
ABSTRACT
Reactivation and expansion of myelin-reactive CD4+ T cells within the central nervous system (CNS) are considered to play a key role in the pathogenesis of multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE). We demonstrated that accumulation of myelin-specific CD4+ T cells within the CNS and subsequent clinical disease development require autophagy related (ATG) protein-dependent phagocytosis in dendritic cells (DCs). Genetic ablation of this pathway impairs presentation of myelin-associated antigen following phagocytosis of injured, phosphatidylserine-exposing oligodendroglial cells. Thus, DCs use ATG-dependent phagocytosis for enhanced presentation of myelin antigen, thereby linking oligodendrocyte injury with antigen processing and T cell-pathogenicity during autoimmune CNS inflammation.
Phagocytosis constitutes a specialized mode of endocytosis, during which cells internalize solid extracellular cargo, and this process can be initiated through triggering of phagocytic receptors such as pattern recognition, opsonic, and phosphatidylserine (Ptd-L-ser) receptors. The emanating phagosome undergoes stepwise maturation and subsequently fuses with lysosomes followed by the enzymatic breakdown of the internalized constituents. In recent years, a novel pathway has been characterized that couples phagocytosis to certain autophagy-related (ATG) proteins. During this ATG-dependent phagocytosis, the ubiquitin-like autophagy protein LC3 is recruited to the outer membrane of single-membrane phagosomes. Whereas this cellular process is independent of the canonical macroautophagy core machinery, certain proteins such as ATG5 and ATG7 exert essential functions. ATG-dependent phagocytosis is involved in the promotion of lysosome-phagosome fusion, targeting of phagosomal cargo to pathogen recognition receptors and the provision of antigenic material to MHC class II-containing compartments. The relative importance of the aforementioned functions appears to be cell specific. Whereas ATG-dependent phagocytosis predominantly supports killing and degradation of engulfed pathogens in macrophages, in professional antigen-presenting cells such as DCs, ATG-dependent phagocytosis is employed to delay hydrolytic cleavage, which results in prolonged antigen storage and sustained antigen presentation via MHC class II.
MHC class II-mediated antigen presentation during CNS autoimmunity
Multiple sclerosis (MS) is a chronic immune-mediated disease of the CNS that develops in young adults. Akin to many autoimmune conditions, HLA-DR and -DQ alleles within the HLA class II region are the strongest risk-conferring genes. Whereas there is a general consensus that MHC class II molecules contribute to autoimmune disease risk by cognate interactions with autoreactive CD4+ T cells, how MHC class II-mediated antigen presentation regulates CD4+ T cell autoreactivity at the molecular level is still not fully understood. The emergence, activation and accumulation of myelin-reactive T cells within the CNS are thought to represent critical processes during the development and progression of MS, and encephalitogenic CD4+ T cells are the main driving forces in the animal model of MS, EAE. The adoptive transfer of primed myelin-specific CD4+ T cells into naïve recipient mice (adoptive transfer EAE, AT-EAE) models this effector phase, which is highly dependent on the presence of ITGAX/CD11c+ DCs within the CNS. How ITGAX/CD11c+ DCs initiate and sustain autoreactive CD4+ T cells within the CNS has not been fully elucidated, partly due to a lack of insight into the detailed antigen-processing route of how myelin self-antigens are processed and presented to CD4+ T cells.
Myelin-specific CD4+ T cells require ATG-dependent phagocytosis in DCs to induce EAE development
To address whether the autophagy machinery modulates EAE development, we generated conditional knockout mice (C57BL/6) for disruption of Atg5 in ITGAX/CD11c+ DCs. We identified a small population of CNS ITGAX/CD11c+ MHC class IIhi DCs that is targeted by Cre-mediated site-specific somatic recombination within the CNS. While ablation of ATG5 in ITGAX/CD11c+ DCs does not impair effector functions of CD4+ T cells nor alleviate peripheral priming of CD4+ T cells following active immunization with myelin protein, it diminishes accumulation of myelin-peptide-specific CD4+ T cells within the CNS and completely prevents clinical disease development following adoptive transfer of primed, encephalitogenic T cells.
Oligodendrocyte injury and concomitant focal demyelination constitute pathological hallmarks of MS lesions and during EAE development, and can even precede the formation of inflammatory infiltrates. Efficient local reactivation of myelin-specific CD4+ T cells in MS and EAE implies preceding phagocytosis, processing and presentation via MHC class II of oligodendrocyte-derived material. ATG-dependent phagocytosis can be initiated by engagement of Ptd-L-ser-recognizing receptors through injured target cells. We observed that, whereas ATG5 in ITGAX/CD11c+ DCs is negligible for the general capacity to phagocytose extracellular constituents, presentation of antigen derived from phagocytosed, Ptd-L-ser-exposing oligodendroglial cells is abrogated in the absence of ATG5.
Conclusions
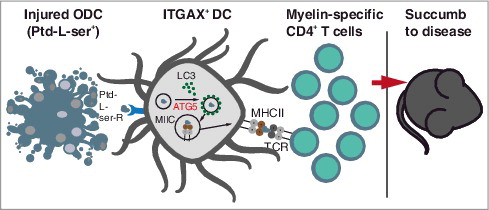
Our data demonstrate that ATG-regulated phagocytosis of injured oligodendrocytes for MHC class II antigen presentation is critical for myelin-specific T cells to accumulate within the CNS and to permit EAE development (). These findings link phagocytosis of oligodendrocyte-derived myelin with antigen processing and T cell-pathogenicity, and identify ATG-dependent phagocytosis in DCs as a key regulator in driving autoimmune CD4+ T cell-mediated CNS damage.
Figure 1. Schematic illustration summarizing how ATG5-dependent phagocytosis may be implicated in the provision of injured oligodendrocyte (ODC)-derived antigenic material to the MHC class II pathway in dendritic cells (DCs) during CNS autoimmunity. ATG5-dependent phagocytosis is initiated by the ingestion of Ptd-L-ser+ ODC fragments upon their binding of Ptd-L-ser receptors (Ptd-L-Ser-R). LC3-I is converted into LC3-II in an ATG5-dependent manner and recruited to the single membrane forming phagosome, which subsequently fuses with MHC class II-containing compartments (MIICs). Myelin-derived antigens are presented to encephalitogenic CD4+ T cells, thus facilitating the development and maintenance of neuroinflammation.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.