
ABSTRACT
TGFB1 (transforming growth factor beta 1) is a potent cytokine playing a driving role in development, fibrosis and cancer. It is synthesized as prodomain-growth factor complex that requires tethering to LTBP (latent transforming growth factor beta binding protein) for efficient secretion into the extracellular space. Upon release, this large latent complex is sequestered by anchorage to extracellular matrix (ECM) networks, from which the mature growth factor needs to be activated in order to reach its receptors and initiate signaling. Here, we uncovered a novel intracellular secretion pathway by which the latent TGFB1 complex reaches the plasma membrane and is released from fibroblasts, the key effector cells during tissue repair, fibrosis and in the tumor stroma. We show that secretion of latent TGFB1, but not of other selected cytokines or of bulk cargo, is regulated by fibroblast-ECM communication through ILK (integrin linked kinase) that restricts RHOA activity by interacting with ARHGAP26/GRAF1. Latent TGFB1 interacts with GORASP2/GRASP55 and is detected inside MAP1LC3-positive autophagosomal intermediates that are secreted by a RAB8A-dependent pathway. Interestingly, TGFB1 secretion is fully abrogated in human and murine fibroblasts and macrophages that lack key components of the autophagic machinery. Our data demonstrate an unconventional secretion mode of TGFB1 adding another level of control of its bioavailability and activity in order to effectively orchestrate cellular programs prone to dysregulation as seen in fibrosis and cancer.
Abbreviations
| ACTB | = | actin, beta |
| ARHGAP26/GRAF1 | = | Rho GTPase activating protein 26 |
| ATG | = | autophagy related |
| BafA | = | bafilomycin A1 |
| CCL2 | = | C-C motif chemokine ligand 2 |
| CFP | = | cerulean or cyan fluorescent protein |
| DAPI | = | 4',6-diamidino-2-phenylindole |
| ECM | = | extracellular matrix |
| GABARAPL2 | = | GABA type A receptor associated protein like 2 |
| GFP | = | green fluorescent protein |
| GORASP1/GRASP65 | = | golgi reassembly stacking protein 1 |
| GORASP2/GRASP55 | = | golgi reassembly stacking protein 2 |
| GST | = | soluble glutathione S-transferase |
| HA | = | hemagglutinin |
| ILK | = | integrin linked kinase |
| IL | = | interleukin |
| LAP | = | latency-associated peptide |
| LIR | = | LC3-interacting region |
| LLC | = | large latent complex |
| LTBP | = | latent transforming growth factor beta binding protein |
| MEF | = | mouse embryonic fibroblast |
| MAP1LC3/LC3 | = | microtubule associated protein 1 light chain 3 |
| Parva | = | murine Parvin alpha gene or RNA |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| ROCK | = | Rho associated coiled-coil containing protein kinase |
| RhoGAP | = | RhoGTPase activating protein |
| SLC | = | small latent complex |
| TGFB | = | transforming growth factor beta |
| TNF | = | tumor necrosis factor |
| VSV-G | = | vesicular stomatitis virus G envelope protein |
| YFP | = | yellow fluorescent protein |
Introduction
TGFB (transforming growth factor beta) comprises a family of growth factors with crucial activities in development, fibrosis and cancer [Citation1–7]. Three different human isoforms have been identified, TGFB1, B2 and B3, and further isoforms exist in chicken and frog. They stimulate growth and production of extracellular matrix in fibroblasts, while potently inhibiting proliferation in epithelial, endothelial and immune cells. Genetic inactivation of the individual Tgfb genes has produced nonoverlapping phenotypes likely reflecting differences in expression patterns and receptor affinities [Citation8–10]. In agreement with its potent biological functions, TGFB activity is controlled at different levels. Exquisite regulation is essential for the maintenance of tissue homeostasis and perturbations have been shown to be involved in a large variety of diseases [Citation2,Citation11–13].
All 3 TGFB isoforms are produced as preproprotein monomers featuring an N-terminal signal peptide, a large N-terminal prodomain (latency-associated peptide, LAP) that is required for proper folding and dimerization, and the smaller C-terminal growth factor. Upon translocation to the ER, proprotein monomers form disulfide-bonded homodimers, producing pro-TGFB [Citation14,Citation15]. In the Golgi, the prodomain LAP is glycosylated and cleaved from the growth factor by furin [Citation16], however, LAP remains noncovalently associated with the growth factor dimer, forming the small latent complex (SLC) [Citation17]., [Citation18] Most cells including fibroblasts are unable to secrete this complex, and efficient secretion requires binding of the SLC by disulfide bridges to latent TGFB binding protein (LTBP), thereby forming the large latent complex (LLC) [Citation19–21]. Once in the extracellular environment, the LLC is anchored to the extracellular matrix (ECM) of connective tissues, e.g. to supramolecular networks of fibronectin or to fibrillin microfibrils, which store and present latent TGFB to cells [Citation22–31]. Activation is then achieved either by proteolytic cleavage of LAP [Citation32-37] or by cell-generated forces transmitted to the matrix via binding of ITGAV/αv integrins to the RGD (arginine-glycine-aspartic acid) motif in LAP, leading to structural deformation of the LLC [Citation38–42]. Both modes of activation result in release of the mature TGFB dimer that is capable of binding to its receptors and thereby eliciting downstream signaling events [Citation2,Citation4].
TGFB1 is the best-characterized isoform of the TGFB superfamily. The structure of the SLC was recently resolved at 3.05 Å, showing that the growth factor dimer is held in a ‘straitjacket’ provided by the LAP dimer, which precludes binding of the growth factor to TGFB receptors [Citation43]. TGFB1 is among the most potent fibrogenic cytokines inducing organ fibrosis [Citation44,Citation45] by promoting the formation of myofibroblasts. These are the key effector cells producing and stiffening excessive amounts of extracellular matrix, resulting in dysfunction of fibrotically altered tissues [Citation46,Citation47].
We have previously shown that conditional ablation of integrin-linked kinase (Ilk) in murine fibroblasts (Ilk cKO) in vivo results in severely attenuated skin fibrosis due to strongly impaired TGFB1 secretion by fibroblasts [Citation48]. ILK is a pseudokinase [Citation49] located in close association with integrin cytoplasmic tails, acting as adaptor and thereby coupling integrin adhesions to the actin cytoskeleton [Citation50–52]. Global ablation of Ilk in mice leads to early embryonic lethality due to severe defects in F-actin organization [Citation53]. Selective loss of ILK in fibroblasts results in defective integrin-ECM adhesions that fail to transmit mechanical forces [Citation48,Citation54]. Ilk-cKO fibroblasts displayed elevated numbers of stress fibers due to enhanced RHOA-ROCK activity accompanied by severely reduced extracellular levels of TGFB1. As Tgfb1 mRNA levels were comparable in Ilk-cKO and control fibroblasts, defects in secretion appeared to be the cause for reduced levels of extracellular TGFB1. Supplementing Ilk-cKO fibroblasts with exogenous TGFB1 rescues their previous inability to convert to myofibroblasts, linking the structural defect in integrin adhesions to impaired myofibroblast formation and to reduced fibrosis in vivo due to loss of force transmission and insufficient fibroblast-generated TGFB1 [Citation47,Citation48]. Notably, reducing ROCK activity leads to normalized TGFB1 secretion. These data clearly indicate that cell-autonomous TGFB1 released from fibroblasts is essential for the formation of myofibroblasts and the development of fibrosis. However, how ILK, RHOA-ROCK activity and TGFB1 secretion are mechanistically linked remains unclear.
Here we investigated this mechanism and show that ILK restricts RHOA activity by binding to ARHGAP26/GRAF1, which together with GORASP2/GRASP55 stimulates autophagy. We further report that TGFB1 secretion is crucially linked to functional autophagy. TGFB1 SLC is selected by GORASP2 in the Golgi and secreted via MAP1LC3/LC3-positive secretory autophagosomes through an unconventional pathway that involves RAB8A activity. This pathway operates in fibroblasts and macrophages and is conserved across human and mice. Mechanistically it differs from reported modes of unconventional secretion and in-depth knowledge of this mechanism may be utilized for the development of novel anti-fibrotic approaches.
Results
Selective TGFB1 secretion defect in murine Ilk-cKO and human ILK-silenced fibroblasts
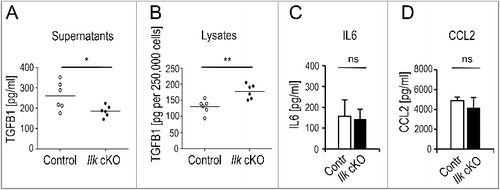
Our previous work demonstrated that production of TGFB1 by fibroblasts is induced by mechanical stress in human and murine skin fibroblasts in a process that requires β1 integrins and the integrin adaptor ILK that mount tensional force to a 3-dimensional (3D) matrix [Citation48,Citation55]. Here we show that exposure of primary murine skin fibroblasts to force in 2-dimensional monolayer cultures (2D) is sufficient to induce TGFB1 secretion. Amounts released by control fibroblasts in 2D cultures were about 50% of those detected in 3D lattice cultures [Citation48]. As in 3D lattices, absence of ILK in mouse fibroblasts (Ilk-cKO or Col1-Ilk [Citation48]) resulted in significant reduction of secreted TGFB1 in culture supernatants (A) and led to significant intracellular retention (B). However, other fibroblast products such as IL6 and CCL2 were secreted at normal levels (C, D), showing that impaired release of TGFB1 was a specific effect. We also confirmed in human fibroblasts that TGFB1 secretion depends on ILK expression. Silencing ILK in human Wi26 fibroblasts also resulted in reduced levels of secreted TGFB1 in comparison to Wi26 fibroblasts transfected with a scrambled control siRNA (si-Scr) (Figure S1A).
Figure 1. Selectively impaired secretion of TGFB1 in ILK-depleted murine fibroblasts. Primary control or Ilk cKO murine fibroblasts were grown for 24 h on fibronectin-coated (10 μg/ml) tissue culture dishes in the absence of serum. Total activatable TGFB1 was determined by ELISA in supernatants (A) and cell lysates (B). Ilk cKO cells released significantly less (*P = 0.0324) TGFB1 into the media than parallel control cultures, but retained significantly higher amounts of TGFB1 intracellularly (**P = 0.0038). Each symbol represents one independently derived fibroblast strain; lines represent mean. Supernatants were further used to determine levels of IL6 (C) and CCL2 (D) by Cytometric Bead Assay. Unlike TGFB1, these fibroblast products were secreted at comparable amounts by control and Ilk cKO fibroblasts. ns: no significant difference. Data are presented as mean ± SD. Three independent experiments, n = 3, each sample was measured 3 times.
In contrast to TGFB1, secretion of bulk cargo was comparable in Ilk-null and control fibroblasts, both in mouse and in human cells. Transfected glycosylphosphatidylinositol-anchored YFP, used as marker for glycosylphosphatidylinositol -anchored proteins [Citation56] was similarly secreted and anchored to the cell surface of control and Ilk-cKO murine fibroblasts (Figure S1B). The same was true for human Wi26 fibroblasts, in which transport of GFP-tagged VSV-G [Citation57] to the cell membrane was comparable in control (si-Scr) and ILK-silenced (si-ILK) fibroblasts (Figure S1C to E). The GFP-tagged VSV-G construct permits visualization of intracellular trafficking owing to its temperature sensitivity: at 40°C, the VSV-G protein is misfolded and retained in the ER (Figure S1C), but shifting cells to the permissive temperature (32°C) resulted in correct folding and transport to the Golgi (Figure S1D) and to the cell membrane within 90 min (Figure S1E). This process was not impaired in ILK-silenced fibroblasts. Together, these results indicated a selective defect in secretion of TGFB1 by human and murine fibroblasts by an ILK-dependent mechanism.
Intracellular retention of pro-TGFB1 in Ilk-cKO fibroblasts
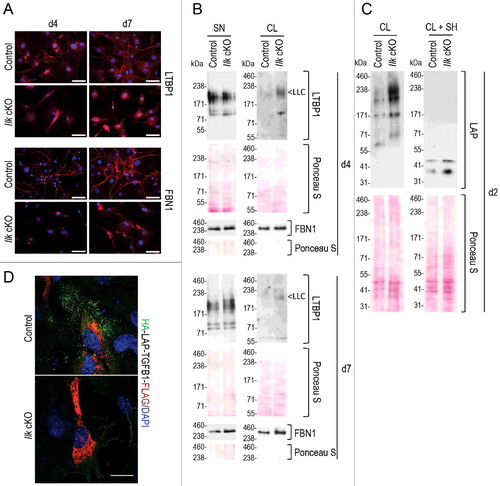
Once released to the extracellular space, the TGFB1 LLC consisting of SLC linked to LTBP1 is targeted to the FN1 (fibronectin) and FBN (fibrillin) microfibril network in a process that is mediated by specific epitopes on the LTBP1 N- and C-terminal ends [Citation22,Citation25]. We observed that Ilk-cKO fibroblasts failed to normally assemble LTBP1 and FBN1 fibers: at 4 and 7 days only rudimentary fibrillar structures were detectable, but clearly present in matrices deposited by control fibroblasts (A). However, FBN1 and LTBP1 free of SLC (184 kDa) were both detected at comparable levels in supernatants of Ilk-null and control fibroblast cultures (B SN), reflecting unimpaired secretion. Accordingly, no free LTBP1 bands were detected in fibroblast lysates (B CL). This indicates that the fibrillar assembly in the extracellular space was affected by the absence of ILK, but synthesis and secretion of FBN1 and LTBP1 remained unchanged. Previously, similar results were obtained for fibronectin, its secretion was unchanged by Ilk ablation, but its extracellular fibrillar assembly was severely compromised, reflecting the important adaptor function that ILK exerts in force-transducing cell-matrix adhesions [Citation48,Citation54]. However, levels of TGFB1 LLC, composed of LTBP1 bound to TGFB1 SLC, were strikingly increased in lysates of Ilk-cKO fibroblasts in comparison to controls at 3 time points (∼240 kDa, LLC in B and C). Detection of LAP confirmed elevated intracellular levels of the LLC in Ilk-null fibroblasts (C). Accordingly, stronger signals of LAP (37 to 41 kDa) were detected in lysates of Ilk-null fibroblasts upon sample reduction (C). Failure to secrete LAP-TGFB in the absence of ILK was further visualized by transfecting control and Ilk-cKO fibroblasts with a tagged HA-LAP-TGFB1-FLAG construct (D) that is described in detail in . These findings demonstrate that synthesis of TGFB1 LLC and its intracellular assembly is normal in Ilk-cKO fibroblasts, however, compared to control fibroblasts, the complex remains retained intracellularly.
Figure 2. Intracellular accumulation of TGFB1 large latent complex in Ilk cKO murine fibroblasts. (A) Control and Ilk cKO primary murine fibroblasts were grown for 4 or 7 d in the absence of serum. Ilk ablation abolished formation of FBN1 (fibrillin 1) and LTBP1 fibers and networks (red) in the extracellular space. Nuclei were stained with DAPI (blue). Scale bars: 50 μm. (B) Levels of LTBP1 (not bound to the TGFB1 SLC and not anchored to ECM) secreted into the supernatant (SN) were comparable in control and Ilk cKO fibroblasts. By contrast, cell lysates (CL) showed an increased presence of shifted signals representing LTBP1 molecules that are disulfide-linked to the TGFB1 prodomain LAP. This complex is indicated as LLC. Levels of FBN1 in supernatants (SN) and cell lysates (CL) were not affected by absence of ILK. (C) Immunoblotting for the TGFB1 prodomain (LAP) confirmed increased intracellular presence (CL) of TGFB1 linked to LTBP1 in Ilk cKO fibroblasts cultured for 2 d. Upon reduction (+SH), the LAP signal shifted down to its monomeric position. Membranes stained by Ponceau indicate comparable loading. (D) Control and Ilk cKO primary murine fibroblasts were transfected with the double-tagged LAP-TGFB1 construct (shown in Figure S2A). After 24 h, HA-LAP-TGFB1 localization was visualized by detection of the tags. First, nonpermeabilized cells were incubated with an antibody specific for the HA-tag to detect secreted HA-LAP-TGFB1 (green). Cells were then permeabilized and incubated with an anti-FLAG antibody to visualize total HA-LAP-TGFB1-FLAG. Abundant intracellularly located, but hardly any extracellular LAP-TGFB1 was detected in Ilk cKO fibroblasts. Scale bar: 10 μm.
ILK regulates activity of RhoGAP ARHGAP26 by interacting with its SH3 domain
Our previous work has clearly linked low TGFB1 secretion to elevated RHOA-ROCK levels by showing that suppressing RHOA-ROCK activity results in normalization of secreted TGFB1 [Citation48]. We therefore asked if high RHOA-ROCK activity resulted from the malfunctioning of a RhoGAP protein, which catalyzes the conversion of RHO-GTP to the inactive RHO-GDP form [Citation58,Citation59]. Among the known RhoGAP proteins, ARHGAP26 stands out as a GAP with prominent localization in matrix adhesion structures [Citation60] with a proposed role in remodeling membrane microdomains at adhesion sites into endocytic carriers [Citation61]. This raised the intriguing question of whether ARHGAP26 may interact with ILK in focal adhesions and contribute to downmodulation of RHOA-ROCK activity. ARHGAP26 is composed of an N-terminal BAR domain, followed by a PH and the catalytic GAP domain, which confer membrane binding, remodeling properties and GTP hydrolysis activities, respectively (A). The C-terminus contains an SH3 domain that is a potent protein-protein interaction domain. Transfection of Wi26 fibroblasts with full-length murine Arhgap26 or with a mutant lacking the SH3 domain (ΔSH3), and coimmunoprecipitation with either anti-ILK or anti-FLAG clearly showed binding of full-length ARHGAP26, but not of the SH3-deletion mutant to ILK that is stabilized by PARVA [Citation54] (B,C), indicating that the SH3 domain in ARHGAP26 acts as the ILK-binding site. We next asked if ILK binding to ARHGAP26 might influence ARHGAP26's GAP activity, thereby modulating RHOA-ROCK activity. Using recombinant proteins in a GAP assay (measuring GTP hydrolysis) confirmed ARHGAP26's GAP activity (D, ARHGAP26 +RHOA) and showed that interaction with ILK significantly enhanced this activity (D, ARHGAP26 +RHOA +ILK). Interestingly, ARHGAP26 also contributed to efficient TGFB1 secretion in fibroblasts as silencing of ARHGAP26 resulted in significantly reduced TGFB1 levels released to fibroblast supernatants (E), while expression of an ARHGAP26 mutant lacking the C-terminal SH3 domain had no influence (F). These results demonstrate that ARHGAP26 limits RHOA-ROCK activity and modulates TGFB1 secretion in fibroblasts.
Figure 3. ARHGAP26 is a RhoGAP that controls TGFB1 secretion by interacting with ILK and both contribute to autophagy. (A) Domain structure of wild-type ARHGAP26 protein (top) and the mutant lacking the SH3 domain (bottom). Red box indicates the N-terminally fused FLAG-tag. BAR, Bin-Amphiphysin-Rvs domain; PH, Pleckstrin homology domain; RhoGAP, RhoGTPase activating domain; SH3, Src homology 3 domain. (B) Human Wi26 fibroblasts were cotransfected with murine HA-Parva (to stabilize ILK [Citation54]) and either wild-type murine FLAG-Arhgap26 or with the mutant lacking the C-terminal SH3 domain (FLAG- Arhgap26 [ΔSH3]). Endogenous ILK was immunoprecipitated, and coimmunoprecipitated PARVA and ARHGAP26 were detected by immunoblotting. The interaction of endogenous ILK with PARVA was used as positive control. Wild-type, but not the mutant ARHGAP26 protein coimmunoprecipitated with ILK. IP, immunoprecipitation; IB, immunoblot; IP neg (IgG), isotype control. (C) In the reciprocal experiment, Wi26 cells were transfected with either wild-type FLAG-Arhgap26 or with the mutant lacking the C-terminal SH3 domain (ΔSH3). ARHGAP26 was immunoprecipitated and ILK coimmunoprecipitated by wild-type, but not the mutant ARHGAP26, confirming the interaction of ILK with ARHGAP26's SH3 domain. (D) RhoGAP assay with recombinant ARHGAP26 and ILK. The hydrolysis of GTP by RHOA and the influence of ARHGAP26 were determined in samples with RHOA or ARHGAP26 alone as controls, with a combination of both and in a sample containing both and added ILK. The basal GAP activity of ARHGAP26 (ARHGAP26 + RHOA) was significantly elevated by the presence of ILK (***P<0.0001). (E) siRNA-mediated silencing of ARHGAP26 (si-ARHGAP26) in Wi26 fibroblasts resulted in significantly reduced TGFB1 amounts released into the medium (**P = 0.0066) compared to controls transfected with scrambled siRNA (si-Scr). Efficient silencing of ARHGAP26 is illustrated in the blot below panel (E). (F) Expression of the ARHGAP26 mutant lacking the C-terminal SH3 domain (ΔSH3) had no influence on TGFB1 release (Mann Whitney U-test: P = 0.9293). Lines indicate means; each symbol represents an individual fibroblast strain; n = 3 experiments. The immunoblot in F illustrates comparable transfection efficiency and expression level of the 2 ARHGAP26 proteins. Untransfected Wi26 fibroblasts (untr.) were used as negative control. (G to I) Autophagy was analyzed in murine (G) and human fibroblasts (H and I) following treatment of cells either with DMSO, or with 20 μg/ml rapamycin or with 100 nM BafA for 4 h. The upper and lower ‘>’ symbols in the immunoblots indicate the position of the MAP1LC3B-I and -II variants, respectively. Autophagy was visualized by presence of MAP1LC3-II bands and actin (ACTB) was used as loading control. Ablation of Ilk in murine fibroblasts (Ilk cKO in G) or siRNA-mediated silencing of ILK (H) or of ARHGAP26 (I) in human Wi26 fibroblasts all resulted in attenuated autophagy. In particular, depletion of ARHGAP26 strongly impaired stimulated (rapamycin) as well as basal autophagy (DMSO), while ILK depletion induced less strong effects, but also affected autophagy maturation (BafA). Signal intensity was quantified densitometrically and the ratio of MAP1LC3-II to ACTB is presented below the blots. Data are representative of 3 (G and I) and 5 experiments (H).
![Figure 3. ARHGAP26 is a RhoGAP that controls TGFB1 secretion by interacting with ILK and both contribute to autophagy. (A) Domain structure of wild-type ARHGAP26 protein (top) and the mutant lacking the SH3 domain (bottom). Red box indicates the N-terminally fused FLAG-tag. BAR, Bin-Amphiphysin-Rvs domain; PH, Pleckstrin homology domain; RhoGAP, RhoGTPase activating domain; SH3, Src homology 3 domain. (B) Human Wi26 fibroblasts were cotransfected with murine HA-Parva (to stabilize ILK [Citation54]) and either wild-type murine FLAG-Arhgap26 or with the mutant lacking the C-terminal SH3 domain (FLAG- Arhgap26 [ΔSH3]). Endogenous ILK was immunoprecipitated, and coimmunoprecipitated PARVA and ARHGAP26 were detected by immunoblotting. The interaction of endogenous ILK with PARVA was used as positive control. Wild-type, but not the mutant ARHGAP26 protein coimmunoprecipitated with ILK. IP, immunoprecipitation; IB, immunoblot; IP neg (IgG), isotype control. (C) In the reciprocal experiment, Wi26 cells were transfected with either wild-type FLAG-Arhgap26 or with the mutant lacking the C-terminal SH3 domain (ΔSH3). ARHGAP26 was immunoprecipitated and ILK coimmunoprecipitated by wild-type, but not the mutant ARHGAP26, confirming the interaction of ILK with ARHGAP26's SH3 domain. (D) RhoGAP assay with recombinant ARHGAP26 and ILK. The hydrolysis of GTP by RHOA and the influence of ARHGAP26 were determined in samples with RHOA or ARHGAP26 alone as controls, with a combination of both and in a sample containing both and added ILK. The basal GAP activity of ARHGAP26 (ARHGAP26 + RHOA) was significantly elevated by the presence of ILK (***P<0.0001). (E) siRNA-mediated silencing of ARHGAP26 (si-ARHGAP26) in Wi26 fibroblasts resulted in significantly reduced TGFB1 amounts released into the medium (**P = 0.0066) compared to controls transfected with scrambled siRNA (si-Scr). Efficient silencing of ARHGAP26 is illustrated in the blot below panel (E). (F) Expression of the ARHGAP26 mutant lacking the C-terminal SH3 domain (ΔSH3) had no influence on TGFB1 release (Mann Whitney U-test: P = 0.9293). Lines indicate means; each symbol represents an individual fibroblast strain; n = 3 experiments. The immunoblot in F illustrates comparable transfection efficiency and expression level of the 2 ARHGAP26 proteins. Untransfected Wi26 fibroblasts (untr.) were used as negative control. (G to I) Autophagy was analyzed in murine (G) and human fibroblasts (H and I) following treatment of cells either with DMSO, or with 20 μg/ml rapamycin or with 100 nM BafA for 4 h. The upper and lower ‘>’ symbols in the immunoblots indicate the position of the MAP1LC3B-I and -II variants, respectively. Autophagy was visualized by presence of MAP1LC3-II bands and actin (ACTB) was used as loading control. Ablation of Ilk in murine fibroblasts (Ilk cKO in G) or siRNA-mediated silencing of ILK (H) or of ARHGAP26 (I) in human Wi26 fibroblasts all resulted in attenuated autophagy. In particular, depletion of ARHGAP26 strongly impaired stimulated (rapamycin) as well as basal autophagy (DMSO), while ILK depletion induced less strong effects, but also affected autophagy maturation (BafA). Signal intensity was quantified densitometrically and the ratio of MAP1LC3-II to ACTB is presented below the blots. Data are representative of 3 (G and I) and 5 experiments (H).](/cms/asset/fa44eead-79df-48b3-8bd7-5591044a759f/kaup_a_1422850_f0003_oc.jpg)
ILK and ARHGAP26 mediate autophagy
Interestingly, ARHGAP26 activity and high RAC1 activity—which is significantly downregulated in Ilk-cKO fibroblasts [Citation48] —have also been implicated in the formation of autophagosomes [Citation62,Citation63]. These reports prompted us to probe the formation of these vesicles and of autophagy in Ilk-cKO fibroblasts, using as readout formation of the lipidated “-II” form of MAP1LC3 (MAP1LC3-II), also called LC3 (and LC3-II in its lipidated form) [Citation64]. Whereas basal autophagy was only slightly reduced in Ilk-cKO fibroblasts (G) and also in ILK-silenced Wi26 human fibroblasts (H), autophagy induced by rapamycin was clearly reduced in both types of fibroblasts (G and H). Accordingly, autophagy was also suppressed in these cells upon inhibition of the vacuolar-type H+-translocating ATPase by bafilomycin A1 (BafA) (G and H), indicating that ILK may be involved in autophagy initiation and maturation. Of note, reducing ARHGAP26 by siRNA strongly impaired both, basal and rapamycin-induced autophagy in Wi26 fibroblasts (I), indicating a role for ARHGAP26 in the initiation step of basal and stimulated autophagy in fibroblasts.
Secretion of latent TGFB1 strictly depends upon autophagosomal intermediates in fibroblasts and macrophages
To follow intracellular trafficking of TGFB1, we generated constructs comprising full-length LAP-TGFB1 with an N-terminal HA-tag linked to LAP and a C-terminal FLAG-tag linked to the growth factor as well as singly tagged constructs (A and S2A). All constructs were translated into proteins that assembled intracellularly in Wi26 fibroblasts to generate the SLC and LLC. All proteins were correctly processed to give rise to LAP and the growth factor (Figure S2B to E).
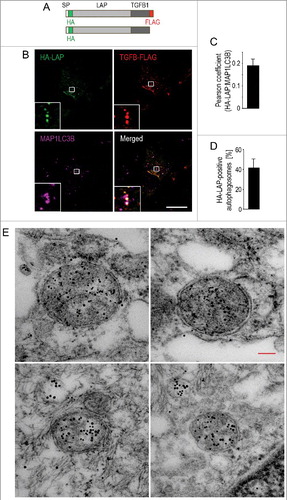
Figure 4. Secretion of latent TGFB1 requires autophagosomal intermediates. (A) Schematic representation of the tagged human LAP-TGFB1 constructs used. (B) Immunofluorescence analysis of human Wi26 fibroblasts that were transfected with the double-tagged construct illustrated in (A) and stained for endogenous MAP1LC3 (using MAP1LC3B antibody, magenta). Both, the prodomain (LAP, green) and the growth factor TGFB1 (red) colocalized with the autophagosomal marker MAP1LC3 (white in merged image). Boxes represent areas shown at higher magnification in insets. Scale bar: 10 μm. (C) The images of 25 cells were used to determine Pearson correlation coefficient, which reflects the extent of colocalization (LAP and MAP1LC3B) used here to indicate the presence of TGFB1 in autophagosomal structures. Values of about 0.2 confirmed selective colocalization under basal conditions. (D) Quantification revealed that about 40% of MAP1LC3-positive autophagosomes were also positive for HA-LAP. (E) Immunoelectron microscopy analysis of human Wi26 fibroblasts that were transfected with the HA-tagged construct illustrated in (A). Colocalization of HA-LAP (marked by small gold particles) and the autophagosomal marker MAP1LC3 (large gold particles) in double-membrane autophagosomes is illustrated in 4 examples. Scale bar: 100 nm.
To determine whether the tagged proteins interact with TGFB receptors and elicit signaling, wild-type murine fibroblasts were incubated with media conditioned by the transfected Wi26 fibroblasts. Nuclear extracts from those mouse fibroblasts were tested for the presence of phosphorylated SMAD2 (p-SMAD2), reflecting canonical TGFB signaling. Clearly only the protein with the N-terminal HA tag linked to LAP-TGFB1 was active and capable of eliciting signaling, whereas proteins carrying the C-terminal FLAG tag were inactive (Figure S2F). This observation correlated with high levels of HA-LAP-TGFB1 secreted to the supernatants of transfected fibroblasts, whereas levels of all secreted FLAG-tagged TGFB1 proteins did not differ from the negative control (Figure S2G). We concluded that the FLAG tag does not interfere with correct folding and secretion of the LAP-TGFB1, but its presence likely results in steric hindrance preventing binding of TGFB to its receptors and signal propagation.
To show if LAP-TGFB1 may be contained in autophagosomal intermediates, HA-LAP-TGFB1-FLAG was expressed in human Wi26 fibroblasts. Staining of the HA-LAP and of the TGFB1-FLAG moieties together with endogenous MAP1LC3 demonstrated colocalization of autophagosomes with LAP and TGFB1, identified via their respective tags (B). Determination of the Pearson correlation coefficient confirmed colocalization (C) in about 40% of the autophagosomes (D). To corroborate our light microscopy data and to demonstrate the presence of LAP-TGFB1 inside autophagosomes, we performed post-embedding immunogold electron microscopy on HA-LAP-TGFB1 expressing Wi26 cells (E). Localization of LAP-TGFB inside double-membrane autophagosomal vesicles was confirmed (small gold particles), and double-immunogold labeling revealed that HA-LAP-TGFB1 colocalized with MAP1LC3 (large gold particles) in these structures. MAP1LC3 was detected mainly inside autophagosomes and rarely seen at the inner or outer membrane.
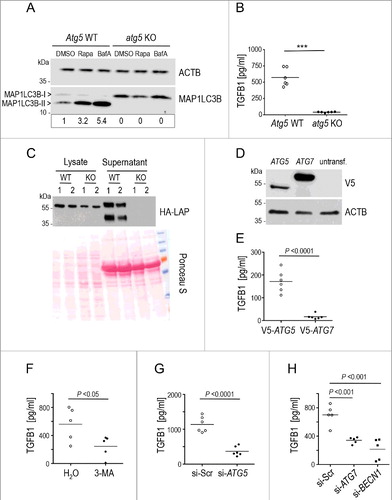
To further validate the link between autophagy and TGFB1 secretion, we probed TGFB1 secretion by fibroblasts that are incapable of executing autophagy due to the complete absence of ATG5, one of the essential proteins assembled onto early autophagosomal intermediates in these cells. As expected, formation of the MAP1LC3-II complex was completely abrogated (A) [Citation65], but of note, so was secretion of TGFB1 (B), which was retained intracellularly (C). This effect was not caused by reduced viability of atg5 KO MEFs, as these cells showed significantly lower cytotoxic stress than wild-type cells in starvation conditions (Figure S3A). Also augmented cell death was not detected (Figure S3B). As autophagosomes were not formed upon deficiency of ATG5, we attempted a rescue by expressing exogenous ATG5 or ATG7 in atg5 KO MEFs. Restoring autophagosome formation by expressing ATG5 but not ATG7 resulted in a partial rescue of TGFB1 release (D and E). To discriminate if TGFB1 secretion depends on an unknown function of ATG5 not related to autophagosome formation or rather to intact autophagy, we further blocked ATG5, ATG7 and BECN1 (beclin 1) by siRNA, and the class III phosphatidylinositol 3-kinase (PtdIns3K) by the inhibitor 3-MA in human Wi26 fibroblasts (Figure S4A to C). All treatments prominently inhibited autophagosome formation especially after autophagy induction (Figure S4D to G). Of note, all treatments interfering with autophagosome formation clearly abrogated TGFB1 release (F to H).
Figure 5. Secretion of latent TGFB1 requires the formation of autophagosomal intermediates by ATG5, ATG7 and BECN1. (A) Mouse embryonic fibroblasts (MEFs) derived from atg5 KO or Atg5 WT control animals were treated either with DMSO, or with 20 μg/ml rapamycin (Rapa) or with 100 nM BafA for 4 h. Immunoblot analysis visualizing the presence of MAP1LC3-II confirmed failure of Atg5-deficient MEFs to execute autophagy. ACTB was used as loading control. Signal intensity was quantified densitometrically and the ratio of MAP1LC3-II to ACTB is presented below the blots. (B) Levels of TGFB1 were assessed by ELISA in supernatants of atg5 KO and control MEFs grown for 24 h in the absence of serum. TGFB1 release into the medium was completely abolished by blocking autophagy (*** P<0.0001). (C) atg5 KO or Atg5 WT MEFs were transfected with the HA-tagged construct illustrated in A and grown for 24 h. The amounts of TGFB1 released to supernatants or retained in cell lysates were determined in 2 independent samples by immunoblotting and staining for LAP (using HA antibody). Ponceau S staining of the membrane was used to confirm equal loading. The upper bands, reflecting the TGFB1 proform, indicate slightly reduced amounts of this form in lysates of atg5 KO cells. By contrast, the secreted TGFB1 proform is present in control supernatants (lower bands) but absent in supernatants of atg5 KO cells. (D) For rescue experiments, atg5 KO MEFs were transfected with V5-tagged constructs of either ATG5 or ATG7 or left untransfected. Immunoblotting confirmed expression of both ATGs in transfected cells. (E) Levels of TGFB1 were assessed by ELISA in supernatants of the transfected atg5 KO MEFs (shown in D) grown for 24 h in the absence of serum. Exogenous ATG5 partially rescued TGFB1 release into the medium, while exogenous ATG7 did not. (F to H) Levels of TGFB1 were analyzed in human Wi26 fibroblasts following treatment with the autophagy inhibitor 3-MA (F), or after siRNA-mediated silencing of ATG5 (G), of ATG7 or of BECN1 (H). All autophagy-inhibiting treatments significantly impaired TGFB1 release. Lines in (B, E, F, G and H) indicate means; each symbol represents an individual fibroblast strain; n = 3 experiments.
Next, we asked if the autophagy-dependent TGFB1 secretion pathway is restricted to fibroblasts or also operating in more ‘professional’ TGFB1 -producing cells. We therefore repeated key experiments in human and murine macrophages. Human THP-1 monocytes were exposed to PMA (phorbol 12-myristate 13-acetate) to trigger differentiation into macrophages, which were subsequently treated with 3-MA. This inhibitor effectively suppressed basal and rapamycin-induced autophagy in THP-1 macrophages (A). Of note, inhibition of autophagy in macrophages resulted in reduced TGFB1 secretion comparable to fibroblasts (B). To exclude that the decrease was caused by the differentiation of THP-1 cells, we repeated the experiment with primary murine peritoneal macrophages. Inhibition of autophagy in these macrophages resulted in an even stronger impairment of TGFB1 secretion than in THP-1 cells or fibroblasts (C). Further, interference with autophagy in THP-1 derived macrophages by silencing ATG7, which effectively impaired formation of the MAP1LC3-II complex [Citation64] in basal and stimulated conditions and after BafA treatment, significantly reduced TGFB1 secretion (D to F).
Figure 6. Attenuation of autophagy impairs secretion of TGFB1 in human and murine macrophages. (A) Autophagy was analyzed in human THP-1-derived macrophages following treatment either with DMSO, or with 20 μg/ml rapamycin (Rapa) for 4 h. Autophagy was visualized by the presence of MAP1LC3-II bands in immunoblots. Inhibition of PtdIns3K by 3-MA (3-MA) strongly attenuated stimulated autophagy. (B and C) TGFB1 secretion was determined by ELISA in supernatants from human THP-1 macrophages (B) or from primary murine peritoneal macrophages (C) after treatment with 3-MA to inhibit the PtdIns3K complex or with water as control. Treatment with 3-MA resulted in significantly reduced TGFB1 levels in supernatants of both, human and murine macrophages (Mann Whitney U-test used in C). (D) Immunoblot illustrating efficient siRNA-mediated knockdown of ATG7 in THP-1 macrophages in comparison to control (si-Scr). ACTB levels were used to indicate comparable loading. (E) TGFB1 secretion was analyzed in supernatants of human THP-1 cells transfected either with siRNA specific for ATG7 or scrambled control. ATG7 depletion resulted in prominently attenuated TGFB1 release. (F) Autophagy was analyzed in THP-1 macrophages by treatment either with DMSO, or with 100 nM BafA, or with 20 μg/ml rapamycin (Rapa) for 4 h. The upper and lower ‘<’ symbols in the immunoblots indicate the position of the MAP1LC3B-I and -II variants, respectively. Autophagy was visualized by the presence of MAP1LC3-II and SQSTM1 bands in immunoblots. Depletion of ATG7 by siRNA clearly impaired basal and stimulated autophagy. Data are representative of 3 experiments. Signal intensity in (A) and (F) were quantified densitometrically; the ratio of MAP1LC3-II to ACTB is presented below the blots.
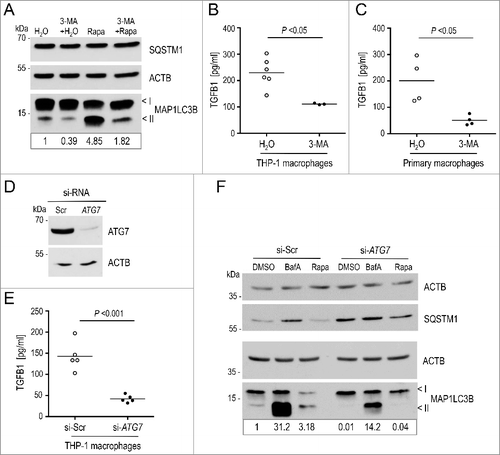
These results clearly implicated the autophagy-dependent pathway for TGFB1 secretion in different cell types.
Secretion of latent TGFB1 requires GORASP2 and its interaction with mammalian Atg8-family proteins
A critical role for autophagy has also been demonstrated in the secretion of IL1B, which further implicated GORASP2/GRASP55 [Citation66], a protein controlling stacking of Golgi ribbons. Hence, we sought to determine if GORASP2 was functionally involved in autophagy induction and TGFB1 secretion.
To uncover a functional role of GORASP2 in autophagy, we silenced GORASP2 in human Wi26 fibroblasts (A). Autophagy stimulated by rapamycin was clearly reduced in these cells while basal autophagy and block of the vacuolar H+-ATPase by (BafA) showed only a minor reduction (B), indicating a role for GORASP2 mainly in stimulated autophagy of fibroblasts.
Figure 7. GORASP2 interacts with TGFB1 and MAP1LC3 and its depletion impairs autophagy and TGFB1 secretion in human fibroblasts. (A) Immunoblot illustrating efficient siRNA-mediated knockdown of GORASP2 in comparison to control in Wi26 fibroblasts. ACTB levels were used to indicate comparable loading. (B) Autophagy was analyzed after treatment of cells either with DMSO, or with 100 nM BafA, or with 20 μg/ml rapamycin (Rapa) for 4 h. The upper and lower ‘<’ symbols in the immunoblots indicate the position of the MAP1LC3B-I and -II variants, respectively. Autophagy was visualized by the presence of MAP1LC3-II and SQSTM1 bands in immunoblots. siRNA-mediated silencing of GORASP2 resulted in a moderate decrease of basal autophagy (DMSO), but prominent attenuation of autophagy after stimulation (Rapa). Data are representative of 3 experiments. Signal intensity was quantified densitometrically and the ratio of MAP1LC3-II to ACTB is presented below the blots. (C) Immunofluorescence micrographs of human Wi26 fibroblasts, expressing HA-LAP-TGFB1 (illustrated in A, green), GORASP2-MYC (red) and endogenous MAP1LC3B (cyan). All 3 proteins colocalized at distinct structures (white in insets). Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (D) Coimmunoprecipitation of HA-LAP-TGFB1 with GORASP2-MYC and the autophagosomal marker MAP1LC3-II (stained by MAP1LC3B antibody) confirmed the close association of the 2 proteins in autophagosomal intermediates. Asterisks in the GORASP2-MYC blot indicate signals obtained with the HA antibody used to probe the same membrane without stripping. (E) In a reciprocal approach, GORASP2-MYC and endogenous MAP1LC3-II were coimmunoprecipitated by HA-LAP-TGFB1. (F-H) siRNA-mediated silencing of GORASP2 (si-GORASP2) in Wi26 fibroblasts resulted in significantly reduced TGFB1 amounts released into the medium (***P = 0.0001) compared to controls transfected with scrambled siRNA (si-Scr) (F), but unchanged amounts of IL6 (G) or CCL2 (H).
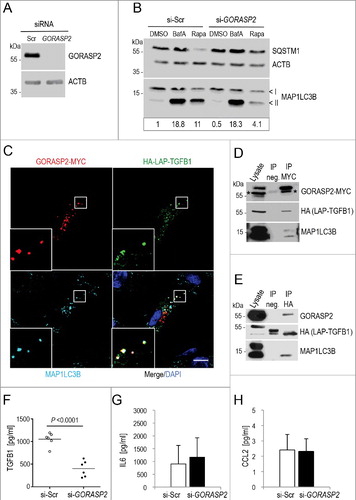
Next, we expressed HA-LAP-TGFB1 together with GORASP2-MYC in human Wi26 fibroblasts. Both proteins clearly colocalized in complexes that also contained MAP1LC3B at Golgi-independent structures (C). Coimmunoprecipitation experiments using recombinant HA-LAP-TGFB1 and GORASP2-MYC confirmed complex formation between LAP-TGFB1 and GORASP2 (D). Presence of MAP1LC3B in this complex was confirmed in HA-LAP-TGFB1-transfected cells using a reciprocal approach by coimmunoprecipitating LAP-TGFB1 with endogenous GORASP2 and MAP1LC3B (E). Moreover, silencing of GORASP2 by siRNA resulted in significantly reduced release of TGFB1 (F), while the release of IL6 and CCL2 were not affected (G and H). These results highlighted the functional involvement of GORASP2 selectively in the TGFB1 secretion process. They further indicated that LAP-TGFB1, the small latent complex, interacts with a subpopulation of GORASP2 at the Golgi and is transported as cargo into autophagosomal intermediates.
As the GORASP2-MAP1LC3B interaction has not been described previously we sought to identify the regions involved in both proteins. GORASP2 is composed of 2 N-terminal PDZ-like domains required for protein interactions, followed by a less well-characterized serine-proline-rich (SPR) sequence (A). Recombinantly expressed full-length GORASP2 as well as mutants comprising either one of the PDZ-like domains (PDZ1 and PDZ2) or the C-terminal SPR region were used for affinity precipitation of MYC-tagged MAP1LC3A from lysates of transfected Wi26 fibroblasts (B). Binding to MAP1LC3A was detected with full-length GORASP2 and with PDZ2 but not with any of the other regions (B), indicating that the second PDZ-like domain in GORASP2 acts as the MAP1LC3A-binding site. To reveal whether GORASP2 interacts specifically with MAP1LC3A or also with other mammalian Atg8-family proteins, we performed comparable experiments with lysates of fibroblasts expressing either MAP1LC3A, MAP1LC3B or GABARAPL2, all of which carried a MYC-tag. GORASP2 precipitated all 3 candidates equally well (D). To determine whether GORASP2 binding to the mammalian Atg8-family proteins depends upon lipidation, we stimulated autophagy in Wi26 fibroblasts with rapamycin and BafA.
Figure 8. Fibroblast secretion of latent TGFB1 requires its interaction with GORASP2 and mammalian Atg8-family proteins. (A) Schematic presentation of GORASP2 fragments used in affinity precipitation assays. PDZ1, N-terminal domain; PDZ2, central domain; SPR, serine/proline-rich. (B to E) Representative results of affinity isolation assays and a co-immunoprecipitation to evaluate the interaction of GORASP2 with MAP1LC3 proteins. (B) Expression of all GORASP2-GST fusion proteins was verified by immunoblotting: control (GST), full-length GORASP2, PDZ1 domain (PDZ1), PDZ2 domain (PDZ2) and SPR region (SPR) (top). To identify which region of GORASP2 interacts with MAP1LC3, GST affinity isolation assays were performed using 20 μg of GORASP2-GST fusion proteins, incubated with lysates of MYC-MAP1LC3A transfected Wi26 fibroblasts. One microgram of lysate was loaded as input control (Lys). MAP1LC3A interacted strongly with full-length GORASP2 and with the PDZ2 domain alone, but not with the PDZ1 domain or the SPR region. (C) Wi26 fibroblasts treated either with rapamycin (20 μg/ml) and BafA (100 nM) or with DMSO (as control) were used to co-immunoprecipitate endogenous GORASP2 and MAP1LC3B. MAP1LC3B interacted with GORASP2 independent of its lipidation state. Arrows indicate the position of bands representing GORASP2, located just above the IgG heavy chains. (D) A comparable affinity isolation assay was performed with the related mammalian Atg8-family proteins MAP1LC3B and GABARAPL2, showing that all 3 interacted with GORASP2 (GO2). (E) To evaluate a potential interaction of MAP1LC3 with the LIR motif found in GORASP2 and its specificity for GORASP2, we performed affinity isolation assays using GST fusion proteins with wild-type GORASP2 (GST-GORASP2 WT), with GORASP2 containing an inactive LIR motif mutant (GST-GORASP2Y196A), wild-type GORASP1 (GST-GORASP1 WT) and GST as control. The LIR motif is essential for the interaction with GORASP2, however, a weak interaction was also found with GORASP1. (F) GORASP2 WT-MYC and GORASP2Y196A-MYC transfected Wi26 fibroblasts were used to confirm the essential role of the LIR motif for MAP1LC3 binding to GORASP2. GORASP2 with mutated LIR motif localized exclusively at the Golgi, and in contrast to the wild-type protein showed no colocalization with MAP1LC3. (G) The images of 41 wild-type and 44 cells expressing mutated GORASP2Y196A were used to determine Pearson correlation coefficient, which reflects the extent of colocalization (GORASP2 and MAP1LC3) used here to indicate the presence of GORASP2 at autophagosomal structures. Values of about 0.25 confirmed the requirement of an intact LIR motif for selective colocalization. (H) GORASP2 WT-MYC (WT) and GORASP2Y196A-MYC (YA) transfected Wi26 fibroblasts were used to detect HA-LAP in cell lysates (CL) and supernatants (SN). Cells expressing the GORASP2 LIR mutant Y196A displayed intracellular retention of and impaired secretion of HA-LAP-TGFB (left panel). The transfection efficiency of both GORASP2 variants was comparable (right panel).
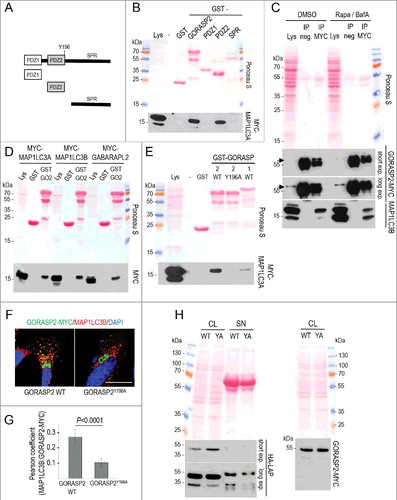
Coimmunoprecipitation showed that endogenous GORASP2 interacts with both unlipidated (MAP1LC3B-I) and lipidated (MAP1LC3B-II) forms of MAP1LC3B, indicating that GORASP2 binds to MAP1LC3B irrespective of its lipidation state (C). Since MAP1LC3-like proteins bind to the LC3-interacting region (LIR) motif (for review, see [Citation67]), which we detected in the PDZ2 domains of both GORASP1 and GORASP2, we asked whether this motif was also required for GORASP2-MAP1LC3 binding. Affinity isolation experiments using GST fusion proteins of either wild-type GORASP2 or the LIR point-mutant GORASP2 Y196A or wild-type GORASP1/GRASP65 clearly showed that MAP1LC3 strongly interacts with GORASP2 and only weakly with GORASP1, and that the intact LIR motif is required (E). Moreover, GORASP2 with an intact LIR motif colocalized extensively with MAP1LC3B at autophagosomal structures in fibroblasts, while the LIR point mutant did not codistribute with MAP1LC3B and was detected almost exclusively at the Golgi (F and G). Finally, the secretion of TGFB was impaired in fibroblasts expressing the LIR point mutant of GORASP2 (H).
Taken together, GORASP2 can bind to diverse mammalian Atg8-family proteins. The interaction requires an intact LIR motif in the PDZ2 domain in GORASP2 and is independent of the lipidation status of mammalian Atg8-family proteins. If this interaction is abrogated, LAP-TGFB is retained intracellularly.
Secretion of TGFB1 involves RAB8A-dependent exophagy
Besides fusion of autophagosomal vesicles with lysosomes and subsequent degradation of content, these vesicles can alternatively be targeted to the cell membrane in a process called regulated secretion or exophagy [Citation66,Citation68]. The decision taken depends on the activity of the small GTPase RAB8. Activation of RAB8A, a regulator of polarized sorting to the plasma membrane, results in the secretion of the content of the vesicles, whereas the closely related RAB8B, involved in the maturation of degradative autophagosomes, directs fusion with the lysosome and degradation [Citation69]. To probe whether exophagy was involved in TGFB1 secretion, Wi26 fibroblasts were transfected to express HA-LAP-TGFB1 and stained for endogenous RAB8A, revealing partial codistribution at vesicular structures (A). Interestingly, Ilk cKO fibroblasts displayed an aberrant localization of RAB8A in perinuclear aggregates (B), suggesting a link between ILK and RAB8A localization. An important role of RAB8A in TGFB1 secretion was further illustrated by silencing of RAB8A and RAB8B in Wi26 fibroblasts ( C and D), which led to a significant reduction of TGFB1 levels in supernatants of RAB8A-silenced cells (E), while silencing of RAB8B had no effect on secreted TGFB1 levels (F). The extent of reduction was comparable to that seen in murine Ilk cKO fibroblasts [Citation48]. Silencing of RAB8A did not affect basal or stimulated autophagy (G and H), suggesting that RAB8A acts downstream of autophagosome formation in the TGFB1 secretion pathway. Accordingly, abrogating autophagy did not significantly alter the distribution of RAB8A-positive structures, however it abolished the colocalization of RAB8A with LAP-TGFB ( I and J).
Figure 9. Transport of latent TGFB1 to the cell surface requires RAB8A-mediated secretion. (A) Immunofluorescence analysis of human Wi26 fibroblasts expressing HA-LAP-TGFB1 (red), stained for endogenous RAB8A (green) and GOLGA2 (magenta) identified colocalization of HA-LAP with RAB8A at Golgi-derived autophagosomal intermediates (yellow in merged inset). Scale bar: 10 μm. (B) Immunofluorescence analysis of murine control and Ilk cKO fibroblasts, transfected with HA-tagged RAB8A (green; DAPI-stained nuclei appear blue) showed relocalization of RAB8A to perinuclear aggregates. Scale bar: 10 μm. (C and D) Immunoblots illustrating knockdown efficiency of RAB8A (C) and RAB8B (D) in comparison to control (si-Scr). ACTB levels were used to indicate comparable loading. (E and F) Secreted TGFB1 levels were significantly reduced upon knockdown of RAB8A (P<0.001) (E); whereas no change in TGFB1 secretion was detected upon knockdown of RAB8B (F). Each symbol represents one independent transfectant. (G and H) Autophagy was analyzed in human Wi26 fibroblasts following treatment either with DMSO, or with 100 nM BafA, or with 20 μg/ml rapamycin (Rapa) for 4 h. The upper and lower ‘<’ symbols in the immunoblots indicate the position of the MAP1LC3B-I and -II variants, respectively. Autophagy was visualized by the presence of MAP1LC3-II and SQSTM1 bands in immunoblots. Depletion of RAB8A (G) and of RAB8B (H) did not affect basal or stimulated autophagy. Data are representative of 3 experiments. Signal intensities were quantified densitometrically and the ratio of MAP1LC3-II to ACTB is presented below the blots. (I) Immunofluorescence analysis of MEFs derived from atg5 KO or Atg5 WT animals expressing HA-LAP-TGFB1 (red) and stained for endogenous RAB8A (green) identified partial colocalization of HA-LAP with RAB8A at Golgi-derived autophagosomal intermediates (yellow in merged inset), which is not observed in atg5 KO cells. Scale bar: 5 μm. (J) The images of 20 Atg5 WT and 26 atg5 KO cells were used to determine the Pearson correlation coefficient, which reflects the extent of colocalization of TGFB1 with RAB8A (LAP and RAB8A). Values of about 0.15 confirmed selective colocalization.
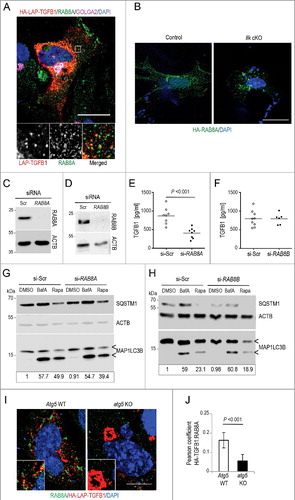
Together, these findings demonstrate that ILK in fibroblast-matrix adhesions enhances the activity of ARHGAP26, which by its RhoGAP activity limits RHOA-ROCK activity. ARHGAP26 and GORASP2 contribute to the formation of secretory autophagosomes, which contain latent TGFB1-GORASP2 complexes and deliver them to the cell membrane in a RAB8A-dependent mechanism. This atypical mechanism used to secrete TGFB1 is conserved across human and mouse fibroblasts and macrophages.
Secretion of TNF also depends on limited RHOA-ROCK activity, but not on autophagosomal structures
To determine whether other inflammatory cytokines besides TGFB1 and IL1B, which require stimulated secretion, are also secreted through exophagy, we analyzed TNF levels in supernatants of control and Ilk-depleted fibroblasts. Notably, Ilk cKO mouse fibroblasts exhibited a severe reduction in TNF secretion (Figure S5A and B). The newly synthesized transmembrane TNF precursor is delivered to the plasma membrane and requires cleavage by the TNF-converting enzyme ADAM17/TACE (ADAM metallopeptidase domain 17) to release soluble TNF [Citation70]. As TNF levels in fibroblasts are low and not easily detectable, we transfected control and murine Ilk cKO fibroblasts with a Tnf-cerulean construct to express TNF fused to CFP. Secreted, membrane-anchored TNF was detected only in control but not in Ilk-deficient fibroblasts, in which TNF was abundantly present intracellularly (Figure S5A). Human Wi26 fibroblasts with ILK knockdown expressing the TNF-CFP protein showed similar accumulation of transfected TNF-CFP in perinuclear Golgi vesicles that were not transported to the cell membrane. Accordingly, TNF-CFP was not anchored to the cell surface, while it was abundantly present at the surface of control fibroblasts (Figure S5C upper panels). Failure to secrete TNF in ILK-knockdown cells was efficiently rescued by simultaneous transfection with murine MYC-Ilk (Figure S5C, bottom panel), indicating that TNF secretion is also controlled by ILK. Of note, secretion of TNF was normalized—as that of TGFB1 [Citation48] —by suppressing RHOA-ROCK activity using Y-27632 (Figure S5D). This result suggested that TNF secretion may also be controlled by ILK binding to ARHGAP26. However, in marked contrast to TGFB1 secretion, TNF was normally secreted by autophagy-deficient atg5 KO fibroblasts, as demonstrated by the surface localization of the transfected TNF (Figure S5E). Accordingly, presence of TNF in supernatants and lack of accumulation in lysates of atg5 KO fibroblasts was confirmed by immunoblotting (Figure S5F). In addition, no colocalization of TNF and MAP1LC3B was detected in wild-type fibroblasts (Figure S5G), together clearly demonstrating that TNF secretion is not coupled to autophagosomal structures. Furthermore, Tnf mRNA levels were comparable in atg5 KO or Ilk cKO cells and wild-type fibroblasts (Figure S5H).
Our findings on the secretion of TGFB1 and TNF highlight the coexistence of different unconventional secretory molecular pathways operating in fibroblasts in the release of potent cytokines.
Discussion
Our current study demonstrates that latent TGFB1 is secreted through an unconventional secretion mode that critically depends on secretory autophagosomes as carriers for latent TGFB1. These carriers form in a process involving GORASP2 and regulated RHOA activity, and their transport to the plasma membrane requires RAB8A activity. We show that this mechanism operates in human and murine skin fibroblasts as well as in MEFs and in human and murine macrophages. As such, secretion via autophagosomal intermediates has been described previously as a mode of secretion of leaderless Golgi-independent cargoes (summarized in [Citation71–74]). However, the pathway that we describe here for TGFB1 secretion represents a novel variant in that we identified TGFB1 as the first cargo harboring a signal peptide that needs to pass the Golgi for maturation and correct folding but still requires the autophagy machinery for export.
This pathway is in part controlled at the cell surface by ILK, an adapter for β1 integrin receptors in cell adhesion structures [Citation75]. We previously demonstrated that ILK deficiency impacts levels of extracellular TGFB1 in murine skin fibroblasts [Citation48]. Data presented here illustrate that the intracellular assembly of the TGFB1 LLC, consisting of LAP-TGFB1 bound to LTBP1, is unaffected by absence of ILK but that its secretion is impeded. Here we show that high RHOA-ROCK activity in Ilk cKO fibroblasts is due to insufficient RHO-GTP hydrolysis catalyzed by the RhoGAP ARHGAP26, whose activity is significantly enhanced by binding to ILK. Our results clearly indicate that one mechanism by which ILK controls RHO-ROCK activity is through binding to ARHGAP26. This mechanism is conserved in fibroblasts derived from humans and mice, but an influence of ILK on other activating proteins or exchange factors involved in regulating RHOA activity has to be considered. Depletion of ILK and even more of ARHGAP26 significantly impairs but does not abolish autophagosome formation, which provides an explanation for the relatively milder reduction in amounts of TGFB secreted by these cells in comparison to autophagy-incompetent cells. Obviously, the ILK-ARHGAP26-RHO pathway is one of several ways regulating autophagosome formation and subsequently TGFB secretion, and it is likely more relevant in adherent cell types than in others.
The presence of autophagosomes carrying LAP-TGFB was confirmed at the ultrastructural level by immunogold labeling of LAP-TGFB in double-membrane autophagosomes that also stained positively for MAP1LC3B. Unexpectedly, MAP1LC3 signals were rarely associated with the outer membranes but were found in the lumen. This feature may distinguish secretory autophagosomes from ER-derived omegasomes and may indicate a different mechanism of their formation. Also, their maturation may correlate with dynamic changes in MAP1LC3 localization and may thereby differ from that of autophagosomes destined for degradation.
We further investigated a role for GORASP2, one of the key proteins involved in controlling the maintenance of stacked Golgi cisternae and ribbons during interphase and their dissolution during cell division [Citation76]. Apart from this structural role, GORASP2 also selects cargo for selective secretory trafficking and can contribute to the initiation of autophagy [Citation66]. Our results show that depletion of GORASP2 in fibroblasts negatively impacts autophagy and strongly reduces TGFB1 secretion. GORASP2 colocalizes with LAP-TGFB1 in Golgi-independent complexes that also contain MAP1LC3. This interaction requires an intact LIR motif that we identified in the PDZ2 domain of GORASP2, and it is functionally relevant for efficient secretion of LAP-TGFB1, but not for the release of IL6 or CCL2. Apart from MAP1LC3A, GORASP2 is able to interact with other mammalian Atg8-family proteins such as MAP1LC3B and GABARAPL2. Their interaction is thought to be direct as they are localized on the cytosolic surface of the Golgi and other vesicular structures, while no direct binding is conceivable between GORASP2 and lumenal LAP-TGFB1. We expect that the complex contains further—likely membrane spanning—proteins that link LAP-TGFB1 in the Golgi lumen to GORASP2 and MAP1LC3, which face towards the cytosolic side. Presence of GORASP2 has been reported previously to be critical for guiding TGFA to the cell surface by a mechanism that depends on direct interaction between PDZ1 in GORASP2 and the C terminus of TGFA [Citation77]. In contrast to TGFB1, the unrelated TGFA is a transmembrane protein and as such, parts of TGFA face inside the Golgi lumen but another part also to the cytosolic compartment where GORASP2 resides. Thus, a direct interaction is feasible between these 2 proteins.
Ilk cKO fibroblasts are characterized by a severely perturbed cytoskeleton [Citation48], an observation that led us to think that vesicle trafficking including those engaged in secretion would result in overall impaired secretion. Surprisingly, however, bulk secretion in these cells was unaffected by the cytoskeletal changes, reflected by normal release of IL6 and CCL2 as well as by normal transport of marker proteins (VSV-G and glycosylphosphatidylinositol-anchored YFP) to the cell surface. These observations clearly indicated that TGFB1 secretion likely involved an unconventional secretion pathway. Our findings discussed here share important steps with the unconventional secretion pathway by which IL1B is delivered by macrophages [Citation66] and by HEK293T cells [Citation78]. Several crucial features of the IL1B pathway are identical to the TGFB1 secretory mode in fibroblasts and include the dependence on GORASP2, the autophagic machinery and RAB8A. In fibroblasts, we can extend this pathway to cell-matrix adhesions and ILK, which contributes to the correct localization of RAB8A. By which mechanism RAB8A-positive vesicles carrying cargo such as LAP-TGFB1 to the cell surface and what controls the release to the extracellular environment remains to be determined.
In the skin, IL1B is released by epidermal keratinocytes in inflammatory conditions [Citation79], however, the levels released by skin fibroblasts were below or approximating detection limits (<0.27 pg/ml), and IL1B secretion could therefore not be used as control for this pathway in fibroblasts. The unconventional pathway whereby IL1B is secreted, termed autosecretion [Citation80] or secretory autophagy [Citation81], has also been described for yeast which secrete proteins that—as with IL1B—are characterized by lack of a signal peptide that guides them across the ER membrane into the secretory pathway [Citation66,Citation82–84]. Unlike IL1B the LAP-TGFB1 sequence does feature a canonical signal peptide enabling its secretion via the conventional pathway, however this would be an unregulated process. In view of the multi-faceted activities elicited by TGFB1 on a broad range of tissues, it is critical to strictly control its release. The unconventional pathway uncovered here provides such a control mechanism to prevent untimely TGFB1 bioavailability.
Our study further uncovered that different inflammatory cytokines are secreted by different unconventional secretory modes that share the control by ILK and RHOA activity. Hence, secretion of TGFB1 and TNF depends on restricted RHOA-ROCK activity by a mechanism dependent on ILK interacting with ARHGAP26 and possibly other factors regulating RHOA, but only TGFB1 secretion requires the autophagic machinery. This is in accordance with reports showing that efficient TNF secretion by macrophages involves the Golgi network and recycling endosomes [Citation85,Citation86].
Recent evidence has established a role for constituents of the extracellular matrix in inducing or suppressing autophagy, depending upon the individual constituent [Citation87]. Although no link to a specific matrix receptor has been established so far, a role for members of the β1 integrin family [Citation88,Citation89] and their organization in functional adhesion structures may be expected. The results presented here clearly indicate a correlation between properly organized cell-matrix adhesions and autophagy, which is impaired in ILK-depleted fibroblasts. Taking into account that ILK plays an important role in force transduction across the cell membrane [Citation54], it is tempting to speculate about a mechanism reported to control autophagy in a process termed tension-induced autophagy [Citation90,Citation91]. This mechanism is considered to be essential for the adaptation of mammalian cells to mechanical strain and integrates tension sensing, autophagosome formation and degradation of tension-damaged proteins, e.g. filamin, with transcriptional regulation to compensate for degraded proteins. It appears conceivable that this mechanism is perturbed in cells with defective tension sensing machinery and would lead to a reduction in autophagosomes and, since TGFB1 secretion depends on secretory autophagosomes, also to reduced TGFB1 secretion.
It remains to be determined whether the mode of TGFB1 secretion demonstrated here that is engaged by different cell types and conserved among humans and mice, is used for all 3 TGFB isoforms present in mammalian cells, and possibly for further members of the large TGFB superfamily.
A number of translational approaches have been initiated to counteract the adverse effects of augmented TGFB1 in the context of stimulating and perpetuating tissue fibrosis [Citation11,Citation92,Citation93]. These focused on ligand capture, receptor decoys, receptor blockade or inhibition of downstream signaling. A better understanding of the secretory pathway may add a novel mechanistic basis to the design of reagents interfering with the detrimental effects of excessive or untimely TGFB1 activity.
Materials and methods
Fibroblast isolation and culture
Primary murine fibroblasts were isolated from the dermis of 3-wk-old Col1-Ilk mice (conditional ablation of ILK specifically in fibroblasts by the Col1a2 promoter driving Cre recombinase) after tamoxifen induction as described [Citation48]. Alternatively fibroblasts were isolated from the dermis of newborn Ilkfloxed/floxed mice, and Cre recombinase was induced by repeated incubation with 4-hydroxytamoxifen (Sigma-Aldrich, T176). Efficient ablation of ILK is shown in C. Such fibroblasts are designated as Ilk cKO. Controls were either Ilkfloxed/+; Cre-positive or Ilkfloxed/+; Cre-negative. Wi26 fibroblasts (SV-40 transformed, from human lung) [Citation94] and murine fibroblasts were cultured in DMEM/F12 GlutaMAX (Thermo Fisher Scientific, 31966-021) and DMEM GlutaMAX (Thermo Fisher Scientific, 31331-028), respectively, with 50 μg/ml Na-ascorbate and antibiotics (Seromed Biochrom, K0282, A2213) in the presence or absence of 10% fetal calf serum (FCS; Life Technologies, 10270) as indicated, at 37°C and 5% CO2 in a humidified atmosphere [Citation48,Citation95,Citation96]. Cells were used between passages 3 and 6.
Macrophage isolation and culture
Five C57BL/6 mice were killed at the age of 8 wk by cervical dislocation. Primary peritoneal macrophages were isolated according to Knipper and coworkers [Citation97]. In brief, cells were collected by peritoneal lavage with phosphate-buffered saline (PBS; Life Technologies, 14190-094), harvested by centrifugation (240 x g, 8 min) and resuspended in RPMI medium (Life Technologies, 61870-010) with 10% FCS and 1% antibiotics. Macrophages were enriched by adherence to tissue culture plastic for 6 h. Confluent macrophage cultures were treated with phorbol myristate acetate (PMA; 100 nM; Sigma-Aldrich, P1585) for 5 d.
Alternatively, the human monocyte THP-1 cell line [Citation98] was used (kindly provided by Dr. T. Langmann, University of Cologne, Germany). The THP-1 cells were maintained in RPMI medium (supplemented with 10% FCS, 1% antibiotics, 25 mM HEPES). Differentiation of confluent monocytes was induced by culture in 100 nM PMA in serum-free macrophage medium for 5 d [Citation99]. Nonadherent cells were removed by aspiration, and differentiated macrophages were washed with RPMI 3 times.
Pharmacological agonists and inhibitors
Autophagy was induced in human Wi26 fibroblasts or MEFs derived from atg5 KO or wild-type embryos by adding 40 μg/ml rapamycin (Alfa Aesar, J62473) or 20 μg/ml for 4 h. Where indicated, cells were treated additionally with BafA (100 nM, Alfa Aesar, J61835) to enrich autophagosomes by inhibiting the fusion of autophagosomes and lysosomes. ROCK inhibitor Y-27632 (Calbiochem, 688001) was added at 5 μM for 4 h, as described previously [Citation48]. PtdIns3K inhibitor 3-methyladenine (3-MA) (Alfa Aesar, J64813) was added at 5 mM for 4 h for immunoblot analysis and for 24 h for ELISA.
Cloning and recombinant protein expression
RNA isolated from mouse wild-type brain and from human Wi26 fibroblasts was converted to cDNA using Superscript II (Thermo Fisher Scientific, 18064014). The following primers were used to amplify the sequences required for transformation, transfection and expression of recombinant proteins:
Arhgap26: sense 5′-acacggatccATGGGGCTCCCGGCGCTCGAG-3′, antisense 5′-atatgtcgacttaGAGGAATTCCACGTAGTTC-3′
ATG5: sense 5′-tctcgatatcaATGACAGATGACAAAGATG-3′, antisense 5′-ctctctcgagtcaATCTGTTGGCTGTGGGATG-3′
ATG7: sense 5′-tatagaattctaATGGCGGCAGCTACGGGG-3′, antisense 5′-tatagcggccgctcaGATGGTCTCATCATCGC-3′
GABARAPL2: sense 5′-TATAGAATTCCCATGAAGTGGATGTTCAAG-3′
antisense 5′-AGAGCTCGAGTCAGAAGCCAAAAGTGTTCTC-3′
GORASP1: sense 5′-tatagaattcgccgccATGGGCCTGGGCGTCAGCGC-3′, antisense 5′-atatgtcgactaTTCTGTGGTAGAGATCTGGG-3′
GORASP2: sense 5′-TCTCGAATTCGCCGCCATGGGCTCCTCGCAAAG-3′, antisense 5′-TATACTCGAGTAAGGTGACTCAGAAGCATTG-3′
Gorasp2: sense 5′-acacggatccATGGGGCTCCCGGCGCTCGAG-3′, antisense 5′-atatgtcgacttaGAGGAATTCCACGTAGTTC-3′
LTBP1: sense 5′-tctcgctagcgccaccATGGATACTAAGCTGATGTG-3′, antisense 5′-gagactcgagCTCCAGGTCACTGTCTTTCTC-3′
MAP1LC3A: sense 5′-TCTCGAATTCCCATGCCCTCAGACCGGCCTTTC-3′, antisense 5′-ATATCTCGAGTCAGAAGCCGAAGGTTTCCTG-3′
Map1lc3b: sense 5′-TCTCGAATTCCCATGCCGTCCGAGAAGACCTTCAAAC-3′, antisense 5′-ATATCTCGAGTCACCCGAACGTCTCCTGGGAGGC-3′
Parva: sense 5′-ctctgaattcATGGCCACATCCCCACAGAAG-3′, antisense 5′-agaggtcgactcaCTCCACATTCCGGTACTTG-3′
TGFB1: sense 5′-TCTCAAGCTTGCCACCATGCCGCCCTCCGGGCTG-3’, antisense 5′-AGAGCTCGAGTCAGCTGCACTTGCAGGAGC-3′
Tagged LAP-TGFB1 constructs were generated by inserting an HA-tag between the signal peptide and LAP of the full-length human pro-TGFB1, acc. no. NM_000660 (sense 5′-CGGATACCCATACGATGTTCCAGATTACGCTCTATCCACCTGCAAGACTATCGACATGGAGCTGGTGAAGCGGAAGCGCATCGAGGCCATCCGC-3′, antisense
5′-GCGGATGGCCTCGATGCGCTTCCGCTTCACCAGCTCCATGTCGATAGTCTTGCAGGTGGATAGAGCGTAATCTGGAACATCGTATGGGTATCCG-3’). Alternatively, a FLAG-tagged construct was generated using sense 5′-TCTCAAGCTTGCCACCATGCCGCCCTCCGGGCTG-3’, antisense with FLAG tag 5′-AGAGCTGCAGTCACTTGTCATCGTCGTCCTTGTAGTCGCTGCACTTGCAGGAGCGCAC-3′.
Recombinant proteins were expressed using the following constructs: Glycosylphosphatidylinositol-anchored YFP [Citation100] and VSV-G-GFP [Citation57]. Full-length murine FLAG-Arhgap26, acc. no. BC141394, and FLAG-Arhgap26 ΔSH3 (murine Arhgap26 lacking aa 667 to 712 corresponding to the SH3 domain) were ligated into pcDNA4TO (Thermo Fisher Scientific, V1020-20) with an N-terminal FLAG tag. Full-length human ATG5, acc. no. NM_004849, and ATG7, acc. no. NM_006395, were ligated into pcDNA4TO (Thermo Fisher Scientific, V1020-20) with an N-terminal V5 tag. Full-length murine Parva, acc. no. NM_020606, was ligated into pcDNA3 (Thermo Fisher Scientific, V790-20) including an HA tag. Murine Ilk, NM_001161724 lacking aa 193 to 452 corresponding to the pseudokinase domain was cloned into pGEX6P2 (GE Healthcare, 28-9546-50), or full-length Ilk was cloned into pCMV-Myc (Clontech, 631604). Full-length human MAP1LC3A, acc. no. NM_032514, and a deletion mutant MAP1LC3A (ΔG120) lacking the last 2 amino acids, full-length rat Map1lc3b, acc. no. NM_022867, and full-length human GABARAPL2, acc. no. 007285, were ligated into pCMV-Myc (Clontech, 631604) adding an N-terminal MYC tag. Full-length human GORASP2, acc. no. 015530, and the point mutant Y196A (LIR mutant) were cloned into pcDNA4TO-MycHis (Thermo Fisher Scientific, V1030-20) adding a C-terminal MYC tag. To express GST fusion proteins for affinity isolation assays, full-length human GORASP1, acc. no. NM_031899, full-length human GORASP2, and GORASP2 domains PDZ1 (aa 14 to 108), PDZ2 (aa 109 to 200) and SPR (aa 201 to 452) as well as the GORASP2Y196A mutant were cloned into pGEX6P1 (GE Healthcare, 28-9546-48). Full-length human RAB8A, acc. no. NM_005370, was cloned into pCMV-HA (Clontech, 631604). Full-length murine Tnf, acc. no. NM_013693, was cloned into pmCerulean-N1 (Addgene, 27795; deposited by Steven Vogel [Citation101]). Tagged LAP-TGFB1 constructs were cloned into pcDNA4TO. Human LTBP1, acc. no. BC130289, was expressed with a C-terminal EGFP tag in pEGFP-N1 (Clontech, PT3027).
Subconfluent fibroblast cultures in 24-well plates were transfected with 1 μg plasmid DNA per well using XtremeGENE HP (Sigma Aldrich, 06366236001) according to the supplier's protocol.
siRNA-mediated gene silencing
Fibroblasts were seeded at 1.5 × 104 cells/well onto glass coverslips in 24-well plates. Transient suppression of target proteins was achieved by adding double-stranded, target-specific Stealth siRNA (Life Technologies) or scrambled control siRNA (si-Scr, med GC, 12935-113), diluted (80 nM) in 100 μl DMEM to 1 × 105 cells/well in 6-well plates in the presence of 8 μl HiPerFect transfection reagent (Qiagen, 301705). Following incubation (12 min) the complex formed was added dropwise to cells and remained for 72 to 96 h. Knockdown efficiency was verified by immunoblotting in each experiment. The following siRNA sequences were used:
In some experiments, silenced fibroblasts were further transfected to express selected proteins 24 h later with 1 μg of plasmid DNA, added in 50 μl of OptiMEM (Thermo Fisher Scientific, 31985-070) and 2 μl XtremeGENE HP (Sigma-Aldrich, 06366236001).
Antibodies
Antibodies directed to the following proteins were used: ACTB/actin (BD Transduction Laboratories, 612656, clone C4); ATG5 (Sigma-Aldrich, A0856); ATG7 (Cell Signaling Technology, 8558P); BECN1 (Novus Biologicals, NB110-87318); cerulean and YFP were detected using a polyclonal antibody against GFP (Abcam, ab290 and ab6556); FLAG M2 (Sigma-Aldrich, F3165); GAPDH (EMD Millipore, ABS16); GFP (Abcam, ab290); GOLGA2/GM130 (BD Transduction Laboratories, 610823); GORASP2 (Proteintech, 10598-1-AP); HA (hemagglutinin; Sigma-Aldrich, 12158167001, clone 3F10); FBN1 (kind gift from the Sakai lab, Shriners Hospital for Children, Portland, OR, USA; 9543); ILK (Sigma-Aldrich, I1907); LAP (R&D Systems, AF-246-NA); LTBP1 (Sakai lab, Shriners Hospital for Children; 8579, for IB); MAP1LC3B (Sigma-Aldrich, L7543); MYC (Santa Cruz Biotechnology, sc40, clone 9E10 and Santa Cruz Biotechnology, sc789, clone A14); RAB8A (Sigma-Aldrich, SAB2500852); RAB8B (Proteintech, 11792-1-AP); SMAD2 (Cell Signaling Technology, 3103, clone L16D3) and phospho-SMAD2 (Cell Signaling Technology, 3101S); SQSTM1 (Enzo Life Sciences, BML-PW9860-0100); TGFB1 (R&D Systems, 3709); V5 (AbD Serotec, MCA1360). F-actin was visualized with Alexa Fluor 488-conjugated phalloidin (Life Technologies, A12379). Nuclei were stained with DAPI (Sigma-Aldrich, D9542). Secondary antibodies were conjugated to Alexa Fluor 488, 555 or 647 (Thermo Fisher Scientific, A-11001, A-21424, A-21235, A-11034, A-21428, A-21245, A-11006, A-21434, A-21247) and cross-adsorbed.
Immunoblotting and detection assays
Cleared culture supernatants or cell lysates were analyzed by immunoblotting. Cells were lysed in HEPES lysis buffer (20 mM HEPES-NaOH, pH 7.4, 150 mM NaCl, 0.5% NP-40 [IGEPAL CA-630; Sigma-Aldrich, I8896], 2 mM EDTA) supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich, P5726), homogenized by 20 strokes (Dounce homogenizer [Braun, Melsungen, Germany], 4°C), centrifuged (12,000 x g, 15 min) to remove debris and separated by SDS-PAGE. To monitor autophagy, cells were lysed in MAP1LC3 lysis buffer (50 mM Tris-HCl, pH 7.5, 150 m MNaCl, 0.5% NP-40) containing 1% SDS, heated for 5 min at 95°C and incubated on ice for 10 min. Samples were sonicated (Fisher Scientific, Vibra-Cell 75115; 65% amplitude, 2 seconds) and subjected to SDS-PAGE. To detect intracellular interactions by coimmunoprecipitation, Wi26 cells were lysed in HEPES lysis buffer (as above) using a glass Dounce homogenizer and incubated for 30 min on ice. Homogenates were cleared by centrifugation (12,000 x g, 10 min) and incubated with protein G-agarose beads (pre-clearing, 1 h at 4°C; Sigma-Aldrich, 11719416001) prior to incubation with specific antibodies (1 μl of crude antiserum or 5 μg of commercial antibody solution) for 3 to 4 h at 4°C under constant agitation. Complexes were harvested by overnight incubation with protein G-agarose beads, from which they were dissociated by heating (95°C) in Laemmli sample buffer and separated by SDS-PAGE. Expression of tagged TGFB1 proteins was determined in supernatants from transfected Wi26 fibroblasts following TCA precipitation. Precipitated proteins were resuspended in 8 M urea. Proteins were transferred to PVDF (Merck/Millipore, IPVH00010), stained with Ponceau S, membranes were blocked with 5% skim milk in TBS-T (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% [vol:vol] Tween-20 [Sigma-Aldrich, P9416]) and incubated with primary antibodies diluted in blocking buffer, followed by incubation with appropriate HRP-conjugated secondary antibodies (Dako, P0399, P0450, P0260). Signals were detected by enhanced chemiluminescence (Thermo Fisher Scientific, 34080). Quantification of band intensities was performed using ImageJ.
Immunofluorescence and microscopy
Cells were grown on glass cover slips, fixed (4% formaldehyde in PBS or methanol), permeabilized with 0.5% NP-40 (Sigma-Aldrich, N-3516) in PBS as indicated, blocked (1% FCS in PBS) and incubated with primary antibodies diluted in 1% FCS in PBS and appropriate secondary antibodies conjugated to Alexa Fluor 488, 555 or 647. Images were acquired by confocal microscopy using a Leica SP5 system controlled by Las AF 3 (Leica Microsystems, Wetzlar, Germany). Image analysis was performed with ImageJ. For calculation of the Pearson correlation coefficient, single cells were marked and analyzed with the JACoP plugin for ImageJ.
Immunoelectron microscopy
To visualize TGFB1 in autophagosomes, human Wi26 fibroblasts transfected with the HA-LAP-TGFB1 construct were harvested by centrifugation (250 x g for 5 min). Samples for immunoelectron microscopy were subsequently prepared by pelleting 5 × 106 cells at 4°C immediately after the addition of fixative (4% paraformaldehyde + 0.1% glutaraldehyde). After incubation at room temperature for 1 h, the fixed pellets were dehydrated in ethanol and further processed for Lowicryl (Agar Scientific, K4M kit, AGR1035) embedding [Citation102]. Sections were cut with a microtome equipped with a diamond knife and mounted on nickel grids.
For immunostaining, the grids were floated on top of 100-μl drops of immune reagents displayed on a sheet of Parafilm in a wet chamber. Free aldehyde groups were blocked with 50 mM glycine, and the grids were then incubated with 5% (vol/vol) donkey serum (Abcam, ab7475) in incubation buffer (0.2% BSA-c [Aurion, 900.022] in PBS, pH 7.6) for 15 min. The blocking procedure was followed by overnight incubation with primary antibodies (monoclonal antibody against HA, dilution1:100; or a polyclonal antiserum specific for MAP1LC3B, dilution 1:50) at 4°C. After washing the grids in a large volume (200 ml) of incubation buffer, floating on drops containing the gold conjugate reagents (diluted 1:20 in incubation buffer) was performed for 60 min at room temperature. After further washes in a large volume of incubation buffer, the sections were postfixed in 2% glutaraldehyde. Finally, sections were washed with distilled water and poststained with uranyl acetate and lead citrate. Specimens were observed in a Philips/FEI CM100 transmission electron microscope (Philips/FEI, Eindhoven, The Netherlands) operated at 80 kV accelerating voltage, and images were recorded with a side-mounted Olympus Veleta camera (Olympus, Solna, Sweden) with a resolution of 2048 × 2048 pixels.
RhoGAP assay
GST fusion proteins were generated from pGEX-6P vectors containing the cDNA of interest (see above) in E. coli BL21 according to standard procedures. Efficiency and purity of each preparation were confirmed by SDS-PAGE. RhoGAP activity of ARHGAP26 was determined using a GAP assay kit (Cytoskeleton, BK105), which measures the amount of inorganic phosphate produced as a result of ARHGAP26-dependent hydrolysis of GTP. The assay was used according to the manufacturer's protocol, with 100 μM recombinant GST-ARHGAP26 and 100 μM GST-ILK (see above) or GST alone as control.
Determination of TGFB1, TNF, IL6 and CCL2 levels
TGFB1 levels were determined in serum-free supernatants or in lysates of fibroblasts after 24 h using Quantikine ELISA (R&D Systems, MB100B) according to the supplier's instructions. Levels of TNF and IL6 were measured using a Mouse Th1/Th2/Th17 Cytokine kit (BD Biosciences, 560485), CCL2 levels by Mouse Flex Set (BD Biosciences, 558342), in culture supernatants by cytometric bead assay (CBA) according to the manufacturer's guidelines. Cell culture supernatants were collected and frozen at -80°C for CBA measurements. After acquisition of sample data using fluorescence-activated cell sorting (FACS Calibur; BD Biosciences, Heidelberg, Germany), concentrations were calculated using the proprietary FCAP Version 1.0 analysis software (BD Biosciences).
Statistical analysis
Results are presented as means ± s.d. Statistical analysis was performed using the unpaired Student t test unless stated differently. *P<0.05 was considered statistically significant; **P<0.01, ***P<0.001. Gaussian distribution was verified by the Kolmogorov-Smirnov test. Calculations were performed using GraphPad Prism (GraphPad Software, La Jolla, USA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KAUP_A_1422850_suppl.zip
Download Zip (27 MB)Acknowledgements
We thank Susanne Neumann, Gabi Scherr and Marina Tauber (Cologne) for excellent technical assistance, Dr. Georg Brunner (Münster) for fruitful discussion, Dr. Lynn Sakai (Portland, OR) for antibodies to FBN1 and LTBP1, Dr. Jennifer Lippincott-Schwartz (NIH, Bethesda) for the YFP-GPI and VSV-G constructs, Dr. Thomas Langmann (Cologne) for THP-1 cells, Dr. Oleg Krut (Cologne) and Dr. Noboru Mizushima (Tokyo) for Atg5-null MEFs and Dr. Angelika Noegel (Cologne) for murine Parva cDNA. We thank the Center for Integrated Microscopy (CFIM) at Panum Institute, University of Copenhagen, for an innovative environment for electron microscopy. This work was supported by Deutsche Forschungsgemeinschaft through SFB 829 (A11 to SAW, B12 to GS, B3 to BE and TK), KR 558 (to TK) and RA 2838 (to AI), by the Koeln Fortune Program, Faculty of Medicine, University of Cologne (216/2016 to AI) and Imhoff-Foundation (to MP).
Additional information
Funding
Related Research Data
References
- Roberts AB, Heine UI, Flanders KC, et al. Transforming growth factor-beta. Major role in regulation of extracellular matrix. Ann New York Acad Sci. 1990;580:225–232. doi:10.1111/j.1749-6632.1990.tb17931.x.
- Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi:10.1038/nrm3434. PMID:22992590
- Moses HL, Yang EY, Pietenpol JA, TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell. 1990;63:245–247. doi:10.1016/0092-8674(90)90155-8. PMID:2208284
- Munger JS, Sheppard D. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harbor Perspect Biol. 2011;3:a005017. doi:10.1101/cshperspect.a005017.
- Kingsley DM. The TGF-beta superfamily: new members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994;8:133–146. doi:10.1101/gad.8.2.133. PMID:8299934
- Li MO, Wan YY, Sanjabi S, et al. Transforming growth factor-beta regulation of immune responses. Ann Rev Immunol. 2006;24:99–146. doi:10.1146/annurev.immunol.24.021605.090737.
- Loeys BL, Gerber EE, Riegert-Johnson D, et al. Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Science Transl Med. 2010;2:23ra0. doi:10.1126/scitranslmed.3000488.
- Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi:10.1038/359693a0. PMID:1436033
- Kaartinen V, Voncken JW, Shuler C, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nature Genet 1995;11:415–421. doi:10.1038/ng1295-415. PMID:7493022
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. PMID:9217007
- Lafyatis R. Transforming growth factor beta–at the centre of systemic sclerosis. Nat Rev Rheumatol. 2014;10:706–719. doi:10.1038/nrrheum.2014.137. PMID:25136781
- Neptune ER, Frischmeyer PA, Arking DE, et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nature Genet. 2003;33:407–411. doi:10.1038/ng1116. PMID:12598898
- Bierie B, Moses HL. Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine & Growth factor Reviews. 2010;21:49–59. doi:10.1016/j.cytogfr.2009.11.008.
- Gentry LE, Lioubin MN, Purchio AF, et al. Molecular events in the processing of recombinant type 1 pre-pro-transforming growth factor beta to the mature polypeptide. Mol Cell Biol. 1988;8:4162–4168. doi:10.1128/MCB.8.10.4162. PMID:3185545
- Gray AM, Mason AJ. Requirement for activin A and transforming growth factor–beta 1 pro-regions in homodimer assembly. Science. 1990;247:1328–1330. doi:10.1126/science.2315700. PMID:2315700
- Dubois CM, Laprise MH, Blanchette F, et al. Processing of transforming growth factor beta 1 precursor by human furin convertase. J Biol Chem. 1995;270:10618–10624. doi:10.1074/jbc.270.18.10618. PMID:7737999
- Miyazono K, Olofsson A, Colosetti P, et al. A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. EMBO J. 1991;10:1091–1101. PMID:2022183
- Walton KL, Makanji Y, Chen J, et al. Two distinct regions of latency-associated peptide coordinate stability of the latent transforming growth factor-beta1 complex. J Biol Chem. 2010;285:17029–17037. doi:10.1074/jbc.M110.110288. PMID:20308061
- Saharinen J, Taipale J, Keski-Oja J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 1996;15:245–253. PMID:8617200
- Todorovic V, Rifkin DB. LTBPs, more than just an escort service. J Cell Biochem. 2012;113:410–418. doi:10.1002/jcb.23385. PMID:22223425
- Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005;280:7409–7412. doi:10.1074/jbc.R400029200. PMID:15611103
- Isogai Z, Ono RN, Ushiro S, et al. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278:2750–2757. doi:10.1074/jbc.M209256200. PMID:12429738
- Hyytiainen M, Penttinen C, Keski-Oja J Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41:233–264. doi:10.1080/10408360490460933. PMID:15307633
- Hildebrand A, Romaris M, Rasmussen LM, et al. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem J. 1994;302:527–534. doi:10.1042/bj3020527. PMID:8093006
- Dallas SL, Sivakumar P, Jones CJ, et al. Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J Biol Chem. 2005;280:18871–18880. doi:10.1074/jbc.M410762200. PMID:15677465
- Ono RN, Sengle G, Charbonneau NL, et al. Latent transforming growth factor beta-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J Biol Chem. 2009;284:16872–16881. doi:10.1074/jbc.M809348200. PMID:19349279
- Olofsson A, Ichijo H, Moren A, et al. Efficient association of an amino-terminally extended form of human latent transforming growth factor-beta binding protein with the extracellular matrix. J Biol Chem. 1995;270:31294–31297. doi:10.1074/jbc.270.52.31294. PMID:8537398
- Horiguchi M, Ota M, Rifkin DB Matrix control of transforming growth factor-beta function. J Biochem. 2012;152:321–329. doi:10.1093/jb/mvs089. PMID:22923731
- Zilberberg L, Todorovic V, Dabovic B, et al. Specificity of latent TGF-beta binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J Cell Physiol. 2012;227:3828–3836. doi:10.1002/jcp.24094. PMID:22495824
- Sengle G, Sakai LY. The fibrillin microfibril scaffold: a niche for growth factors and mechanosensation? Matrix Biol. 2015;47:3–12. doi:10.1016/j.matbio.2015.05.002. PMID:25957947
- Robertson IB, Horiguchi M, Zilberberg L, et al. Latent TGF-beta-binding proteins. Matrix Biol. 2015;47:44–53. doi:10.1016/j.matbio.2015.05.005. PMID:25960419
- Mu D, Cambier S, Fjellbirkeland L, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi:10.1083/jcb.200109100. PMID:11970960
- Lyons RM, Gentry LE, Purchio AF, et al. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J Cell Biol. 1990;110:1361–1367. doi:10.1083/jcb.110.4.1361. PMID:2139036
- Jenkins RG, Su X, Su G, et al. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J Clin Invest. 2006;116:1606–1614. doi:10.1172/JCI27183. PMID:16710477
- Tatti O, Vehvilainen P, Lehti K, et al. MT1-MMP releases latent TGF-beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp Cell Res. 2008;314:2501–2514. doi:10.1016/j.yexcr.2008.05.018. PMID:18602101
- Taipale J, Lohi J, Saarinen J, et al. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem. 1995;270:4689–4696. doi:10.1074/jbc.270.9.4689. PMID:7876240
- Koli K, Saharinen J, Hyytiainen M, et al. Latency, activation, and binding proteins of TGF-beta. Microscopy Res Tech. 2001;52:354–362. doi:10.1002/1097-0029(20010215)52:4%3c354:AID-JEMT1020%3e3.0.CO;2-G.
- Annes JP, Chen Y, Munger JS, et al. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol. 2004;165:723–34. doi:10.1083/jcb.200312172. PMID:15184403
- Aluwihare P, Mu Z, Zhao Z, et al. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci. 2009;122:227–232. doi:10.1242/jcs.035246. PMID:19118215
- Buscemi L, Ramonet D, Klingberg F, et al. The single-molecule mechanics of the latent TGF-beta1 complex. Curr Biol. 2011;21:2046–2054. doi:10.1016/j.cub.2011.11.037. PMID:22169532
- Hinz B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015;47:54–65. doi:10.1016/j.matbio.2015.05.006. PMID:25960420
- Wipff PJ, Rifkin DB, Meister JJ, et al. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi:10.1083/jcb.200704042. PMID:18086923
- Shi M, Zhu J, Wang R, et al. Latent TGF-beta structure and activation. Nature. 2011;474:343–349. doi:10.1038/nature10152. PMID:21677751
- Roberts AB, Sporn MB, Assoian RK, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA. 1986;83:4167–4171. doi:10.1073/pnas.83.12.4167. PMID:2424019
- Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi:10.1056/NEJM199411103311907. PMID:7935686
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. doi:10.1056/NEJMra0806188. PMID:19420368
- Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi:10.1038/nrm809. PMID:11988769
- Blumbach K, Zweers MC, Brunner G, et al. Defective granulation tissue formation in mice with specific ablation of integrin-linked kinase in fibroblasts – role of TGFbeta1 levels and RhoA activity. J Cell Sci. 2010;123:3872–3883. doi:10.1242/jcs.063024. PMID:20980390
- Lange A, Wickstrom SA, Jakobson M, et al. Integrin-linked kinase is an adaptor with essential functions during mouse development. Nature. 2009;461:1002–1006. doi:10.1038/nature08468. PMID:19829382
- Ghatak S, Morgner J, Wickstrom SA. ILK: a pseudokinase with a unique function in the integrin-actin linkage. Biochem Soc Trans. 2013;41:995–1001. doi:10.1042/BST20130062. PMID:23863169
- Morgner J, Wickstrom SA. The weakest link: a new paradigm for stabilizing the integrin-actin connection. Cell Cycle. 2013;12:2929–2930. doi:10.4161/cc.26213. PMID:23974089
- Wickstrom SA, Radovanac K, Fassler R. Genetic analyses of integrin signaling. Cold Spring Harbor Perspect Biol. 2011;3. doi:10.1101/cshperspect.a005116.
- Sakai T, Li S, Docheva D, et al. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 2003;17:926–940. doi:10.1101/gad.255603. PMID:12670870
- Radovanac K, Morgner J, Schulz JN, et al. Stabilization of integrin-linked kinase by the Hsp90-CHIP axis impacts cellular force generation, migration and the fibrotic response. EMBO J. 2013;32:1409–1424. doi:10.1038/emboj.2013.90. PMID:23612611
- Kessler D, Dethlefsen S, Haase I, et al. Fibroblasts in mechanically stressed collagen lattices assume a “synthetic” phenotype. J Biol Chem. 2001;276:36575–36585. doi:10.1074/jbc.M101602200. PMID:11468280
- Polishchuk R, Di Pentima A, Lippincott-Schwartz J. Delivery of raft-associated, GPI-anchored proteins to the apical surface of polarized MDCK cells by a transcytotic pathway. Nat Cell Biol. 2004;6:297–307. doi:10.1038/ncb1109. PMID:15048124
- Presley JF, Cole NB, Schroer TA, et al. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi:10.1038/38001. PMID:9288971
- Guilluy C, Garcia-Mata R, Burridge K. Rho protein crosstalk: another social network? Trends Cell Biol. 2011;21:718–726. doi:10.1016/j.tcb.2011.08.002. PMID:21924908
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Ann Rev Cell Dev Biol. 2005;21:247–269. doi:10.1146/annurev.cellbio.21.020604.150721.
- Taylor JM, Macklem MM, Parsons JT. Cytoskeletal changes induced by GRAF, the GTPase regulator associated with focal adhesion kinase, are mediated by Rho. J Cell Sci. 1999;112:231–242. PMID:9858476
- Doherty GJ, Ahlund MK, Howes MT, et al. The endocytic protein GRAF1 is directed to cell-matrix adhesion sites and regulates cell spreading. Mol Biol Cell. 2011;22:4380–4389. doi:10.1091/mbc.E10-12-0936. PMID:21965292
- Aguilera MO, Beron W, Colombo MI. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy 2012;8:1590–1603. doi:10.4161/auto.21459. PMID:22863730
- Moreau K, Ravikumar B, Puri C, et al. Arf6 promotes autophagosome formation via effects on phosphatidylinositol 4,5-bisphosphate and phospholipase D. J Cell Biol. 2012;196:483–496. doi:10.1083/jcb.201110114. PMID:22351926
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12:1–222. doi:10.1080/15548627.2015.1100356. PMID:26799652
- Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi:10.1038/nature03029. PMID:15525940
- Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30:4701–4711. doi:10.1038/emboj.2011.398. PMID:22068051
- Birgisdottir AB, Lamark T, Johansen T. The LIR motif – crucial for selective autophagy. J Cell Sci. 2013;126:3237–3247. PMID:23908376
- Ejlerskov P, Rasmussen I, Nielsen TT, et al. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013;288:17313–17335. doi:10.1074/jbc.M112.401174. PMID:23629650
- Pilli M, Arko-Mensah J, Ponpuak M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–234. doi:10.1016/j.immuni.2012.04.015. PMID:22921120
- Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi:10.1038/385729a0. PMID:9034190
- Zhang M, Schekman R. Cell biology. Unconventional secretion, unconventional solutions. Science. 2013;340:559–561. doi:10.1126/science.1234740. PMID:23641104
- Jiang S, Dupont N, Castillo EF, et al. Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Inn Immun. 2013;5:471–479. doi:10.1159/000346707.
- Rabouille C, Malhotra V, Nickel W, et al. Diversity in unconventional protein secretion. J Cell Sci. 2012;125:5251–5255. doi:10.1242/jcs.103630. PMID:23377655
- Rabouille C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2017;27:230–240. doi:10.1016/j.tcb.2016.11.007. PMID:27989656
- Wickstrom SA, Lange A, Montanez E, et al. The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J. 2010;29:281–291. doi:10.1038/emboj.2009.376. PMID:20033063
- D'Angelo G, Prencipe L, Iodice L, et al. GRASP65 and GRASP55 sequentially promote the transport of C-terminal valine-bearing cargos to and through the Golgi complex. J Biol Chem. 2009;284:34849–34860. doi:10.1074/jbc.M109.068403. PMID:19840934
- Kuo A, Zhong C, Lane WS, et al. Transmembrane transforming growth factor-α tethers to the PDZ domain-containing, Golgi membrane-associated protein p59/GRASP55. EMBO J. 2000;19:6427–6439. doi:10.1093/emboj/19.23.6427. PMID:11101516
- Zhang M, Kenny SJ, Ge L, et al. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. eLife. 2015;4.
- Feldmeyer L, Werner S, French LE, et al. Interleukin-1, inflammasomes and the skin. Eur J Cell Biol. 2010;89:638–644. doi:10.1016/j.ejcb.2010.04.008. PMID:20605059
- Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012;22:397–406. doi:10.1016/j.tcb.2012.04.008. PMID:22677446
- Ponpuak M, Mandell MA, Kimura T, et al. Secretory autophagy. Curr Op Cell Biol. 2015;35:106–116. doi:10.1016/j.ceb.2015.04.016. PMID:25988755
- Giuliani F, Grieve A, Rabouille C. Unconventional secretion: a stress on GRASP. Curr Op Cell Biol. 2011;23:498–504. doi:10.1016/j.ceb.2011.04.005. PMID:21571519
- Duran JM, Anjard C, Stefan C, et al. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. 2010;188:527–536. doi:10.1083/jcb.200911154. PMID:20156967
- Manjithaya R, Anjard C, Loomis WF, et al. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010;188:537–546. doi:10.1083/jcb.200911149. PMID:20156962
- Murray RZ, Kay JG, Sangermani DG, et al. A role for the phagosome in cytokine secretion. Science. 2005;310:1492–1495. doi:10.1126/science.1120225. PMID:16282525
- Lieu ZZ, Lock JG, Hammond LA, et al. A trans-Golgi network golgin is required for the regulated secretion of TNF in activated macrophages in vivo. Proc Natl Acad Sci USA. 2008;105:3351–3356. doi:10.1073/pnas.0800137105. PMID:18308930
- Gubbiotti MA, Iozzo RV. Proteoglycans regulate autophagy via outside-in signaling: an emerging new concept. Matrix Biol. 2015;48:6–13. doi:10.1016/j.matbio.2015.10.002. PMID:26462577
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi:10.1016/S0092-8674(02)00971-6. PMID:12297042
- Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi:10.1126/science.1176009. PMID:19965464
- Ulbricht A, Eppler FJ, Tapia VE, et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol. 2013;23:430–435. doi:10.1016/j.cub.2013.01.064. PMID:23434281
- Ulbricht A, Hohfeld J. Tension-induced autophagy: may the chaperone be with you. Autophagy. 2013;9:920–922. doi:10.4161/auto.24213. PMID:23518596
- Denton CP. Systemic sclerosis: from pathogenesis to targeted therapy. Clin Exp Rheumatol. 2015;33:S3–S7. PMID:26457375
- Palumbo-Zerr K, Zerr P, Distler A, et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-beta signaling and fibrosis. Nature Med. 2015;21:150–158. doi:10.1038/nm.3777. PMID:25581517
- Zhang ZG, Lambert CA, Servotte S, et al. Effects of constitutively active GTPases on fibroblast behavior. Cell Mol Life Sci. 2006;63:82–91. doi:10.1007/s00018-005-5416-5. PMID:16378244
- Schulz JN, Zeltz C, Sorensen IW, et al. Reduced granulation tissue and wound strength in the absence of alpha11beta1 integrin. J Invest Dermatol. 2015;135:1435–1444. doi:10.1038/jid.2015.24. PMID:25634355
- Schulz JN, Nuchel J, Niehoff A, et al. COMP-assisted collagen secretion–a novel intracellular function required for fibrosis. J Cell Sci. 2016;129:706–716. doi:10.1242/jcs.180216. PMID:26746240
- Knipper JA, Willenborg S, Brinckmann J, et al. Interleukin-4 receptor alpha signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity. 2015;43:803–816. doi:10.1016/j.immuni.2015.09.005. PMID:26474656
- Tsuchiya S, Yamabe M, Yamaguchi Y, et al. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer. 1980;26:171–176. doi:10.1002/ijc.2910260208. PMID:6970727
- Takashiba S, Van Dyke TE, Amar S, et al. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kappaB. Infection Immun. 1999;67:5573–5578.
- Nichols BJ, Kenworthy AK, Polishchuk RS, et al. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J Cell Biol. 2001;153:529–541. doi:10.1083/jcb.153.3.529. PMID:11331304
- Koushik SV, Chen H, Thaler C, et al. Cerulean, Venus, and VenusY67C FRET reference standards. Biophys J. 2006;91:L99–L101. doi:10.1529/biophysj.106.096206. PMID:17040988
- Carlemalm E, Villiger W. Low temperature embedding. In: Bullbock GR, Petrucz P (eds). Techniques in immunocytochemistry. London: Academic Press; 1989, p. 29–45.