control or csnk2b∆/∆, HSA-Cre muscles were compared. In all muscle fibers, the myosin signal was used as a gauge for muscle fiber volume and detected by second harmonic generation microscopy, and mitochondrial EGFP was imaged by 2-photon microscopy and related to fiber volume (B). Third, the amount of several endogenous mitochondrial proteins in muscle lysates from control and csnk2b∆/∆, HSA-Cre mice was compared by western blot and protein bands were detected and quantified by densitometric scanning (C, D). All mitochondrial protein amounts were significantly reduced in csnk2b∆/∆, HSA-Cre in comparison with control muscle cells (C, D). Fourth, because lower mitochondrial activity and higher number of central nuclei in csnk2b∆/∆, HSA-Cre muscles (B, C) might indicate a higher vulnerability of mutant fibers, we examined sensitivity of csnk2b∆/∆, HSA-Cre muscle cells towards oxidative stress. Mitochondria are known to provide the energy for most cell functions, but at the same time senescent or damaged mitochondria influence the amount of toxic reactive oxygen species.Citation42 Muscle cells from neonatal limbs of wild-type or csnk2b∆/∆, HSA-Cre litters were cultured, differentiated to myotubes to induce cre recombinase expression, and exposed to oxidative stress by adding hydrogen peroxide to their culture medium. In comparison with wild-type muscle cells, csnk2b∆/∆, HSA-Cre myotubes are more sensitive to a dose-dependent increase of hydrogen peroxide as demonstrated by significantly higher numbers of TUNEL-positive cell nuclei (E). Fifth, we compared mitochondrial function in control and csnk2b∆/∆, HSA-Cre muscle fibers by epifluorescence microscopy based on the accumulation of TMRM signal and sequential addition of the ATP synthase inhibitor oligomycin and the ionophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP).Citation43 After addition of oligomycin to muscle fibers, only damaged mitochondria fail to keep mitochondrial membrane potential, an event that is reflected by a decrease of TMRM signal intensity. csnk2b∆/∆, HSA-Cre fibers from 6- to 8-mo-old mice were not able to keep mitochondrial membrane potential over time, unlike muscle fibers from control mice (F). Next, the respiratory control rates were measured and found to be significantly lower in mitochondria isolated from csnk2b∆/∆, HSA-Cre muscles in comparison with mitochondria from controls indicating a lower capacity for substrate oxidation and ATP turnover and confirming an eventual increase in proton leak (F, G). Altogether, our data suggest that number and functionality of mitochondria are affected in csnk2b∆/∆, HSA-Cre muscle fibers of adult mice of 6 to 8 mo of age.
Figure 2. csnk2b∆/∆, HSA-Cre muscles possess fewer and functionally abnormal mitochondria. (A) The relative mitochondrial genome copy numbers of adult 6- to 8-mo-old mice of control and csnk2b∆/∆, HSA-Cre muscles were determined by qPCR. Values were normalized to PECAM1 and are presented as relative amount of control mitochondrial genome copy number (control is set to 1.0). N = 3 mice per genotype. (B) A Mito-EGFP allele was introduced into csnk2b∆/∆, HSA-Cre mice. Fluorescence intensities of Mito-EGFP of control and csnk2b∆/∆, HSA-Cre diaphragm muscles from 4- to 6-mo-old mice were detected by 2-photon microscopy and related to the sum of second-harmonic generation and Mito-EGFP signal intensities. This sum correlates with muscle fiber volume. Note, less EGFP fluorescence signal was detected in csnk2b∆/∆, HSA-Cre muscles in comparison with controls which points to a lower mitochondrial amount. N = 3 mice per genotype. (C) Representative immunoblot images of mitochondrial proteins (TOMM20, a subunit of the mitochondrial outer membrane translocase; VDAC1/2 and VDAC3 (voltage dependent anion channel 1/2/3); SLC25A members 4/5/31 (solute carrier family 25 members 4/5/31); SDHA (succinate dehydrogenase complex flavoprotein subunit A); MDH2 (malate dehydrogenase 2) prepared from control and csnk2b∆/∆, HSA-Cre soleus and tibialis anterior muscles of adult 6 to 8-mo-old mice. Histone H3 was used as a control representing a protein, which belongs to a different organelle than mitochondria. ACTN2 was probed as loading control. (D) Protein bands shown in (C) were quantified using ImageJ, normalized to ACTN2, and their expression was plotted relative to control (set to 1.0). N = 5 mice per genotype. Note, there is a significant reduction of mitochondrial proteins in csnk2b∆/∆, HSA-Cre muscles. (E) The total number of TUNEL-positive cultured myotubes was determined after incubation of the cells with increasing amounts of hydrogen peroxide. Graph shows a comparison between wild-type and csnk2b∆/∆, HSA-Cre TUNEL-positive cells. (F) Mitochondrial membrane potential was measured in isolated fibers from old flexor digitorum brevis muscles of adult 6 to 8-mo-old mice of control and csnk2b∆/∆, HSA-Cre mice. Fibers were isolated, placed in cell culture, and loaded with TMRM. TMRM accumulates in the mitochondria that are able to maintain mitochondrial membrane potential. Oligomycin and the protonophore FCCP were added at the indicated time points. TMRM staining was monitored in at least 10 fibers per genotype. N = 3 mice per genotype. Note, mitochondria from csnk2b∆/∆, HSA-Cre mice were not able to hold inner mitochondrial membrane potential, like control mitochondria. (G) Isolated mitochondria from control and csnk2b∆/∆, HSA-Cre muscles were used to measure oxygen consumption after incubation with the indicated substrates and inhibitors, and calculate respiratory control rates which are summarized by the graph. N = 6 mice per genotype. Note, mitochondria of csnk2b∆/∆, HSA-Cre mice show lower respiratory control rates in comparison with mitochondria isolated from control muscles.
CSNK2 phosphorylates mouse TOMM complex receptor TOMM22
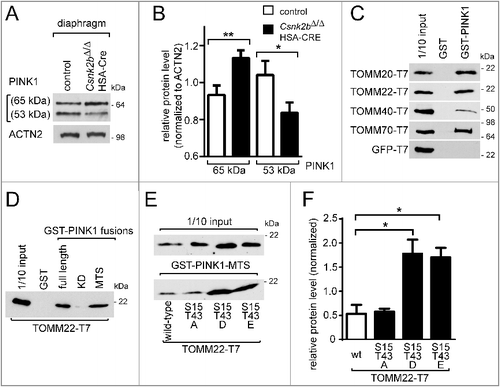
At this point, our data point to an impairment of oxidative metabolism and mitochondrial number and functionality (B, A to D, F, G). One reason might be impaired mitochondrial protein import. In fact, yeast CK1, CK2 and PKA regulate the import and assembly of Tom22, demonstrating that this central receptor is a target for the posttranslational regulation of mitochondrial protein import.Citation9,Citation10 In yeast, CK2 constitutively phosphorylates the cytosolic precursor of Tom22 at S44 and S46.Citation10 Strikingly, the yeast and mouse Tom22/TOMM22 primary structures have only 24.2%, identity and mouse TOMM22 does not contain S44 and S46, not to mention the absence of any CSNK2 target site within the amino acid stretch aligning with yeast Tom22 S44 and S46 (C). Still, we asked whether CSNK2-dependent TOMM22 phosphorylation also occurs in mice. We performed in vitro phosphorylation assays and demonstrated that recombinant mouse TOMM22 is phosphorylated by purified CSNK2 in vitro but not by CSNK1 or PKA as in yeast (A). Moreover, phosphorylation of recombinant TOMM22 also occurred after incubating it with control muscle lysates from 4 different mouse hind limb muscles (B). On the other hand, recombinant TOMM22 was significantly less phosphorylated using corresponding muscle lysates from csnk2b∆/∆, HSA-Cre litter mice (B). Then, we scanned the primary structure of mouse TOMM22 to identify potential CSNK2 target motifs. Three amino acid residues were predicted by different algorithms as being targets of CSNK2 in mice, namely S15, T43, and S45 (C). We substituted all 3 residues independently by alanine and subjected the resulting TOMM22 mutant proteins to in vitro phosphorylation by recombinant CSNK2 (D). S15 and T43 of TOMM22 turned out to be specifically phosphorylated by CSNK2, but that was not the case with S45 (D). Next, we confirmed phosphorylation of TOMM22 at S15 by transfecting the T7-tagged wild-type TOMM22, and TOMM22 containing the S15A or T43A single nonphosphorylatable mutations (TOMM22S15A or TOMM22T43A), as well as the combined TOMM22S15A,T43A nonphosphorylatable mutations, individually into cultured cells, preparing cell lysates, and analyzing phosphorylation of TOMM22 S15 with a TOMM22-phosphoserine-15 -specific antibody by western blot (E). Previously, ablation of Csnk2b in muscle fibers has been associated with both, higher CSNK2 catalytic activity, and impaired phosphorylation of a CSNK2 target protein,Citation35 arguing that changed protein amounts of subunits CSNK2A1 or CSNK2A2 in muscles might indicate affected phosphorylation pattern. We asked whether the absence of Csnk2b has any effect on the endogenous protein amount of the catalytic activity-containing subunits CSNK2A1 or CSNK2A2 in csnk2b∆/∆, HSA-Cre muscle (F). By western blots, csnk2b∆/∆, HSA-Cre soleus and tibialis anterior muscle lysates showed different patterns for CSNK2A subunits; CSNK2B was in both muscles not detectable (F to H). Interestingly, csnk2b∆/∆, HSA-Cre soleus in comparison with control muscle lysates, regardless whether from approximately 2- or 6- to 8-mo-old adult mice, contained significantly less CSNK2A2 protein, (F). Soleus muscles from mice (6 to 8 mo) possess more CSNK2A1, but less CSNK2A2 protein levels, in csnk2b∆/∆, HSA-Cre compared to controls (F, G). In the csnk2b∆/∆, HSA-Cre tibialis anterior in young and old mice significantly more CSNK2A1, but less CSNK2A2 protein levels were detected in comparison with controls (F, H).
Figure 3. CSNK2 phosphorylates TOMM22 and the absence of Csnk2b compromises CSNK2 catalytical activity and protein amount, but neither is required for TOMM complex biogenesis nor mitochondrial protein import. (A) Mouse TOMM22 was purified and used for in vitro radio-isotope-assisted phosphorylation with different kinases. The image of the autoradiogram shows that mouse TOMM22 was only phosphorylated in the presence of protein kinase CSNK2 and is not phosphorylated by any of the other kinases used. The amount of histidine-tagged TOMM22 served as loading control. (B) In vitro radio-isotope-assisted phosphorylation of TOMM22 is less efficient by muscle lysates of csnk2b∆/∆, HSA-Cre mice in comparison with muscles of control litters. The amount of recombinant TOMM22 was adjusted by measuring the total protein amount and verified by Coomassie-stained SDS-PAGE. (C) Alignment of mouse and yeast TOMM22/Tom22 primary structure stretches. Potential mouse TOMM22 phosphosites in comparison with phosphosites in yeast Tom22 are depicted by asterisks. The panel also depicts kinase prediction scores for mouse phosphosites serine 15, threonine 43 and serine 45, obtained as potential CSNK2 target sites with ScanSite 3, Disphos 1.3, NetPhosK 1.0 and NetPhos 2. The target sequence of CSNK2 is known to be represented by [S-X-X-(D/E/pS/pY)].Citation39 (D) In vitro phosphorylation experiments were performed with purified mouse TOMM22 wild-type protein and its alanine mutants together with recombinant CSNK2 using radiolabeled ATP. (E) T7 tagged TOMM22 wild-type and alanine-mutant expression plasmids were transfected into cultured cells, protein lysates immunprecipitated by a T7-specific antibody, precipitates were resolved by SDS-PAGE, and western blot membranes incubated with either a T7 or a TOMM22-p-S15-specific antibody. Note, the TOMM22-p-S15-specific antibody detects wild-type TOMM22, but not the TOMM22S15A or TOMM22S15A,T43A mutants. (F) The amount of catalytic activity-containing CSNK2A1 and CSNK2A2 subunits was determined in the absence of Csnk2b in skeletal muscle fiber lysates. Skeletal muscles soleus and tibialis anterior were used from approximately 2- (n = 3 mice per genotype) and 6- to 8-mo-old (n = 3 mice per genotype) mice. Obviously, CSNK2B protein is absent in csnk2b∆/∆, HSA-Cre muscle lysates. ACTN2 served as loading control. (G and H) Graphs represent protein amounts of CSNK2 subunits which were analyzed before by western blot (F). Note, in response to the absence of Csnk2b, protein amounts of CSNK2A1 and CSNK2A2 subunits are delicately balanced and seem to be adjusted in a muscle-type specific manner. (I) To determine the capacity of mitochondria for importing precursor proteins the in organello import assay with radiolabeled precursor proteins was used.Citation72 [35S]-radiolabeled yeast proteins Cox4, Mdh1 and Atp2 were individually imported into mitochondria (Δψ, membrane potential). Mitochondria were treated with proteinase K and analyzed by SDS-PAGE. p, precursor; m, mature. Import into mitochondria after the longest import time was set to 100% (control). Note, all mitochondrial precursor (p) proteins are imported into mitochondria and processed to shorter mature (m) size with similar time kinetic between control and the csnk2b∆/∆, HSA-Cre indicating mitochondrial protein import in vitro being unaffected. (J) Mitochondria were isolated from skeletal muscles of adult wild-type or csnk2b∆/∆, HSA-Cre mice and equal amounts of native TOMM complexes were resolved by blue-native gels (BN) which are used to separate native protein complexes. After western blot, different TOMM family members within native TOMM complexes were detected by specific antibodies as shown by representative images. The amount of these TOMM family members is similar between TOMM complexes from mitochondria of wild-type or csnk2b∆/∆, HSA-Cre muscles, indicating proper biogenesis of TOMM complexes.
![Figure 3. CSNK2 phosphorylates TOMM22 and the absence of Csnk2b compromises CSNK2 catalytical activity and protein amount, but neither is required for TOMM complex biogenesis nor mitochondrial protein import. (A) Mouse TOMM22 was purified and used for in vitro radio-isotope-assisted phosphorylation with different kinases. The image of the autoradiogram shows that mouse TOMM22 was only phosphorylated in the presence of protein kinase CSNK2 and is not phosphorylated by any of the other kinases used. The amount of histidine-tagged TOMM22 served as loading control. (B) In vitro radio-isotope-assisted phosphorylation of TOMM22 is less efficient by muscle lysates of csnk2b∆/∆, HSA-Cre mice in comparison with muscles of control litters. The amount of recombinant TOMM22 was adjusted by measuring the total protein amount and verified by Coomassie-stained SDS-PAGE. (C) Alignment of mouse and yeast TOMM22/Tom22 primary structure stretches. Potential mouse TOMM22 phosphosites in comparison with phosphosites in yeast Tom22 are depicted by asterisks. The panel also depicts kinase prediction scores for mouse phosphosites serine 15, threonine 43 and serine 45, obtained as potential CSNK2 target sites with ScanSite 3, Disphos 1.3, NetPhosK 1.0 and NetPhos 2. The target sequence of CSNK2 is known to be represented by [S-X-X-(D/E/pS/pY)].Citation39 (D) In vitro phosphorylation experiments were performed with purified mouse TOMM22 wild-type protein and its alanine mutants together with recombinant CSNK2 using radiolabeled ATP. (E) T7 tagged TOMM22 wild-type and alanine-mutant expression plasmids were transfected into cultured cells, protein lysates immunprecipitated by a T7-specific antibody, precipitates were resolved by SDS-PAGE, and western blot membranes incubated with either a T7 or a TOMM22-p-S15-specific antibody. Note, the TOMM22-p-S15-specific antibody detects wild-type TOMM22, but not the TOMM22S15A or TOMM22S15A,T43A mutants. (F) The amount of catalytic activity-containing CSNK2A1 and CSNK2A2 subunits was determined in the absence of Csnk2b in skeletal muscle fiber lysates. Skeletal muscles soleus and tibialis anterior were used from approximately 2- (n = 3 mice per genotype) and 6- to 8-mo-old (n = 3 mice per genotype) mice. Obviously, CSNK2B protein is absent in csnk2b∆/∆, HSA-Cre muscle lysates. ACTN2 served as loading control. (G and H) Graphs represent protein amounts of CSNK2 subunits which were analyzed before by western blot (F). Note, in response to the absence of Csnk2b, protein amounts of CSNK2A1 and CSNK2A2 subunits are delicately balanced and seem to be adjusted in a muscle-type specific manner. (I) To determine the capacity of mitochondria for importing precursor proteins the in organello import assay with radiolabeled precursor proteins was used.Citation72 [35S]-radiolabeled yeast proteins Cox4, Mdh1 and Atp2 were individually imported into mitochondria (Δψ, membrane potential). Mitochondria were treated with proteinase K and analyzed by SDS-PAGE. p, precursor; m, mature. Import into mitochondria after the longest import time was set to 100% (control). Note, all mitochondrial precursor (p) proteins are imported into mitochondria and processed to shorter mature (m) size with similar time kinetic between control and the csnk2b∆/∆, HSA-Cre indicating mitochondrial protein import in vitro being unaffected. (J) Mitochondria were isolated from skeletal muscles of adult wild-type or csnk2b∆/∆, HSA-Cre mice and equal amounts of native TOMM complexes were resolved by blue-native gels (BN) which are used to separate native protein complexes. After western blot, different TOMM family members within native TOMM complexes were detected by specific antibodies as shown by representative images. The amount of these TOMM family members is similar between TOMM complexes from mitochondria of wild-type or csnk2b∆/∆, HSA-Cre muscles, indicating proper biogenesis of TOMM complexes.](/cms/asset/09be7583-a709-4f1c-b799-7fdb4755a5d7/kaup_a_1403716_f0003_b.gif)
Neither mitochondrial protein import, nor TOMM complex biogenesis is impaired in mitochondria isolated from Csnk2b ablated skeletal muscles
After we showed that CSNK2 phosphorylates TOMM22, we expected to detect impairments of mitochondrial protein import and TOMM complex biogenesis in mice similar to yeast.Citation9 We examined mitochondrial protein import by monitoring the fate of 3 different radiolabeled yeast mitochondrial precursor proteins, Cox4, Mdh1, and Atp2, after incubation with isolated functional mitochondria from adult control and csnk2b∆/∆, HSA-Cre muscles (I). To our surprise, mitochondrial protein import was not affected by the absence of Csnk2b (I).
In yeast, TOM complex biogenesis was also impaired in the absence of CK2.Citation9 Accordingly, we looked for TOMM complex biogenesis in mice by analyzing the amount of different TOMM complex protein members, TOMM20, TOMM22, and TOMM40 (J). To this end, we analyzed the TOMM complex from mitochondria of skeletal muscles of 2 and 6 to 8-mo-old wild-type or csnk2b∆/∆, HSA-Cre mice by blue native polyacrylamide gel electrophoresis (BN-PAGE) upon solubilization of mitochondria with Triton X-100 and compared the amount of specific TOMM proteins within the native TOMM complex by western blot (J). We did not detect any difference of protein band intensities for TOMM proteins 20, 22, and 40, between mitochondria originating from wild-type or csnk2b∆/∆, HSA-Cre muscles (J).
Removal of mitochondria in csnk2b∆/∆, HSA-Cre muscle fibers occurred by autophagy
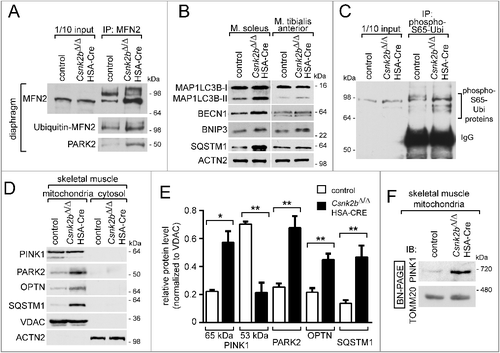
Our data indicate that protein homeostasis in mitochondria of csnk2b∆/∆, HSA-Cre muscle fibers from adult 6 to 8-mo-old mice is affected (C, D). We asked whether in the csnk2b∆/∆, HSA-Cre fibers defective mitochondria are selectively degraded by autophagy, a process termed mitophagy. This might explain the lower amount of mitochondrial proteins in csnk2b∆/∆, HSA-Cre muscle lysates from adult mice in comparison with controls (C, D). A marker for mitophagy, PINK1, is imported through the TOMM complex in healthy mitochondria and known to bind to the OMM of damaged mitochondria.Citation44 We analyzed whether PINK1 is involved in labeling of mitochondria in csnk2b∆/∆, HSA-Cre muscle cells. In fact, we observed an accumulation of the full-length PINK1 (65 kDa) in csnk2b∆/∆, HSA-Cre compared with control muscles, indicating that PINK1 labeled mitochondria for removal in csnk2b∆/∆, HSA-Cre muscle fibers (A, B). Accordingly, the amount of the processed PINK1 (53 kDa) was reduced in csnk2b∆/∆, HSA-Cre muscles (A, B). So far it is unclear, to which of the TOMM receptors PINK1 binds.Citation23,Citation33 Here, recombinant GST-tagged PINK1 was expressed in bacteria, purified, and used to pull down TOMM20, TOMM22, TOMM70, or TOMM40 (C). Importantly, PINK1 pulled down all of these TOMM proteins, albeit to a lower extent even TOMM40 (C). Regarding its topology, PINK1 has a predicted mitochondrial targeting signal (MTS) at its amino-terminal end, a transmembrane (TM) domain in its central part, and a kinase domain (KD) at its carboxy terminus.Citation45 Mapping the interacting epitopes of PINK1, we detected only its mitochondrial targeting signal (MTS) interacting with TOMM22 protein (D). We used PINK1-MTS to analyze its binding to different TOMM22 variants. We wondered whether the interaction between PINK1 and TOMM22 is influenced by the phosphorylation status of TOMM22 S15 and T43, as these residues are part of the cytosolic domain of TOMM22 and phosphorylated by CSNK2 (D). In fact, wild-type TOMM22 and TOMM22S15A,T43A bound similar amounts, while the phosphomimetic TOMM22 mutants bound almost 4-fold more PINK1 (E, F).
Figure 4. PINK1 accumulates as a full-length 65-kDa protein in csnk2b∆/∆, HSA-Cre diaphragms and preferentially binds to phosphomimetic TOMM22. (A) Representative immunoblot images showing that diaphragms of csnk2b∆/∆, HSA-Cre mice in comparison with controls contain less of processed PINK1 (53 kDa) and more of the full-length PINK1 (65 kDa). (B) Protein levels of unprocessed and processed PINK1 as shown in (A) were quantified by ImageJ and normalized to ACTN2. N = 3 mice per genotype. SDS-PAGE and western blot was repeated 3 to 5 times per protein lysate. (C) Representative western blot membrane images demonstrated interaction of PINK1 with all TOMM receptors, TOMM20, 22, 70, and the import channel TOMM40. GFP-T7 was used as a negative control. (D) Western blot membrane image demonstrated that the mitochondrial targeting sequence (MTS, spanning the area from amino acid 1 to 94) of PINK1 interacts with TOMM22. (E) Representative TOMM22 immunoblot images of GST affinity isolations. GST-PINK1-MTS was utilized to affinity isolate individually wild-type, inactive alanine- or phosphomimetic-TOMM22 mutants. Note, TOMM22S15D,T43D or TOMM22S15E,T43E bound significantly stronger to PINK1-MTS in comparison with wild-type TOMM22 or TOMM22S15A,T43A. N = 3 independent experiments. (F) Protein levels of wild-type and mutant TOMM22 as shown in (E) were quantified using ImageJ, normalized to 1/10 input. Note, PINK1 bound significantly more phosphomimetic TOMM22, than wild-type or alanine-mutant TOMM22 proteins.
Removal of mitochondria in csnk2b∆/∆, HSA-Cre muscle fibers might be monitored by several key mitophagy markers. As known, the dissipation of the mitochondrial membrane potential and labeling of impaired mitochondria by PINK1 and PRKN/PARK2 ensures ubiquitination of mitochondrial outer membrane proteins, like MFN2.Citation2 Significantly higher amounts of MFN2, PARK2, and ubiquitinated MFN2, were detected in diaphragm muscle protein lysates of csnk2b∆/∆, HSA-Cre compared with control 6 to 8-mo-old littermates (A). During autophagy, autophagosomes engulf cytoplasmic components, including cytosolic proteins and organelles. MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) is involved during autophagosome formation and recruited to phagophore membranes.Citation46 MAP1LC3B exists in 2 forms, the nonlipidated form I and the lipidated form II, the latter is anchored in phagophore and autophagosomal membranes. Here, we analyzed both MAP1LC3B forms and detected more of them in csnk2b∆/∆, HSA-Cre in comparison with control muscles (B). In accordance with these MAP1LC3B data, significantly higher BECN1/Beclin 1 amounts, a marker for localization of autophagic proteins to phagophores,Citation47 were detected in csnk2b∆/∆, HSA-Cre soleus muscle lysates (B). Yet another mitophagy marker family, BNIP3 (BCL2 interacting protein 3) and BNIP3L (BCL2 interacting protein 3 like), are proteins involved in induction of cell death and autophagy.Citation48 Indeed, BNIP3 levels were increased in csnk2b∆/∆, HSA-Cre soleus muscles (B). We also found SQSTM1, an autophagic receptor protein which is a well-known substrate of the autophagy-lysosome system, to be increased in muscles of csnk2b∆/∆, HSA-Cre mice (B). The increased amounts of these markers in csnk2b∆/∆, HSA-Cre mice fed ad libitum suggested an impaired autophagy eventually due to a block of the autophagic flux. Ubiquitin is phosphorylated at S65 by PINK1 to activate PRKN/PARK2.Citation28,Citation29 By analyzing muscle lysates of control and csnk2b∆/∆, HSA-Cre mice significant higher amount of phospho-S65 ubiquitinated proteins were detected in csnk2b∆/∆, HSA-Cre lysates (C). To further focus on PINK1- and PARK2-mediated autophagy mitochondria were isolated from skeletal muscles of adult control and csnk2b∆/∆, HSA-Cre mice, lysed, and analyzed by SDS-PAGE and western blot (D, E). Accumulated amounts of PARK2, the mitophagy receptor OPTN, and SQSTM1 were detected and quantified in mitochondria which were isolated from the csnk2b∆/∆, HSA-Cre (D, E). It is known that mitochondria sustaining damage, accompanied by loss of membrane potential, accumulate PINK1 on their outer membrane rather than import and proteolyse PINK1. In this context, a 700-kDa PINK1 complex was previously reported.Citation33 We analyzed the 700-kDa PINK1-complex formation in mitochondria isolated from skeletal muscles of control and csnk2b∆/∆, HSA-Cre mice. BN-PAGE immunoblot analysis demonstrated significantly more PINK1 accumulated on the outer membrane of mitochondria (700-kDa PINK1 complex) isolated from csnk2b∆/∆, HSA-Cre muscles (F). Taken together, these results reveal that endogenous imported PINK1 accumulates into a 700-kDa PINK1 complex on the outer membrane of mitochondria which were isolated from adult 6- to 8-mo-old csnk2b∆/∆, HSA-Cre mice.
Figure 5. Mitochondria from csnk2b∆/∆, HSA-Cre muscles display impaired mitophagy and accumulation of endogenous and radiolabeled PINK1 at mitochondrial outer membranes. (A) Representative immunoblot images of muscle lysates after immunoprecipitation of MFN2, SDS-PAGE, and western blot are shown. Note, more ubiquitinated MFN2 was detected in csnk2b∆/∆, HSA-Cre diaphragm, and more PARK2 coprecipitated with MFN2 from csnk2b∆/∆, HSA-Cre muscle lysates. (B) Representative immunoblot images of muscle lysates from control and csnk2b∆/∆, HSA-Cre mice probed with antibodies labeling proteins implicated in autophagy, are shown. (C) Phospho-S65-ubiquitinated proteins from diaphragm muscle lysates from control and csnk2b∆/∆, HSA-Cre mice were immunoprecipitated with a phospho-S65-ubiquitin-reactive antibody. The representative immunoblot image shows higher amounts of phospho-S65 ubiquitinated proteins in muscle lysates from csnk2b∆/∆, HSA-Cre mice in comparison with control controls. (D) Mitochondria were isolated from skeletal muscle, lysed and subjected to immunoblot analysis. Cytosolic lysates served as controls, are shown on the same blots, and demonstrate the quality of the mitochondrial fraction. Note, these data confirm the increase of 65-kDa PINK1 in csnk2b∆/∆, HSA-Cre like shown in A where whole diaphragm lysates instead of isolated mitochondria were used. (E) Quantifications of immunoblots, like the representative one shown in (D). N = 3 mice per genotype. Note, isolated mitochondria of csnk2b∆/∆, HSA-Cre muscles possess more of the 65 kDa PINK1, PARK2, OPTN, and SQSTM1. (F) Immunoblot analysis of PINK1 isolated from mitochondria and resolved by BN-PAGE. Note, significantly higher amounts of PINK1 are part of a quaternary 700-kDa PINK1 complex in mitochondria isolated from csnk2b∆/∆, HSA-Cre muscle.
Autophagic flux is blocked in skeletal muscle Csnk2b cKO muscles
Autophagy is also detectable by formation of SQSTM1-associated cytosolic aggregates.Citation49,Citation50 To find out whether csnk2b∆/∆, HSA-Cre muscle cells contain aggregates associated with SQSTM1, we immunostained frozen muscle cross sections of oxidative soleus and glycolytic tibialis anterior muscles of adult 6- to 8-month-old control and csnk2b∆/∆, HSA-Cre mice with a SQSTM1-reactive antibody (A). SQSTM1-associated aggregates were significantly accumulated in the cytosol of csnk2b∆/∆, HSA-Cre muscle cells (A, B). Additional evidence for the occurrence of mitophagy in csnk2b∆/∆, HSA-Cre in comparison with control muscle fibers was supported by colocalization of the markers SQSTM1 with TOMM20 on cross sectioned muscle fibers (Fig. S1). The occurrence of mitophagy in the absence of Csnk2b in muscle fibers was further confirmed by electron microscopy (C). Autophagosomes containing mitochondria were accumulated in csnk2b∆/∆, HSA-Cre muscle fibers in comparison with controls (C). In fact, in both csnk2b∆/∆, HSA-Cre soleus and extensor digitorum longus muscles, the number of autophagosomes that contained mitochondria was significantly higher compared to controls (D). Further, mitochondria were rarely localized in lysosomes of csnk2b∆/∆, HSA-Cre muscle fibers by detecting colocalization of LAMP2 (lysosomal-associated membrane protein 2) with TOMM20 by immunofluorescence staining of muscle cross sections (Fig. S1).
Figure 6. A block of mitophagy flux is involved in the accumulation of SQSTM1-associated cytosolic aggregates in csnk2b∆/∆, HSA-Cre muscles of adult mice. (A) Representative images of SQSTM1-immunostained soleus and tibialis anterior muscle cross-sections. High-resolution images of individual muscle fibers are shown on the right. Note, a higher number of SQSTM1-associated accumulations in cross-sectioned csnk2b∆/∆, HSA-Cre muscle fibers were visible. N = 3 mice per genotype. (B) Graph shows the quantification of grey sum fluorescence intensities of SQSTM1-immunostained cross-sections using ImageJ. (C) Representative electron microscopy images of csnk2b∆/∆, HSA-Cre muscle fibers showing autophagosomes containing mitochondria (capital ‘M’). Mitochondria outside of vacuoles are labeled by a lowercase ‘m’. (D) Autophagosomes were quantified in control and csnk2b∆/∆, HSA-Cre muscle fibers and are represented as a graph. N = 3 mice per genotype; 5 to 10 fibers were used for quantification for each of the muscles. Data are presented as mean ± s.e.m. (E) Flux of mitophagy was analyzed by electroporation of a reporter plasmid (mt-mKEIMA) into flexor digitorum brevis muscles of adult control and csnk2b∆/∆, HSA-Cre mice, changes of fluorescent spectra were detected and summarized by the presented graph. (F) Representative immunoblot images of muscle lysates from fed and starved adult 6- to 8-month-old control and csnk2b∆/∆, HSA-Cre mice. After SDS-PAGE and western blot, membranes were probed with antibodies labeling proteins implicated in autophagy, namely MAP1LC3B and SQSTM1. (G) Quantification of protein bands as seen in (F) using Image J. For normalization, ACTN2 was used. N = 3 mice per genotype. (H) Representative immunoblot images of mitochondria isolated from muscles from 3 different mice per genotype after colchicine treatment. After SDS-PAGE and western blot, membranes were probed with antibodies labeling proteins implicated in autophagy, namely MAP1LC3B-II, SQSTM1, and PARK2. Note, the increased amount of PARK2 refers to impaired mitophagy in csnk2b∆/∆, HSA-Cre muscles, while high amounts of SQSTM1, even in the absence of colchicine, indicate a block of mitophagy flux. (I) Quantification of protein bands as seen in (H) by ImageJ. VDAC1/2 served as a loading control.
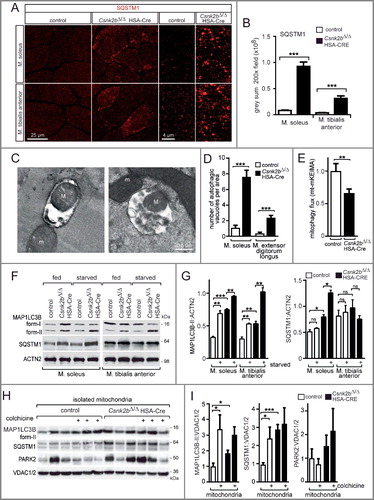
To monitor mitophagy-driven entry of damaged mitochondria into autolysosomes and their degradation, we used a mitochondrial targeted Keima (mt-mKEIMA) fluorescent protein which is sensitive to low pH and resistant to degradation by lysosomal hydrolases. Mt-mKEIMA fluoresces green at neutral pH in the cytosol, and red upon entry into acidic autolysosomes.Citation51 Importantly, electroporation of a plasmid encoding mt-mKEIMA into flexor digitorum brevis muscles of control and csnk2b∆/∆, HSA-Cre mice revealed a significant decrease of mitophagy flux in csnk2b∆/∆, HSA-Cre muscle fibers thereby indicating a block of autophagy flux (E). To understand more about the mitophagy in csnk2b∆/∆, HSA-Cre muscles, we decided to apply established paradigms, starvation and colchicine treatment to control and csnk2b∆/∆, HSA-Cre mice. First, we starved control and csnk2b∆/∆, HSA-Cre mice for 24 h, a well-characterized stimulus able to induce the formation of autophagosomes in muscles.Citation52 Fasting prompted autophagosome formation reflected by higher amounts of MAP1LC3B-II in control and, even more, in csnk2b∆/∆, HSA-Cre muscles which is more evident after normalization to ACTN2 (actinin alpha 2) and presentation of MAP1LC3B protein amounts by a graph (F, G). We assessed variations in protein amounts of SQSTM1 in the fed condition and during fasting (F, G).Citation49,Citation50 The increase of SQSTM1 in soleus muscle of fed CSNK2 mutant mice mirrored MAP1LC3B-I and -II levels, while in the glycolytic tibialis muscle an increase of SQSTM1 was not detectable (F, G). In csnk2b∆/∆, HSA-Cre muscles after starvation MAP1LC3B-I levels significantly decrease pointing to an increase of autophagosome formation (F, G). Second, we treated mice with colchicine, a drug that blocks the fusion of the autophagosome with the lysosome and thereby prevents the degradation of the autophagosome contents, including MAP1LC3B.Citation53 Mitochondria were isolated from 3 different mice per genotype and analyzed by SDS-PAGE and western blot for autophagy markers, MAP1LC3B-II, SQSTM1, and mitophagy marker PRKN/PARK2 (H, I). Colchicine treatment led to a marked increase in MAP1LC3B-II in isolated mitochondria of control and csnk2b∆/∆, HSA-Cre mice, while SQSTM1 levels in mitochondria isolated from csnk2b∆/∆, HSA-Cre mice were comparably high, like in colchicine-treated control and did not further change upon treatment, indicating a block of autophagy flux (H, I).
Phosphomimetic TOMM22 expression in skeletal muscle Csnk2b cKO muscle cells rescues mitochondrial protein amounts, restores oxygen consumption rates, and diminishes the number of SQSTM1-associated aggregates
In order to confirm the role of CSNK2-dependent mouse TOMM22 phosphorylation for mitochondrial homeostasis in vivo, we asked whether the introduction of phosphomimetic TOMM22 in csnk2b∆/∆, HSA-Cre muscle might reduce the number of SQSTM1-associated aggregates. At the same time, the expression of endogenous Tomm22 in the same muscle cells was knocked down by appropriate shRNAs complementary to Tomm22 3’-UTR (Fig. S2A). The efficiency of Tomm22 knockdown was measured with the help of a reporter plasmid, which was generated for this purpose and contained a luciferase gene fused to Tomm22 3’-UTR (Fig. S2A). Accordingly, decreased luciferase activity reflects the efficiency of Tomm22 knockdown (Fig. S2A). Additionally, we electroporated a CMV-driven Tomm22-T7 expression plasmid. After electroporation of this plasmid together with the shRNA-containing plasmid (Fig. S2A, no. 2), the expression of Tomm22-T7 was still detectable in hind limb muscle fibers (Fig. S2B). Next, a Tomm22-specific shRNA was electroporated together with a Tomm22 expression plasmid into the soleus muscle of mice. The electroporated areas of muscle fibers were detected by coelectroporation of a RFP-nls plasmid, which stained the nuclei of transfected cells (A). Electroporation of a wild-type Tomm22 expression plasmid into soleus muscle did not change the number of SQSTM1-associated aggregates (A). However, electroporation of the phosphomimetic TOMM22 protein did not change SQSTM1 immunostaining pattern in control, but significantly reduced number of SQSTM1-associated aggregates in csnk2b∆/∆, HSA-Cre soleus muscle (A, B).
Figure 7. Phosphomimetic and inactive TOMM22 proteins correlate with the number of SQSTM1-associated accumulations and oxygen consumption rates in Csnk2b ablated muscle cells. (A) Representative images of SQSTM1 immunostain of longitudinal sections of soleus muscle which were electroporated in vivo with phosphomimetic TOMM22 and shRNA transcribing plasmids to knockdown endogenous Tomm22. Note, a higher number of SQSTM1-associated accumulations was visible in csnk2b∆/∆, HSA-Cre soleus muscle fibers. This number decreased in csnk2b∆/∆, HSA-Cre muscle fibers which were electroporated with a phosphomimetic TOMM22 mutant (TOMM22S15E,T43E); but not after electroporation with wild-type TOMM22. Contemporary electroporation of an RFP-nuclear localization signal (nls) expression plasmid served for identification of the electroporated muscle fiber areas. (B) Quantification of the number of SQSTM1-associated accumulations in control or csnk2b∆/∆, HSA-Cre muscle cells which were electroporated with either wild-type or phosphomimetic TOMM22. N = 3 mice per genotype. Note that expression of phosphomimetic TOMM22 lowered the number of SQSTM1-associated accumulations in csnk2b∆/∆, HSA-Cre muscle fibers to almost wild-type levels. (C and D) Cultured C2C12 muscle cells were transfected with shRNA to knock down endogenous Tomm22 expression and with either wild-type or inactive Tomm22 expression plasmids. Cells were then immunostained for SQSTM1, DAPI and monitored for GFP expression encoded by shRNA transcribing plasmids. N = 3 independent experiments. (C). Note, the number of SQSTM1-associated aggregates is significantly higher in muscle cells expressing TOMM22S15A,T43A in comparison with wild-type TOMM22 (C). Number of SQSTM1-associated aggregates per cell was quantified and is depicted as graph (D). (E) Cultured primary muscle cells were untransfected or transfected with Tomm22 wild-type or Tomm22S15E,T43E expression plasmids, both together with shRNA plasmid to reduce endogenous Tomm22 expression, lysed, resolved by SDS-PAGE, western blotted, and membranes were immunostained with antibodies specific for different mitochondrial proteins. Representative images of immunostains demonstrated that the decreased mitochondrial protein amount in csnk2b∆/∆, HSA-Cre muscle cells, in comparison with controls, was rescued to normal values by transfection of a phosphomimetic TOMM22 mutant. (F) Quantification of protein bands as seen in (E) using ImageJ. All numbers were normalized to ACTN2. N = 3 independent experiments. (G) Graphs show oxygen consumption rates (OCR) in C2C12 cells which were transfected with expression plasmids encoding wild-type Tomm22 or inactive Tomm22S15A,T43A, together with GFP, and FACS sorted prior to OCR measurement by Seahorse methodology. N = 3 independent experiments. (H) OCRs measured with cultured primary muscle cells from wild-type or csnk2b∆/∆, HSA-Cre mice. Cells were transfected with expression plasmids as indicated, together with a GFP expression plasmid. Like in (G), transfected cells were FACS sorted by their fluorophore prior to Seahorse measurements. Note, constitutively expressed Cre recombinase was transfected to all cells to ensure deletion of floxed Csnk2b in csnk2b∆/∆, HSA-Cre muscle cells which occurred 48 to 72 h prior to the seahorse measurements.
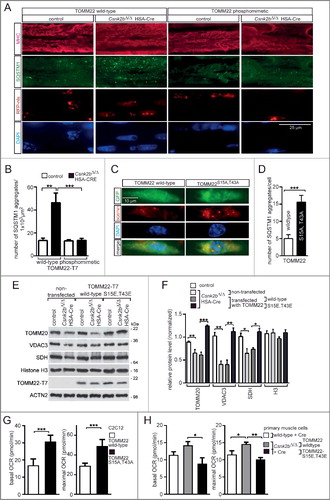
Vice versa, we would now expect that transfection of inactive TOMM22S15A,T43A should result in the increase of SQSTM1-associated aggregates in muscle cells. To this end, we cultured C2C12 cells and transfected them with shRNA to knockdown endogenous wild-type TOMM22 expression, and with an expression plasmid encoding either wild-type or inactive TOMM22 (C). Importantly, only after transfection of a plasmid encoding inactive, not wild-type, TOMM22 a significant increase of SQSTM1-associated aggregates was observed (C, D).
We asked whether phosphomimetic TOMM22 mutant protein is able to rescue the reduction of mitochondrial protein amount in csnk2b∆/∆, HSA-Cre muscle cells. Primary myoblasts were isolated from skeletal muscle tissue of control and csnk2b∆/∆, HSA-Cre mice, cultured, differentiated to myotubes, lysed, and monitored for expression of several mitochondrial proteins by SDS-PAGE and western blot (E). Differentiation of myoblast to myotubes was maintained for more than 10 days to ensure induction of HSA-Cre recombinase in cultured cells (data not shown). By comparison with control muscle cells, the amount of mitochondrial proteins was reduced in csnk2b∆/∆, HSA-Cre myotubes (E, F, 2C). After transfection of cultured myoblasts with phosphomimetic, but not wild-type, Tomm22 expression plasmid in csnk2b∆/∆, HSA-Cre cells and differentiation to myotubes no reduction of mitochondrial proteins in csnk2b∆/∆, HSA-Cre myotubes was observed (E, F).
Finally, we asked whether oxygen consumption rates (OCRs) are changed in muscle cells depending on whether they express wild-type, inactive, or phosphomimetic TOMM22 proteins; together with a GFP plasmid. First, OCRs from transfected and FACS sorted C2C12 cells were measured and compared depending on whether transfected expression plasmids encode wild-type TOMM22 or an inactive TOMM22S15A,T43A. OCRs were increased in C2C12 cells expressing TOMM22S15A,T43A in comparison with wild-type TOMM22 (G). Second, OCRs were measured using cultured primary muscle cells from wild-type or csnk2b∆/∆, HSA-Cre mice (H). csnk2b∆/∆, HSA-Cre muscle cells were also transfected with expression plasmids encoding either wild-type or the double-mutant, phosphomimetic TOMM22S15E,T43E. Additionally, all primary cultured muscle cells were cotransfected with GFP- and ubiquitous Cre- recombinase expressing plasmids prior to FACS sort. Importantly, while csnk2b∆/∆, HSA-Cre cells in comparison with the wild-type control showed the same OCR profile like C2C12 cells transfected with TOMM22S15A,T43A (G, H), csnk2b∆/∆, HSA-Cre cells OCR profile was restored and similar to wild-type cells by expression of phosphomimetic TOMM22 (H).
Altogether, introduction of phosphomimetic TOMM22 into csnk2b∆/∆, HSA-Cre muscle cells rescues both the increased number of SQSTM1-associated aggregates, the reduction of mitochondrial protein markers back to the physiological levels detected in control muscle cells, and their OCR profile.
Discussion
Previously, protein kinase CSNK2 has been identified as being crucial for the maintenance of NMJs.Citation35,Citation36 Both subunits of CSNK2 holoenzyme bind several protein members at NMJs and some of them are phosphorylated by CSNK2 catalytical activity.Citation35,Citation36 CSNK2 likely contributes to the maintenance of pretzel-shaped aggregated nicotinic acetylcholine receptors in adult skeletal muscles of mice.Citation35,Citation36 In the absence of Csnk2b in skeletal muscles, mice lose muscle grip strength in an age-dependent manner and neuromuscular junctions are fragmented.Citation35 But the severity and extent of muscle weakness of conditional muscle-specific Csnk2b knockout mice cannot be solely explained by impaired neural transmission because electrophysiological measurements detected small changes by recording strongly fragmented NMJs (A). Histological and functional studies of the csnk2b∆/∆, HSA-Cre mice demonstrated changes of oxidative metabolism (B, F, G, 7G, H).Citation40 COX staining showed that oxidative fibers were less intensely colored (B) and we wondered whether a muscle fiber type switch might have occurred in csnk2b∆/∆, HSA-Cre muscles. We stained muscles of soleus or tibialis anterior from control and csnk2b∆/∆, HSA-Cre mice with different marker proteins, but did not detect any fiber type change, neither by myosin heavy chain isoform composition, nor by using antibodies that are specific for different troponins (unpublished data). Accordingly, an extensive fiber type switch in csnk2b∆/∆, HSA-Cre muscles was unlikely.
Alternatively, less contractile activity might be the reason for the lower amount of mitochondria, but we failed to detect differences in voluntary walking distances between control and csnk2b∆/∆, HSA-Cre mice arguing against changes of contractile activity (E). Still, the amount of mitochondrial DNA and proteins were reduced in csnk2b∆/∆, HSA-Cre muscles of adult 6 to 8-mo-old mice (A to D). In addition, mitochondria of csnk2b∆/∆, HSA-Cre muscle cells were functionally compromised and unable to hold mitochondrial membrane potential after inhibiting their ATP synthase with oligomycin thereby blocking the generation of new ATP, and displayed lower respiratory control rates (F, G). Further evidence for dysfunctional mitochondria in csnk2b∆/∆, HSA-Cre muscles was provided by seahorse experiments demonstrating changes of oxygen consumption rates (H) and higher proton leak (data not shown). Of course, respiratory control rates and oxygen consumption rates are not comparable, the former refers to the respiratory control ratio (state 3/state 4), while the latter serves like an indicator for mitochondrial respiration. Both strategies confirmed proton leak as demonstrated by inability of csnk2b∆/∆, HSA-Cre fibers to hold mitochondrial membrane potential after inhibiting their ATP synthase with oligomycin (F, G, 7H). In fact, our data indicate that in some heavily impaired muscle fibers in csnk2b∆/∆, HSA-Cre muscles mitochondrial ATP synthase might function in reverse mode to ensure proper mitochondrial membrane potential, like it is reported in other mitochondrial pathologies.Citation54,Citation55
Generally, CSNK2 is known to phosphorylate many different proteins in cells.Citation39 Intriguingly, CK2 plays a role in mitochondrial protein import and TOM complex biogenesis in yeast by constitutively phosphorylating the main mitochondrial import receptor Tom22 at S44 and S46.Citation9,Citation10 We wondered whether CSNK2 plays a similar role in mice, like in yeast, regarding mitochondrial biology of skeletal muscle fibers. In fact, alignment of yeast and mouse Tom22/TOMM22 primary structures does not allow functional correlations, because the degree of identity is only ∼25% between these Tom22/TOMM22 proteins (C). Further, mouse TOMM22 does not possess serine residues at positions S44 and S46, like yeast Tom22 (C). Different algorithms predicted amino acid residues S15 and T43 as CSNK2 targets within mouse TOMM22 primary structure. We showed that recombinant mouse TOMM22 was phosphorylated (1) by CSNK2 in vitro (A), (2) by skeletal muscle lysates of control mice, but not by csnk2b∆/∆, HSA-Cre muscle lysates (B), and (3) at S15 in lysates of cultured cells (E). Previously, we observed an increased catalytical CSNK2 activity by incubating a CSNK2-specific target peptide with csnk2b∆/∆, HSA-Cre muscle lysates.Citation35 However, both in mutants and wild-type skeletal muscles, CSNK2 activity declined with age.Citation35 It has been speculated whether CSNK2B recruits CSNK2A1 to its target proteins to be phosphorylated.Citation35 Importantly, we now show that protein amounts of CSNK2A1 and CSNK2A2 are modulated in a muscle-type specific pattern in the absence of Csnk2b (F to H). Our data indicate that a changed CSNK2 protein amount correlates with different phosphorylation of CSNK2 targets.
Interestingly and in agreement with our data on phosphorylation of TOMM22 in mice, S15 was reported being both phosphorylated by phosphoproteomic studies, and a potential target of CSNK2.Citation56 Furthermore, quantitative phosphoproteomics studies suggest that the phosphorylation of S15 may be modulated when cells are treated with inhibitors of CSNK2 (D. Litchfield and A.J. Rabalski, unpublished data).
Obviously, we first supposed that CSNK2-dependent phosphorylation of TOMM22 might have similar roles as those in yeast, regulating TOMM complex biogenesis and mitochondrial protein import.Citation9,Citation10 But at second glance, Tom22 in yeast is also phosphorylated by CK1/CSNK1, and to lower degree by mammalian GSK3B and PRKACA/PKA, neither CK1 nor PKA are able to phosphorylate mouse TOMM22 (A),Citation9 and might indicate different regulation of Tom22/TOMM22 in yeast and mice. In accordance and in contradiction to the role of CSNK2-dependent TOMM22 phosphorylation in yeast, we demonstrated that neither TOMM complex biogenesis was impaired, nor mitochondrial protein import affected, in csnk2b∆/∆, HSA-Cre mice (I, J).
In Parkinson disease, PINK1 labels damaged mitochondria, to recruit PRKN/PARK2 and direct it to ubiquitinate MFN2 and VDAC1 (voltage dependent anion channel 1), with the consequence of increased mitophagic elimination.Citation20 Here, lack of CSNK2 in skeletal muscle cells changed the processing pattern of PINK1 (A, B; 5D, E). Moreover, we show that PINK1 interacts with all receptors (TOMM20, 22, 70) of the TOMM complex and also with the import channel protein TOMM40 (C). During mitochondrial protein import through the OMM, upon interaction with the initial receptors TOMM20 and TOMM70, the protein precursors designated for mitochondrial import are transferred to the central receptor TOMM22 and from there to the import channel TOMM40.Citation4 Apparently, the interaction between PINK1 and TOMM40 is less prominent, obviously because this is the import channel and not one of the exposed receptor proteins, which are likely required at early import steps. It is tempting to speculate that the interaction of PINK1 with the TOMM complex initiates by binding of PINK1 to phosphorylated TOMM22. The interaction of PINK1 with TOMM20 and TOMM70 might represent later steps during mitochondrial PINK1 protein import. Here, we present a model where weaker binding of PINK1 to TOMM22 eventually does not permit further steps of PINK1 protein import ending up in labeling of mitochondria for mitophagy (). PINK1-labeled mitochondria were removed by mitophagy involving typical markers, like PARK2, MFN2, MAP1LC3B, BECN1, BNIP3, SQSTM1, OPTN (A, B; 5A, B, D, E). The enrichment of MAP1LC3B-II was more prominent in the soleus muscle of 6 to 8-month-old csnk2b∆/∆, HSA-Cre mice (B), which fits to our assumption that mitochondria rich fibers, like type I and type IIa muscle fibers, are more affected in csnk2b∆/∆, HSA-Cre muscle cells. Further, our data indicate an impairment of the delivery of autophagosomes to lysosomes or autophagosome lysosome fusion in adult 6 to 8-mo-old csnk2b∆/∆, HSA-Cre muscle fibers because of the higher MAP1LC3B-II levels in comparison with controls. The additional increase of SQSTM1 levels in csnk2b∆/∆, HSA-Cre muscles might reflect early or late step block of the autophagic flux.Citation57 Altogether, increased MAP1LC3B-II, OPTN, and SQSTM1 protein amounts point to impairments in autophagosomes degradation in adult csnk2b∆/∆, HSA-Cre muscles. The decrease in mitophagy flux (F), the appearance of protein aggregates associated with SQSTM1 (A, B), and the increase of autophagosomes by electron microscopy (C, D) confirmed a defect of the fusion of autophagosomes with lysosomes. The decrease of mt-mKEIMA signal in csnk2b∆/∆, HSA-Cre mice indicates again a block in autophagic flux or dysfunctional lysosomes (E). Consistently, a higher number of autophagosomes detectable by electron microscopy usually points to a block of autophagic flux due to a failure to fuse autophagosomes with lysosomes. In summary, ablation of Csnk2b in skeletal muscle fibers causes autophagosome formation on mitochondria, mitophagy, but during time a suppression of autophagosome degradation. The decrease in mitochondrial content is due to a decrease in mitochondrial biogenesis (data not shown).
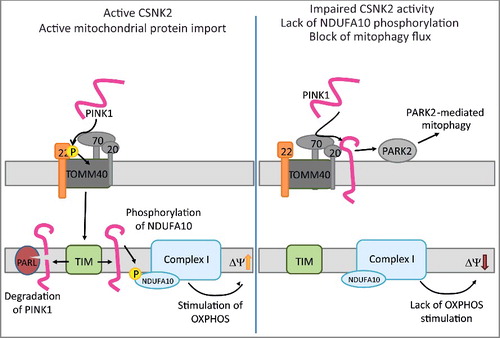
Figure 8. Sketch hypothesizes a mechanism for impaired mitochondrial homeostasis and mitophagy in wild type and csnk2b∆/∆, HSA-Cre muscle fibers. (Left) TOMM22 in its phosphorylated state facilitates import of PINK1 into the inner mitochondrial membrane. PINK1 in the inner membrane will be degraded by PARL, but also lead via phosphorylation of NDUFA10 to the stimulation of oxidative phosphorylation (OXPHOS) activity. Note, it remains to be determined whether PINK1 directly phosphorylates NDUFA10. (Right) In csnk2b∆/∆, HSA-Cre cells, lack of phosphorylated TOMM22 impairs PINK1 import and induce PARK2-mediated mitophagy. In addition, the lack of NDUFA10 phosphorylation will reduce OXPHOS activity and therefore lower the membrane potential of mitochondria. We speculate that accordingly loss of TOMM22 phosphorylation fosters PINK1 to accumulate on the outer membrane of mitochondria.
Our data might raise the question how mitophagy in adult csnk2b∆/∆, HSA-Cre muscles might cause mitochondrial dysfunction. Elevated mitophagy solely due to PINK1 accumulation via changes in TOMM22 phosphorylation appears insufficient to explain why the mitochondria are dysfunctional in csnk2b∆/∆, HSA-Cre muscle fibers. An alternative interpretation might be that functionally important mitochondrial proteins, not identified in this study, fail to import properly causing mitochondrial dysfunction. This might additionally promote PINK1 accumulation in adult csnk2b∆/∆, HSA-Cre mice. In accordance, the mitochondrial membrane potential deficits in csnk2b∆/∆, HSA-Cre muscle (F, G, 7H) might contribute to PINK1 accumulation.
We speculate to consider consecutive stages in csnk2b∆/∆, HSA-Cre muscles. First, up to 2 or 3 mo of age, csnk2b∆/∆, HSA-Cre mice do not show any phenotype (B).Citation35,Citation40 This might be because csnk2b∆/∆, HSA-Cre muscle fibers are likely obtaining functional Csnk2b by frequently occurring fusions of existing muscle fibers with muscle satellite cells at young age. Note, HSA-Cre recombinase in csnk2b∆/∆, HSA-Cre mice is expressed in adult striated muscle fibers and embryonic striated muscle cells of the somites and heart, and not in muscle satellite cells.Citation37,Citation38,Citation58 Second, at adulthood (6- to 8-mo of age) and due to lack of Csnk2b, the CSNK2-dependent phosphoproteome profile of skeletal muscles of csnk2b∆/∆, HSA-Cre mice will be affected. Further, muscle mitophagy is not compromised by lack of CSNK2-dependent TOMM22 phosphorylation, but the degradation of the cargo is impaired suggesting an eventual lysosomal dysfunction or a problem in docking of the autophagosome to lysosome. Interestingly, fasting was able to reactivate autophagy flux in tibialis anterior muscle similar to COL6 (collagen type VI) muscular dystrophies (F, G), suggesting that some stress conditions are able to reactivate autophagy in csnk2b∆/∆, HSA-Cre muscle.Citation59
We asked whether dysfunctional mitochondria and impaired mitophagy due to lack of CSNK2-dependent TOMM22 phosphorylation in csnk2b∆/∆, HSA-Cre muscle fibers might be restorable by phosphomimetic TOMM22 mutants. As proof of concept, the muscular SQSTM1-associated aggregates which accumulate in the absence of Csnk2b disappeared after electroporation of phosphomimetic TOMM22 into csnk2b∆/∆, HSA-Cre soleus muscle in vivo (A, B). Importantly, the simultaneous introduction of inactive TOMM22S15A,T43A into skeletal muscle cells and knockdown of endogenous wild-type TOMM22 expression induced SQSTM1-associated aggregate formation (C, D). These data argue that changes of CSNK2-dependent phosphorylation status of TOMM22 are involved in mitophagy; and may be TOMM22 even plays a role in general autophagy by yet unknown mechanisms because phosphomimetic TOMM22 is able to rescue csnk2b∆/∆, HSA-Cre muscle cell phenotype (see above). Recently, it has been shown that PINK1 loss-of-function mutations affect mitochondrial complex I activity via NDUFA10 (NADH:ubiquinone oxidoreductase subunit A10).Citation60 Here, we present a model summarizing our data and we speculate that impaired PINK1 mitochondrial import in csnk2b∆/∆, HSA-Cre muscle fibers might be additionally affecting mitochondrial protein import for some proteins, not investigated within this study, due to lack of PINK1-dependent NDUFA10 phosphorylation and decreasing mitochondrial membrane potential (). Interestingly, respiratory control rates from mitochondria in the muscle fibers of csnk2b∆/∆, HSA-Cre in comparison with control mice were significantly decreased after addition of substrates glutamate and malate entering respiratory chain via complex I, but the decrease was less prominent after block of complex I by rotenone and addition of substrate succinate for complex II further supporting a potential involvement of NDUFA10 (G, ).
It has been reported that CK2 mediates mitophagy and mitochondrial fragmentation in yeast by phosphorylation of Atg32, the yeast ortholog of BCL2L13 (Bcl2-like 13) in mammals,Citation61,Citation62 increasing complexity and offering yet another mechanism responsible for the control of mitophagy by CSNK2. Even more important was the demonstration that in vitro in HeLa cells CSNK2 phosphorylates FUNDC1 (FUN14 domain containing 1), an important mitophagy receptor to reverse the effect of the PGAM5 phosphatase in the activation of mitophagy.Citation63 Although these reports are very interesting, their value for understanding of impairments in csnk2b∆/∆, HSA-Cre mice remains to be investigated. Since Tom22/TOMM22 of yeast and mice are phosphorylated at different residues and result in different functional roles, CSNK2 might not phosphorylate BCL2L13 at the same residues, like in mice. Importantly, neither BCL2L13 nor FUNDC1 were quantified in phosphoproteomic studies to identify the CSNK2-dependent phosphoproteome (D. Litchfield and A.J. Rabalski, unpublished data). Further experiments are required to understand whether these findings play a role in vivo and whether FUNDC1 activity is changed in murine csnk2b∆/∆, HSA-Cre muscle.
To our knowledge, this is the first study demonstrating that in mammalian skeletal muscle cells protein kinase CSNK2 is involved in the regulation of mitochondrial homeostasis by being involved in the removal of damaged mitochondria through mitophagy. Up to now, PINK1 has been mainly studied in relation with Parkinson disease in the central nervous system. Our data presented here, might allow approaching principal questions about the function of TOMM proteins and PINK1 using skeletal muscle cells.
Materials and methods
Plasmids, primers, mutagenesis
Plasmids pRFP-nls or pMAX-GFP (Lonza, VPD-1001) were used as controls for transfection. RFP-nls was cloned into pCMV5-T7b (derivative of pCMV5 with T7 tag, D. Russell, University of Texas Southwestern) by using NotI and EcoRI to introduce the 3’ nuclear localization signals of RPS6/S6 ribosomal subunit, and EcoRI and NheI to place the RFP (pDS-Red N1; Clontech Laboratories, 632429) in frame 5’ of the nuclear localization signal, similarly previously GFP-nls was generated.Citation64 Cloning of mouse Tomm22 (NM_172609.3) was performed by extracting RNA from wild-type hindlimb muscle using TRIzol Reagent (Thermo Fisher Scientific, 15596026) according to the manufacturer's instructions, reverse transcribed with M-MuLV Reverse Transcriptase (New England Biolabs, M0253), amplified by PCR (for primers see below), and cloned in EcoRV-digested pBluescript SK+ (Agilent Technologies, 212205). After digestion of murine wild-type Tomm22 using primer-specific restriction sites, it was recloned in pET19b (Merck, 69677). Mutations for alanine, aspartate, or glutamate substitutions of TOMM22 amino acid residues serine 15, threonine 43 and serine 45 were introduced by Q5 Site-Directed Mutagenesis Kit (New England Biolabs, E0554S). Plasmids were transformed in E. coli bacteria NEB 5-alpha (New England Biolabs, C2992) or Xl10-GOLD (Agilent Technologies, 200314) and extracted by alkaline lysis with Nucleobond PC100 Midiprep kits (Macherey-Nagel, 740573). For generation of GST fusion proteins, full-length mouse Vdac1 (NM_011694.4), Tomm7 (NM_025394.3), Slc25a31 (NM_178386.3), Cox5b (NM_009942.2), Mdh2 (NM_008617.2), Pink1 (NM_026880.2) and Pink1 epitopes were amplified by PCR and cloned in pGEX 4T1 (GE Healthcare Life Sciences, 28-9545-49). Mouse full-length T7-tagged Tomm22 was amplified by PCR and cloned in pcDNA3 (currently available as pcDNA3.1; ThermoFisher Scientific, V79020). Mouse Tomm20 (NM_024214.2), Tomm40 (NM_001109748.1) and Tomm70 (NM_138599.5) were amplified by PCR and cloned into pCMV5 with T7 tags. To knock down gene expression of endogenous Tomm22, 3 different shRNA were designed complementary to 3’ UTR of mouse Tomm22 using BLOCK-iT™ RNAi Designer (Thermo Fisher Scientific; https://rnaidesigner.thermofisher.com /rnaiexpress/) and subcloned in pSUPERneoGFP (Oligoengine, VEC-PBS-0004) using restriction enzyme sites HindIII and BglII. To check efficiency of knock down, full size 3’UTR of mouse Tomm22 was amplified by PCR (for primers see below) and subcloned in pCMX pl1 luciferase at the 3’ end of the luciferase gene using restriction enzymes XhoI and KpnI.Citation35 For constitutive Cre recombinase expression pPGK-Cre-bpA (addgene, 11543) was nucleofected into csnk2b∆/∆, HSA-Cre primary muscle cells.
All genes and proteins names conform to the recommendations of the HUGO (human) gene nomenclature committee, the Mouse Genome Informatics guidelines, or the SGD (yeast) database.
DNA preparation and qPCR
To detect relative mitochondrial copy number, total DNA was extracted from hind limb muscles.Citation65 Lysis of the tissue was performed by adding the same volume of a solution composed of 2 mg/ml proteinase K (Carl Roth, 7528.3), 0.5% sodium dodecyl sulfate (Carl Roth, CN30) and 10 mM EDTA, and incubating at 37°C for 2 h. The genomic DNA was precipitated with isopropanol, dissolved in H20, and subjected to quantitative RT-PCR with mouse-specific primers as listed below.
Tissue culture, culturing of primary muscle cells, transfection, luciferase assay, immunocytochemistry, oxygen consumption rate (Seahorse)
HEK 293 cells were cultured in DMEM/F-12 (Thermo Fisher Scientific, 31331-028) with 10% fetal calf serum (FCS; Thermo Fisher Scientific, 10270-106) and 1% penicillin/streptomycin (Thermo Fisher Scientific, 15140-163). Cells were transfected with 8 µg DNA, 400 µl DMEM and 30 µl 1xPEI (Sigma-Aldrich Chemie, 408727). Forty-eight h after transfection cells were lysed in 10 mM HEPES, 400 mM NaCl, 10 mM KCl, 1% NP 40 (AppliChem, A1694), 0.2 mM EDTA, 2 mM DTT (Applichem, A1101), 10 µg/ml leupeptin (AppliChem, A2183), 10 µg/ml aprotinin (Carl Roth, A162.3).
To check knockdown efficiency of Tomm22 shRNAs, HEK293 cells extracts were prepared 48 h after transfection and luciferase assays performed, as described previously.Citation35
Primary skeletal muscle cells were prepared from leg muscles of neonatal mice. The tissue was minced with a razor blade, dissociated with collagenase type IV (Sigma-Aldrich Chemie, C5138) and Dispase II (Roche Diagnostics, 04942078001) in Mg2+- and Ca2+-free DPBS buffer (Thermo Fisher Scientific, 14190). Digestion was stopped by adding FCS. After filtering through a cell strainer (Thermo Fisher Scientific, 352350) and centrifugation at 350 x g for 10 min, cells were resuspended in 80% Ham F10 (Ca2+-free) (PAN-Biotech, P04-13251), 20% FCS, 1% penicillin/streptomycin, and recombinant human FGF (fibroblast growth factor) (5 ng/ml; Promega Corporation, G507A). Subsequently, cells were seeded on Matrigel-coated plates (Thermo Fisher Scientific, CB-40234). After 24 h, the culture medium was replaced with 40% DMEM (Thermo Fisher Scientific, 61965), 40% Ham F10, 20% FCS, 1% penicillin/streptomycin, and recombinant human FGF (5 ng/ml). For nucleofections, 5 × 105 primary skeletal myoblasts were used according to the manufacturer's instructions (Lonza, VPD-1001). Transfection efficiency was determined after 24 h. After primary skeletal muscle cells reached confluency their medium was replaced for differentiation medium (98% DMEM, 2% horse serum [Gibco, 26050-088], 1% penicillin/streptomycin).
For Seahorse experiments, EGFP-positive C2C12 and primary muscle cells were sorted 48 h after nucleofection with a MoFlo cell sorter (Dako, Glostrup, Denmark) and kept in the proliferation medium for another 72 h prior to the assay. Bioenergetics of C2C12 and primary muscle cells were determined using the XFe96 Extracellular Flux Analyzer (Seahorse Bioscience/Agilent Technologies, North Billerica, MA, USA). Cells were seeded in cell culture microplates coated with Cell-TAK (Sigma Aldrich Chemie, DLW354240) at a density of 20,000 cells/well and adhered for 90 min. One h before the measurement cells were incubated at 37°C in a CO2-free atmosphere. For the determination of respiratory parameters basal oxygen consumption rate (OCR, indicator for mitochondrial respiration) was measured. Next, responses toward the subsequent addition of oligomycin (1 µM; Sigma Aldrich Chemie, 75351), FCCP (1 µM; Sigma Aldrich Chemie, C2920) and the combination of antimycin A (3 µM) and rotenone (3 µM; Agilent Technologies, 103015-100; XF Cell Mito Stress Test Kit) were evaluated allowing for the calculation of basal and maximal respiration as well as proton leak.
To generate oxidative stress, primary myoblast cells from wild-type and csnk2b∆/∆, HSA-Cre murine muscles were differentiated to myotubes and treated with H2O2 for 30 min at 37°C. The number of apoptotic cells was counted after TUNEL assays were performed with the ApopTag® Red In Situ Apoptosis Detection Kit (Merck, S7165). Cells were fixed in 4% paraformaldehyde for 10 min at room temperature, permeabilized with 0.5% Triton X-100 (Carl Roth, 3051) in PBS (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4), incubated in a solution of ethanol:acetic acid (2:1) for 5 min at -20°C, incubated 10 sec with equilibration buffer, incubated for 1 h at 37°C with the TdT Enzyme, and the reaction was then stopped with the Stop/Wash Buffer. Finally, the cells were incubated overnight at 4°C with the anti-digoxigenin-rhodamine conjugate. Next day, cells were washed, stained with DAPI, and mounted with Mowiol (Merck, 475904).
Mice, genotyping, tissue sections, histochemical stainings, immunohistochemistry, fasting, autophagic flux, in vivo electroporation
Mouse mating and genotyping were performed as described previously.Citation35,Citation66 In brief, the generation of conditional knockout Csnk2b mice and their genotyping by PCR were reported by Buchou et al.Citation66 Reporter mice expressing bacterial cre recombinase under the control of the human skeletal actin (ACTA1) promoter were reported by Leu et al.Citation38 Striated muscle conditional knockout Csnk2b mice were generated by introducing HSA-Cre reporter into homozygous mice with floxed Csnk2b alleles and genotyped by PCR as described by Cheusova et al.Citation35 Conditional Mito-EGFP (R26R-Mito-EGFP) mice (RIKEN Center for Developmental Biology, CDB0216K; http://www.cdb.riken.jp/arg/reporter_mice.html) were already described.Citation41 For histochemical and immunofluorescence analysis, all muscles were quick-frozen in prechilled isopentane.
Muscles were cryotome-sectioned (10 μm), permeabilized for 5 to 10 min in PBS with 0.1 to 0.3% Triton X-100, blocked in 10% FCS, 1% BSA (Carl Roth, 8076) for 1 h. Cryotome sections were either used for histochemical or for immunofluorescence stainings. Sections were embedded in DPX or mowiol.
Hematoxylin and eosin staining: sections were incubated 15 min in Mayer hemalum solution (Merck, 109249), washed 10 min in tap water, dipped 6 times in a solution containing 96% ethanol and 4% HCl, 10 min in tap water, 1 min in 70% ethanol, 2 min in Eosin (Merck, 115935), 1 min in 100 % ethanol. COX: Sections were incubated 60 min at 37°C in a solution containing 50 mM phosphate buffer, pH 7.4, 3,3-di-aminobenzidinetetrahydrochloride (DAB; Sigma Aldrich Chemie, D8001), catalase (20 μg/ml; Sigma Aldrich Chemie, S41168), sucrose and CYCS/cytochrome c (Sigma-Aldrich Chemie, C2037). Afterwards they were washed in H2O and embedded.
The following antibodies were used for staining at indicated dilutions: anti-SQSTM1 (PROGEN Biotechnik, GP62-C; 1:500), anti-T7 (Merck, 69522; 1:1,000), anti-LAMP2 (Developmental Studies Hybridoma Bank, ABL-93; 1:1,000), anti-MYH7/slow skeletal myosin heavy chain (Abcam, clone NOQ7.5.4D; 1:2,000). Secondary antibodies conjugated to Cy5, Cy3, and Alexa Fluor 488 immunofluorescent dyes (Dianova, 111-165-144, 115-165-146, 112-165-143) were used for detection. Staining with the secondary antibody without previous incubation with the primary antibody served as a negative control.
For starvation experiments control mice had free access to food and water while food pellets were removed for starved mice. Autophagic flux was monitored in fed condition using colchicine (Sigma-Aldrich Chemie, C9754) as previously described.Citation67 Briefly, control and csnk2bΔ/Δ HSA-Cre mice were treated, by intraperitoneal injection, with vehicle or with 0.4 mg/kg colchicine. The treatment was administered twice, at 24 h and at 12 h before muscle dissection and isolation of mitochondria.
In vivo electroporation was adopted to the soleus muscle, but in principle performed as described previously.Citation68 In brief, the shaved skin of the left lower hind limb of the anesthetized animal was opened at the border between anterior and posterior hind limb muscles with a longitudinal cut of ∼1-cm length. The fascia was cut between the border of soleus and gastrocnemius muscle and an electrode was inserted below the soleus muscle. With a 10-µl microsyringe (World Precision Instruments, Nanofil), in total 5 µg of expression plasmids were injected into the exposed soleus muscle before placing the second electrode on top of the muscle. With an Electro Square Porator (ECM 830; Harvard Apparatus, MA, USA), 5 unipolar pulses with a voltage of 20 V, each with 20 ms duration and an interval of 221 ms, were applied. The electrodes were removed, and the wound was closed (Silkam DSM11, Braun, Melsungen, Germany) and sterilized. Antibiotic was injected as antiinflammatory prevention.
Mitochondria isolation, BN PAGE, muscle protein lysates, western blots, GST affinity isolation, immunoprecipitation, antibodies
Mice were sacrificed and different muscles (hind limb, diaphragm) dissected and used for mitochondrial isolation or frozen in liquid nitrogen and used later for muscle lysates preparation and immunoblotting. For mitochondrial isolation and BN PAGE for the members of the TOMM complex we used previously reported protocols.Citation69,Citation70 Pelleted mitochondria were solubilized in 50 µl ice-cold buffer (50 mM NaCl, 50 mM Imidazole-HCl, 1% Triton X-100, 5% [v:v] glycerol, 2 mM 6-aminohexanoic acid [Merck, 800145], 1 mM EDTA, pH 7.4 at 4°C) prior to addition of 5% (w:v) Coomassie Brilliant Blue G-250 (Serva Electrophoresis, 17524). BN PAGE was performed at 4°C using 4–13% gradient gels with 50 µg mitochondria loaded per lane. For SDS PAGE and immunoblotting, isolated mitochondria or muscles were homogenized in lysis buffer (10 mM HEPES, 400 mM NaCl, 10 mM KCl, 0.2 mM EDTA, 1% NP 40, 2 mM DTT, pH 7.9) with cOmplete protease inhibitors (Sigma-Aldrich Chemie, 04693116001) and phosphatase inhibitors (Sigma-Aldrich Chemie, P5726 and P0044). Skeletal muscle homogenates were sonicated for 10 sec and centrifuged at 16,100 g at 4°C for 5 min. Cleared lysate was used for immunoblotting experiments. Aliquots of mitochondrial or muscle lysates were solubilized in Laemmli buffer (150 mM Tris, pH 6.8, 6% SDS, 30% glycerol, 0.3% bromophenol blue, 3% ß-mercaptoethanol), boiled at 95°C, and loaded on 8% or 10% SDS-PAGE. Proteins were transferred to nitrocellulose membrane (Sigma Aldrich Chemie, Protran BA 85), blocked in 5% BSA (Carl Roth, 8076.4) or 5% nonfat dry milk (Heirler Cenovis, 3030) in PBS, 0.1% Tween20 (Carl Roth, 9127.1) for 1 h at room temperature.
For GST affinity-isolation assay, PINK1 and its epitopes were fused to GST and overexpressed in E. coli BL21-CodonPlus (DE3)-RP (Agilent Technologies, 230255). GST fusion proteins were harvested in 50 mM NaCl, 50 mM Tris, pH 7.6, 1 mM EDTA, 10% glycerol (Carl Roth, 3783), 2 mM DTT, 10 µg/ml leupeptin (AppliChem, A2183), 10 µg/ml aprotinin (Carl Roth, A162) and purified with GST beads (GE Healthcare Life Sciences, 17075601). Tom20, Tom22 (wild-type or mutant proteins), Tom40, and Tom70, all Tom proteins with T7 tag, were overexpressed in HEK293 cells and their interaction with recombinant GST fusion proteins was studied in 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.0, 300 mM NaCl, 2.7 mM KCl, 1% Triton X-100.
Immunoprecipitation was done with specific antibodies for overexpressed proteins from cell lysates or endogenous proteins from diaphragm muscle lysates. Antibodies were incubated with lysates overnight at 4°C. The next day, protein A or protein G Sepharose beads (GE Healthcare Bio-Sciences AB, 10009441 and 17061801) were added and incubation was continued for another 2 h. Afterwards, beads were washed 3 times, boiled in Laemmli buffer and loaded on SDS PAGE gel.
Primary antibodies were incubated at 1:1,000 dilution or as mentioned. Following antibodies were purchased from Cell Signaling Technology: Histone H3 (4499; 1:5,000), MAP1LC3B (2775), MDH2 (8610). Additionally used antibodies: from Santa Cruz Biotechnology Inc.: TOMM20 (sc-11415; 1:3,000), TOMM40 (sc-11414), SLC25A31/ANT4 (detects in mouse also SLC25A4/ANT1, SLC25A5/ANT2) (sc-11433), PRKN/PARK2 (sc-32282); from Sigma-Aldrich Chemie; ACTN2 (A 7811; 1:10,000); from Abcam: PRKN/PARK2 (ab15954), SQSTM1 (ab56416; 1:5,000), MFN2 (ab56889; 1:5,000), VDAC1/2/3 (ab15895; 1:5,000), SDHA (ab14715; 1:20,000); from Abnova/Biozol: TOMM22 (H00056993-M01); from Novus Biologicals: PINK1 (BC100-494); from Proteintech: OPTN (10837-1-AP); from Merck: anti-phospho-S65 Ubiquitin (ABS1513); from Enzo Life Sciences: Mono- and polyubiquitinated conjugates (FK2); from Novagen: T7 (69522; 1.10,000); anti-p-S15-AA (generated in the lab of Dr. Michael Marber, UK); anti-CSNK2A1 (1:500) and anti-CSNK2A2 (1:100), both generated in the lab of Dr. Olaf-Georg Issinger (Odense, Denmark). Note, the VDAC antibody detects all three VDAC isoforms. VDAC1/2 show a very similar migration electrophoretic pattern and are indistinguishable in the immunoblot. VDAC3 migrates faster and separately detectable by with the VDAC antibody by immunoblot.Citation71 Corresponding secondary antibodies conjugated with horseradish peroxidase (Cell Signaling Technology, 7074 and 7076; 1:3,000) were used for 2 h at room temperature. Protein bands were detected either by SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, 34095) or by homemade chemiluminescence reagent composed of 50 mg Luminol (Sigma Aldrich Chemie, A-4685) in 200 ml 0.1 M Tris, pH 8.6, combined with 11 mg para-hydroxy-cumarinic acid (Sigma Aldrich Chemie, C-9008) in 10 ml DMSO. For that, 3 ml of the first solution and 40 µl of the second solution were mixed with 3 ml of PBS and 1.2 μl 30% H2O2. Western Blot results were quantified by densitometric analysis using ImageJ software (NIH, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/). The background was subtracted with rolling ball radius 1,000 pixels and disabled smoothing option. Afterwards, protein bands of interest were labeled and measured. If not described specifically, values in mutant mice were expressed as relative values to control mice normalized to ACTN2 and set to 1.0.
In vitro phosphorylation assays, mitochondrial import assay
His-tagged yeast Tom22 was purified as described,Citation5 his-tagged cytosolic domain of mouse TOMM22 was produced in BL21 E.coli and the bacterial pellet was lysed in 20 mM Tris/HCl, pH 7.9, 300 mM NaCl, 10 mM β-mercaptoethanol, 2 mM MgCl2, 10 mM imidazole, 2 mM phenylmethylsulfonyl fluoride with addition of lysozyme, DNaseI and cOmplete protease inhibitors (Sigma Aldrich Chemie, 04693116001), and lysed using 3 freeze-thaw cycles in liquid nitrogen and pulsed sonication on ice using a Branson sonifier (G. Heinemann Ultraschall- und Labortechnik, model 250) (3/3/1 min at 80% duty). Filtered lysates were incubated with Ni-NTA (QIAGEN, 30210), washed with wash buffer (20 mM Tris-HCl, pH 7.9, 300 mM NaCl, 10 mM β-mercaptoethanol, 10 mM imidazole) and the bound protein was stepwise eluted with increasing amount of imidazole in the elution buffer (20 mM Tris-HCl, pH 7.9, 300 mM NaCl, 10 mM β-mercaptoethanol, 20–300 mM imidazole). Elutions with high protein content were determined by SDS-PAGE, pooled and concentrated using the Amicon filter system (Merck, UFC201024). Using the Amicon filter, the buffer was exchanged to contain 20 mM Tris-HCl, pH 7.2, 30 mM NaCl, 10% glycerol, and the purified proteins stored in aliquots at −80°C.
Muscle lysates were adjusted with lysis buffer (10 mM HEPES, pH 7.0, 400 mM NaCl, 10 mM KCl, 20 mM EDTA, 1% [v:v] NP 40, 2 mM DTT) to similar protein concentrations using adsorption at 280 nm and immunoblotting and quantification of ACTB (MultiGauge software, Fujifilm, Kleve, Germany).
For in vitro phosphorylation 6 µg TOMM22 were incubated with 20 µl of muscle lysate in assay buffer 20 mM Tris-HCl, pH 7.2, 30 mM NaCl, 10% (v:v) glycerol, 1 mM phenylmethylsulfonyl fluoride, 1x PhosStop (Sigma Aldrich Chemie, 04906845001) supplemented with 10 mM MgCl2 and 10 µCi [γ-33P]ATP for 30 min at 37°C. The reaction was stopped by the addition of 4x Laemmli buffer and analyzed by SDS-PAGE, autoradiography and western blot. Mammalian kinase PRKACA/PKA (P6000S) was purchased from New England Biolabs.
[35S]Methionine-labeled yeast precursor proteins (Cox4, Mdh1, Atp2) were synthesized in rabbit reticulocyte lysate (Promega Corporation, L4960), and levels were adjusted as necessary. Yeast Cox4, Mdh1 and Atp2 precursor proteins were incubated with isolated mitochondria in import buffer (20 mM HEPES, pH 7.4, 250 mM sucrose, 80 mM potassium acetate, 5 mM magnesium acetate, 10 mM sodium succinate) with 5 mM ATP (Carl Roth, K054).Citation72 Mitochondria were washed, treated with proteinase K, and analyzed by SDS-PAGE and digital autoradiography. Mitochondrial membrane potential was dissipated with the addition of 1 µM FCCP. Import reactions were performed at 24°C for 60 min. For BN PAGE, mitochondrial pellet (50 µg protein) was solubilized in 1% digitonin (Sigma Aldrich Chemie, 11024-24-1) solution containing 50 mM NaCl, 10% (v:v) glycerol, 20 mM Bis-Tris (Sigma Aldrich Chemie, B9754), pH 7.0 and loaded on 4–13% gradient gels. Radiolabeled proteins were detected by phosphorimaging (STORM 860, Molecular Dynamics, Ramsey, MN, USA).
Electron microscopy, fluorescence microscopy, confocal microscopy, 2-photon-microscopy, second harmonic generation microscopy
For electron microscopy, mice were fixed by perfusion with 2.5% glutaraldehyde in 100 mM phosphate buffer, pH 7.4. After dissection muscles were additionally fixed in osmium tetroxide. Muscles were then embedded in Epon (Serva Electrophoresis, 21045), stained with lead citrate and uranyl acetate. Electron microscopy of ultrathin sections was done using a ZEISS EM 6 microscope (Carl Zeiss MicroImaging, Göttingen, Germany). The number of autophagic vacuoles was quantified per surface area of a muscle fiber (200-µm fiber length and per average 20-µm fiber diameter).
For fluorescence microscopy, stained cryosections or muscle bundles were analyzed and documented using a Zeiss Axio Examiner Z1 microscope equipped with an AxioCam MRm camera and software ZEISS AxioVision Release 4.8 (Carl Zeiss MicroImaging, Göttingen, Germany). For quantitative analysis of SQSTM1, images of muscles were taken and analysed using ‘Automatic Measurement’. With the Command Manager a script was created for quantitative analysis (lower threshold level 0 and upper threshold level 1.190). Gray sum values were quantified for each genotype as mean value of >50 images for 3 mice per genotype (no grey sum value without SQSTM1 antibody was detected meaning that background subtraction was not necessary). Quantitative analysis of SQSTM1-associated aggregates on electroporated soleus longitudinal sections was performed by ImageJ. Electroporated muscle fiber area was determined, image was thresholded and number of SQSTM1-associated aggregates was calculated by the option ‘analyze particles’ of ImageJ.
For confocal microscopy, a Leica TCS-AS (Leica Microsystems, Wetzlar, Germany) confocal system with an inverted DM IRE2 microscope equipped with 2 spectral 12-bit PMT detectors and Leica Confocal software (version 2.6.1, build 1537) was used. Single muscle fibers were imaged using a multiphoton microscope (TriMScope II, LaVision BioTech, Bielefeld, Germany) with a mode-locked fs-pulsed Ti:Sa-laser (Chameleon Vision II, Coherent, CA, USA) at 900 nm and a combination of 2 water immersion objectives (LD C-APO 40x/1.1 W Corr M27 on the excitation side, W Plan-APO 20x/1.0 DIC M27 on transmission side, Zeiss). The SHG signal from myosinCitation73 and EGFP signal (2PE) are separated by a band pass filter (450/30 for SHG and 525/50 for EGFP, Semrock, NY, USA) and detected by a nondescanned photomultiplier (H7422-40, Hamamatsu Photonics, Hamamatsu, Japan). EGFP signal is measured by thresholding. Image processing was performed in MATLAB (MathWorks, Natick, MA, USA) and Fiji based on ImageJ (NIH, Bethesda, MD, USA).
Behavioral studies, mitochondrial membrane potential, mitophagy flux measurements
TSE ActiMot/MoTil TSE system (Bad Homburg, Germany) was used for activity analysis as described before.Citation74
Mitochondrial membrane potential was measured in isolated fibers from flexor digitorum brevis (FDB) muscles. FDB fibers were obtained as described previously.Citation75,Citation76 Mitochondrial membrane potential was measured by epifluorescence microscopy based on the accumulation of tetramethyl rhodamine methyl ester (TMRM) fluorescence as described before.Citation54 Briefly, FDB myofibers were placed in 1 ml Tyrode buffer (135 mM NaCl, 4 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 0.33 mM KH2PO4, 10 mM glucose, 10 mM HEPES, pH 7.3, supplemented with 0.3% collagenase type I [Sigma Aldrich Chemie, C01030] and 0.2% BSA) and loaded with 2.5 nM TMRM (Thermo Fisher Scientific, T668) supplemented with 1 µM cyclosporine H (a P-glycoprotein inhibitor; Enzo Life Sciences, ALX-380-28) for 30 min at 37°C. Myofibers were then observed using an Olympus IMT-2 inverted microscope (Melville, NY, USA) equipped with a CellR imaging system. Sequential images of TMRM fluorescence were acquired every 60 s with a 20 × 0.5, UPLANSL N A objective (Olympus, Hamburg, Germany). At specific times, oligomycin (5 µM) or the protonophore FCCP (4 µM) were added to the cell culture medium. Images were acquired, stored and analysis of TMRM fluorescence over mitochondrial regions of interest was performed using ImageJ software.
Mitophagy flux was measured in isolated transfected fibers from FDB muscles. In vivo muscle transfection was achieved by injection with mitochondria targeted-mKeima (mt-mKeima) (a gift from A. Miyawaki, RIKEN, Brain Science Institute, Japan) followed by in vivo electroporation as described earlier.Citation76 Twelve days after transfection, FDB fibers were obtained as described earlier.Citation75,Citation76 Measurements of mt-mKeima were made using dual-excitation ratiometric measurements at 488 and 561 nm lasers with 620/29 nm and 614/20 nm emission filters, respectively.
Muscle mitochondria were isolated as described.Citation69 Mitochondrial oxygen consumption was measured with a Clark-type oxygen electrode (Hansatech Instruments, Norfolk, UK) as described in the same protocol. Briefly, mitochondria (1 mg/ml) were incubated in Experimental Buffer (150 mM KCl, 10 mM Tris [pH adjusted with MOPS to pH 7.4], 10 μM EGTA-Tris, 10 μM ATP). When indicated, mitochondria were transferred into a Clark-type oxygen electrode chamber (Hansatech Instruments, model: Oxygraph) and 5 mM glutamate-2.5 mM malate or 2 μM rotenone-10 mM succinate were added.
Nerve-muscle preparations
Isolated diaphragm-phrenic nerve preparations were maintained in Liley solution (137,8 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1 mM KH2PO4, 2 mM CaCl2, 11 mM glucose) gassed with 95% O2-5% CO2 at room temperature.Citation77 The recording chamber had a volume of approximately 1 ml and was perfused at a rate of 1 ml/min. The nerve was drawn up into a suction electrode for stimulation with pulses of 0.1 ms duration. The preparation was placed on the stage of a Zeiss Axio Examiner Z1 microscope fitted with incident light fluorescence illumination with filters for red (Zeiss filter set 20) fluorescing fluorophore (Carl Zeiss MicroImaging, Göttingen, Germany). At the beginning of the experiment the compound muscle action potential was recorded using a micropipette with a tip diameter of ca. 10 μm, filled with bathing solution. The electrode was positioned so that the latency of the major negative peak was minimized. The electrode was then positioned 100 μm above the surface of the muscle and CMAP was recorded.
Intracellular recording and data analysis
To block muscle action potentials, so that EPCs could be recorded μ-conotoxin GIIIB (μ-CTX; Peptide Institute, 4217-v; 2 μM) was added to Liley solution.Citation78,Citation79 CHRNs were labeled by adding 0.5 × 10−8 M of rhodamine-α-bungarotoxin (Thermo Fisher Scientific, T1175) to the same solution. In some experiments, the effect of the toxin wore off after 1 to 2 h and contractions resumed in response to nerve stimulation. These preparations were then exposed a second time to the toxin.
Two intracellular electrodes (resistance 10 to 15 MΩ) were inserted within 50 μm of the NMJs under visual inspection for rhodamine-α-bungarotoxin-labeled CHRNs.Citation79 Current was passed through one electrode to maintain the membrane potential within 2 mV of -75 mV while voltage transients were recorded with the other. Signals were amplified by an Axoclamp 900A and digitized at 40 kHz by a Digidata 1440A under the control of pCLAMP 10 (Molecular Devices, Sunnyvale, CA, USA). Voltage records were filtered at 3 kHz and current records at 1 kHz (8-pole Bessel filter). Current transients were recorded using the 2-electrode voltage-clamp facility of the Axoclamp 900A. Clamp gains were usually 300 to 1,000, reducing the voltage transients to <3% of their unclamped amplitudes. At most NMJs, 50 to 100 spontaneous quantal events were recorded during a period of 1 min. Records were analyzed using pCLAMP 10. Spontaneous events were extracted using the ‘template search’ facility and edited by eye to remove obvious artefacts. Events recorded from each NMJ were averaged and the amplitude, rise time and single exponential decay time constant determined.
Statistical analysis
Data are presented as the mean values, and the error bars indicate ±s.e.m.. The number of biological replicates per experimental variable (n) is usually n>5 or as indicated in the figure legends. The significance is calculated by unpaired 2-tailed student t test, or as indicated by the figure legends, and provided as real P values that are believed to be categorized for different significance levels, ***P <0.001, **P <0.01, or *P <0.05.
Disclosure of potential conflicts of interest
The authors declare that they have no competing financial interests.
supp_data_1403716.zip
Download Zip (5.2 MB)Acknowledgments
We are grateful to Dr. Asushi Miyawaki for the kind gift of mt-mKeima. We thank Alexander Frick, Tassilo Wachsmann, and Nicolas Lamoureux for help regarding cloning of Tomm22, some of its mutants, and RFP-nls. BK received a scholarship as part of the program to promote equal opportunities for women in research and teaching (FFL) from the Friedrich-Alexander University of Erlangen-Nürnberg.
Additional information
Funding
Related Research Data
References
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi:10.1016/j.cell.2012.02.035. PMID:22424226
- Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014;19:357–372. doi:10.1016/j.cmet.2014.01.010. PMID:24561263
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi:10.1016/j.cell.2009.08.005. PMID:19703392
- Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. doi:10.1146/annurev.biochem.76.052705.163409. PMID:17263664
- Brix J, Dietmeier K, Pfanner N. Differential recognition of preproteins by the purified cytosolic domains of the mitochondrial import receptors Tom20, Tom22, and Tom70. J Biol Chem. 1997;272:20730–20735. doi:10.1074/jbc.272.33.20730. PMID:9252394
- Brix J, Rudiger S, Bukau B, Schneider-Mergener J, Pfanner N. Distribution of binding sequences for the mitochondrial import receptors Tom20, Tom22, and Tom70 in a presequence-carrying preprotein and a non-cleavable preprotein. J Biol Chem. 1999;274:16522–16530. doi:10.1074/jbc.274.23.16522. PMID:10347216
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi:10.1016/S0092-8674(02)01250-3. PMID:12526792
- Dolezal P, Likic V, Tachezy J, Lithgow T. Evolution of the molecular machines for protein import into mitochondria. Science. 2006;313:314–318. doi:10.1126/science.1127895. PMID:16857931
- Schmidt O, Harbauer AB, Rao S, Eyrich B, Zahedi RP, Stojanovski D, Schönfisch B, Guiard B, Sickmann A, Pfanner N, et al. Regulation of mitochondrial protein import by cytosolic kinases. Cell. 2011;144:227–239. doi:10.1016/j.cell.2010.12.015. PMID:21215441
- Gerbeth C, Schmidt O, Rao S, Harbauer AB, Mikropoulou D, Opalinska M, Guiard B, Pfanner N, Meisinge C. Glucose-Induced Regulation of Protein Import Receptor Tom22 by Cytosolic and Mitochondria-Bound Kinases. Cell Metab. 2013;18:578–587. doi:10.1016/j.cmet.2013.09.006. PMID:24093680
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi:10.1126/science.1219855. PMID:22936770
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi:10.1038/emboj.2012.38. PMID:22354038
- Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J. 2012;31:3038–3062. doi:10.1038/emboj.2012.170. PMID:22735187
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi:10.1038/nrm3028. PMID:21179058
- de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol. 2006;5:525–535. doi:10.1016/S1474-4422(06)70471-9. PMID:16713924
- Bekris LM, Mata IF, Zabetian CP. The genetics of Parkinson disease. J Geriatr Psychiatry Neurol. 2010;23:228–242. doi:10.1177/0891988710383572. PMID:20938043
- Burbulla LF, Kruger R. Converging environmental and genetic pathways in the pathogenesis of Parkinson's disease. J Neurol Sci. 2011;306:1–8. doi:10.1016/j.jns.2011.04.005. PMID:21513949
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Turco DD, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi:10.1126/science.1096284. PMID:15087508
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi:10.1038/33416. PMID:9560156
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi:10.1371/journal.pbio.1000298. PMID:20126261
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi:10.1093/hmg/ddq419. PMID:20871098
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012;13:378–385. doi:10.1038/embor.2012.14. PMID:22354088
- Kato H, Lu Q, Rapaport D, Kozjak-Pavlovic V. Tom70 is essential for PINK1 import into mitochondria. PLoS One. 2013;8:e58435. doi:10.1371/journal.pone.0058435. PMID:23472196
- Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9:1758–1769. doi:10.4161/auto.24633. PMID:24121706
- Fedorowicz MA, de Vries-Schneider RL, Rub C, Becker D, Huang Y, Zhou C, Alessi Wolken DM, Voos W, Liu Y, Przedborski S. Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep. 2013. PMID:24357652
- Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman-Ackah SM, Hessa T, Guha R, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013;504:291–295. doi:10.1038/nature12748. PMID:24270810
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou Y-S, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi:10.1083/jcb.200910140. PMID:20404107
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. PMID:24784582
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–153. doi:10.1083/jcb.201402104. PMID:24751536
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi:10.1083/jcb.201008084. PMID:21115803
- Becker D, Richter J, Tocilescu MA, Przedborski S, Voos W. Pink1 kinase and its membrane potential (Deltapsi)-dependent cleavage product both localize to outer mitochondrial membrane by unique targeting mode. J Biol Chem. 2012;287:22969–22987. doi:10.1074/jbc.M112.365700. PMID:22547060
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14:3477–3492. doi:10.1093/hmg/ddi377. PMID:16207731
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22:320–333. doi:10.1016/j.devcel.2011.12.014. PMID:22280891
- Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi:10.1042/bj20021469. PMID:12396231
- Cheusova T, Khan MA, Schubert SW, Gavin AC, Buchou T, Jacob G, Sticht H, Allende J, Boldyreff B, Brenner HR, et al. Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction. Genes Dev. 2006;20:1800–1816. doi:10.1101/gad.375206. PMID:16818610
- Herrmann D, Straubinger M, Hashemolhosseini S. Protein kinase CK2 interacts at the neuromuscular synapse with Rapsyn, Rac1, 14-3-3gamma, and Dok-7 proteins and phosphorylates the latter two. J Biol Chem. 2015;290:22370–22384. doi:10.1074/jbc.M115.647610. PMID:26198629
- Escher P, Lacazette E, Courtet M, Blindenbacher A, Landmann L, Bezakova G, Lloyd KC, Mueller U, Brenner HR. Synapses form in skeletal muscles lacking neuregulin receptors. Science. 2005;308:1920–1923. doi:10.1126/science.1108258. PMID:15976301
- Leu M, Bellmunt E, Schwander M, Farinas I, Brenner HR, Muller U. Erbb2 regulates neuromuscular synapse formation and is essential for muscle spindle development. Development. 2003;130:2291–2301. doi:10.1242/dev.00447. PMID:12702645
- Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? Faseb J. 2003;17:349–368. doi:10.1096/fj.02-0473rev. PMID:12631575
- Eiber N, Simeone L, Hashemolhosseini S. Ablation of protein kinase CK2beta in skeletal muscle fibers interferes with their oxidative capacity. Pharmaceuticals (Basel). 2017;10:13.
- Abe T, Kiyonari H, Shioi G, Inoue K, Nakao K, Aizawa S, Fujimori T. Establishment of conditional reporter mouse lines at ROSA26 locus for live cell imaging. Genesis. 2011;49:579–590. doi:10.1002/dvg.20753. PMID:21445964
- Schapira AH. Mitochondrial disease. Lancet. 2006;368:70–82. doi:10.1016/S0140-6736(06)68970-8. PMID:16815381
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Piccolo PD, Foretz M, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. Embo J. 2010;29:1774–1785. doi:10.1038/emboj.2010.60. PMID:20400940
- Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci. 2012;125:795–799. doi:10.1242/jcs.093849. PMID:22448035
- Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A. 2008;105:12022–12027. doi:10.1073/pnas.0802814105. PMID:18687899
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi:10.1093/emboj/19.21.5720. PMID:11060023
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi:10.1038/45257. PMID:10604474
- Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi:10.1038/cdd.2009.16. PMID:19229244
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi:10.1038/ncb2012. PMID:20098416
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6:1090–1106. doi:10.4161/auto.6.8.13426. PMID:20890124
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18:1042–1052. doi:10.1016/j.chembiol.2011.05.013. PMID:21867919
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi:10.1016/j.cell.2007.12.018. PMID:18191218
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi:10.1016/j.cell.2010.01.028. PMID:20144757
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet. 2003;35:367–371. doi:10.1038/ng1270. PMID:14625552
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi:10.1016/S0166-2236(99)01534-9. PMID:10717676
- Gnad F, Forner F, Zielinska DF, Birney E, Gunawardena J, Mann M. Evolutionary constraints of phosphorylation in eukaryotes, prokaryotes, and mitochondria. Mol Cell Proteomics. 2010;9:2642–2653. doi:10.1074/mcp.M110.001594. PMID:20688971
- Zhang XJ, Chen S, Huang KX, Le WD. Why should autophagic flux be assessed? Acta Pharmacol Sin. 2013;34:595–599. doi:10.1038/aps.2012.184. PMID:23474710
- Miniou P, Tiziano D, Frugier T, Roblot N, Le Meur M, Melki J. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 1999;27:e27. doi:10.1093/nar/27.19.e27. PMID:10481039
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16:1313–1320. doi:10.1038/nm.2247. PMID:21037586
- Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, Aerts L, Overbergh L, Grünewald A, Seibler P, et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science. 2014;344:203–207. doi:10.1126/science.1249161. PMID:24652937
- Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527. doi:10.1038/ncomms8527. PMID:26146385
- Kanki T, Kurihara Y, Jin X, Goda T, Ono Y, Aihara M, Hirota Y, Saigusa T, Aoki Y, Uchiumi T, et al. Casein kinase 2 is essential for mitophagy. EMBO Rep. 2013. doi:10.1038/embor.2013.114. PMID:23897086
- Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, et al. A Regulatory Signaling Loop Comprising the PGAM5 Phosphatase and CK2 Controls Receptor-Mediated Mitophagy. Mol Cell. 2014;54:362–377. doi:10.1016/j.molcel.2014.02.034. PMID:24746696
- Hashemolhosseini S, Moore C, Landmann L, Sander A, Schwarz H, Witzemann V, Sakmann B, Brenner HR. Electrical activity and postsynapse formation in adult muscle: gamma-AChRs are not required. Mol Cell Neurosci. 2000;16:697–707. doi:10.1006/mcne.2000.0911. PMID:11124891
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi:10.1016/j.cell.2010.02.026. PMID:20403324
- Buchou T, Vernet M, Blond O, Jensen HH, Pointu H, Olsen BB, Cochet C, Issinger OG, Boldyreff B. Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol Cell Biol. 2003;23:908–915. doi:10.1128/MCB.23.3.908-915.2003. PMID:12529396
- Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015;6:6670. doi:10.1038/ncomms7670. PMID:25858807
- Rudolf R, Mongillo M, Magalhaes PJ, Pozzan T. In vivo monitoring of Ca(2+) uptake into mitochondria of mouse skeletal muscle during contraction. J Cell Biol. 2004;166:527–536. doi:10.1083/jcb.200403102. PMID:15314066
- Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi:10.1038/nprot.2006.478. PMID:17406588
- Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi:10.1038/nprot.2006.62. PMID:17406264
- Messina A, Reina S, Guarino F, De Pinto V. VDAC isoforms in mammals. Biochim Biophys Acta. 2012;1818:1466–1476. doi:10.1016/j.bbamem.2011.10.005. PMID:22020053
- Stojanovski D, Pfanner N, Wiedemann N. Import of proteins into mitochondria. Methods Cell Biol. 2007;80:783–806. doi:10.1016/S0091-679X(06)80036-1. PMID:17445722
- Buttgereit A, Weber C, Garbe CS, Friedrich O. From chaos to split-ups–SHG microscopy reveals a specific remodelling mechanism in ageing dystrophic muscle. J Pathol. 2013;229:477–485. doi:10.1002/path.4136. PMID:23132094
- Guth SI, Schmidt K, Hess A, Wegner M. Adult-onset degeneration of adipose tissue in mice deficient for the Sox8 transcription factor. J Lipid Res. 2009;50:1269–1280. doi:10.1194/jlr.M800531-JLR200. PMID:19286648
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi:10.1016/j.cmet.2007.11.001. PMID:18054315
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi:10.1016/j.cmet.2007.11.004. PMID:18054316
- Liley AW. An investigation of spontaneous activity at the neuromuscular junction of the rat. J Physiol. 1956;132:650–666. doi:10.1113/jphysiol.1956.sp005555. PMID:13332600
- Plomp JJ, van Kempen GT, Molenaar PC. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol. 1992;458:487–499. doi:10.1113/jphysiol.1992.sp019429. PMID:1302275
- Rogozhin AA, Pang KK, Bukharaeva E, Young C, Slater CR. Recovery of mouse neuromuscular junctions from single and repeated injections of botulinum neurotoxin A. J Physiol. 2008;586:3163–3182. doi:10.1113/jphysiol.2008.153569. PMID:18467364