
ABSTRACT
Although antiretroviral therapy is highly effective in suppressing human immunodeficiency virus type-1 (HIV) replication, treatment has failed to eliminate viral reservoirs and discontinuation of treatment results in viral reactivation. Here, we demonstrate that peptides Tat-vFLIP-α2 and Tat-Beclin 1/BECN1 which have been shown to induce a Na+/K+-ATPase- and a macroautophagy/autophagy-dependent form of cell death, autosis, can preferentially kill HIV-infected macrophages while preventing virological rebound. To improve bioavailability and drug delivery, Tat-vFLIP-α2 was encapsulated into biodegradable PLGA (poly lactic-co-glycolic acid)-lipid-PEG (polyethylene glycol) nanoparticles for long-lasting intracellular delivery. After a single dose of NP-vFLIP-α2, HIV-infected macrophages were preferentially killed in a dose-dependent manner compared to uninfected or untreated HIV-infected cells with complete inhibition of HIV infection at 10 μM of peptide. HIV-infected macrophages treated with NP-vFLIP-α2 exhibited increased markers of autophagy including LC3B lipidation, SQSTM1/p62 degradation and Na+/K+-ATPase expression compared to untreated uninfected or infected cells. Moreover, the increased cell death observed in HIV-infected cells was not altered by treatment with bafilomycin A1 (BAF) or the caspase inhibitor Z-VAD-FMK, but could be reversed following treatment with the Na+/K+-ATPase inhibitor, digoxin, or knockdown of ATG5 or ATG7. NP-vFLIP-α2 induced preferential killing was also detected in HIV-infected macrophages under antiretroviral suppression without inducing viral reactivation. Additionally, we found that Na+/K+-ATPase was upregulated in HIV-infected cells, which enhanced NP-vFLIP-α2 induced cell death. These findings provide a novel strategy to eradicate HIV-infected macrophages by selectively killing infected cells through the induction of Na+/K+-ATPase dependent autophagy, while preventing reactivation of virus and new infection of uninfected bystander cells.
Introduction
Antiretroviral therapy (ART) for persons infected with human immunodeficiency virus type-1 (HIV) has greatly progressed since the initial studies demonstrating that a combination of drugs from at least 2 different classes can provide sustained viral suppression [Citation1]. The combination of drugs into a single formulation has enabled older children and adults to take one pill once daily to control viral replication, restore immune function and greatly extend life expectancy [Citation2–Citation4]. Despite this remarkable achievement, HIV remains an incurable infection that invariably reactivates if antiretroviral drugs are discontinued [Citation5]. Much research has identified long-lived CD4+ T memory cells, tissue macrophages and brain microglia as important sites of HIV persistence during sustained viral suppression in patients receiving ART [Citation6–Citation11]. Thus, there remains a need for the development of novel strategies to control and preferably eradicate HIV such that what is now life-long treatment can be discontinued.
Macroautophagy (hereafter referred to as autophagy) is a highly conserved cellular catabolic pathway that regulates homeostasis, innate and adaptive immunity, and cell death [Citation12–Citation14]. HIV utilizes autophagy proteins to promote its own replication [Citation15]. However, during permissive infection, HIV downregulates autophagy to avoid proteolytic degradation [Citation16,Citation17]. Thus, HIV modulates autophagy to promote its own replication while prolonging cell survival. Whereas in activated CD4+ T cells [Citation18,Citation19], HIV infection ultimately kills the cell, in resting memory CD4+ cells, macrophages and microglia HIV persists without leading to cell death [Citation20,Citation21].
Autophagy is also considered an intrinsic immune mechanism against pathogen invasion. Induction of autophagy is proposed to target and restrict intracellular pathogens through autolysomal degradation. However, some viruses including HIV evade autophagy and have evolved to use the autophagy-associated proteins to promote their own replication [Citation15,Citation22,Citation23]. Additionally, although autophagy is essential for maintenance of cell survival, excessive or persistent digestion of intracellular nutrients, organelles and proteins will lead to autophagic cell death [Citation24,Citation25].
A selective form of autophagy leading to cell death has been identified by the Levine laboratory, termed autosis, which is dependent on the induction of Na+/K+-ATPase [Citation26,Citation27]. Using a Tat-Beclin 1/BECN1 peptide derived from the portion of BECN1 that binds to HIV Nef, they found a concentration-dependent induction of autotic cell death. In conjunction with the Levine lab, we found that pretreatment of macrophages with the Tat-Beclin 1 peptide efficiently inhibited HIV replication. An additional novel autophagic-inducing peptide, Tat-vFLIP-α2 peptide, which targets ATG3 binding protein cellular FLICE inhibitory protein (CFLAR), has been found to activate ATG3 dependent autophagy [Citation28], and to induce cell death regulated by the Na+/K+-ATPase [Citation26]. In the research presented here, we hypothesized that excessive autophagy induced by Tat-Beclin 1 and Tat-vFLIP-α2 would suppress HIV replication and promote the selective killing of HIV-infected macrophages. To achieve this goal, we developed biodegradable PLGA (poly lactic-co-glycolic acid)-lipid-PEG (polyethylene glycol) nanoparticles to improve intracellular delivery of peptides and demonstrate that these nanopeptides can preferentially kill HIV-infected macrophages and have the potential to be used as part of an HIV cure strategy.
Results
Autophagy inducing peptides, Tat-Beclin 1 and Tat-vFLIP-α2, preferentially kill HIV chronically infected human macrophages
Based on our previous studies showing that pretreatment of macrophages with Tat-Beclin 1 inhibits HIV replication through induction of autophagy [Citation27], we suspected that a similar inhibition would occur following treatment with Tat-vFLIP-α2. Thus in our initial set of experiments, we confirmed that pretreatment of macrophages with Tat-vFLIP-α2 peptide would provide similar inhibition as with Tat-Beclin 1 (Figure S1).
Having demonstrated that pretreatment provided significant inhibition, we next examined if viral replication could be inhibited in macrophages with an established HIV productive infection. In these experiments, macrophages were infected with HIV for 10 days which consistently resulted in > 90% of cells being HIV-infected (Figure S2) [Citation16,Citation29–Citation31]. Macrophages were then treated with either Tat-Beclin 1 or Tat-vFLIP-α2 at increasing concentrations of peptide. To maintain effective peptide drug concentrations, media were changed and new peptide was replenished every other day. Collected supernatants were tested for HIV p24 antigen release into culture supernatants at baseline and days 2, 4 and 6 of treatment. By day 6 of treatment, HIV p24 antigen was decreased by 24.0%, 88.6% and 96.5% following 1, 5 and 10 μM of Tat-vFLIP-α2 peptide (Figure 1(a,b)). Similarly, Tat-Beclin 1 at 1, 5 and 10 μM exhibited a −7.3%, 55.0% and 73.7% reduction in p24 antigen, respectively ((,)).
Figure 1. Autophagy inducing peptides Tat-vFLIP-α2 and Tat-Beclin 1 inhibit HIV replication in chronically infected macrophages. (a) Human macrophages were infected with HIV and cultured for 10 days to establish a chronic infection. (b) At day 10 postinfection (p.i.), macrophages were treated with 1, 5 and 10 μM Tat-vFLIP-α2 or Tat-Beclin 1 peptides. At day 12 and day 14 p.i., media were changed and peptides replenished. The collected cell culture supernatants were measured for HIV p24 antigen by ELISA. (c and d) Cytotoxicity of Tat-vFLIP-α2 and Tat-Beclin 1 was tested by LDH assay. Data are derived from 4 different donors and reported as means ± s.e.m. * P < 0.05, **P < 0.01, *** P < 0.001.
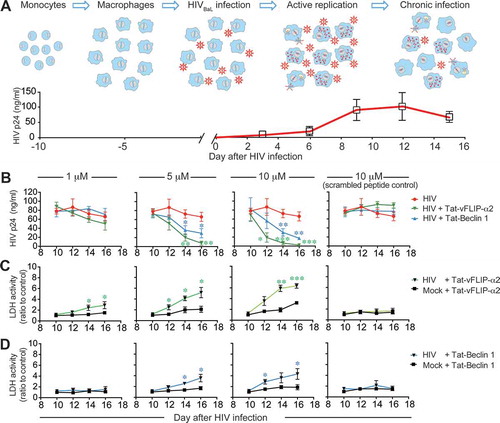
Since in earlier studies we had found that pretreatment with either Tat-Beclin 1 or Tat-vFLIP-α2 had little effect on cell viability in HIV-infected or uninfected cells, we examined the viability of HIV chronically infected macrophages following peptide treatment. For these experiments, cytotoxicity was assessed using a LDH (lactate dehydrogenase) release assay as a marker for plasma membrane damage. Of Interest, we found that both of Tat-vFLIP-α2 and Tat-Beclin 1 treatment specifically increased cytotoxicity in HIV-infected macrophages with increasing concentrations of peptides and duration of treatment. The maximum cytotoxicity for Tat-vFLIP-α2 was observed at 6 days with a mean 6.3-fold increase in cell death compared to uninfected cells at 10 μM treatment (()). Following 6 days of treatment with Tat-Beclin 1, maximum cell death at 10 µM of peptide was a mean 4.3-fold increase compared to uninfected controls (()). In contrast, there was no detectable antiviral activity or cytotoxicity in the scrambled peptide control groups. Thus, prolonged exposure to both peptides led to inhibition HIV infection and preferential killing of productively infected macrophages while scrambled peptide had no virological effect or cell toxicity. Additionally, because in these and in additional experiments described below, we consistently observed increased or similar activity for the Tat-vFLIP-α2 peptide formulation compared to the Tat-Beclin 1 peptide, the data presented below will be for only for the Tat-vFLIP-α2 peptide.
Formulation and characterization of Tat-vFLIP-α2 loaded lipid nanoparticle (NP-vFLIP-α2)
To improve the bioavailability of the Tat-vFLIP-α2, we encapsulated the peptide into a biocompatible lipid-coated polymeric nanoparticle, which is optimized from a combination of liposomes and polymeric (PLGA) nanoparticles. This nanoformulation contains a hydrophobic polymeric core, a lipid shell surrounding the polymeric core, and a hydrophilic polymer stealth layer outside the lipid shell. After loading with the Tat-vFLIP-α2 peptide, transmission electron microscopy (TEM) of negative staining revealed a size of ~ 147 nm for the nanoformulation which was confirmed by dynamic light scattering ((,)). The nanoparticles exhibited a + 30 mV zeta potential with maximal loading capacity observed at ~ 15% (wt:wt) ((,)). Thus, the final peptide-loaded formulation was engaged at 20% input. Nanoparticle stability was evaluated over time in both water and phosphate buffered saline (()). No size change was detected under either condition over 96 h. The peptide release kinetics was plotted as the weight ratio of the accumulative released peptides to the total peptide payload against time. The initial release was observed within 1 hr with 50% of the peptide released at 34 h (()).
Figure 2. Synthesis of PLGA-lecithin-PEG loaded Tat-vFLIP-α2 nanoparticles. (a) The representative image of lipid PLGA nanoparticles encapsulated with Tat-vFLIP-α2 was captured by transmission electron microscopy. Scale bar equals 100 nm. (b) Size distribution of nanoparticles was measured by dynamic light scattering (DLS). (c) Surface charge of nanoparticles was represented by DLS. (d) The loading capacity and efficiency of Tat-vFLIP-α2. (e) Stability of nanoparticles over 96 h in vitro. (f) Controlled and sustained Tat-vFLIP-α2 released from nanoparticles in PBS over 96 h. (g) Fluorescent tag TAMRA-labelled Tat-vFLIP-α2 and its nanoformulation were loaded into human macrophages for 8 h. After PBS wash, the intracellular retention of fluorescence-labelled peptides was monitored by fluorescent microplate reader. a.u., arbitrary units. (h) At day 6 post treatment, with 10 μM peptides or nanopeptides, the representative cell images were captured by confocal microscopy. Scale bar: 50 µm. All cell related experiments are derived from 4 different donors and reported as means ± s.e.m.
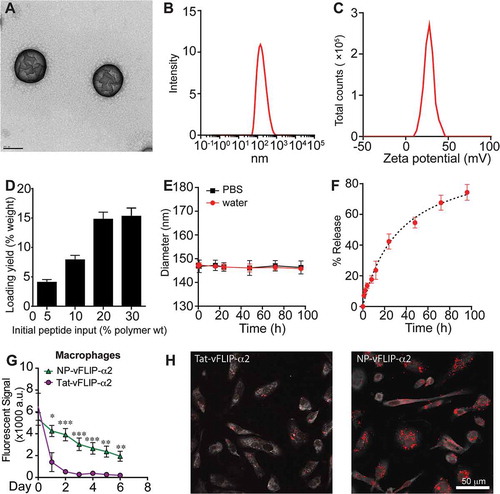
Intracellular drug delivery and retention were evaluated in human primary macrophages following loading with NP-vFLIP-α2 for 8 h. After replacement with fresh medium, the uptake and retention of intracellular NP-vFLIP-α2 was evaluated over 4 or 6 days post loading. NP-vFLIP-α2 showed similar uptake compared to the free Tat-vFLIP-α2 peptide. However, the intracellular retention of NP-vFLIP-α2 was increased 4-fold in macrophages compared to free peptide ((,)).
During autophagy, cytosolic LC3B-I is converted to its lipidated form (LC3B-II) by an ubiquitin-like system involving ATG7, ATG3 and the ATG12–ATG5 complex. The ATG12–ATG5 complex ligates LC3B-II to the nascent phagophore membrane through phosphatidylethanolamine. LC3B-II is then degraded after fusion of the autophagosome with a lysosome. Therefore, the conversion of LC3B-I to LC3B-II and its turnover is an indicator of autophagy induction and flux. Additionally, the degradation of the polyubiquitin-binding protein SQSTM1/p62 occurs when autophagy goes to completion. To confirm that the nanopeptides induce autophagy, following treatment of macrophages, cell lysates were collected and assessed for LC3B-I to LC3B-II lipidation and SQSTM1/p62 degradation. As expected, there was an increase in LC3B-II lipidation and SQSTM1/p62 degradation verifying that the NP-vFLIP-α2 peptides induce autophagy that goes to completion (Figure S3). Thus, the biodegradable NP-vFLIP-α2 was efficiently taken up and retained in macrophages, and effectively induced autophagy.
NP-vFLIP-α2 inhibits HIV replication and promotes killing of chronically infected macrophages
Our initial experiments were designed to assess the anti-HIV activity of controlled release nanoparticles loaded with NP-vFLIP-α2 in chronically infected macrophages. For these experiments, macrophages were infected with HIV for 10 days followed by treatment with NP-vFLIP-α2 and monitored for HIV p24 antigen release into supernatant every 48 h as described above. Following a single treatment with NP-vFLIP-α2, there was a gradual decline in HIV p24 antigen. At 6 days post treatment, HIV p24 declined to 45.7%, 77.6% and 98.2% of control untreated cells at nanopeptide concentrations of 1, 5 and 10 μM, respectively (()). The inhibition was specific to the NP-vFLIP-α2 since the nanoparticle formulation containing scrambled peptide had no effect on viral replication.
Figure 3. NP-vFLIP-α2 inhibits HIV replication through the preferential killing of chronically infected macrophages. (a) At day 10 p.i., chronically infected macrophages were treated with a single dose of NP-vFLIP-α2. HIV infection was measured in cell culture supernatants every other day for 6 days post treatment by HIV p24 antigen ELISA. (b) NP-vFLIP-α2 induced cytotoxicity was assessed by LDH assay from the collected supernatants. (c) At day 6 post treatment, NP-vFLIP-α2-treated cells were stained with the cell-death marker nuclear dye SYTOX. The percentage of dead cells was plotted. (d) Representative images of SYTOX staining at day 16 p.i. with NP-vFLIP-α2 treatment captured by fluorescence microscopy (200X). Scale bar: 50 ±m. Data are derived from 4 different donors and reported as means ± s.e.m. * P < 0.05, ** P < 0.01.
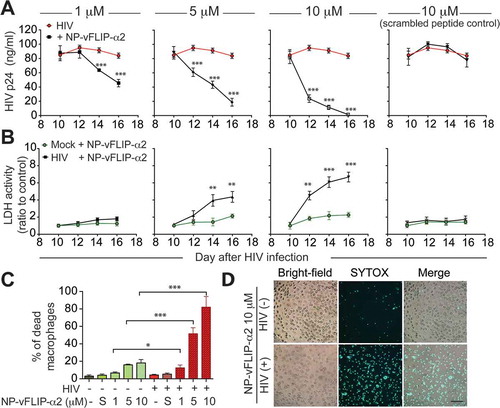
Next, we examined if the decrease observed in HIV replication might be associated with an increase in cytotoxicity assessed using the LDH release assay. Comparing HIV-infected macrophages to uninfected cells, LDH was increased 3-fold in the infected cells with increasing evidence of cytotoxicity through day 6 (()). To further assess cell damage, dsDNA was stained using SYTOX green. At 6 days following a single treatment of NP-vFLIP-α2, 12.5%, 51.6% and 89.0% of HIV-infected macrophages were SYTOX green-positive at NP-vFLIP-α2 concentrations of 1, 5 and 10 μM approximately 5-fold greater than that observed in uninfected cells ((,)). Since not all macrophages would be expected to be infected, the 89% cell death represents most, if not all, infected macrophages.
NP-vFLIP -α2 induces preferential killing of HIV-infected macrophages through autophagy-dependent cell death
To address the mechanism of NP-vFLIP-α2 induced cell death, cell lysates were harvested after 24 h of NP-vFLIP-α2 treatment. Western blot analysis showed that NP-vFLIP-α2 activated autophagy in a dose dependent manner with an increase in LC3B-II lipidation and SQSTM1/p62 degradation (()). Of interest, the induction of autophagy was consistently observed to be significantly increased in HIV-infected macrophages compared to mock infected cells treated with the same concentrations of peptide.
Figure 4. NP-vFLIP-α2 induces autophagy-dependent cell death in chronically infected macrophages. (a) At day 10 p.i., macrophages were treated with NP-vFLIP-α2 for 8 h. To remove nanoparticles that were not internalized, cells were washed extensively with PBS. After an additional 24 h in culture, macrophages were harvested, proteins extracted and assessed for LC3B-II lipidation and SQSTM1/p62 degradation by western blot. (b) Macrophages were transduced with nonspecific scrambled shRNA (shNS), shATG5 and shATG7, and monitored for 10 days. Cell lysates were assessed for ATG5 and ATG7 expression by western blot using ImageJ software. (c and d) At day 10 p.i., shATG5 and shATG7-transduced macrophages were treated with a single dose of NP-vFLIP-α2 for 8 h and cultured for an additional 24 h. Harvested cells were assessed by western blot for markers of autophagy. (e and f) The collected supernatants were assessed by LDH assay for cytotoxicity of NP-vFLIP-α2. All densitometric quantifications were collected and plotted based on 4 different donors that were normalized to ACTB as means ± s.e.m. S stands for 10 µM nanoformulated Tat-vFLIP-α2 scrambled peptides. Representative immunoblots are shown. * P < 0.05, ** P < 0.01, *** P < 0.001.
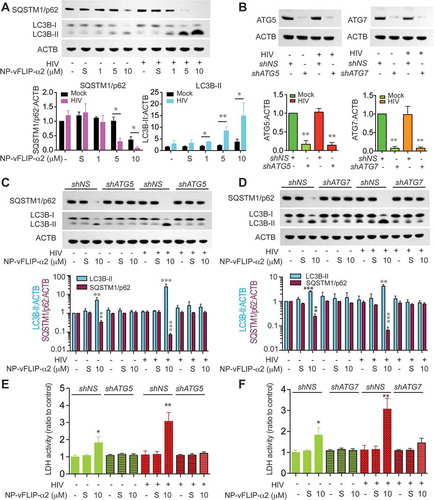
To further characterize NP-vFLIP-α2 induced cell death, small hairpin RNA (shRNA) was employed to investigate the molecular mechanism(s) associated with the preferential killing of HIV-infected macrophages over uninfected cells. Following knockdown of autophagy elongation proteins ATG5 and ATG7 (()), NP-vFLIP-α2 treatment failed to induce LC3B-I to II lipidation and SQSTM1/p62 degradation, and there was no increase in cytotoxicity in mock- or HIV-infected macrophages ((-)).
Although our findings strongly suggested that the overinduction of autophagy is the mechanism of cell death for NP-vFLIP-α2-treated HIV-infected macrophages, we further examined if other forms of cell death (i.e. necroptosis or apoptosis) might also be involved. Because CASP3 (caspase 3) and CASP8 (caspase 8) are activated during apoptotic cell death, we examined cleavage of these proteins following NP-vFLIP-α2 treatment and found no increase in cleavage of either caspase suggesting that apoptosis was not occurring in treated cells ((,)). To identify if necroptosis might be occurring, macrophages were pretreated with the pan-caspase inhibitor Z-VAD-FMK and necroptosis inhibitor necrostatin-1 followed by exposure to NP-vFLIP-α2. Again, there was no effect on cell death with either pretreatment (())
Figure 5. Characterization of NP-vFLIP-α2-induced cell death in chronically infected macrophages. (a) After a single treatment with NP-vFLIP-α2 for 8 h, chronically infected macrophages were cultured for an additional 6 days. Cell lysates were assessed for cleaved CASP3 by western blot and analyzed using ImageJ software. (b) Cleaved CASP8 was also measured in the same cell lysates. Staurosporine 1 µM treatment for 6 h was used as a positive control. (c) Chronically infected macrophages were pretreated with 1 mM 3-methyladenine, 20 µM Z-VAD-FMK, 50 μM necrostatin-1 or 200 nM BAF for 2 h, followed by treatment with 10 µM NP-vFLIP-α2 for an additional 24 h. Cell culture supernatants were tested for cytotoxicity. Data are derived from 4 different donors and reported as means ± s.e.m. Representative immunoblots are shown. * P < 0.05, ** P < 0.01, *** P < 0.001.
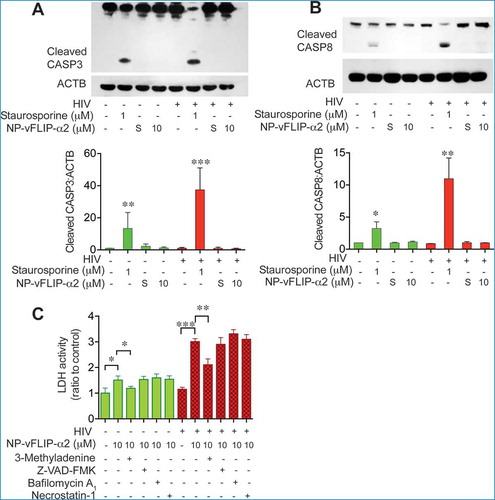
To further explore the role of autophagy in the preferential killing of HIV-infected macrophages, cells were pretreated with the vacuolar-type H+-ATPase (V-ATPase) inhibitor BAF following treatment with NP-vFLIP-α2. Under these conditions, a high level of cell death continued to be observed. In contrast, the class III phosphatidylinositol 3-kinase inhibitor (and also a class I phosphoinositide 3-kinase inhibitor) 3-methyladenine (3-MA) partially blocked NP-vFLIP-α2 induced cell death (()). In total, these findings indicate that the cell death associated with NP-vFLIP-α2 is dependent on a unique form of autophagy.
Na+/K+-ATPase dependent cell death, autosis, is induced by NP-vFLIP -α2 in chronically infected macrophages
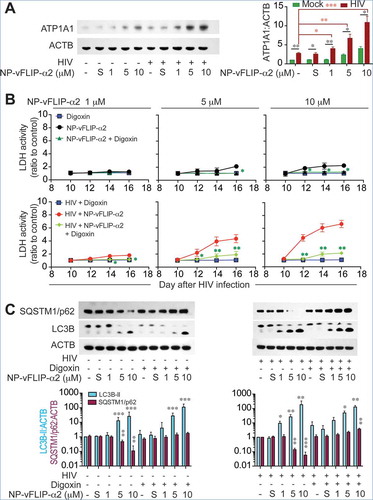
Recent studies have demonstrated that Tat-vFLIP-α2 and Tat-Beclin 1 peptides induce autophagy through a unique Na+/K+-ATPase dependent mechanism, autosis [Citation32]. Thus, we reasoned that the Na+/K+-ATPase might be involved with the Tat-vFLIP-α2 induced preferential killing of HIV-infected cells. To this end, we tested Na+/K+-ATPase expression in HIV-infected and mock-infected macrophages when treated with cationic nanoformulation NP-vFLIP-α2. For these experiments, macrophages were infected with HIV for 12 days and evaluated for expression of ATP1A1 (ATPase Na+/K+ transporting subunit alpha 1), which is the critical catalytic component of the Na+/K+-ATPase. As HIV infection progressed, there was an increase in ATP1A1 which by day 12 was 3-fold higher in infected macrophages compared to mock-infected controls (()). Using confocal microscopy, following HIV infection, ATP1A1 was found to be upregulated intracellularly and at the plasma membrane ((,)). We next examined the expression of ATP1A1 following increasing treatment with NP-vFLIP-α2 at 1, 5 or 10 μM. When compared to mock-infected macrophages, NP-vFLIP-α2 specifically increased ATP1A1 expression in infected macrophages by 4.0-, 6.7- and 10.8-fold, respectively (()). As expected, we also observed, 1.0-, 2.1- (P < 0.01) and 3.8- (P < 0.001) fold increase of ATP1A1 in mock-infected cells after treatment with the same doses of NP-vFLIP-α2. To confirm the action of Na+/K+-ATPase, the classical Na+/K+-ATPase inhibitor cardiotonic steroid, digoxin, was selected to inhibit the activity of Na+/K+ ATPase. Digoxin treatment inhibited NP-vFLIP-α2 induced cell death and preferential killing (() and Figure S4). Parallel with the reduction in cell death, digoxin also substantially inhibited autophagic flux with inhibition of SQSTM1/p62 degradation (). These results were further verified by RNA interference of ATP1A1 (). Knockdown of the ATP1A1 subunit inhibited NP-vFLIP-α2-induced cell death and blocked autophagic flux.
Figure 6. HIV upregulates Na+/K+-ATPase expression in macrophages. (a) Macrophages were infected with HIV for 12 days. The collected cell lysates were measured for ATP1A1 expression, a critical catalytic component of Na+/K+-ATPase. (b) At day 12 p.i., macrophages were fixed with 4% paraformaldehyde in PBS, and permeabilized with 0.5% Triton X-100 for intracellular immunofluorescence staining. The representative images of ATP1A1 expression were captured using confocal microscopy. (c) The non-permeabilized macrophages were stained for ATP1A1 expression on plasma membrane. Representative images are shown. Scale bar: 50 μm. Data are derived from 4 different donors and reported as means ± s.e.m. Representative immunoblots are shown. * P < 0.05.
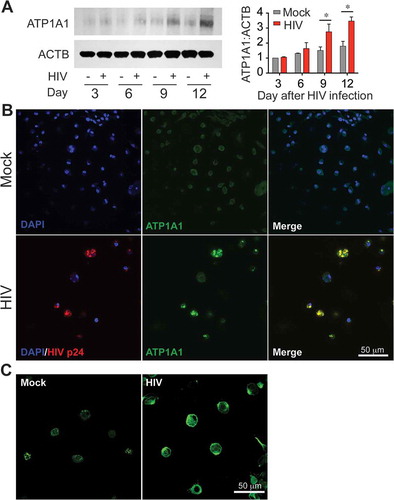
Figure 7. NP-vFLIP-α2 kills HIV-infected macrophages through a Na+/K+-ATPase dependent mechanism. (a) Chronically HIV-infected macrophages were treated with NP-vFLIP-α2 for 8 h and further cultured for an additional 24 h. The collected cell lysates were analyzed by western blot. A representative image is shown on left with densitometric quantification shown on right. (b) Chronically HIV-infected macrophages were treated with 50 nM digoxin and a single dose of NP-vFLIP-α2, and cultured for 6 additional days. The collected cell culture supernatants were measured by LDH assay. (c) Cell lysates harvested from digoxin and NP-vFLIP-α2-treated macrophages were verified for autophagy activity. Data are derived from 4 different donors and reported as means ± s.e.m. S stands for 10 µM nanoformulated Tat-vFLIP-α2 scrambled peptides. * P < 0.05, ** P < 0.01, *** P < 0.001.
Because digoxin has been reported to inhibit HIV replication in 293 T cells, HeLa rtTA-HIV-ΔMls cells, and activated PBMCs from HIV patients [Citation33,Citation34], we examined if digoxin inhibited HIV replication in primary macrophages. At the concentrations studied, we found that digoxin alone had little effect on detectable release of p24 Ag into culture supernatants (Figure S5). Interestingly, we observed that knockdown of ATP1A1 partially decreased HIV replication, but did not eliminate HIV persistence (Figure S5). Thus, inhibition of Na+/K+-ATPase reversed the anti-HIV activity and preferential killing of HIV-infected macrophages treated with NP-vFLIP-α2.
In additional experiments, we also assessed the direct interaction between Na+/K+-ATPase and autophagy. We observed that knockdown of ATG5 decreased ATP1A1 expression in NP-vFLIP-α2-treated macrophages (Figure S6). Thus, the cumulative data support that Na+/K+-ATPase has a central role in NP-vFLIP-α2 induced cell death and autophagic activity.
NP-vFLIP -α2 preferentially kills chronically HIV-infected macrophages with antiretroviral suppressed viral replication
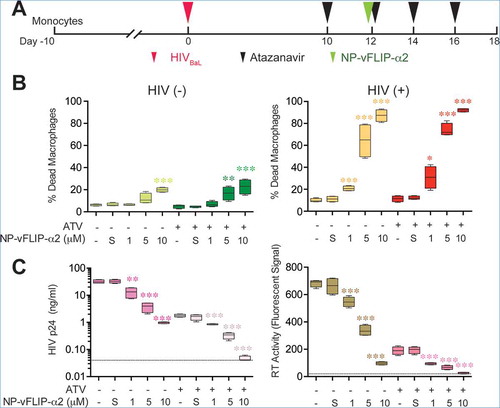
To evaluate the translational potential of NP-vFLIP-α2 use as part of a cure strategy in patients receiving suppressive ART, we assessed the cytotoxicity and virological control of the nanopeptide on HIV-infected macrophages under antiretroviral suppression. For these studies, macrophages were infected with HIV for 10 days and then treated with the protease inhibitor atazanavir to block the proteolytic cleavage of HIV Gag and Pol polyproteins preventing HIV particles to mature into infectious virus. Following antiretroviral suppression, a single treatment of NP-vFLIP-α2 was added to the cell cultures and followed for an additional 6 days (()). At the end of the observation period, there was a dose-dependent response to NP-vFLIP-α2 with 30.8%, 73.8% and 92.1% of dead cells detected at 1, 5 and 10 μM (()) similar to what was observed in chronically infected macrophages treated with NP-vFLIP-α2. In contrast, the maximum cell death observed in uninfected macrophages treated with 10 µM NP-vFLIP-α2 was 22.6% and 19.9% with or without antiretroviral suppression, respectively. Importantly, there was no evidence of viral reactivation as monitored by release of p24 antigen into culture supernatants and reverse transcriptase activity under any of NP-vFLIP-α2 treatment conditions (()). Of note, under these conditions, the antiretroviral drug atazanavir itself had no effect on autophagy (Figure S7).
Figure 8. RNA interference of ATP1A1 inhibits NP-vFLIP-α2-induced cell death in chronically infected macrophages. (a) At 5 days p.i., macrophages were transduced with shATP1A1 and nonspecific shRNA. The efficiency of RNAi knockdown was assayed by western blot analysis for ATP1A1 expression. A representative western blot and quantification are shown. (b) Chronically HIV-infected macrophages were treated with a single dose of NP-vFLIP-α2 and cultured for an additional 24 h. Culture supernatants were collected and analyzed for cytotoxicity using LDH assay. (C) Representative image of harvested cell lysates evaluated for autophagy by western blot. Data are derived from 4 different donors and reported as means ± s.e.m. S stands for 10 µM nanoformulated Tat-vFLIP-α2 scrambled peptides. * P < 0.05, ** P < 0.01, *** P < 0.001.
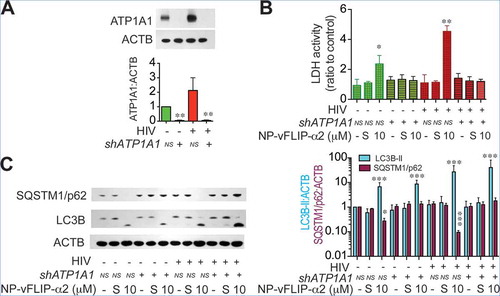
Figure 9. NP-vFLIP-α2 preferentially kills HIV-infected macrophages with antiretroviral suppressed viral replication. (a) Chronically HIV-infected macrophages were treated with 500 nM atazanavir for 2 days. At day 12 p.i., cells were treated with NP-vFLIP-α2 for 8 h. After washing with PBS, cells were replenished with 500 nM atazanavir in cell culture media every other day and cultured for an additional 6 days. (b) The collected culture supernatants were analyzed for cell death signaling through SYTOX staining. (c) Antiretroviral activity was evaluated using HIV p24 ELISA and reverse transcriptase activity assay in collected culture supernatants. Dashed line represents the detection limit. Data are derived from 4 different donors and reported as means ± s.e.m. S = 10 µM nanoformulated Tat-vFLIP-α2 scrambled peptides. * P < 0.05, ** P < 0.01, *** P < 0.001.
To verify the selectivity of NP-vFLIP-α2 induced cell death, we developed a mixed culture of HIV-positive and -negative macrophages using visible HIV-GFP-pSF162R3Nef+ infection (Figure S8). NP-vFLIP-α2-induced cell death was detected by live cell membrane impermeant dye DAPI, and analyzed by live cell imaging. Under these conditions, a single treatment with NP-vFLIP-α2 at 5 and 10 µM killed 92.2% and 93.3% of HIV-infected cells, respectively. In contrast, only 2.4% and 13.2% of bystander cells were killed at the 5 and 10 µM concentrations NP-vFLIP-α2, respectively (Figures S8 and S9). Importantly, we found no evidence of viral rebound following treatment with NP-vFLIP-α2 (Figure S9). Thus, under all conditions studied, NP-vFLIP-α2 demonstrated the potential to preferentially kill HIV-infected macrophages either chronically infected with HIV or during antiretroviral viral suppression.
Discussion
Although current antiretroviral therapy results in inhibition of HIV replication, treatment fails to eliminate virus from latently infected cell reservoirs. Thus, there remains an urgent need to develop novel strategies to eliminate HIV from persistent reservoirs. In the data presented here, we demonstrate that preferential killing of HIV-infected macrophages can be achieved through the induction of a specific form of cell death that is dependent on the autophagy and Na+/K+-ATPase, autosis.
Since the initial articles identifying a role for autophagy during HIV infection, an expanding literature has demonstrated that autophagy plays a central role in HIV pathogenesis [Citation15–Citation17,Citation35–Citation41]. The data presented here using nanoformulated autophagy-inducing peptides represent a novel approach to exploiting the interaction between HIV and autophagy that leads to the preferential killing of infected macrophages. We have used lipid-coated PLGA nanoparticles because they significantly improve the intracellular delivery and bioavailability of our peptides. The biocompatible nanoparticle platform used combines the positive attributes of both liposomes and PLGA nanoparticles while excluding some of their deficiencies [Citation42–Citation44]. This nanoparticle system consists of a hydrophobic polymeric core, a lipid shell surrounding the polymeric core, and a hydrophilic polymer stealth layer outside the lipid shell. By varying the concentrations of the building blocks and preparation conditions, the nanoparticle’s size can be tuned between tens of nm to a few hundred nm. The versatility of this nanoparticle platform allows for the loading of autosis-inducing peptides using a variety of methods, ranging from chemical conjugation to physical encapsulation via emulsion or nanoprecipitation methods. Per mg of polymer, up to 150 μg of peptide was able to be loaded. The loading yield can be adjusted by controlling the initial input amount of the peptide. The maximally loaded nanoparticles used in our experiments were approximately 147 nm in diameter with uniform size, size distribution, and surface charge and demonstrated sustained release of the peptides; ~ 70% of the peptide was released in 96 h with no evident burst release from the nanoparticles. This nanoparticle-based delivery approach allowed for the single administration of NP-Tat-vFLIP-α2 to be released over time resulting in killing of infected cells and a marked decline in HIV p24 antigen released into culture supernatants. The size of nanoparticles can greatly impact their distribution throughout the body. Nanoparticles > 200 nm are readily cleared by the liver and spleen, while nanoparticles < 5 nm are filtered by the kidney [Citation45–Citation47]. Recently, we have synthesized sub-25 nm lipid-polymer hybrid nanoparticles that can potentially be loaded with enhanced targeting through the addition of a targeting ligand on the nanoparticle surface[Citation47]. These sub-25 nm nanoparticles are particularly suitable for facilitating drug delivery to the brain [Citation47–Citation49]. These nanoparticles are synthesized from FDA-approved material. Thus, it is possible to synthesize a nanoparticle containing Tat-vFLIP-α2 that can enter the CNS and potentially be used in humans. Additionally, these lipid coated polymeric nanoparticles can be readily functionalized with targeting ligands using a half-antibody technique for cell-specific drug delivery [Citation50].
We have focused the results presented on NP-Tat-vFLIP-α2 because at equivalent concentrations we consistently found increased or equivalent activity of the NP-Tat-vFLIP-α2 peptide compared to the NP-Tat-Beclin 1 peptide. However, both peptides were found to induce a Na+/K+-ATPase dependent form of cell death, autosis, that preferentially killed HIV-infected cells. We have studied macrophages because they are permissive to HIV infection, establish persistent infection without cell death, infection is more resistant to antiretroviral therapy, and they constitute an important HIV reservoir within tissues and the CNS where they have been implicated as having an important role in HIV-associated dementia and other HIV-related disorders [Citation16,Citation51]. Moreover, despite sustained virological suppression on ART, as defined by undetectable virus within plasma, as many as 25% of HIV-infected person still exhibit mild cognitive impairment. Of importance, antiretroviral drugs are frequently less active in macrophages as nucleoside reverse transcriptase inhibitors and non-nucleoside reverse transcriptase inhibitors have been found not to be effective in macrophages after initial HIV infection [Citation52,Citation53]. Thus, it is not surprising that HIV has been identified in macrophages within the brain, lungs and other tissues despite suppressive treatment [Citation54]. For these reasons, the experiments performed in macrophages persistently infected with HIV and treated with combination antiretrovirals are of particular importance. Despite suppression of virus in these cultures, treatment with NP-vFLIP-α2 peptide resulted in the preferential killing of infected cells with potent inhibition of HIV replication supporting the potential use of this formulation as part of a cure strategy in patients receiving antiretroviral therapy. Studies are currently in progress in our laboratory that are examining the potential of NP-vFLIP-α2 to preferentially kill HIV-infected resting memory CD4+ lymphocytes in a similar manner as described here for macrophages.
Several viruses, including HIV, have been demonstrated to control autophagy in order to promote their own replication. Consistent with these findings, the induction of autophagy inhibits many of these viruses including HIV [Citation27,Citation55–Citation63]. Thus, macrophages with chronic infection or antiretroviral suppressed chronic infection when treated with NP-vFLIP-α2 resulted in killing of infected cells without viral reactivation. The preferential killing of HIV-infected cells without viral reactivation is critical to the potential application of this approach to HIV eradication strategies. First described by the Levine laboratory using the Tat-Beclin 1 peptide, autophagy inducing peptides were identified as potent inhibitors of several viruses as well as the bacterium, Listeria monocytogenes [Citation27]. Additionally, in neonatal mouse models of infection with chikungunya virus and West Nile virus, Tat-Beclin 1 significantly reduced mortality. In subsequent studies, Tat-Beclin 1 was found to induce cell death in a dose-dependent manner regulated by induction of the Na+/K+-ATPase. In earlier research, Lee et al identified that CFLAR/FLIP proteins interact with the FADD (Fas associated via death domain)-CHUK/IKKα-IKBKB/IKKβ-IKBKG/IKKγ complex, TRAFs and SFN/14-3-3σ [Citation28]. Additionally, the FLIP protein was found to compete with LC3 binding of ATG3 and that FLIP peptide bound sufficiently to ATG3 facilitating the ATG3-LC3 interaction with the induction of high levels of autophagy and killing of cancer cells. The Levine group extended these findings demonstrating that Tat-vFLIP-α2 induced autophagy through the same Na+/K+-ATPase-dependent pathway and similarly resulted in a dose-dependent cell death. Moreover, Pennarun et al show that Tat bound to a peptide corresponding to the β5-strand of CFLAR/cFLIP results in the preferential killing of cancer cells when compared to noncancerous cells [Citation64]. In particular, they have found that the E fragment (termed killerFlip) which constitutes the active site of full-length CFLAR-derived peptide increases preferential killing of cancer cells which appears to be independent from its sequence similarity with CFLAR. They further suggest that the cell killing observed is independent of apoptosis and necroptosis, but do not examine the potential role of autophagy. Of interest, the preferential ratio of killing cancer cells to noncancerous cells is similar the ratio observed in our studies of HIV-infected and -uninfected macrophages.
In a high-throughput screen for inhibitors of HIV, Laird et al identify that pretreatment of a 293T indicator cell line with cardiac glycosides including digoxin inhibits HIV gene expression [Citation33]. In another study, Wong et al. have found that digoxin alters HIV RNA splice site usage resulting in the loss of Rev function [Citation34]. Thus, it would appear that alterations in the Na+/K+-ATPase can affect HIV replication through a number of different mechanisms. In the data presented here, HIV infection was found to increase Na+/K+-ATPase expression in macrophages. Na+/K+-ATPase is a membrane pump involved with the transport sodium and potassium ions across the plasma membrane [Citation65]. HIV disturbs plasma membrane permeability and ion transport through alterations induced by Nef resulting in increased intracellular K+ without affecting intracellular pH [Citation66,Citation67]. Because the enhanced expression of Na+/K+-ATPase is central to the unique mechanism of autosis, the data presented here strongly suggest that the combination of HIV infection and NP-vFLIP-α2 induce the over-induction of Na+/K+-ATPase leading to the preferential killing of HIV-infected cells ().
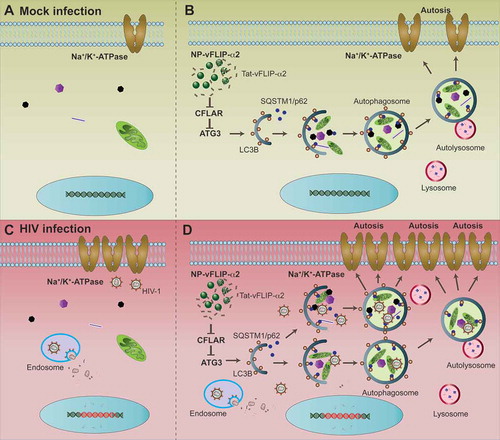
Figure 10. NP-vFLIP-α2-induced preferential killing of HIV-infected macrophages: a Na+/K+-ATPase dependent form of autophagy. (a and b) Autophagy, as an essential survival mechanism, controls cellular homeostasis in macrophages. Controlled-release PLGA nanoparticles intracellularly deliver autophagy-inducing peptides Tat-vFLIP-α2 into the cytoplasm of mock-infected macrophages. The released Tat-vFLIP-α2 targets the ATG3 inhibitor CFLAR protein resulting in the release of ATG3 protein and induction of autophagy. The accumulated autophagy-inducing peptides activate Na+/K+-ATPase-dependent autophagic cell death, autosis. (c and d) HIV infection increases the basal level of Na+/K+-ATPase expression in human macrophages. In HIV chronically infected macrophages, NP-vFLIP-α2 treatment results in an over induction of autophagy with increased expression of Na+/K+-ATPase compared to uninfected cells leading to the preferential killing of HIV-infected cells through a Na+/K+-ATPase form of autophagic cell death, autosis.
In summary, we have synthesized an autophagy-inducing peptide incorporated in a PLGA-nanoparticle comprised of FDA approved materials that efficiently delivers peptide intracellularly, which is slowly released over time. This NP-vFLIP-α2 peptide induces Na+/K+-ATPase and preferentially kills HIV chronically infected macrophages with and without antiretroviral suppression. Our findings are consistent with studies that have examined human cancer cell lines and suggest that cellular alterations can be exploited to preferentially kill HIV-infected cells. These data further support the over-induction of autophagy as one approach that should be considered as part of a cure strategy for HIV.
Materials and methods
Ethics statement
Venous blood was obtained from HIV-seronegative subjects. All blood donors were adult. The protocol was reviewed and approved by the Human Research Protection Program of the University of California, San Diego in accordance with the requirements of the Code of Federal Regulation on the Protection of Human Subjects (45 CFR 46 and 21 CFR 50 and 56). Written informed consent was obtained from all blood donors.
Human monocytes derived macrophages
Peripheral blood mononuclear cells (PBMC) were isolated from buffy coats of healthy HIV-seronegative blood donors using Ficoll-PaqueTM density centrifugation (GE Healthcare,17-1441-03) as previously described [Citation16]. PBMC were seeded in 24- or 96-well plates. Nonadherent cells were removed by gentle pipetting and washed after 24 h in culture at 37°C in a humidified atmosphere containing 5% CO2 [Citation16,Citation68]. Adherent cells were grown in RPMI 1640 medium (Life Technologies, 11875–093) containing penicillin/streptomycin (1%; Life Technologies, 15140-122), L-glutamine (1%; Life technologies, 25030-081), heat-inactivated fetal bovine serum (10%; Sigma-Aldrich, F0925-500ML) and CSF1/MCSF (colony stimulating factor 1) (Humanzyme Inc, HZ-1193) 10 ng/ml for 10 days. The adherent monocyte-derived macrophages (macrophages) were infected with HIVBaL and HIVGFP-pSF162R3Nef+ (a gift from Dr. Amanda M. Brown, Johns Hopkins University) at a multiplicity of infection of 0.1 [Citation69].
Cell staining for immunofluorescence microscopy
Macrophages were washed with phosphate-buffered salien (PBS; Life Technologies, 14190–144) and fixed with 4% paraformaldehyde (Affymetrix Inc, 19943) at room temperature for 20 min. After permeabilizing the cell membrane with 0.5% Triton X-100 (Sigma-Aldrich, X100) in PBS, macrophages were sequentially incubated with primary antibody anti-ATP1A1 (Santa Cruz Biotechnology, sc-28800) and HIVp24 (Dako, M0857), and secondary antibody goat anti-mouse and goat anti-rabbit (Thermo Fisher Scientific, A11004 and A11034) at room temperature. Macrophage culture slides were covered in ProLong Gold antifade reagent with DAPI (4ʹ, 6-diamidino-2-phenylindole) (Thermo Fisher Scientific, P36931) [Citation70–Citation72]. The fluorescent images were captured with an FV1000 Olympus confocal microscope (Olympus Corporation of Americas, Waltham, MA, USA).
Western blot
Macrophages were lysed in 20 mM HEPES, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 1 mM EDTA supplemented with 1% (v:v) 4-(1,1,3,3-tetramethylbutyl) phenyl-polyethylene glycol (Sigma-Aldrich, H3375,746398, M8266 and 798681) and 1% (v:v) Halt protease and phosphatase inhibitor mixture (Thermo Fisher Scientific, 78440). Protein concentrations were measured by MicroBCA protein assay kit (Thermo Fisher Scientific, 23235). Equal amounts of proteins were electrophoresed using XCell SureLockTM mini-cell electrophoresis system (Thermo Fisher Scientific, EI0001), followed by transfer to nitrocellulose membranes (Bio-Rad laboratories, 1620115). Blots were blocked with 5% nonfat dry milk (wt:wt) or bovine serum albumin in Tris-buffered saline (Thermo Fisher Scientific, 28358) with 0.1% (v:v) Tween 20 (Sigma-Aldrich, P1379) buffer for 1 h at room temperature, and probed with LC3B antibody (Novus Biologicals, NB100-2220), SQSTM1/p62 (Abcam, ab56416), ATG5 (Cell Signaling Technology, 2630), ATG7 (Cell Signaling Technology, 2631), CASP3 (Cell Signaling Technology, 9662), CASP8 (Cell Signaling Technology, 9746) for determination of the targeted protein. The immunoreactive protein signals were identified using Western-SuperStarTM immunodetection system (Thermo Fisher Scientific, T2147) [Citation31,Citation73,Citation74]. The signal intensity of detected protein was normalized to that of ACTB (Sigma-Aldrich, A5441) and analyzed using ImageJ analysis software [Citation75,Citation76]. All targeted proteins and internal loading controls were detected and verified within the same linear range.
Measurement of cell death
The LDH (lactate dehydrogenase) cytotoxicity assay kit (Clontech Laboratories, MK401) was used to measure cell damage following the manufacturers recommended procedures. The report signal was detected by measurement of absorbance at 490 nm wavelength. Dead cells were quantified using SYTOX® green nucleic acid stain (ThermoFisher, S7020). The positive staining cells were determined at fluorescent excitation/emission 504/523 nm wavelength.
Measurement of Na+/K+-ATPase
HIV-infected human macrophages were collected at day 3, 6, 9 and 12 post infection. The harvested whole cell lysates were tested for expression of ATP1A1 using western blotting. Primary rabbit anti-ATP1A1 (Cell Signaling Technology, 23565), mouse anti-ACTB and secondary goat anti-rabbit and goat anti-mouse (Santa Cruz Biotechnology, sc-2004 and sc-2005) were applied for detection of protein signaling.
RNAi knockdown
Knockdown ATG5, ATG7 and ATP1A1 expression in macrophages was achieved using MISSIONpLK0.1-puro lentiviral transduction particles kits, containing shRNAs. Following the manufacturer’s recommended protocol, macrophages were transfected with shRNA (Sigma-Aldrich, ATG5-TRCN0000151963, ATG7-TRCN0000435480 and ATP1A1-TRCN0000424769) and non-target shRNA control vector (Sigma-Aldrich, SHC002), and selected with 0.15 μg/ml puromycin.
Synthesis of Tat-vFLIP-α2-loaded lipid-polymer nanoparticles
The retro-inverso peptides (D-isomer) Tat-vFLIP-α2 (RRRQRRKKRGY-G-FVNLLFLVVE), Tat-vFLIP-α2 scramble peptide (RRRQRRKKRGY-G-FVNLAAAVVE) and Tat-vFLIP-α2 conjugated with C-terminal 5-carboxytetramethylrhodamine (TAMRA) fluorescent tag were synthesized by BIOMATIK (BIOMATIK). The Tat-Beclin 1 peptide (YGRKKRRQRRR-GG-TNVFNATFEIWHDGEFGT) and Tat-Beclin 1 scrambled peptide (YGRKKRRQRRR-GG-VGNDFFINHETTGFATEW) were also obtained from BIOMATIK. Lipid-polymer nanoparticles were formulated through a single step of nanoprecipitation. Ester-ended Poly (D,L-lactide-co-glycolide) (PLGA) with a 50:50 monomer ratio (Sigma-Aldrich, P2191) was dissolved at 10 mg/ml in acetonitrile. 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(carboxy[polyethylene glycol]2000) (DSPE-PEG-carboxy; Avanti Polar Lipids, 880135P) was dissolved in chloroform at 20% of the PLGA polymer weight and evaporated to form a film. The PLGA-acetonitrile solution was then added to the lipid film and vortexed vigorously for 3 min. The PLGA-lipid solution was then added to 10 mM Tris-HCl, pH 8 solution and sonicated for 2 min in a bath sonicator. After the nanoparticles were formed through self-assembly of the polymers and lipids, the remaining acetonitrile and free molecules were removed by washing the nanoparticles 3 times using an Amicon Ultra-4 centrifugal filter with a MW cut-off of 10,000 Da (EMD Millipore, UFC801096). To encapsulate Tat-vFLIP-α2 peptides into the lipid-coated polymeric nanoparticles, the peptide was dissolved in the organic phase along with the PLGA polymer. This combined solution was transferred dropwise along with the lipids into the aqueous solution and sonicated for 2 min. After the nanoparticles were formed through self-assembly of the polymers and lipids, the resulting nanoparticles were washed using repeated centrifugation steps.
Characterization of Tat-vFLIP-α2 loaded lipid-polymer PLGA nanoparticles
Nanoparticle size (diameter, nm), polydispersity index (PDI), and surface charge (zeta potential, mV) were determined by dynamic light scattering using a Malvern Zetasizer Nano ZS analyzer (Malvern Panalytical, Malvern, UK) at room temperature. The drug-loading yield and peptide release profile was measured by Slide-A-Lyzer MINI dialysis microtube with a molecular weight cutoff of 3,500 Da (Thermofisher, 88400). The nanoparticles were dialyzed against 4 L of PBS buffer (pH 7.4) at 37ºC. At each predetermined time point, nanoparticle solutions from 3 mini dialysis units were collected separately for drug quantification by absorbance. The drug-loading yield was defined as the weight ratio of the peptide payload to the nanoparticles. The peptide release kinetics was used to demonstrate the release of peptide from nanoparticles over time, which was plotted as the weight ratio of the accumulative released peptides to the total peptide payload against time.
Transmission electron microscopy (TEM)
TEM experiments were performed on a JEOL 1200 EX II Gatan instrument (JEOL, Peabody, MA, USA) at an acceleration voltage of 120 kV. The nanoparticles were applied onto formvar and carbon-coated copper grids for sample preparation, and negatively stained for 10 min at room temperature with freshly prepared and sterile-filtered 2% (w/v) uranyl acetate aqueous solution. The grids were then washed twice with distilled water and air dried before for TEM imaging.
Statistics
Data were assessed for symmetry or skewness using the Pearson skewness coefficient. Fold change data were log2 transformed for statistical analyses. Comparisons between groups were performed using the two-tailed Student t test, ANOVA, Pearson correlation and Wilcoxon rank test where appropriate. P values less than 0.05 two-tailed were considered statistically significant.
Abbreviations
| ACTB: | = | actin beta; |
| ART: | = | antiretroviral therapy; |
| ATG3: | = | autophagy related 3; |
| ATG5: | = | autophagy related 5; |
| ATG7: | = | autophagy related 7; |
| ATP1A1: | = | ATPase Na+/K+ transporting subunit alpha 1; |
| BAF: | = | bafilomycin A1; BECN1: beclin 1; |
| CASP3; | = | caspase 3; |
| CASP8: | = | caspase 8; |
| CFLAR: | = | CASP8 and FADD like apoptosis regulator; |
| HIV: | = | human immunodeficiency virus type-1; |
| LDH: | = | lactate dehydrogenase; |
| MAP1LC3B/LC3B: | = | microtubule associated protein 1 light chain 3 beta; |
| PLGA: | = | poly lactic-co-glycolic acid; |
| SQSTM1/p62: | = | sequestosome 1 |
Supplemental Material
Download MS Word (23.1 MB)Acknowledgments
We thank Sophie Chen, Columbus Zhang, and Bella Zhang for figure organization. We thank Simson Hon, Pratima Rawat and Rodney Trout for experimental assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Gulick RM, Hirsch MS, Lane HC, et al. Guidelines for the use of antiretroviral agents in adults and adolescents living with HIV. Department of health and human services. AIDSinfonihgov. 2018 Mar 27. Available from: https://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf
- Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013 Nov 7;369(19):1807–1818. PubMed PMID: 24195548.
- Group ISS, Lundgren JD, Babiker AG, et al. Initiation of antiretroviral therapy in early asymptomatic HIV infection. N Engl J Med. 2015 Aug 27;373(9):795–807. PubMed PMID: 26192873; PubMed Central PMCID: PMCPMC4569751.
- Le T, Wright EJ, Smith DM, et al. Enhanced CD4+ T-cell recovery with earlier HIV-1 antiretroviral therapy. N Engl J Med. 2013 Jan 17;368(3):218–230. PubMed PMID: 23323898; PubMed Central PMCID: PMCPMC3657555.
- International ASSWGoHIVC, Deeks SG, Autran B, et al. Towards an HIV cure: a global scientific strategy. Nat Rev Immunol. 2012 Jul 20;12(8):607–614. PubMed PMID: 22814509; PubMed Central PMCID: PMCPMC3595991.
- Gama L, Abreu CM, Shirk EN, et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS. 2017 Jan 2;31(1):5–14. PubMed PMID: WOS:000390240700002; English.
- Spector SA, Rappaport J. HIV cure strategists: ignore the central nervous system at your patients’ peril. AIDS. 2017 Jan 2;31(1):167–168. PubMed PMID: WOS:000390240700018; English.
- Barton K, Winckelmann A, Palmer S. HIV-1 reservoirs during suppressive therapy. Trends Microbiol. 2016 May;24(5):345–355. PubMed PMID: 26875617; PubMed Central PMCID: PMC5319871.
- Lorenzo-Redondo R, Fryer HR, Bedford T, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature. 2016 Feb 4;530(7588):51–56. PubMed PMID: 26814962; PubMed Central PMCID: PMCPMC4865637.
- Martinez-Picado J, Deeks SG. Persistent HIV-1 replication during antiretroviral therapy. Curr Opin HIV AIDS. 2016 Jul;11(4):417–423. PubMed PMID: 27078619; PubMed Central PMCID: PMCPMC4900428.
- Chun TW, Stuyver L, Mizell SB, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997 Nov 25;94(24):13193–13197. PubMed PMID: 9371822; PubMed Central PMCID: PMC24285.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013 Oct;13(10):722–737. PubMed PMID: 24064518; PubMed Central PMCID: PMC5340150.
- Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007 Jul;27(1):11–21. PubMed PMID: 17663981.
- Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005 Nov;12(Suppl 2):1528–1534. PubMed PMID: 16247500.
- Kyei GB, Dinkins C, Davis AS, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009 Jul 27;186(2):255–268. PubMed PMID: 19635843; PubMed Central PMCID: PMCPMC2717652.
- Campbell GR, Rawat P, Bruckman RS, et al. Human immunodeficiency virus type 1 nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015 Jun;11(6):e1005018. PubMed PMID: 26115100; PubMed Central PMCID: PMC4482621.
- Zhou D, Spector SA. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS. 2008 Mar 30;22(6):695–699. PubMed PMID: 18356598; PubMed Central PMCID: PMCPMC2764485.
- Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J Gen Virol. 2003 Jul;84(Pt 7):1649–1661. PubMed PMID: 12810858.
- Doitsh G, Greene WC. Dissecting how CD4 T cells are lost during HIV infection. Cell Host Microbe. 2016 Mar 9;19(3):280–291. PubMed PMID: WOS:000372457500006; English.
- Dahabieh MS, Battivelli E, Verdin E. Understanding HIV latency: the road to an HIV cure. Annu Rev Med. 2015;66:407–421. PubMed PMID: 25587657; PubMed Central PMCID: PMCPMC4381961.
- Castellano P, Prevedel L, Eugenin EA. HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci Rep. 2017 Oct 9;7(1):12866. PubMed PMID: 28993666; PubMed Central PMCID: PMCPMC5634422.
- Lennemann NJ, Coyne CB. Catch me if you can: the link between autophagy and viruses. PLoS Pathog. 2015 Mar;11(3):e1004685. PubMed PMID: 25811485; PubMed Central PMCID: PMC4374752.
- Jackson WT, Giddings TH Jr., Taylor MP, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005 May;3(5):e156. PubMed PMID: 15884975; PubMed Central PMCID: PMC1084330.
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008 Dec;9(12):1004–1010. PubMed PMID: 18971948; PubMed Central PMCID: PMCPMC2727358.
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. PubMed PMID: 19653858; PubMed Central PMCID: PMCPMC2831538.
- Liu Y, Shoji-Kawata S, Sumpter RM Jr., et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013 Dec 17;110(51):20364–20371. PubMed PMID: 24277826; PubMed Central PMCID: PMCPMC3870705.
- Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013 Feb 14;494(7436):201–206. PubMed PMID: 23364696; PubMed Central PMCID: PMC3788641.
- Lee JS, Li Q, Lee JY, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009 Nov;11(11):1355–1362. PubMed PMID: 19838173; PubMed Central PMCID: PMCPMC2802862.
- Ho DD, Rota TR, Hirsch MS. Infection of monocyte/macrophages by human T lymphotropic virus type III. J Clin Invest. 1986 May;77(5):1712–1715. PubMed PMID: 2422213; PubMed Central PMCID: PMCPMC424579.
- Campbell GR, Spector SA. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012;8(5):e1002689. PubMed PMID: 22589721; PubMed Central PMCID: PMCPMC3349755.
- Zhang G, Guo D, Dash PK, et al. The mixed lineage kinase-3 inhibitor URMC-099 improves therapeutic outcomes for long-acting antiretroviral therapy. Nanomedicine. 2016 Jan;12(1):109–122. PubMed PMID: 26472049; PubMed Central PMCID: PMC4728028.
- Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015 Mar;22(3):367–376. PubMed PMID: 25257169; PubMed Central PMCID: PMCPMC4326571.
- Laird GM, Eisele EE, Rabi SA, et al. A novel cell-based high-throughput screen for inhibitors of HIV-1 gene expression and budding identifies the cardiac glycosides. J Antimicrob Chemother. 2014 Apr;69(4):988–994. PubMed PMID: 24275119; PubMed Central PMCID: PMCPMC3956374.
- Wong RW, Balachandran A, Ostrowski MA, et al. Digoxin suppresses HIV-1 replication by altering viral RNA processing. PLoS Pathog. 2013 Mar;9(3):e1003241. PubMed PMID: 23555254; PubMed Central PMCID: PMCPMC3610647.
- Spector SA, Zhou D. Autophagy: an overlooked mechanism of HIV-1 pathogenesis and neuroAIDS? Autophagy. 2008;4(5):704–706. 6105 [pii]. PubMed PMID: 18424919.
- Zhou D, Masliah E, Spector SA. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J Infect Dis. 2011 Jun 1;203(11):1647–1657. PubMed PMID: 21592995; PubMed Central PMCID: PMC3096793. eng.
- Borel S, Robert-Hebmann V, Alfaisal J, et al. HIV-1 viral infectivity factor interacts with microtubule-associated protein light chain 3 and inhibits autophagy. AIDS. 2015 Jan 28;29(3):275–286. PubMed PMID: 25490467.
- Espert L, Denizot M, Grimaldi M, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest. 2006 Aug;116(8):2161–2172. PubMed PMID: 16886061; PubMed Central PMCID: PMC1523410.
- Nardacci R, Amendola A, Ciccosanti F, et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy. 2014 Jul;10(7):1167–1178. PubMed PMID: 24813622; PubMed Central PMCID: PMC4203545.
- Sagnier S, Daussy CF, Borel S, et al. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J Virol. 2015 Jan;89(1):615–625. PubMed PMID: 25339774; PubMed Central PMCID: PMC4301118.
- Valera MS, De Armas-Rillo L, Barroso-Gonzalez J, et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology. 2015 Jun 24;12:53. PubMed PMID: 26105074; PubMed Central PMCID: PMC4479245.
- Gao W, Zhang L. Coating nanoparticles with cell membranes for targeted drug delivery. J Drug Target. 2015 Aug;23(7–8):619–626. PubMed PMID: 26453159.
- Luk BT, Zhang L. Current advances in polymer-based nanotheranostics for cancer treatment and diagnosis. ACS Appl Mater Interfaces. 2014 Dec 24;6(24):21859–21873. PubMed PMID: 25014486; PubMed Central PMCID: PMC4278687.
- Zhao L, Li N, Wang K, et al. A review of polypeptide-based polymersomes. Biomaterials. 2014 Jan;35(4):1284–1301. PubMed PMID: 24211077.
- Alexis F, Pridgen E, Molnar LK, et al. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008 Jul-Aug;5(4):505–515. PubMed PMID: 18672949; PubMed Central PMCID: PMC2663893.
- Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015 Sep;33(9):941–951. PubMed PMID: 26348965; PubMed Central PMCID: PMC4978509.
- Dehaini D, Fang RH, Luk BT, et al. Ultra-small lipid-polymer hybrid nanoparticles for tumor-penetrating drug delivery. Nanoscale. 2016 Aug 14;8(30):14411–14419. PubMed PMID: 27411852; PubMed Central PMCID: PMC4977227.
- Bramini M, Ye D, Hallerbach A, et al. Imaging approach to mechanistic study of nanoparticle interactions with the blood-brain barrier. ACS Nano. 2014 May 27;8(5):4304–4312. PubMed PMID: 24773217.
- Jo DH, Kim JH, Lee TG, et al. Size, surface charge, and shape determine therapeutic effects of nanoparticles on brain and retinal diseases. Nanomedicine. 2015 Oct;11(7):1603–1611. PubMed PMID: 25989200.
- Hu CM, Kaushal S, Tran Cao HS, et al. Half-antibody functionalized lipid-polymer hybrid nanoparticles for targeted drug delivery to carcinoembryonic antigen presenting pancreatic cancer cells. Mol Pharm. 2010 Jun 07;7(3):914–920. PubMed PMID: 20394436; PubMed Central PMCID: PMC2884057.
- Rawat P, Spector SA. Development and characterization of a human microglia cell model of HIV-1 infection. J Neurovirol. 2017 Feb;23(1):33–46. PubMed PMID: 27538994; PubMed Central PMCID: PMCPMC5593752.
- Aquaro S, Svicher V, Schols D, et al. Mechanisms underlying activity of antiretroviral drugs in HIV-1-infected macrophages: new therapeutic strategies. J Leukoc Biol. 2006 Nov;80(5):1103–1110. PubMed PMID: 16931601.
- Gavegnano C, Schinazi RF. Antiretroviral therapy in macrophages: implication for HIV eradication. Antivir Chem Chemother. 2009 Oct 19;20(2):63–78. PubMed PMID: 19843977; PubMed Central PMCID: PMC2978531.
- Cribbs SK, Lennox J, Caliendo AM, et al. Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res Hum Retroviruses. 2015 Jan;31(1):64–70. PubMed PMID: 25134819; PubMed Central PMCID: PMC4287110.
- Liang XH, Kleeman LK, Jiang HH, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998 Nov;72(11):8586–8596. PubMed PMID: 9765397; PubMed Central PMCID: PMC110269.
- Liu Y, Schiff M, Czymmek K, et al. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005 May 20;121(4):567–577. PubMed PMID: 15907470.
- Shelly S, Lukinova N, Bambina S, et al. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. 2009 Apr 17;30(4):588–598. PubMed PMID: 19362021; PubMed Central PMCID: PMC2754303.
- Talloczy Z, Virgin H, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006 Jan-Mar;2(1):24–29. PubMed PMID: 16874088.
- Orvedahl A, MacPherson S, Sumpter R Jr., et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010 Feb 18;7(2):115–127. PubMed PMID: 20159618; PubMed Central PMCID: PMC2860265.
- Won M, Jun EJ, Khim M, et al. Antiviral protection against enterovirus 71 mediated by autophagy induction following FLICE-inhibitory protein inactivation. Virus Res. 2012 Oct;169(1):316–320. PubMed PMID: 22960766.
- Moy RH, Gold B, Molleston JM, et al. Antiviral autophagy restrictsRift Valley fever virus infection and is conserved from flies to mammals. Immunity. 2014 Jan 16;40(1):51–65. PubMed PMID: 24374193; PubMed Central PMCID: PMC3951734.
- Kobayashi S, Orba Y, Yamaguchi H, et al. Autophagy inhibits viral genome replication and gene expression stages in West Nile virus infection. Virus Res. 2014 Oct;13(191):83–91. PubMed PMID: 25091564.
- Griffin LM, Cicchini L, Pyeon D. Human papillomavirus infection is inhibited by host autophagy in primary human keratinocytes. Virology. 2013 Mar 01;437(1):12–19. PubMed PMID: 23290079; PubMed Central PMCID: PMC3615978.
- Pennarun B, Gaidos G, Bucur O, et al. killerFLIP: a novel lytic peptide specifically inducing cancer cell death. Cell Death Dis. 2013 Oct;31(4):e894. PubMed PMID: 24176852; PubMed Central PMCID: PMC3920952.
- Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem. 2002;71:511–535. PubMed PMID: 12045105.
- Choi B, Fermin CD, Comardelle AM, et al. Alterations in intracellular potassium concentration by HIV-1 and SIV Nef. Virol J. 2008 May;19(5):60. PubMed PMID: 18489774; PubMed Central PMCID: PMCPMC2396157.
- Voss TG, Fermin CD, Levy JA, et al. Alteration of intracellular potassium and sodium concentrations correlates with induction of cytopathic effects by human immunodeficiency virus. J Virol. 1996 Aug;70(8):5447–5454. PubMed PMID: 8764056; PubMed Central PMCID: PMCPMC190502.
- Yang J, Hu D, Xia J, et al. Enhancement of NMDA receptor-mediated excitatory postsynaptic currents by gp120-treated macrophages: implications for HIV-1-associated neuropathology. J Neuroimmune Pharmacol. 2013 Sep;8(4):921–933. PubMed PMID: 23660833; PubMed Central PMCID: PMC3740075.
- Brown AM. Use of a macrophage-tropic GFP-tagged human immunodeficiency virus type 1 (HIV-1) to study viral reservoirs. Methods Mol Biol. 2009;515:165–175. PubMed PMID: 19378134.
- Li T, Gendelman HE, Zhang G, et al. Magnetic resonance imaging of folic acid-coated magnetite nanoparticles reflects tissue biodistribution of long-acting antiretroviral therapy. Int J Nanomedicine. 2015;10:3779–3790. PubMed PMID: 26082630; PubMed Central PMCID: PMC4461087.
- Puligujja P, Balkundi SS, Kendrick LM, et al. Pharmacodynamics of long-acting folic acid-receptor targeted ritonavir-boosted atazanavir nanoformulations. Biomaterials. 2015 Feb;41:141–150. PubMed PMID: 25522973; PubMed Central PMCID: PMC4272445.
- Zhang G, Kiyota T, Batrakova EV, et al. Inhibitory effect of nanozymes on amyloid beta aggregation and oxidative stress: a new therapeutic implication for alzheimer’s disease. J Neuroimmune Pharm. 2012 Apr;7:S61–S61. PubMed PMID: WOS:000302288800143; English.
- Guo D, Zhang G, Wysocki TA, et al. Endosomal trafficking of nanoformulated antiretroviral therapy facilitates drug particle carriage and HIV clearance. J Virol. 2014 Sep 01;88(17):9504–9513. PubMed PMID: 24920821; PubMed Central PMCID: PMC4136325.
- Kiyota T, Zhang G, Morrison CM, et al. AAV2/1 CD74 gene transfer reduces beta-amyloidosis and improves learning and memory in a mouse model of alzheimer’s disease. Mol Ther. 2015 Nov;23(11):1712–1721. PubMed PMID: 26227349; PubMed Central PMCID: PMC4817947.
- Kiyota T, Gendelman HE, Weir RA, et al. CCL2 affects beta-amyloidosis and progressive neurocognitive dysfunction in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2013 Apr;34(4):1060–1068. PubMed PMID: 23040664; PubMed Central PMCID: PMC4011558.
- Kiyota T, Morrison CM, Tu G, et al. Presenilin-1 familial Alzheimer’s disease mutation alters hippocampal neurogenesis and memory function in CCL2 null mice. Brain Behav Immun. 2015 Oct;49:311–321. PubMed PMID: 26112421; PubMed Central PMCID: PMC4567522.