
ABSTRACT
During macroautophagy/autophagy, mammalian Atg8-family proteins undergo 2 proteolytic processing events. The first exposes a COOH-terminal glycine used in the conjugation of these proteins to lipids on the phagophore, the precursor to the autophagosome, whereas the second releases the lipid. The ATG4 family of proteases drives both cleavages, but how ATG4 proteins distinguish between soluble and lipid-anchored Atg8 proteins is not well understood. In a fully reconstituted delipidation assay, we establish that the physical anchoring of mammalian Atg8-family proteins in the membrane dramatically shifts the way ATG4 proteases recognize these substrates. Thus, while ATG4B is orders of magnitude faster at processing a soluble unprimed protein, all 4 ATG4 proteases can be activated to similar enzymatic activities on lipid-attached substrates. The recognition of lipidated but not soluble substrates is sensitive to a COOH-terminal LIR motif both in vitro and in cells. We suggest a model whereby ATG4B drives very fast priming of mammalian Atg8 proteins, whereas delipidation is inherently slow and regulated by all ATG4 homologs.
Abbreviations
| ATG | = | autophagy related |
| BafA1 | = | bafilomycin A1 |
| CM | = | complete media |
| GABARAP | = | GABA type A receptor-associated protein |
| GABARAPL1 | = | GABA type A receptor-associated protein like 1 |
| GABARAPL2 | = | GABA type A receptor-associated protein like 2 |
| IF | = | immunofluorescence |
| LIR | = | LC3-interacting region |
| MAP1LC3/LC3B | = | microtubule associated protein 1 light chain 3 beta |
| PE | = | phosphatidylethanolamine |
| PS | = | phosphatidylserine |
| PtdIns | = | phosphatidylinositol |
| YFP | = | yellow fluorescent protein |
Introduction
The attachment of the Atg8 family of proteins (including Atg8 in yeast and both LC3 and GABARAP subfamilies in mammals) to a growing phagophore membrane is a critical event in autophagosome maturation.Citation1 Atg8-family proteins undergo 2 distinct proteolytic processing steps during autophagy (A): First, in support of phagophore growth, each protein is cleaved by ATG4 to expose a glycine at the COOH-terminus. The exposed glycine is then utilized in a series of ubiquitin-like reactions culminating in the covalent attachment of the Atg8-family protein to the phospholipid phosphatidylethanolamine (PE). This Atg8–PE conjugate is then involved in membrane expansion, maturation, and eventual closure into a double-membrane vesicle. Second, following autophagosome maturation, each protein is recycled off the membrane by an ATG4-mediated cleavage that breaks the bond between glycine and PE. Thus, ATG4 is responsible for both early and late cleavage events on distinct pools of Atg8-related proteins (soluble and lipid-attached respectively).
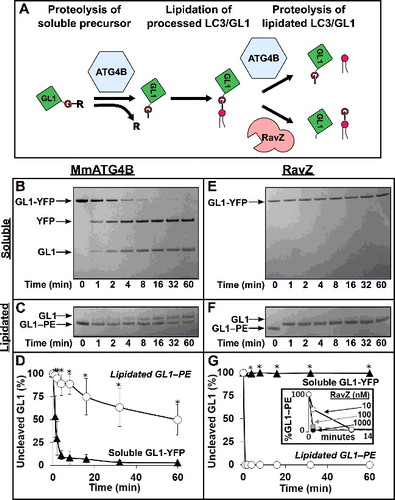
Figure 1. Proteases are naturally selective for either soluble GABARAPL1 (GL1) or GABARAPL1–PE (GL1–PE). (A) Role of ATG4 in the lipidation cycle of LC3. ATG4 primes LC3 by removing the C-terminal tail to reveal the glycine residue necessary for lipidation by ATG7 and ATG3. Later, ATG4 cleaves LC3 from the PE lipid allowing it to be recycled. Conversely, the Legionella effector protease, RavZ, cleaves lipidated LC3 at the peptide bond immediately upstream from the glycine residue, resulting in an LC3 lacking the necessary glycine residue. MmATG4B (1 µM) was incubated with either GABARAPL1-YFP (GL1-YFP) (B) or GABARAPL1-conjugated liposomes (C) for 1 h at 37°C (∼ 7 µM total protein in both experiments). Samples were removed at the noted time points and then subjected to SDS-PAGE. RavZ (1 µM) was also tested against the same substrates (E and F). Gels were then quantified using densitometry and the percent of remaining uncleaved GABARAPL1 substrate was determined (D and G). Inset of (G) further displays RavZ delipidation activity at very low concentrations. Graphs show the average of at least 3 independent proteolysis experiments. Error bars display ± 1 standard deviation. Asterisks denote p-values <0.05 between delipidation and soluble cleavage at each time point as determined by a Student t test.
Lipidated Atg8/LC3 proteins are involved in an uncertain way in virtually every aspect of autophagosome maturation. In the absence of these proteins, open-cup shaped autophagosome intermediates (i.e., phagophores) accumulate,Citation2,Citation3 and mature autophagosomes are present at lower numbers (mammals) or are very small (yeast).Citation4,Citation5 Recent work strongly suggests these proteins govern a kinetic event late in autophagosome maturation;Citation6 complete knockout of the genes encoding the mammalian Atg8-family proteins does not stop autophagy, but dramatically slows delivery into the lysosome, implying a direct role in lysosome-autophagosome fusion.Citation7 Likewise, block of lipidation dramatically slows the closing of the phagophore, a process that may be temporally linked to SNARE-recruitment ahead of lysosome delivery.Citation8 Thus, both yeast and mammalian forms of lipidated Atg8-family proteins may be important for membrane expansion and the final fission/fusion event.
Within the cell, lipidated and soluble Atg8-family proteins are involved in separate functions with distinct temporal demands, but how the large number of Atg8-family protein interactors distinguishes between the soluble and lipidated pools is poorly understood. Previous work from many groups has suggested that the natural processing of soluble mammalian Atg8-family proteins is fast and essentially constitutive. In cells, ATG4 targets and cleaves new LC3 and GABARAP proteins immediately after translation.Citation9,Citation10 In fact, unprocessed forms of mammalian and yeast Atg8-family proteins do not ordinarily accumulate even when ATG4/Atg4 levels are diminished by protein knockdown or drug treatment (e.g. [Citation11,Citation12]). Instead, ATG4 expression must be eliminated by genetic knockoutCitation13,Citation14 or tagged Atg8/LC3 proteins must be significantly overexpressed (e.g. [Citation15]) to find uncleaved proteins. Thus, the resting levels of enzyme and substrate are maintained to keep a steady pool of available processed Atg8-family proteins. From in vitro proteolysis kinetics, it has been suggested that in mammals this fast constitutive priming of the soluble Atg8-family proteins is likely controlled by ATG4B.Citation16–Citation18 In contrast, other mammalian homologs are much slower at this priming event in vitro (ATG4A) or nearly inactive (ATG4C and ATG4D) suggesting reduced roles or a need for additional regulated activation.Citation16,Citation17,Citation19–Citation21
The second Atg4/ATG4-dependent proteolysis event, release of lipidated Atg8-family proteins from the mature autophagosome, is also essential,Citation22,Citation23 but cannot be fast or constitutive. Autophagic membranes formed during starvation persist for at least 10–20 min from the time at which LC3 begins to associate with the structures (e.g. [Citation24–Citation26]). Very rapid release of LC3 would run counter to all of the LC3–PE-dependent maturation events during phagophore growth. Indeed, we have described a bacterially expressed, anti-autophagy protease called RavZ that functions exactly by driving the accelerated and irreversible release of LC3 and GABARAP from autophagic membranesCitation27 and thereby blocking autophagic progression at the ATG12–ATG5-ATG16L1-positive cup-shaped intermediate of autophagosome growth. Thus, to support full autophagosome maturation, the delipidation event must be delayed or regulated to restrict the release of the lipidated form to only the final stages of autophagosome maturation. Elazar and colleagues noted that in cells undergoing starvation, globally elevated reactive oxygen species (ROS) can chemically inactivate ATG4 and, in this way, could allow lipidation to persist.Citation9,Citation10 However, in mammalian cells, autophagosomes form throughout the cytoplasm and are not limited to periods of prolonged stress; indeed basal autophagy appears to occur more or less constitutively. Thus, how LC3–PE and GABARAP–PE proteins persist long enough to support autophagosome maturation when ATG4 activity is otherwise normal has been an area of intensive investigation. Several recent reports have identified additional post-translational modifications that limit or alter ATG4 function, including phosphorylation.Citation28,Citation29 In addition, working with whole cells, groups have identified LC3/Atg8 interaction domains on ATG4 proteins that increase the overall efficiency of substrate cleavageCitation30,Citation31 and which could combine with posttranslational modifications to potentially affect the relative delipidation and priming rates.
The vast majority of Atg8-family protein biochemistry has thus far been conducted in solution, with soluble forms of yeast and mammalian Atg8-family proteins interacting with soluble pools or detergent lysates of other proteins. These studies have been tremendously valuable in establishing fundamental interactions (e.g. [Citation32–Citation35]) and have also led to the discovery of many novel effectors (e.g [Citation36]). Nonetheless, the Atg8 family of proteins is unlikely to utilize most of these interactions in the free cytosol, but rather becomes functionally relevant to macroautophagy only after its recruitment to a membrane and its covalent attachment to a lipid. Thus, here we test the model that soluble and lipidated mammalian Atg8-family proteins may be intrinsically distinct substrates independent of post-translational modifications, and are processed with separate kinetics and ATG4 homolog specificity. We discover that the physical anchoring of Atg8-family proteins in the membrane strongly determines the rate of delipidation suggesting that local cues augment or mitigate ATG4 activity. Furthermore, these assays reveal that among the mammalian proteases, ATG4B is a functional outlier, capable of processing both soluble and lipidated forms of the Atg8-family proteins, whereas the other ATG4 proteins are most active only on membrane-associated lipidated proteins. This specific recognition of LC3–PE or its homologs depends upon COOH-terminal LC3-interacting regions (LIRs) on the ATG4 proteins, which are necessary to support efficient delipidation both in vitro and in cells.
Results
ATG4B release of lipidated proteins is an intrinsically slow process
To directly establish the relative efficiency of priming and delipidation by ATG4B, we turned to a reconstituted system. In vitro assays have been remarkably effective at establishing the intrinsic rate at which different mammalian ATG4 homologs proteolyze the soluble forms of mammalian Atg8-family proteins.Citation16–Citation21 Because the ATG4 proteolysis event generally releases a very short COOH-terminal peptide from these substrates (in some cases only a single amino acid), most in vitro assays use recombinant Atg8-family proteins with an extended COOH-terminal protein tag to facilitate easy detection of proteolysis. The overall conclusion from these studies is that ATG4B-mediated proteolysis is fast and constitutive in simple systems. How ATG4B processes lipidated substrates has received much less attention in part because similar in vitro substrates can be more difficult to produce. Intuitively, one might expect that as both substrates are cleaved at the peptide bond on the COOH-side of glycine, ATG4B activity against soluble substrates would be a meaningful predictor of its activity against lipidated Atg8 proteins. However, we have already shown that the Legionella pneumophila protease, RavZ, which cleaves Atg8-family proteins just one amino acid over from the ATG4 cleavage site, exhibits a dramatic and nearly specific preference for lipidated substrates.Citation27,Citation37 Whether ATG4 proteases exhibit similar substrate preferences is not clear.
Using a modified version of the protocol first published by the Kominami group,Citation38 we have optimized the ATG7- and ATG3-dependent coupling of mammalian Atg8-family proteins to PE head groups on synthetic liposomes of different sizesCitation39 and enriched for the fully-lipidated product by employing a density centrifugation float-up assay that separated LC3–PE- or GABARAP–PE-decorated liposomes from uncoupled protein (Figure S1). In this way, we are able to accumulate high concentrations of LC3–PE or any of its mammalian homologs. In particular, the lipidation of GABARAPL1 to PE is highly efficient and thus we have established many of the basic kinetics of delipidation below with this substrate before comparing general efficiencies of delipidation across multiple LC3 homologs. To put our delipidation results into some context with other published studies, we have also generated soluble tagged LC3-family substrates. For each of these soluble substrates we have extended the sequence beyond the reactive glycine used in lipidation with an extended peptide or protein motif (see methods for sequence details). For GABARAPL1, we have fused YFP to the COOH terminus (forming GABARAPL1-YFP).
With both soluble and lipidated substrates in hand, we could quantify how fast and efficiently each is proteolyzed by following the differences in electrophoretic mobility of the substrate and product(s) of the reaction (B,C and E,F). In we compared mouse ATG4B (MmATG4B) to Legionella pneumophila RavZ at equal concentrations to ascertain their relative effectiveness against soluble and lipidated substrates. As we noted previously,Citation27 RavZ did not cleave a soluble substrate (E, G), even when present at stoichiometric concentrations with the substrate and even after overnight incubation at 37 degrees (Figure S2A). In contrast, RavZ completely processed lipidated substrate (F). Incubating 1 μM RavZ with 10 µM GABARAPL1–PE, led to complete separation of GABARAPL1 and PE within just 1 min, the shortest time point we can effectively measure in our electrophoresis-based assay (G). When GABARAPL1–PE was present at a 1000-fold excess over RavZ, we still detected nearly 50% cleavage in one min, consistent with a Vmax of ∼ 500 GABARAPL1–PE/min/RavZ (G inset). Thus, RavZ is an absolutely selective and efficient protease against LC3-related lipidated but not soluble substrates.
Similar to other published work, we observed that MmATG4B cleaves soluble GABARAPL1 very efficiently. MmATG4B cleaved an approximately 10-fold excess of GABARAPL1-YFP about as fast as we could detect in this assay (B,D), in line with published values of ATG4B when processing other LC3 family members (28-39 per min for LC3B and GABARAPL2Citation16,Citation17), and consistent with the idea that ATG4B is ordinarily a constitutively active protease. However, when mixed with GABARAPL1–PE-decorated liposomes, MmATG4B proteolysis was ∼100 times slower, with a Vmax of 0.1 GABARAPL1–PE/min/MmATG4B (C,D). Thus, MmATG4B exhibits an opposite selectivity to RavZ; delipidation of GABARAPL1–PE is intrinsically slow and occurs with a time constant on the order of minutes. Remarkably, in a reaction with nothing but MmATG4B and substrate, we recapitulate the normal cellular processing of Atg8-family proteins; fast cleavage of the unprimed full-length protein and slow release of the lipidated Atg8.
Other ATG4 homologs drive delipidation faster than soluble substrate cleavage
In humans, there are 4 different ATG4 homologs (HsATG4A, HsATG4B, HsATG4C, HsATG4D) and at least 7 different Atg8 homologs, including 3 LC3 subfamily members (LC3A, LC3B, LC3B2 and LC3C) and 3 GABARAP subfamily members (GABARAP, GABARAPL1 and, GABARAPL2). HsATG4B can proteolyze the soluble form of every human Atg8 homolog.Citation40 The kinetics of this reaction have been determined in vitro by multiple labs. ATG4A is 3–10 times slower and is limited to only the GABARAP subfamily.Citation17 ATG4C and ATG4D encode an additional large amino-terminal domain and are essentially inactive in proteolysis assaysCitation16,Citation17 although removal of the ATG4D amino-terminal domain restores some GABARAPL1-specific activity.Citation19 The rates of delipidation have not been well established for any ATG4 protein.
Human ATG4B is mostly identical to MmATG4B, with the long isoforms differing at only 19 amino acids. To assess HsATG4B activity against a range of appropriate LC3 homologs, we used our lipidation systems to make PE-anchored forms of LC3B, GABARAPL1, and GABARAPL2. For soluble substrates, we tested the impact of extending the LC3 protein family sequence with either full protein motifs (GABARAPL1-YFP and GABARAPL2-YFP) or short peptide extensions (LC3B-tag; includes the peptide ALGFSDDLPRAFR immediately after the reactive glycine). Similar to MmATG4B, HsATG4B cleaved soluble GABARAPL1-YFP faster than liposome-associated lipidated GABARAPL1–PE; thus, the intrinsically slow delipidation appears to be an evolutionarily conserved aspect of ATG4B function. Furthermore, very fast cleavage of soluble proteins and slow proteolysis of lipidated LC3-related proteins by HsATG4B was conserved across all 3 human ATG8 homologs we have tested (GABARAPL1, LC3B, and GABARAPL2; and Figure S3).
Figure 2. Human ATG4A, ATG4C and ATG4D are predominantly delipidation enzymes. (A) Diagram of the priming and delipidation of human Atg8 homologs by ATG4 isoforms. (B) Lipidated or soluble forms of GABARAPL1 (GL1), GABARAPL2 (GL2), and LC3B were incubated with 150 nM of the indicated ATG4 protease for one h at 37°C. Samples were removed at the noted times and then analyzed by SDS-PAGE. Gels were then quantified using densitometry and the percent of remaining uncleaved substrate was determined. Graphs display the average of 3 independent proteolysis experiments. Error bars display ± 1 standard deviation. Cartoon alignment shows the domains of the 4 human ATG4 proteases including the caspase cleavage site of HsATG4C and HsATG4D. Asterisks denote p-values <0.05 between delipidation and soluble cleavage at each time point as determined by a Student t test.
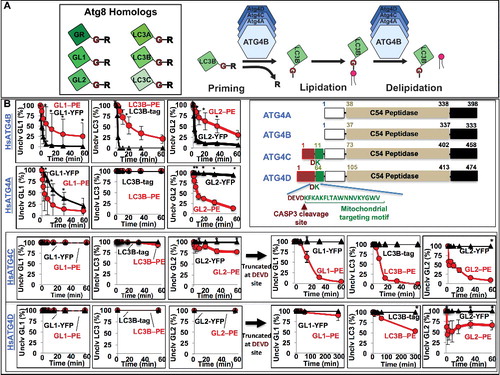
As expected, HsATG4A was active against only the GABARAP subfamily and proteolyzed the soluble substrates GABARAPL1-YFP or GABARAPL2-YFP much more slowly than HsATG4B. In contrast, HsATG4A delipidated GABARAPL1–PE about twice as fast as HsATG4B. The selectivity for GABARAPL2 was even more stark. HsATG4A dramatically favored the turnover of GABARAPL2–PE over GABARAPL2-YFP in vitro. This suggests that the GABARAP subfamily may be particularly susceptible to altered delipidation conditions in cells, due to both its sensitivity to each of the 2 major ATG4 isoforms and to the relatively high efficiency of HsATG4A.
HsATG4C and HsATG4D did not exhibit significant catalytic activity against any substrate, although very modest delipidation was suggested for HsATG4C and GABARAPL2–PE. At much longer time points, this activity became more apparent (Figure S2B and S2C), suggesting that HsATG4C was naturally specific for delipidation. The general lack of activity from HsATG4C and HsATG4D is consistent with work suggesting they are ordinarily maintained in an auto-inhibited state by the presence of an amino-terminal inhibitory domain. This domain can be removed by caspase cleavage at a conserved DEVD motif in each protein.Citation19 To establish the selectivity of the “activated” proteins, we expressed deletion mutants of HsATG4C and HsATG4D in which the sequence up to the DEVD motif is missing (ATG4C[Δ10aa] and ATG4D[Δ63aa]). Strikingly, these “activated” proteins each gained hydrolysis activity against all 3 Atg8 homologs, but only for their lipidated forms; we could not detect any significant cleavage of soluble proteins. Thus, following activation, HsATG4C and HsATG4D are delipidation-specific enzymes.
Protease recognition of lipidated proteins is sensitive to membrane organization
How is selectivity for or against lipidated LC3–PE and GABARAPL1–PE protein determined? In principle, lipidated proteins are distinguished from nonlipidated proteins by both the physical presence of the PE lipid on the protein-lipid adduct, and by the localization of this protein to a lipid membrane. Recognition of the lipid-modified protein as different from the unmodified protein would be an example of isopeptidase activity, analogous to how some deubiquitinases distinguish unprimed ubiquitin from ubiquitin attached to other proteins. Alternatively, recognition that one protein resides in a membrane while the other does not, would be an example of interfacial regulation, analogous to how lipase activity is restricted to lipids residing in intact membranes.Citation41
We recently solved the structure of RavZCitation37 and speculated that potential membrane-binding motifs near the active site might be able to confer interfacial activation upon the enzyme. In this model, these motifs would normally inhibit access to the active site, but upon interaction with an intact membrane, this inhibition would be relieved, and local, lipidated substrates could now be engaged. To test whether an intact membrane influences the selectivity of RavZ, we repeated our delipidation assays in the presence of increasing amounts of the nonionic detergent Triton X-100, in order to convert the GABARAPL1–PE decorated liposomes into micelles ( cartoon). The loss of bilayer integrity dramatically reduced RavZ-mediated GABARAPL1–PE proteolysis (A), such that at 3% Triton X-100, GABARAPL1–PE was cleaved nearly 3 orders of magnitude slower than without any detergent. We cannot formally rule out that RavZ is inactivated in detergent directly, but importantly, transient treatment with detergent did not lead to a general loss of enzyme activity (Figure S3A). Furthermore, inhibition of GABARAPL1–PE cleavage was also observed with another non-ionic detergent (octyl-glucoside; Figure S4B), consistent with a general requirement for membrane integrity. Thus, RavZ is very likely an interfacially activated enzyme.
Figure 3. Protease selectivity is driven predominantly by interfacial recognition of intact membranes. RavZ (A) and MmATG4 (B) delipidation of GABARAPL1–PE (GL1–PE) in the presence of the nonionic detergent Triton X-100. Liposomes were incubated with the indicated percent of Triton X-100 for 10 min on ice before the addition of protease (1 µM). Delipidation assays were then conducted as in . Graphs display the average delipidation rate of 3 independent proteolysis experiments. Error bars display ± 1 standard deviation.
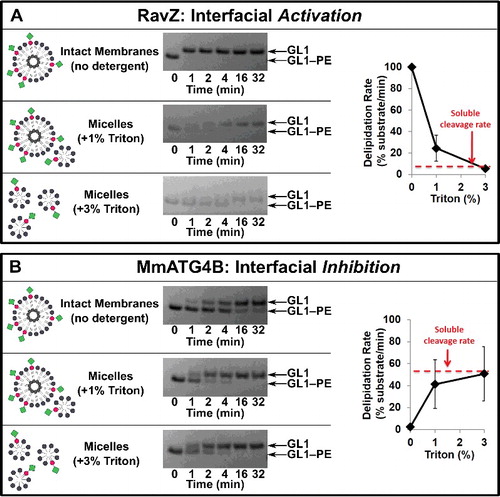
In contrast, in the presence of either detergent, GABARAPL1–PE was proteolyzed by MmATG4B much faster (B and Figure S4B). At 3% Triton X-100, the Vmax of MmATG4B was approximately 50 GABARAPL1–PE/min/MmATG4B, making processing of detergent solublized GABARAPL1–PE almost indistinguishable from the processing of the fully-proteinaceous soluble GABARAPL1-YFP construct. Thus, the presence of the PE itself is not a contributing factor to the selectivity and instead implies that MmATG4B is interfacially inhibited on intact GABARAPL1–PE-containing bilayers.
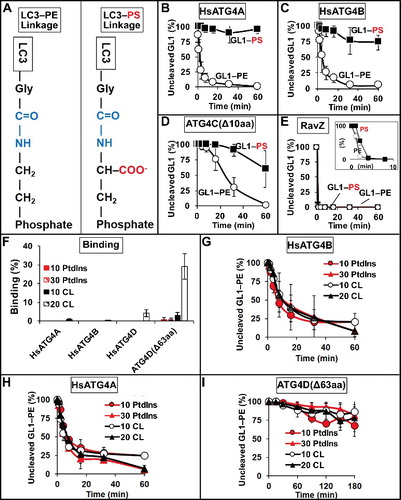
Figure 4. Effect of membrane electrostatics on ATG4 activity. (A) Differences in the LC3-lipid adduct when bound to PE lipid headgroups versus PS headgroups. (B to E) Protease activity against GABARAPL1–PS (GL1–PS). HsATG4A (B), HsATG4B (C), and ATG4C(Δ10aa) (D) cleave GABARAPL1–PS more slowly than GABARAPL1–PE (GL1–PE), whereas RavZ (E) cleaves both very quickly. Inset in (E) uses only 1.5 nM RavZ. (F) Binding of HsATG4A, HsATG4B, HsATG4D and ATG4D(Δ63aa) to liposomes with and without cardiolipin (CL). “10 PtdIns” liposomes were composed of 10 mol percent PtdIns, 55 mol percent DOPE and 35 mol percent POPC. “30 PtdIns” liposomes were composed of 30 mol percent PtdIns, 55 mol percent DOPE, and 15 mol percent POPC. “10 CL” liposomes were composed of 10 mol percent cardiolipin, 10 mol percent PtdIns, 55 mol percent DOPE, 25 mol percent POPC. “20 CL” liposomes contained 20 mol percent cardiolipin, 10 mol percent PtdIns, 55 mol percent DOPE, and 15 mol percent POPC. Liposomes were extruded to a size of 100 nm. Delipidation of GL1 from the liposome compositions in (F) by HsATG4B (G), HsATG4A (H) and ATG4D(Δ63aa) (I). All graphs display the average of 3 independent experiments. Error bars display ± 1 standard deviation.
HsATG4A, ATG4C(Δ10aa), and ATG4D(Δ63aa) each preferentially delipidated LC3 homologs (), suggesting these proteins may also be subject to interfacial regulation. Indeed, both HsATG4A and ATG4C(Δ10aa) lost delipidation activity in detergent (Figure S4C and S4D, respectively), consistent with such a model. However, HsATG4A also lost activity against soluble substrates when incubated in detergent (Figure S4C), and HsATG4C had no activity against soluble substrates (as a control for detergent inhibition), thus whether either protein is truly subject to interfacial control cannot be ascertained.
It is not clear which elements of the membrane control ATG4B activity. The fact that ATG4B-mediated delipidation was fast in detergent suggests that the PE lipid head group is not a significant determinant in the kinetics. This result is consistent with numerous earlier studies on ATG4B soluble protein processing where it was established that the peptide sequence after the reactive glycine residue is unimportant. Likewise, the ATG4B crystal structure only describes ATG4-LC3 interactions on LC3 sequences upstream of the glycine. However, changing this attached lipid to a phosphatidylserine (PS), which harbors a negative charge, dramatically impaired ATG4 function for both yeast Atg4Citation42,Citation43 and mammalian ATG4B.Citation38 Given that both RavZ and the other mammalian ATG4 proteins appear to gain function on membranes, we tested whether conjugation to PS affects the way GABARAPL1 is recognized by these proteases.
In cells, LC3 proteins have only been shown to lipidate on PE head groups, but in vitro, the free amine on PS could also be used as a substrate (A and [Citation38]). We generated liposomes with 50 mol percent PS and then used lipidation enzymes and floatation to isolate GABARAPL1–PS-decorated membranes (as in Figure S1). Surprisingly, GABARAPL1–PS was removed by RavZ with nearly the same kinetics as GABARAPL1–PE (E), indicating that this enzyme is not strongly sensitive to the nature of the attached lipid. However, GABARAPL1–PS was not a substrate for ATG4B (mouse or human; C, S5A and S6), consistent with previous publications, nor for HsATG4A (B). ATG4C(Δ10aa) was much less active against GABARAPL1–PS, but retained some slow proteolytic activity (D).
The loss of ATG4-mediated proteolysis was not simply a consequence of liposome charge density, because HsATG4B could still cleave GABARAPL1–PE when it was embedded in liposomes with high concentrations of phosphatidylinositol (PtdIns) (Figure S5B). Instead, it appears as though PS is directly inhibitory when included as part of the protein-lipid adduct, implying a very local impact of the presence of this anionic head group. Indeed, whereas detergent solubilization increased ATG4B delipidation rates of GABARAPL1–PE to approximately the same as soluble cleavage, there was a more modest increase in the rate of GABARAPL1–PS cleavage (Figure S6).
Thus, both the organization of the proteins into an intact membrane, and the composition of the substrate lipid are important, but beyond these features, we have not been able to identify a membrane composition that dramatically affects ATG4 activity. We considered whether membrane curvature, or the proportion of conical lipids in the membrane might influence activity, because the forward lipidation reaction is dramatically increased in vitro by the high affinity of ATG3 for these surfacesCitation39 and because RavZ exhibits some curvature sensitivity.Citation37 However, neither strident curvature (50-nm diameter liposomes compared to 400-nm diameter liposomes) nor high levels of the conical lipid DOPE (30 mol percent vs 55 mol percent) influenced HsATG4B-mediated delipidation kinetics (Figure S5C). We also tested membrane binding of each protease, using a nonequilibrium floatation assay we have previously published with both ATG3 and RavZ. We did not observe any significant binding of HsATG4A, HsATG4B, HsATG4D or ATG4D(Δ63aa) to the liposomes used in or to liposomes with increased PtdIns surface densities (F; 10% PtdIns lipid composition). Thus, these proteins do not harbor exposed motifs that favor lipid interactions. ATG4D has been shown to target mitochondria in cells and to have enhanced targeting after removal of the auto-inhibitory domainCitation44 because this exposes an amino-terminal mitochondrial targeting motif. Thus, we also tested whether any of these proteases bind the mitochondria-specific lipid cardiolipin. There was virtually no binding of any protein to liposomes with low (10 mol percent) cardiolipin, but ATG4D(Δ63aa) bound very efficiently to liposomes with 20 mol percent cardiolipin, and full-length HsATG4D bound weakly. However, we could not detect any cardiolipin-dependent changes in the rate of delipidation of GABARAPL1–PE for any of the homologs (G-I), suggesting this increased binding of ATG4D(Δ63aa) does not affect proteolysis activity. Thus, membrane-dependent regulation of ATG4 proteins either involves motifs that do not contribute significantly to membrane binding affinity, or motifs that are specific for LC3/GABARAP-decorated proteo-membranes.
Interaction with multiple substrate proteins likely confers interfacial regulation
Another feature unique to an intact phagophore membrane is the presence of a high density of lipidated LC3/GABARAP. Several proteins target phagophores through low-affinity interactions with multiple LC3–PEs decorating the membrane, via LIRs. ATG4B has a conserved N-terminal LIR that engages a nonsubstrate LC3 protein in the co-crystal of the protease and substrate.Citation45 In addition, the entire ATG4 family has a conserved COOH-terminal LIR motif. In yeast, this motif is a general modifier of Atg4-Atg8 interaction in solution or on membranes.Citation30 On mammalian ATG4B, this LIR controls 2 separate functions; 1) it stabilizes the pool of soluble GABARAP-family proteins from an autophagy-independent turnover pathway, and 2) it supports efficient ATG4B-mediated proteolysis of soluble LC3 proteins in vitro, and efficient proteolysis of soluble and lipidated substrates in atg4b knockout MEFs.Citation31 Thus, we tested whether either of these 2 LIR motifs influences ATG4B substrate selectivity in our reconstituted system.
To disrupt either the NH or COOH terminal LIRs (A), we mutated the conserved aromatic residue and leucine each to alanine (ATG4B-NHmut and ATG4B-COOHmut, respectively). For the COOH-terminal LIR we also truncated the protein just before (ATG4B[1-384]) or within the LIR (ATG4B[1-388]). Finally, we also made double mutants in which both LIRs were disrupted (NHmut -ATG4B[1-384] and NHmut-COOHmut). We then tested these ATG4B proteins against both soluble and lipidated substrates ().
Figure 5. COOH-terminal LIR motif on HsATG4B controls delipidation specificity in vitro. (A) Diagram of LIR mutants used in vitro. (B and C) Activity of LIR mutants in proteolysis assays of LC3 and GABARAPL1 substrates. Insets magnify soluble protease kinetics during the first 10 min. Assays conducted as in . Error bars represent ± 1 standard deviation.
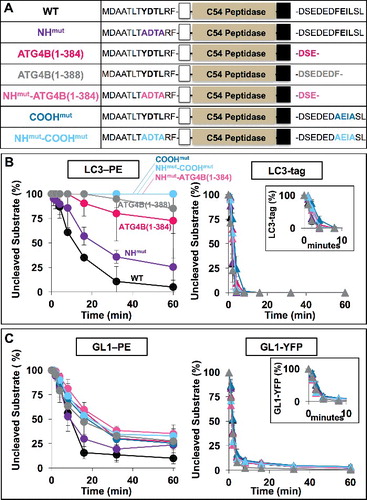
We could not detect a significant impact on the soluble protein cleavage rate for any of the mutants when tested against either soluble LC3 or GABARAPL1, suggesting these motifs are not required for substrate recognition in solution. Likewise, the NH-terminal mutation had only a modest (∼2 fold) impact on delipidation kinetics for LC3–PE and an insignificant impact on GABARAPL1–PE.
In contrast, mutation or deletion of the COOH-terminal LIR specifically affected delipidation. The rate of GABARAPL1–PE turnover was reduced by about a factor of ∼4 when the COOH terminal motif was mutated or deleted, and was not any more severe when the NH-terminal motif was also mutated, suggesting essentially all of the specificity for delipidation resides in the COOH terminus. Disruption of the COOH-terminal motif led to a complete loss of LC3–PE delipidation even out to 60 min in 3 of the 5 mutants tested. In the LIR deletion mutants, ATG4B[1-384] and ATG4B[1-388], delipidation was severely affected but some weak activity was observed.
ATG4B-mediated priming and delipidation in mammalian cells
Thus, efficient delipidation in vitro requires the COOH-terminal LIR and this motif appears to be selective for only delipidation activity. To test the impact of these mutations in vivo, we wanted to compare ATG4B activity with and without this motif. Reduction of ATG4B expression levels, either by genetic knockdown or by chemical inhibition, leads to an overall reduction in autophagy flux (e.g. see Citationrefs. 11,Citation15,Citation46,Citation47), consistent with a broad role for this enzyme in the regulation of each of the 6 to 8 ATG8 proteins present in mammals. To explore the relative contributions of ATG4B at both the priming step and the delipidation event, we set up a knockout and rescue system in HEK293 cells, using the CRISPR/Cas9 system to knock out ATG4B.
In resting wild-type cells, LC3B-I and LC3B-II were both apparent, while each of the 3 GABARAP homologs we tested (GABARAP, GABARAPL1, and GABARAPL2) appeared only as a single band likely representing the primed but unlipidated soluble forms of these proteins (A and S7A). Following treatment with bafilomycin A1 (bafA1) to prevent lysosome-dependent clearance, lipidated forms of all 4 tested Atg8 homologs accumulated, and the effect was even stronger when cells were both starved and treated with bafA1. This bafA1-dependence is indicative of autophagic flux.
Figure 6. ATG4 QKO shows complete loss of LC3/GABARAP-family priming. (A) Western blot reveals the accumulation of unprimed LC3B and GABARAPL1 (GL1) in ATG4B KO and ATG4 QKO cells. (B) Western blots of individual ATG4 homologs show complete loss of ATG4 at the protein level. (C) Representative anti-LC3B immunofluorescence images show loss of LC3B puncta formation in ATG4B KO and ATG4 QKO cells. N = 47, 50, 83, 85, 65 and 42, respectively. (D) Representative anti-FLAG immunofluorescence images of cells stably expressing 3xFLAG-GABARAPL1 show loss of puncta formation in ATG4B KO and ATG4 QKO cells. N = 67, 81, 76, 57, 95 and 66, respectively. Puncta number per cell is quantified blindly, and shown by median with 95% confidence interval. Statistical significance is assessed by ranked Mann-Whitney test. ns, not significant; *, p < 0.05; ****, p < 0.0001. CM, complete medium. (E) In ATG4B KO cells, LC3B and GL1 processing can be partially or fully rescued by high (CMV) or low (PGK1) overexpression of ATG4B (left). Western blot of ATG4B KO cells stably expressing ATG4B driven by each promoter (right).
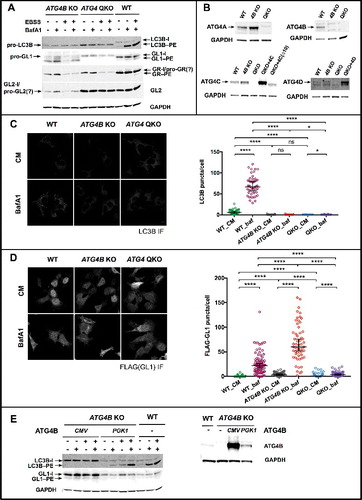
In contrast, in cells where ATG4B had been removed by gene editing (ATG4B knockout [KO]), neither LC3-I nor LC3-II were formed, and instead the unprimed LC3B accumulated ([Citation14] and A). The levels of this unprimed protein were not sensitive to autophagic induction (starvation) or to flux (bafA1). Furthermore, LC3 puncta formation was lost (C). Thus, at physiological expression levels, ATG4B is necessary to support unprimed LC3 to LC3-I conversion in HEK293 cells and in its absence, no lipidated LC3 can be produced. In this context, the role of ATG4B in LC3–PE delipidation cannot be assessed.
Among GABARAP homologs, the role of ATG4B is more complicated. For GABARAP or GABARAPL2, we could still detect weak bafA1-dependent accumulation of lipidated material, suggesting priming is still active. Total levels of these 2 proteins appeared to be reduced, however, consistent with the previous observation that ATG4B protects these proteins from degradation.Citation31 Immunoblotting for GABARAPL1 revealed a slower migrating band that was likely the unprimed form implying that GABARAPL1 priming was incomplete in the absence of ATG4B. In addition, the lipidated form of GABARAPL1 accumulated to very high levels. This accumulation might reflect a direct role for ATG4B in GABARAPL1–PE delipidation. Alternatively, GABARAPL1–PE accumulation may be a secondary consequence to the loss of LC3–PE consistent with the accumulation of some lipidated GABARAP protein-family members in LC3 family knockouts.Citation7 In either event, FLAG-tagged GABARAPL1 could still decorate puncta in cells, and total levels of these puncta were actually elevated in the ATG4B KO (D). The bafA1-dependent accumulation of each of the GABARAP-family members and the presence of FLAG-GABARAPL1-positive puncta suggest that autophagic flux is at least partially intact in the ATG4B KO.
To confirm the specificity of these phenotypes, we rescued ATG4B expression in the knockout cells using lentiviral expression of ATG4B from both low and high-expressing promoters (E). Expression from either promoter eliminated the accumulation of unprimed LC3B or GABARAPL1 (E). However, as others have noted previously, rescue/overexpression experiments are complicated because at high expression levels ATG4 proteins exhibit a dominant negative effect that is unrelated to priming or delipidation but instead stems from the sequestration of free LC3-I.Citation2,Citation40 Indeed, from the high-expressing promoter, LC3 lipidation was inhibited and there was no apparent flux of the protein (E). Our “low” expression promoter also resulted in overexpression of ATG4B (about 4 fold over endogenous levels), but under these conditions, LC3-II formation was rescued and the dramatic accumulation of GABARAPL1-II observed in the knockout was reversed. The control “rescue” with either empty vector did not reverse or alter LC3 levels (Figure S7C). Thus, ATG4B contributes to the priming of at least LC3B and GABARAPL1, and possibly to delipidation activity of GABARAPL1.
Further interpretation in this simple knockout/rescue system is complicated by the redundancy of both the ATG4- and Atg8-protein families, and by the partial compensation in our KO line, where levels of ATG4A and ATG4C were each elevated (B). To more closely align with our in vitro experiments where we can interrogate single ATG4 proteins, we next produced HEK293 cells that did not express any ATG4 proteases using CRISPR/Cas9 to knock out all 4 ATG4 genes (A,B). In this quadruple knockout (QKO), the cells were completely incapable of priming and only the unprimed forms of LC3B and GABARAPL1 accumulated (A and S7B). We also detected only a single band in the GABARAP and GABARAPL2 blots, which likely represents the unprimed form of each; however, these run very close to form I making absolute identification uncertain. This also means that we cannot definitively establish whether priming is complete in the wild-type or ATG4B KO lines for these 2 proteins. The overall expression level of each of the Atg8-family proteins was similar to the ATG4B KO condition, including the overall reduction in GABARAPL1 and GABARAP levels. Surprisingly however, GABARAPL2 levels were restored to near wild type in the QKO.
Importantly, no lipidated material was observed for any Atg8 homolog (A) and both LC3 puncta (C) and FLAG-GABARAPL puncta (D) were lost. In this context, we could test the sufficiency of ATG4B and the biological significance of the LIR. Rescue with wild-type ATG4B alone was nearly sufficient to restore normal processing of the Atg8-family proteins (). We no longer detected any accumulated unprimed LC3 or GABARAPL1. Furthermore all 4 homologs existed at least partially in a lipidated state, and LC3, GABARAP and GABARAPL2 all showed evidence of bafA1 sensitivity. There is one subtle but reproducible difference; LC3-II was elevated in the nonstressed full medium condition (A and Figure S8) and LC3 puncta levels were also marginally elevated (about 2 fold; compare C and B). As this is the opposite effect ordinarily observed with ATG4B overexpression, it suggests that ATG4B may be insufficient to maintain normal delipidation of LC3-II when other ATG4 proteases are absent. Thus, ATG4B alone provides a nearly complete rescue of Atg8-family protein processing events.
Figure 7. ATG4 QKO cells stably expressing ATG4B without its COOH-terminal LIR motif accumulate lipidated LC3/GABARAP-family proteins. (A) ATG4 QKO cells stably expressing either ATG4B or ATG4B without a C-terminal LIR motif (ATG4B[1-388]) under a low (PGK1)-expression promoter. (B) Representative anti-LC3B immunofluorescence images of ATG4 QKO expressing either construct. N = 19, 30, 30 and 18, respectively. (C) Representative anti-FLAG immunofluorescence images of ATG4 QKO cells expressing either construct, and stably expressing 3xFLAG-GABARAPL1 (GL1). N = 54, 79, 46 and 49, respectively. Puncta number per cell is quantified blindly, and shown by median with 95% confidence interval. Statistical significance is assessed by ranked Mann-Whitney test. ns, not significant; *, p < 0.05; ****, p < 0.0001. CM, complete medium. Representative western blots of 3xFLAG-GABARAPL1 processing are shown in Figure S9.
![Figure 7. ATG4 QKO cells stably expressing ATG4B without its COOH-terminal LIR motif accumulate lipidated LC3/GABARAP-family proteins. (A) ATG4 QKO cells stably expressing either ATG4B or ATG4B without a C-terminal LIR motif (ATG4B[1-388]) under a low (PGK1)-expression promoter. (B) Representative anti-LC3B immunofluorescence images of ATG4 QKO expressing either construct. N = 19, 30, 30 and 18, respectively. (C) Representative anti-FLAG immunofluorescence images of ATG4 QKO cells expressing either construct, and stably expressing 3xFLAG-GABARAPL1 (GL1). N = 54, 79, 46 and 49, respectively. Puncta number per cell is quantified blindly, and shown by median with 95% confidence interval. Statistical significance is assessed by ranked Mann-Whitney test. ns, not significant; *, p < 0.05; ****, p < 0.0001. CM, complete medium. Representative western blots of 3xFLAG-GABARAPL1 processing are shown in Figure S9.](/cms/asset/af85afc6-789f-418d-9a20-b783354666a8/kaup_a_1437341_f0007_oc.jpg)
To test the role of the LIR, we deleted the last 3 amino acids from ATG4B, disrupting the COOH-terminal LIR but leaving the NH-terminal LIR intact (ATG4B[1-388]). Expression of ATG4B(1-388) in the QKO cells rescued the priming defect of both LC3B and GABARAPL1; no unprimed protein was detected (A). However, there was almost no LC3-I present, all of the LC3B we could detect was in the lipidated form and we observed no bafA1-dependent change in LC3-II levels (A and Figure S8). This suggests that LC3–PE delipidation is impaired in this system.
Furthermore, disruption of the COOH-LIR also clearly impaired regulation of GABARAPL1 and GABARAP. GABARAPL1-I and GABARAP-I levels were reduced and both GABARAPL1-II and GABARAP-II accumulated in the LIR-deletion (). As with LC3B, GABARAPL1 did not show a strong bafA1-dependent accumulation (Figure S8), again suggesting that delipidation was uniquely impaired in the absence of the LIR. ATG4B(1-388) expression also could not restore GABARAP, GABARAPL1 or GABARAPL2 total levels to wild-type conditions, further confirming the critical role for the LIR in protecting these proteins.Citation31
Finally, we tested whether ATG4A, ATG4C, ATG4C(Δ10aa), ATG4D or ATG4D(Δ63aa) proteins can restore LC3 or GABARAP turnover in our QKO cells (). Despite overexpressing each of these ATG4 proteins ( and Figure S9), none of our 5 constructs restored any LC3 priming. Likewise, we could not detect lipidation of GABARAPL2. Each of these results were consistent with our in vitro system. We also could not detect processing of GABARAP.
Figure 8. Systematic analysis of ATG4 QKO rescued with a single ATG4 homolog. (A) ATG4 QKO stably expressing ATG4A driven by the CMV promoter. (B) ATG4 QKO stably expressing ATG4C or ATG4C without N-terminal extension (ATG4C[∆10]) driven by the CMV promoter. (C) ATG4 QKO stably expressing ATG4D or ATG4D without N-terminal extension (ATG4D[∆63]) driven by the CMV promoter.
![Figure 8. Systematic analysis of ATG4 QKO rescued with a single ATG4 homolog. (A) ATG4 QKO stably expressing ATG4A driven by the CMV promoter. (B) ATG4 QKO stably expressing ATG4C or ATG4C without N-terminal extension (ATG4C[∆10]) driven by the CMV promoter. (C) ATG4 QKO stably expressing ATG4D or ATG4D without N-terminal extension (ATG4D[∆63]) driven by the CMV promoter.](/cms/asset/1015773b-7cb6-4638-9651-d2bf2331752b/kaup_a_1437341_f0008_b.gif)
All 5 constructs led to at least partial processing of GABARAPL1. ATG4A was by far the most efficient and resulted in an accumulation of GABARAPL1-II to higher than wild-type levels. However, no GABARAP-I was detected and significant unprimed GABARAPL1 remained. Collectively, this suggests that priming is still very slow and that delipidation is either also slow or that the recycled GABARAPL1-I is immediately redeposited as GABARAPL1-II. The 4 ATG4C and ATG4D constructs also failed to prime most of the GABARAPL1 pool, but trace amounts of GABARAPL1-II could be detected. Interestingly, these amounts were independent of whether we used “activated” or full-length ATG4 enzymes, suggesting they have equal potency in our cells.
Overall, our 2 experimental systems largely agree. In each case ATG4B is sufficient for efficient priming of all Atg8-family proteins but is less active in delipidation. Likewise, both in vitro and in cells, disrupting the COOH-LIR leads to more dramatic impairments of the LC3 pool than the GABARAPL1 pool and in both cases the effects are specific for delipidation rather than priming. Of the other 3 ATG4s, only ATG4A shows significant priming and only of GABARAPL1 both in vitro and in cells. Thus, we conclude that delipidation and priming are largely separable activities, encoded within ATG4B through motifs that include the COOH-terminal LIR, and each present to different degrees for different ATG4 homologs.
Discussion
Through interfacial regulation of enzyme activity at membrane-water interfaces, substrate recognition can be enhanced or depressed simply by sequestering substrates at the membrane surface. Such a mechanism allows a small fraction of a substrate pool to become bioactive and to engage in productive downstream events on the membrane while the larger soluble pool is essentially ignored by the same sets of downstream factors. These mechanisms have wide applicability in intracellular trafficking pathways where membrane cues that include unique lipid distributions and localized accumulations of specific membrane proteins define “landing sites” for peripheral enzymes.
The surface of the growing phagophore is an extreme version of such a site, as it is nearly devoid of integral membrane proteins and yet develops a complex local biochemistry built upon several dynamic elements of membrane character.Citation48 These include the local accumulation of PtdIns3P, the formation of distinct membrane architectures including both convex and concave curvatures and the covalent modification of the soluble protein Atg8 into a lipid-anchored form that accumulates on the growing membrane. Reductionist approaches have revealed how many proteins in the macroautophagy pathway rely upon intact membranes to drive meaningful protein-protein interactions.Citation49,Citation50 For example, intact membranes present curvatures that promote LC3/Atg8 lipidation,Citation39,Citation51 and also scaffold the formation of large protein complexes that engage and organize Atg8–PE and its associated enzymes.Citation52–Citation54 Each of these processes imply that LC3 and Atg8 are treated differently on and off membranes.
The most extreme example of a protein making this distinction is RavZ, a protease specific for only lipidated Atg8-family proteinsCitation27 through a mechanism that we now formally establish as an interfacial activation (). RavZ engages the membrane through multiple motifs, including a short hydrophobic stretch near and possibly occluding the active site,Citation37 which likely limits RavZ to activity on membranes. In addition, RavZ encodes multiple LIR sequences that would likely favor targeting of LC3–PE-rich membranes,Citation55,Citation56 akin to how LIR-rich Atg8-associated cargo adaptors naturally target areas where Atg8-family proteins are present at high density.Citation57 These interactions provide both specificity and may also directly contribute to catalysis by promoting accessibility of the cleavage site on LC3–PE.Citation56
Unlike RavZ, ATG4 proteolysis needs to be selective rather than specific; soluble targets are primed constitutively whereas delipidation is limited to late stages of autophagosome maturation. We suggest this selectivity arises in part due to differences in the kinetics with which soluble and membrane-bound LC3 proteins are recognized and is a natural feature of the ATG4 protein family. From our in vitro assays we describe the following first principles of ATG4 function. First, and consistent with many other groups, efficient priming is an activity that is unique to ATG4B. Although ATG4A can also prime the GABARAP family, it is an order of magnitude slower than ATG4B. Neither ATG4C nor ATG4D contribute significantly. Second, the levels of priming activity for any ATG4 protein do not correlate with its delipidation activity. Thus, the presence of a covalently attached lipid on LC3 proteins fundamentally changes its recognition by ATG4 proteins and, critically, this involves anchoring of the LC3 protein in intact membranes. Third, within our simple system, the rates of delipidation are remarkably similar between ATG4A and ATG4B and even when compared to the “activated” forms of ATG4C and ATG4D. This suggests that all 4 proteins engage lipidated substrates in a similar manner and perhaps the real functional outlier is simply that ATG4B has gained the ability to recognize and proteolyze soluble proteins. Fourth, delipidation is inherently slow for all 4 proteases, consistent with the need to limit this activity under ordinary autophagic induction. Like RavZ, the selectivity depends at least in part on a LIR sequence outside of the normal ubiquitin-like interaction of the substrate and protease. Although early crystal structures of ATG4B complexed to LC3 suggested an NH-terminal LIR might be critical to its activity,Citation45 we do not detect major changes in the recognition of soluble or lipidated substrates when the NH-terminal LIR is mutated. Instead, we observe substrate-specific consequences to the deletion or mutation of a COOH-terminal LIR recently described by others as a key regulator of ATG4B function.Citation31 The major effect of altering this LIR is to reduce or eliminate processing of lipidated substrates in vitro. In our quadruple knockout cell line, expression of ATG4B without this motif leads to accumulations of lipidated LC3B and GABARAPL1 also consistent with a role in delipidation.
Our LIR results have many features in common with the recent earlier work of Skytte-Rasmussen et al. On the one hand, both groups identified an impact on ATG4B activity from mutation or deletion of this motif, and in many cases used the exact same deletions. Both groups used a knockout and rescue strategy to evaluate the biological relevance of this motif and we confirm their unique observation that the GABARAP proteins are particularly susceptible to turnover when this motif is missing. On the other hand, we differ in a few critical observations. We do not detect significant effects on priming in the absence of the LIR either in cells or in vitro. In cells, because our knockout includes all 4 ATG4 proteins, we can monitor the extent of priming for endogenous LC3B and GABARAPL1, and this may differ from the priming of overexpressed tagged-LC3 proteins used in their single ATG4B KO. Further, our HEK293 cells were generated with CRISPR/Cas9 editing and they used mouse embryonic fibroblasts from a whole animal knockout, so compensation is likely different in each of the 2 systems. The design of our in vitro experiments testing soluble cleavage is fundamentally similar to theirs and yet we see no impact on deletion of the LIR motif in this event. Most of our reactions include only 1/15 the total enzyme concentration in their study (and about 50-fold less enzyme relative to substrate), but are conducted at a higher temperature (37 vs 30) and result in faster overall cleavage. Whether these reaction conditions or differences in specific activity of our purified proteins contribute to our differences in detection is uncertain. Importantly, in every case we directly compare cleavage of soluble and lipidated material and thus we are confident that the processing of lipidated material is much more sensitive to the presence of this motif.
The role and limits of ATG4B activity in cells
In vitro assays have long established that ATG4B is the most efficient protease on a variety of soluble substrates ranging from whole Atg8-family proteins to partial peptides.Citation16–Citation18,Citation40,Citation58 Likewise, complete knockout of ATG4B from several cell lines is sufficient to eliminate all primed LC3B ([Citation14] and ), suggesting this protease has a uniquely privileged role in the priming of the LC3 family. To what extent ATG4B contributes to delipidation has been harder to judge. Very strong but incomplete reduction of ATG4B expression does not seem to result in the accumulation of any unprimed endogenous LC3B (e.g. [Citation12]), but a reduced capacity to turn over exogenous overexpressed LC3 proteins can sometimes be detected.Citation15 Instead, reduced ATG4B expression or activity (by pharmacological regulation) can lead to changes in autophagic flux.Citation11,Citation15
If endogenous priming is not being affected, this implies that a crucial role for ATG4B in these systems is regulating delipidation and further that this role is sensitive to small changes in ATG4B availability. ATG4B overexpression studies have not clarified its cellular role because although these experiments generally lead to less total lipidated LC3, overexpressed ATG4 proteins inhibit the forward lipidation reaction by binding to and sequestering primed LC3 proteins independent from their role in proteolysis.Citation2,Citation40 Likewise, in vitro assays using LC3–PE isolated from whole cells have suggested significant delipidation activity for ATG4 proteins,Citation40,Citation59 but as these results were collected on samples that were fully solubilized in detergent we conclude here that this assay condition is biochemically akin to priming rather that delipidation (). Where groups have tested delipidation on intact cellular membranes, the activity is not kinetically robust.Citation19 Thus, to establish the capacity of ATG4B in delipidation, we developed both in vitro proteolysis assays of delipidation as well as quadruple knockouts of ATG4 protein to provide a cellular context where ATG4B activity could be studied. Taking the existing literature and our in vitro results as a whole, we would argue that ATG4B is naturally capable of limited constitutive delipidation. As such, ATG4B is positioned to be sensitive to either up or down regulation within the cell. Under conditions where delipidation activity is selectively activated, ATG4B might become a major player in autophagosome maturation (i.e., activation by phosphorylation,Citation28). Likewise, activation of ATG4A or especially relief of the autoinhibitory domains in ATG4C and ATG4D would dramatically support the overall delipidation activity in the cell. Potentially, this increased delipidation could even become inhibitory, akin to the way RavZ limits phagophore growth. Negative control of delipidation activity could occur locally or globally. Locally, the presence of other LC3–PE binding proteins will likely limit access of ATG4 to the autophagosome (similar to how Atg8–PE is broadly protected by Atg8-binding proteinsCitation52). Both ATG4A and ATG4B are sensitive to broad cytoplasmic regulation by the production of reactive oxygen species in support of increased autophagy during starvationCitation9 and metabolic stress.Citation60 Likewise, the targeted degradation of membrane-associated ATG4 proteins could also prolong the lifetime of lipidated LC3.Citation61 Finally, although ATG4B is the major priming protease in cell culture, other ATG4 proteins are likely to compensate in whole animal knockouts, as atg4b knockout mice are viable and fertile, exhibiting only subtle defects including equilibrioception impairmentCitation47 and increased susceptibility to some chemical challenges.Citation46
Segregation of isopeptidase and endopeptidase activities in organisms with multiple ATG4 proteins
For other ubiquitin-like molecules, isopeptidase and endopeptidase activities are often encoded in separate deubiquitinase molecules such that preprocessing of the ubiquitin is handled separately from release of ubiquitin on a modified protein. Clearly, this model is not used by autophagy in S. cerevisiae because there is only one Atg4 protein and it handles both jobs with approximately equal effectiveness.Citation50 Likewise, such a model is not a strictly accurate depiction of the mammalian ATG4s, because although we can detect clear preferences for substrates, there is generally a basal level of processing for both lipidated and soluble proteins (i.e. ). Thus, mammalian ATG4 proteins are selective rather than specific for different forms of each substrate. The concept of separating target selectivity across multiple ATG4 proteins may be a common feature as similar selectivity is detected in a variety of lower organisms where one ATG4 is predominantly responsible for soluble preprocessing and a second, almost completely inactive ATG4 (based on in vitro measures of soluble substrates) plays a critical role in delipidation. This paradigm has been described in Leishmania major,Citation62 in C. elegans,Citation63 and in T. cruzi.Citation64 We do not yet understand how selectivity is encoded, but, interestingly, despite the apparent segregation of responsibilities in their host organism, overexpression of the delipidation-specific ATG4 can still rescue ATG4 deletions in S. cerevisiae for many of these examples (i.e., ATG4CCitation20 and T. cruzi TcATG4.2Citation64). This suggests that either yeast Atg8 is processed in a nonselective way or that in cellular models, ATG4 activity can be turned “on” and is perhaps constitutively “on” in yeast. To that end, an “on” switch for ATG4B has recently been described; phosphorylation of ATG4B at sites proximal to the COOH-LIR increases its overall activity in mammalian cell culture studies and the biggest impact appears to be on its delipidation activity.Citation28 O-GlcNAcylation of ATG4B also increases protease activity, suggesting there may be several ways to activate the protein.Citation65 It will be interesting to establish whether these regulatory mechanisms specifically control delipidation, perhaps by modifying the interfacial regulation of ATG4 proteins, or instead represent broad manipulation of basic catalytic activity.
Materials and methods
Materials
All lipids were purchased predissolved in chloroform from Avanti Polar Lipids. Lipids used in this study include:1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE; 850725C), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; 850457C), L-α-phosphatidylinositol (Liver, Bovine) (PtdIns; 840042C), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS; 840035C), and cardiolipin (heart, bovine) (CL; 840012C). Sodium chloride (AB1915), Tris-HCl (AB02005), dithiothreitol (DTT; AB00490), and ATP (AB00162) were purchased from AmericanBio. Magnesium chloride, 6-hydrate (2444) and calcium chloride, dehydrate (1332) were purchased from JT Baker. Nycodenz (1002424) was purchased from Accurate Chemical and Scientific Corp. Octyl-glucoside (O8001) was purchased from Sigma-Aldrich.
Production of recombinant proteins
RavZ, ATG3 and ATG7 were purified as previously described ([Citation37,Citation39]).
A bacterial expression plasmid for human ATG4A in the pGex-4T backbone (HsATG4A; Q8WYN0) was a gift from Z. Elazar (Weizmann Institute). Sequences encoding human ATG4B (Q9Y4P1: Variant p.Leucine354Gln), ATG4C (Q96DT6), and ATG4D (Q86TL0) as well as mouse ATG4B (Q8BGE6) were each also cloned into pGex-2T (GE Healthcare, 28954653) for expression. To create ATG4C and ATG4D mutants mimicking the caspase-cleaved forms, proteins were cloned into the pGex-2T vector without the initial N-terminal domain. RavZ was cloned into the pGex6P plasmid (GE Healthcare) for expression.
LC3-family proteins were each cloned into pGex-2T for expression (RnLC3B from rat and HsGABARAP HsGABARAPL1 and HsGABARAPL2 from human). The final sequences adjacent to the thrombin cut site for each protein are depicted in the . LC3 proteins used for lipidation were designed in truncated forms such that the glycine residue necessary for lipidation is the terminal amino acid (i.e., GABARAPL1[G116], GABARAPL2[G116], and LC3B[G120]). Soluble ATG8 proteins were expressed with carboxyl terminal protein extensions to monitor proteolytic release (i.e., GABARAPL1-YFP, GABARAPL2-YFP, RnLC3B-tail). For these 3 constructs, the final protein sequences immediately adjacent to the glycine are depicted in .
Table 1. N-termini of the proteins used in this study.
Table 2. Sequences immediately adjacent to the glycine in Atg8-family fusion proteins.
Each GST-tagged protein was expressed and purified in the same manner. Briefly, plasmids were transformed into BL21-Gold(DE3) E. coli (Agilent Technologies, 230132). Cells were grown at 37°C to an OD of 0.6-0.8 before induction with 0.5 mM IPTG (AmericanBio, AB00841). Cells were then grown for an additional 3 h before they were collected by centrifugation. Cells were resuspended in thrombin buffer (20 mM Tris, pH 7.6, 100 mM NaCl, 5 mM MgCl2, 2 mM CaCl2, 1 mM DTT), which was supplemented with a cOmplete™, EDTA-free Protease inhibitor cocktail tablet (Roche, 11873580001). Cells were lysed using a cell disruptor and cleared by centrifugation. The supernatant was incubated with glutathione beads (Sigma Aldrich, G4501) for 4 h at 4°C. Beads were collected and washed twice with thrombin buffer before thrombin (Sigma Aldrich, T6884) was added. Thrombin was allowed to cut at 4°C overnight. The next morning, fractions of protein were collected and stored at -80 in 20% glycerol.
Liposome and proteoliposome preparation
To prepare liposomes, lipids were mixed in the noted compositions and dried to a thin film under chloroform. Unless otherwise noted, the composition of liposomes for delipidation assays was 55 mol percent DOPE, 35 mol percent POPC, and 10 mol percent PI. The lipid film was further dried under vacuum for one h. The lipids were reconstituted in SN buffer (100 mM NaCl, 20 mM Tris-HCl, pH 7.4, 5 mM MgCl2) and subjected to 7 cycles of flash freezing in liquid nitrogen and thawing in a 37°C bath. After freeze-thaw, liposomes were extruded 21 times through 2 polycarbonate membranes. For cardiolipin binding assays, liposomes were extruded to 100 nm. For lipidation reactions, liposomes were first extruded to 400 nm and then were sonicated to a size of ≤ 50 nm immediately prior to the lipidation reaction.
GABARAPL1, LC3B, and GABARAPL2 were coupled to liposomes as described previously. Because these Atg8-family proteins were designed such that they already contained the terminal glycine residue necessary for lipidation (GABARAPL1[G116], GABARAPL2[G116], and LC3B[G120]), the initial priming activity of ATG4 is not needed. Atg8-family proteins (15 µM), Atg3 (2 µM), Atg7 (2 µM), and sonicated liposomes (3 mM) were mixed in SN buffer containing 1 mM DTT. Lipidation was initiated by adding 1 mM ATP and reactions were incubated at 37°C for 90 min. After the reaction was complete, the lipidation reaction was run on a Nycodenz density gradient to remove uncoupled substrate and other reaction components from the proteoliposomes. The bottom layer of the gradient consisted of 150 µL of 80% Nycodenz and 150 µL of the lipidation reaction. The second layer consisted of 250 µL of 30% Nycodenz while the top layer was 50 µL of SN buffer. Gradients were spun at 48000 rpm at 4°C for 4 h in a Beckman SW55 rotor (average RCF of 218,438 according to manufacturer's calculations). Liposomes with the conjugated Atg8-family protein were collected from the 30% Nycodenz-buffer interface and were stored at 4°C before use in subsequent delipidation experiments.
In vitro proteolysis assays
To measure the activity of ATG4 proteases, 2 sets of substrates were used. The proteoliposomes from above were used to determine the delipidation activity whereas GABARAPL1-YFP, GABARAPL2-YFP, and rLC3-tag were used to follow priming activity. Proteoliposomes or 7 µM of the protein substrate were mixed with SN buffer and kept on ice until activity assays were initiated by the addition of the stated concentration of ATG4. Reactions were incubated at 37°C for the stated time. Samples of 10 µL were removed at the noted time, mixed with LDS loading buffer (ThermoFisher Scientific, NP007) and immediately boiled to stop proteolysis. Time course samples were then visualized using SDS-PAGE and Coomassie Brilliant Blue staining and analyzed with densitometry using ImageJ. Graphs display the average of at least 3 independent assays with standard deviation.
For interfacial activation assays, the substrate mixtures were further incubated with the stated detergent for 10 min on ice before the addition of ATG4 or RavZ.
Membrane binding assays
ATG4 proteins (10 µM) were incubated with liposomes (2 mM lipid) in SN buffer with 1 mM DTT at 37°C for 20 min. To separate free protein from protein bound to liposomes, the mixture was then subjected to a Nycodenz density gradient as described above. Liposomes and bound protein were recovered from the top 80 µl of the gradient. To determine the percentage of bound protein, 1/8 of the volume of the recovered liposomes and 1/20 of the initial amount of ATG4 protein were then visualized through SDS-PAGE and Imperial™ Protein Stain (Thermo Scientific, 24615). Gels were analyzed with densitometry using ImageJ software.
CRISPR/Cas9 knockout of human ATG4 genes
To knock out the 4 ATG4 homologs in HEK293 cells, several independent sgRNA guides were designed against multiple sites within each of the 4 human ATG4 genes using the CRISPR Design Tool through MIT (crispr.mit.edu) (Table below). To generate and insert guides into pX459 (Addgene, 62988), sgRNA guides were synthesized (Yale Keck Facility, New Haven, CT, USA) containing the targeting sequence as well as overhangs that were complementary to overhangs in BbsI-digested pX459. Guides were then phosphorylated using T4 PNK (NEB, M0201S) and annealed using cooling from 95°C to 25°C in 10°C steps. Finally, guides were ligated into the pSpCas9 (BB)-2A-Puro (pX459) V2.0 plasmid (Addgene, 62988; Zhang Lab) using QuickLigase (NEB, M2200S). HEK293 cells were transfected using Lipofectamine 3000 (Thermo Fisher Scientific, L3000008) using the manufacturer's protocol. After 24 h, cells were treated with selection medium containing 2–5 μg/ml puromycin (Clontech, 631306). Individual clones were isolated by serial dilution and validated by western blotting. In initial screenings, single knockouts were created and validated, before combining guides to make the quadruple knockout. Ultimately, 6 guides were used together to eliminate all ATG4 protein expression and activity (noted in Bold in ).
Table 3. CRISPR gRNAs tested and used for gene editing.
Lentivirus production
293FT cells (Thermo Fisher Scientific, R70007) cells were seeded onto a 10-cm plate at a concentration of 4 × 106. Plates were grown for 24 h at 37°C with 5% CO2 in DMEM (Thermo Fisher Scientific, 11965092) containing 10% FBS (Thermo Fisher Scientific, 10438062) and 1% PS (Thermo Fisher Scientific, 15140122) until confluency reached 60%. Cells were then transfected with psPAX2 (from Didier Trono; Addgene, 12260), pCMV-VSV-G (from Bob Weinberg; Addgene 8454), and target plasmids using Lipofectamine 3000. After transfection, media was collected every 24 h for 3 days into 50-ml tubes, and stored at 4°C. To obtain virus, the collected media was filtered with a 0.45-µm filter. Lenti-X Concentrator (Clontech, 631232) was added to the clarified supernatant at a ratio of 1:3 and mixed by gentle inversion before incubation at 4°C for at least 2 h. The mixture was centrifuged at 1500 g for 45 min at 4°C. Supernatant was carefully removed. The pellet was gently resuspended in 1/1000 of the original volume using complete DMEM and stored at −80°C.
Western blot protocols for each antibody
Antibodies used in this manuscript:
ATG4A: Cell Signaling Technology, 7613
ATG4B: Cell Signaling Technology, 13507
ATG4C: Abcam, ab183516
ATG4D: MilliporeSigma, ABC22
LC3B: CST, 3868S
GABARAP: Cell Signaling Technology, 13733
GABARAPL1: Cell Signaling Technology, 26632
GABARAPL2: Cell Signaling Technology, 14256
GAPDH: Thermo Fisher Scientific, MA5-15738
FLAG: Sigma, F1804
IRDye® 800CW Donkey anti-Rabbit IgG (H + L): LICOR, 925–32213
IRDye® 680RD Donkey anti-Mouse IgG (H + L): LICOR, 925–68072
To collect cell lysates for western blotting, working on ice, the cell medium was replaced with 1XPBS (Thermo Fisher Scientific, 14190144) and cells were scraped from the plates. After centrifugation at 700 g for 3 min at 4°C, the supernatant was removed and replaced with lysis buffer (300 mM NaCl, 10 mM HEPES, 1% Triton X-100 [American Bioanalytical, AB02025-00500], pH 7.5). Cells were resuspended and incubated on ice for 5 min before centrifugation at 20,000 g for 10 min at 4°C. The supernatant, containing the cell lysate, was removed and the protein concentration was determined using a BCA assay (Thermo Fisher Scientific, 23225). Lysate samples were electrophoresed on Bis-Tris gels. LC3/GABARAP family proteins were resolved with precast 12% Bis-Tris protein gels (Thermo Fisher Scientific, NP0341BOX or NP0342BOX), whereas ATG4 family proteins were resolved using precast Bolt™ 8% Bis-Tris Plus protein gels (Thermo Fisher Scicentific, NW00082BOX). Gels were then transferred to Immobilon-FL PVDF membranes (MilliporeSigma, IPFL00010).
When blotting proteins (except LC3B and GABARAPL1), membranes were washed with 1XPBS and blocked with 1% casein (Sigma Aldrich, C7078) in 1X PBS with 0.2% sodium azide (Sigma Aldrich, S2002) for one h at room temperature (RT). Blocked membranes were then washed 3 times with PBS-T (1X PBS containing 0.02% Tween 20 (AmericanBio, AB02038) and incubated with the primary antibody (diluted 1:1000 in PBS-T containing 5% BSA (Sigma Aldrich, A9647) and 0.005% sodium azide overnight at 4°C. Membranes were then washed 3 times before incubation with the IRDye secondary antibody (diluted 1:10,000 in PBS containing 0.5% casein, 0.1% sodium azide, 0.1% SDS and 0.25% Tween 20) for one h at RT. Membranes were then washed again before imaging with the Li-COR Odyssey system. In the case of LC3B, membranes were first dried thoroughly after transfer, activated with methanol and washed in PBS 3 times prior to blocking.
In the case of GABARAPL1, membranes were blocked with 1% BSA in PBS-T for 1 h at RT before being washed 3 times with PBS-T. Membranes were then incubated with the primary antibody (antibody diluted 1:1000 in PBS-T with 5% BSA and 0.005% sodium azide) overnight at 4°C. Membranes were washed another 3 times with PBS-T before incubation with the secondary antibody (GE Healthcare Life Sciences, NA934; 1:5000 diluted in PBS-T with 1% BSA). Membranes were then incubated with SuperSignal West Femto (Thermo Fisher Scientific, 34095) and imaged with a VersaDoc Imaging System (BioRad, Hercules, CA, USA).
Immunofluorescence and confocal microscopy
Cells seeded on FN1/fibronectin (MP Biomedicals, 02150025)-coated coverslips were treated with complete medium (DMDM + 10% FBS), 100 nM bafilomycin A1 (Enzo Life Sciences, BML-CM110-0100), EBSS (Thermo Fisher Scientific, 24010043) or EBSS + bafA1 for 2 h. Cells were then washed with PBS and fixed in 4% paraformaldehyde (Electron Microscopy Sciences, 15710) at room temperature for 15 min. After 3 PBS washes, cells were permeabilized with PBS with 0.05% saponin (Sigma Aldrich, 47036) for 10 min, then quenched in 50 mM NH4Cl. Samples were then incubated with primary antibody solution for one h at room temperature, washed with PBS containing 0.05% saponin 3 times, then incubated with secondary antibody for one h. After 3 washes with PBS containing 0.05% saponin, coverslips were rinsed in ddH2O once, and mounted on slides in ProLong Gold antifade reagent (Thermo Fisher Scientific, P36930). Images were obtained with a 63x oil immersion objective on a Zeiss 710Duo confocal microscope supported by the Center for Cellular and Molecular Imaging facility at Yale. LC3B antibody (MBL, PM036) was used 1:500. Anti-rabbit secondary antibody conjugated with Alexa Fluor 488 or 594 (Thermo Fisher Scientfic, A11034 or R37117) was used at 1:500.
For experiments following overexpressed 3xFLAG-GABARAPL1 in cells, conditions were the same as above except cells were permeabilized in PBS with 0.1% Triton X-100 and subsequent washes also included 0.1% Triton-X100. FLAG antibody (Sigma Aldrich, F1804) was used at 1:500. Anti-mouse secondary antibody conjugated with Alexa Fluor 488 (Thermo Fisher Scientific, A32723) was used 1:500.
Puncta number per cell is quantified blindly, and counted manually using Cell Counter plugin in ImageJ (NIH). Data are presented in scatter dot plots, with each dot representing one cell. n numbers are shown in the figure legends. Any 2 samples with the same genotype or treatment are subject for ranked Mann-Whitney test. Results were compiled in Prism 7 and statistical significance labeled in the figure.
Disclosure of potential conflicts of interest
The authors declare to have no competing financial interests.
1437341-supp_data.zip
Download Zip (10.4 MB)Acknowledgements
This work was supported by grants from the NIH (GM1000930 and NS063973; TJM), the pre-doctoral program in Cellular and Molecular Biology (T32GM007223; supporting KJK and NN), a postdoctoral fellowship (Norwegian Cancer Society; AL), a student exchange fellowship and pre-doctoral training grant (Chinese Scholarship Council; JJ and SY respectively) and a Yale Undergraduate Fellowship to BM (STARS II program). We also thank Yale undergraduates, Theodore Agbi and Peter Suwondo for helpful contributions along the way.
Additional information
Funding
Related Research Data
References
- Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol. 2014;21:336–45. doi:10.1038/nsmb.2787. PMID:24699082
- Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–9. Epub 2008/09/05. doi:10.1091/mbc.E08-03-0312. PMID:18768752
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. The EMBO journal. 2010;29:1792–802. Epub 2010/04/27. doi:10.1038/emboj.2010.74. PMID:20418806
- Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–76. Epub 2000/10/19. doi:10.1083/jcb.151.2.263. PMID:11038174
- Abeliovich H, Dunn WA, Jr., Kim J, Klionsky DJ. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol. 2000;151:1025–34. Epub 2000/11/22. doi:10.1083/jcb.151.5.1025. PMID:11086004
- Yu S, Melia TJ. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr Opin Cell Biol. 2017;47:92–8. doi:10.1016/j.ceb.2017.03.010. PMID:28463755
- Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, Lazarou M. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016;215:857–74. PMID:27864321
- Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science (New York, NY.) 2016;354:1036–41. doi:10.1126/science.aaf6136.
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. The EMBO journal. 2007;26:1749–60. Epub 2007/03/10. doi:10.1038/sj.emboj.7601623. PMID:17347651
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–8. Epub 2010/08/24. doi:10.1016/j.tibs.2010.07.007. PMID:20728362
- Bortnik S, Choutka C, Horlings HM, Leung S, Baker JH, Lebovitz C, Dragowska WH, Go NE, Bally MB, Minchinton AI, et al. Identification of breast cancer cell subtypes sensitive to ATG4B inhibition. Oncotarget. 2016;7:66970–88. doi:10.18632/oncotarget.11408. PMID:27556700
- Yoshimura K, Shibata M, Koike M, Gotoh K, Fukaya M, Watanabe M, Uchiyama Y. Effects of RNA interference of Atg4B on the limited proteolysis of LC3 in PC12 cells and expression of Atg4B in various rat tissues. Autophagy. 2006;2:200–8. doi:10.4161/auto.2744. PMID:16874114
- Nakatogawa H, Ishii J, Asai E, Ohsumi Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy. 2012;8:177–86. Epub 2012/01/14. doi:10.4161/auto.8.2.18373. PMID:22240591
- Wang W, Chen Z, Billiar TR, Stang MT, Gao W. The carboxyl-terminal amino acids render pro-human LC3B migration similar to lipidated LC3B in SDS-PAGE. PLoS One. 2013;8:e74222. doi:10.1371/journal.pone.0074222. PMID:24040206
- Akin D, Wang SK, Habibzadegah-Tari P, Law B, Ostrov D, Li M, Yin XM, Kim JS, Horenstein N, Dunn WA, Jr. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10:2021–35. doi:10.4161/auto.32229. PMID:25483883
- Li M, Chen X, Ye QZ, Vogt A, Yin XM. A high-throughput FRET-based assay for determination of Atg4 activity. Autophagy. 2012;8:401–12. Epub 2012/02/04. doi:10.4161/auto.18777. PMID:22302004
- Li M, Hou Y, Wang J, Chen X, Shao ZM, Yin XM. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. The J Biol Chem. 2011;286:7327–38. Epub 2010/12/24. doi:10.1074/jbc.M110.199059. PMID:21177865
- Shu CW, Madiraju C, Zhai D, Welsh K, Diaz P, Sergienko E, Sano R, Reed JC. High-throughput fluorescence assay for small-molecule inhibitors of autophagins/Atg4. J Biomol Screen. 2011;16:174–82. Epub 2011/01/20. doi:10.1177/1087057110392996. PMID:21245471
- Betin VM, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. Journal of cell science. 2009;122:2554–66. Epub 2009/06/25. doi:10.1242/jcs.046250. PMID:19549685
- Marino G, Uria JA, Puente XS, Quesada V, Bordallo J, Lopez-Otin C. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J Biol Chem. 2003;278:3671–8. Epub 2002/11/26. doi:10.1074/jbc.M208247200. PMID:12446702
- Scherz-Shouval R, Sagiv Y, Shorer H, Elazar Z. The COOH terminus of GATE-16, an intra-Golgi transport modulator, is cleaved by the human cysteine protease HsApg4A. J Biol Chem. 2003;278:14053–8. Epub 2002/12/11. doi:10.1074/jbc.M212108200. PMID:12473658
- Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou S, Chen Y, Zheng XL, Klionsky DJ, Liang Y, et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy. 2012;8:883–92. doi:10.4161/auto.19652. PMID:22652539
- Nair U, Yen WL, Mari M, Cao Y, Xie Z, Baba M, Reggiori F, Klionsky DJ. A role for Atg8-PE deconjugation in autophagosome biogenesis. Autophagy. 2012;8:780–93. doi:10.4161/auto.19385. PMID:22622160
- Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19:3290–8. Epub 2008/05/30. doi:10.1091/mbc.E07-12-1292. PMID:18508918
- Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147:435–46. doi:10.1083/jcb.147.2.435. PMID:10525546
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–67. Epub 2010/05/19. doi:10.1016/j.cell.2010.04.009. PMID:20478256
- Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, Roy CR. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science (New York, NY. 2012;338:1072–6. Epub 2012/11/01. doi:10.1126/science.1227026. PMID:23112293
- Yang Z, Wilkie-Grantham RP, Yanagi T, Shu CW, Matsuzawa S, Reed JC. ATG4B (Autophagin-1) phosphorylation modulates autophagy. J Biol Chem. 2015;290:26549–61. doi:10.1074/jbc.M115.658088. PMID:26378241
- Sanchez-Wandelmer J, Kriegenburg F, Rohringer S, Schuschnig M, Gomez-Sanchez R, Zens B, Abreu S, Hardenberg R, Hollenstein D, Gao J, et al. Atg4 proteolytic activity can be inhibited by Atg1 phosphorylation. Nat Commun. 2017;8:295. doi:10.1038/s41467-017-00302-3. PMID:28821724
- Abreu S, Kriegenburg F, Gomez-Sanchez R, Mari M, Sanchez-Wandelmer J, Skytte Rasmussen M, Soares Guimaraes R, Zens B, Schuschnig M, Hardenberg R, et al. Conserved Atg8 recognition sites mediate Atg4 association with autophagosomal membranes and Atg8 deconjugation. EMBO Rep. 2017;18:765–80. doi:10.15252/embr.201643146. PMID:28330855
- Skytte Rasmussen M, Mouilleron S, Kumar Shrestha B, Wirth M, Lee R, Bowitz Larsen K, Abudu Princely Y, O'Reilly N, Sjottem E, Tooze SA, et al. ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy. 2017;13:834–53. doi:10.1080/15548627.2017.1287651. PMID:28287329
- Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjorkoy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–69. Epub 2010/01/27. doi:10.1083/jcb.200907015. PMID:20100911
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi:10.1074/jbc.M702824200. PMID:17580304
- Lystad AH, Ichimura Y, Takagi K, Yang Y, Pankiv S, Kanegae Y, Kageyama S, Suzuki M, Saito I, Mizushima T, et al. Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep. 2014;15:557–65. doi:10.1002/embr.201338003. PMID:24668264
- Abert C, Kontaxis G, Martens S. Accessory Interaction Motifs in the Atg19 Cargo Receptor Enable Strong Binding to the Clustered Ubiquitin-related Atg8 Protein. J Biol Chem. 2016;291:18799–808. doi:10.1074/jbc.M116.736892. PMID:27402840
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. Epub 2010/06/22. doi:10.1038/nature09204. PMID:20562859
- Horenkamp FA, Kauffman KJ, Kohler LJ, Sherwood RK, Krueger KP, Shteyn V, Roy CR, Melia TJ, Reinisch KM. The Legionella Anti-autophagy Effector RavZ Targets the Autophagosome via PI3P- and Curvature-Sensing Motifs. Developmental Cell. 2015;34:569–76. doi:10.1016/j.devcel.2015.08.010. PMID:26343456
- Sou YS, Tanida I, Komatsu M, Ueno T, Kominami E. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J Biol Chem. 2006;281:3017–24. doi:10.1074/jbc.M505888200. PMID:16303767
- Nath S, Dancourt J, Shteyn V, Puente G, Fong WM, Nag S, Bewersdorf J, Yamamoto A, Antonny B, Melia TJ. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol. 2014;16:415–24. Epub 2014/04/22. doi:10.1038/ncb2940. PMID:24747438
- Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsATG4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem. 2004;279:36268–76. doi:10.1074/jbc.M401461200. PMID:15187094
- van Tilbeurgh H, Egloff MP, Martinez C, Rugani N, Verger R, Cambillau C. Interfacial activation of the lipase-procolipase complex by mixed micelles revealed by X-ray crystallography. Nature. 1993;362:814–20. Epub 1993/04/29. doi:10.1038/362814a0. PMID:8479519
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. Epub 2007/11/08. doi:10.1074/jbc.C700195200. PMID:17986448
- Oh-oka K, Nakatogawa H, Ohsumi Y. Physiological pH and acidic phospholipids contribute to substrate specificity in lipidation of Atg8. J Biol Chem. 2008;283:21847–52. Epub 2008/06/12. doi:10.1074/jbc.M801836200. PMID:18544538
- Betin VM, MacVicar TD, Parsons SF, Anstee DJ, Lane JD. A cryptic mitochondrial targeting motif in Atg4D links caspase cleavage with mitochondrial import and oxidative stress. Autophagy. 2012;8:664–76. Epub 2012/03/24. doi:10.4161/auto.19227. PMID:22441018
- Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. The EMBO Journal. 2009;28:1341–50. Epub 2009/03/27. doi:10.1038/emboj.2009.80. PMID:19322194
- Cabrera S, Maciel M, Herrera I, Nava T, Vergara F, Gaxiola M, Lopez-Otin C, Selman M, Pardo A. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy. 2015;11:670–84. doi:10.1080/15548627.2015.1034409. PMID:25906080
- Marino G, Fernandez AF, Cabrera S, Lundberg YW, Cabanillas R, Rodriguez F, Salvador-Montoliu N, Vega JA, Germana A, Fueyo A, et al. Autophagy is essential for mouse sense of balance. J Clin Invest. 2010;120:2331–44. Epub 2010/06/26. doi:10.1172/JCI42601. PMID:20577052
- Nguyen N, Shteyn V, Melia TJ. Sensing Membrane Curvature in Macroautophagy. J Mol Biol. 2017;429:457–72. doi:10.1016/j.jmb.2017.01.006.
- Rao Y, Matscheko N, Wollert T. Autophagy in the test tube: In vitro reconstitution of aspects of autophagosome biogenesis. The FEBS journal. 2016;283:2034–43. doi:10.1111/febs.13661. PMID:26797728
- Zens B, Sawa-Makarska J, Martens S. In vitro systems for Atg8 lipidation. Methods. 2015;75:37–43. doi:10.1016/j.ymeth.2014.11.004. PMID:25461810
- Knorr RL, Nakatogawa H, Ohsumi Y, Lipowsky R, Baumgart T, Dimova R. Membrane morphology is actively transformed by covalent binding of the protein Atg8 to PE-lipids. PLoS One. 2014;9:e115357. doi:10.1371/journal.pone.0115357. PMID:25522362
- Kaufmann A, Beier V, Franquelim HG, Wollert T. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell. 2014;156:469–81. Epub 2014/02/04. doi:10.1016/j.cell.2013.12.022. PMID:24485455
- Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy. 2013;9:424–5. doi:10.4161/auto.22931. PMID:23321721
- Fracchiolla D, Sawa-Makarska J, Zens B, Ruiter A, Zaffagnini G, Brezovich A, Romanov J, Runggatscher K, Kraft C, Zagrovic B, et al. Mechanism of cargo-directed Atg8 conjugation during selective autophagy. Elife. 2016;5:e18544.
- Kwon DH, Kim S, Jung YO, Roh KH, Kim L, Kim BW, Hong SB, Lee IY, Song JH, Lee WC, et al. The 1:2 complex between RavZ and LC3 reveals a mechanism for deconjugation of LC3 on the phagophore membrane. Autophagy. 2017;13:70–81. doi:10.1080/15548627.2016.1243199. PMID:27791457
- Yang A, Pantoom S, Wu YW. Elucidation of the anti-autophagy mechanism of the Legionella effector RavZ using semisynthetic LC3 proteins. Elife. 2017;6: e23905.
- Sawa-Makarska J, Abert C, Romanov J, Zens B, Ibiricu I, Martens S. Cargo binding to Atg19 unmasks additional Atg8 binding sites to mediate membrane-cargo apposition during selective autophagy. Nat Cell Biol. 2014;16:425–33. Epub 2014/04/08. doi:10.1038/ncb2935. PMID:24705553
- Ni Z, Gong Y, Dai X, Ding W, Wang B, Gong H, Qin L, Cheng P, Li S, Lian J, et al. AU4S: a novel synthetic peptide to measure the activity of ATG4 in living cells. Autophagy. 2015;11:403–15. Epub 2015/04/02. doi:10.1080/15548627.2015.1009773. PMID:25831015
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. Journal of cell science. 2004;117:2805–12. Epub 2004/06/01. doi:10.1242/jcs.01131. PMID:15169837
- Qiao S, Dennis M, Song X, Vadysirisack DD, Salunke D, Nash Z, Yang Z, Liesa M, Yoshioka J, Matsuzawa S, et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. 2015;6:7014. doi:10.1038/ncomms8014. PMID:25916556
- Kuang E, Okumura CY, Sheffy-Levin S, Varsano T, Shu VC, Qi J, Niesman IR, Yang HJ, Lopez-Otin C, Yang WY, et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS genetics. 2012;8:e1003007. doi:10.1371/journal.pgen.1003007. PMID:23093945
- Williams RA, Woods KL, Juliano L, Mottram JC, Coombs GH. Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy. 2009;5:159–72. Epub 2008/12/11. doi:10.4161/auto.5.2.7328. PMID:19066473
- Wu F, Li Y, Wang F, Noda NN, Zhang H. Differential function of the two Atg4 homologues in the aggrephagy pathway in Caenorhabditis elegans. J Biol Chem. 2012;287:29457–67. Epub 2012/07/07. doi:10.1074/jbc.M112.365676. PMID:22767594
- Alvarez VE, Kosec G, Sant'Anna C, Turk V, Cazzulo JJ, Turk B. Autophagy is involved in nutritional stress response and differentiation in Trypanosoma cruzi. J Biol Chem. 2008;283:3454–64. doi:10.1074/jbc.M708474200. PMID:18039653
- Jo YK, Park NY, Park SJ, Kim BG, Shin JH, Jo DS, Bae DJ, Suh YA, Chang JH, Lee EK, et al. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget. 2016;7:57186–96. doi:10.18632/oncotarget.11083. PMID:27527864